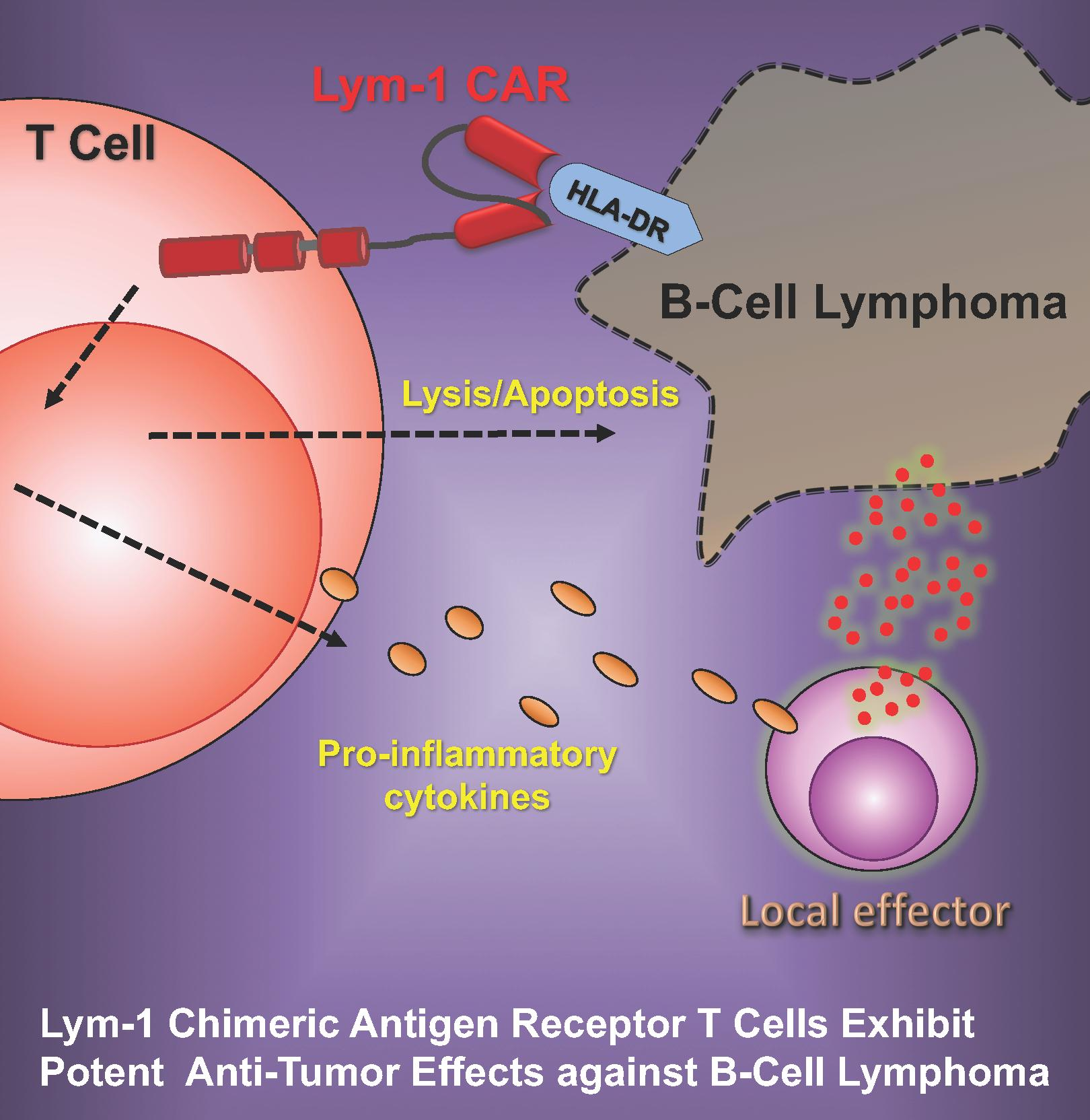
Lym-1 Chimeric Antigen Receptor T Cells Exhibit Potent Anti-Tumor Effects against B-Cell Lymphoma


 , and
, and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
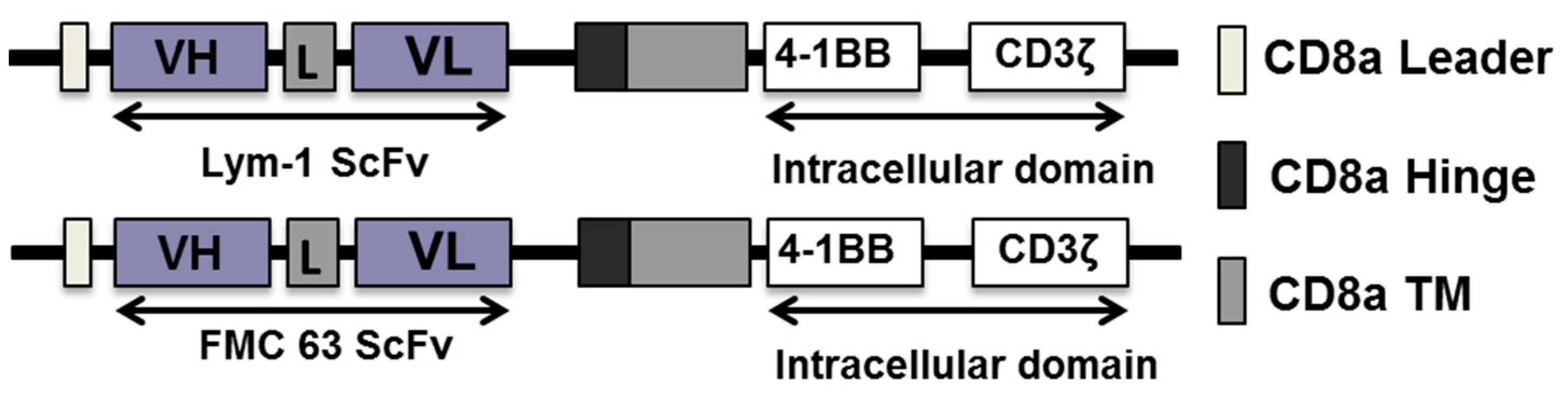
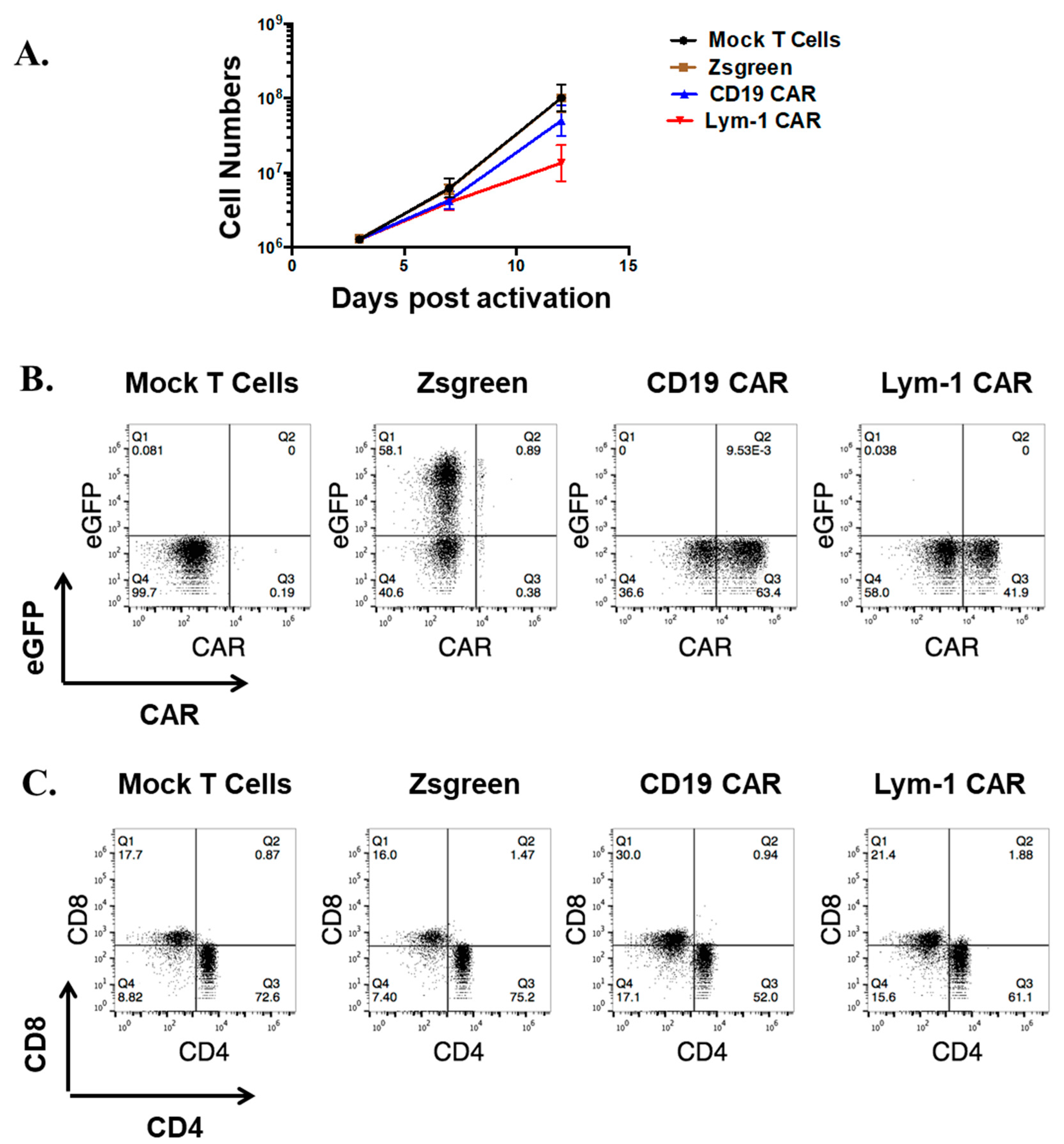
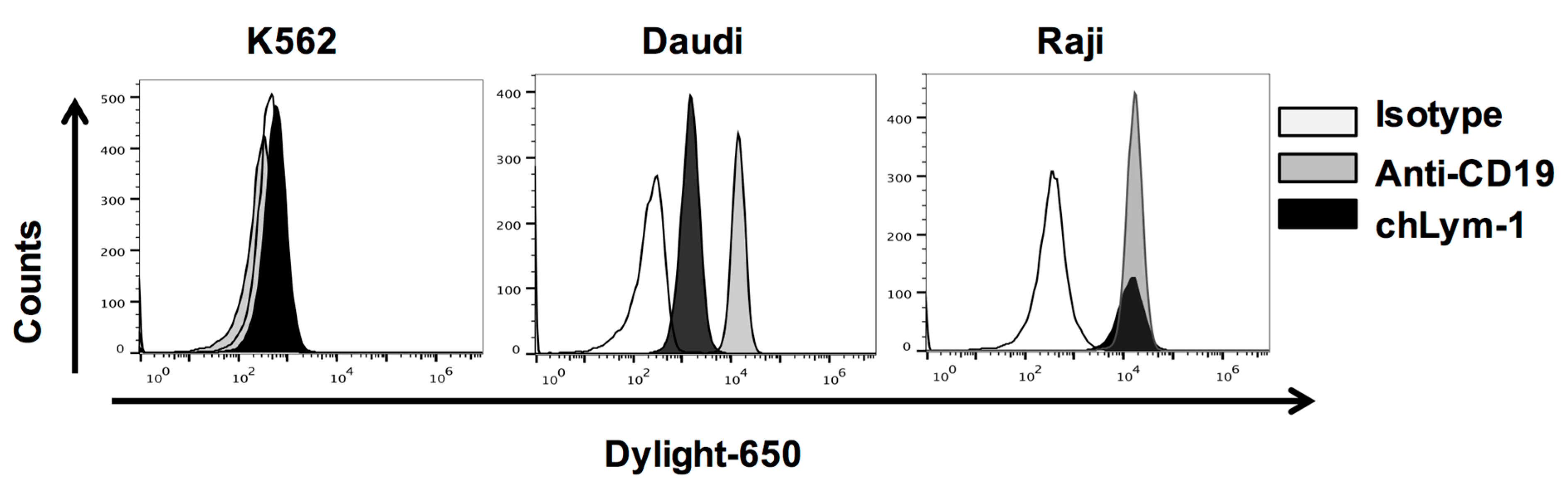
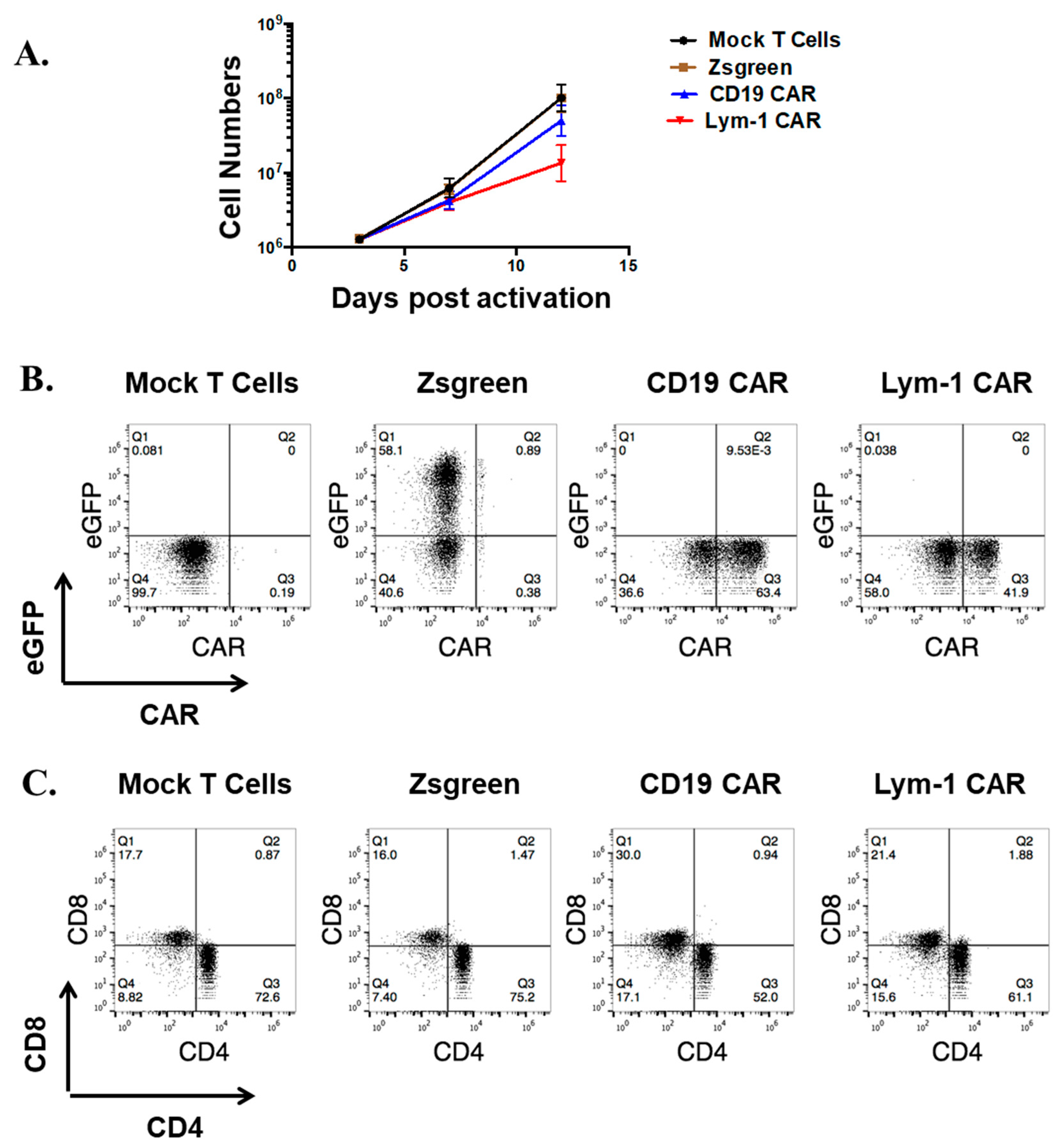
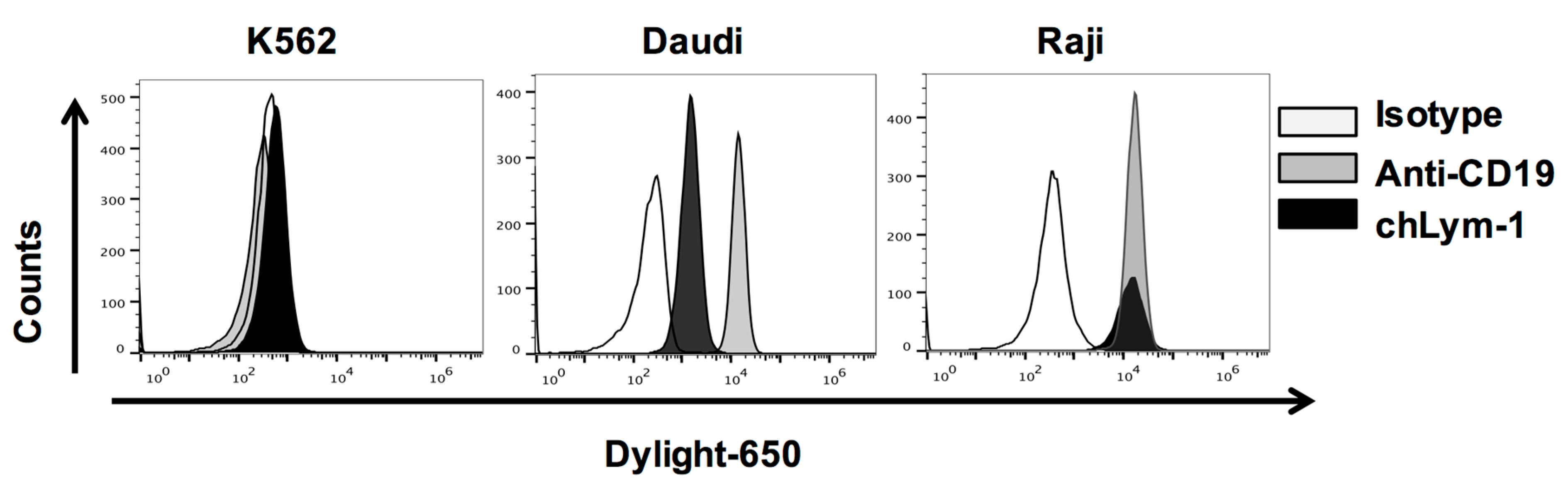
2.1. Efficient Expression of Lym-1 CAR on Transduced Primary Human T Cells
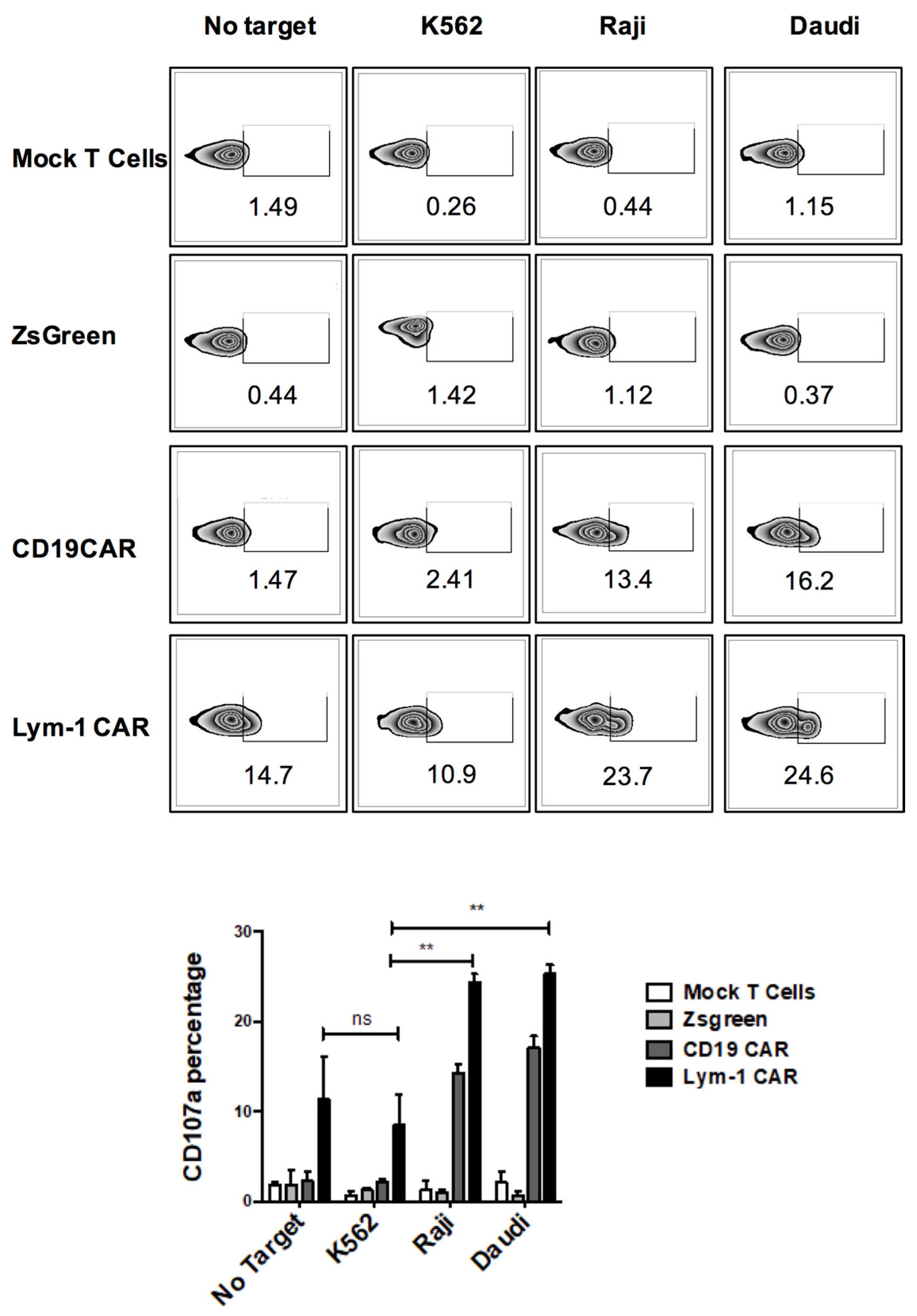
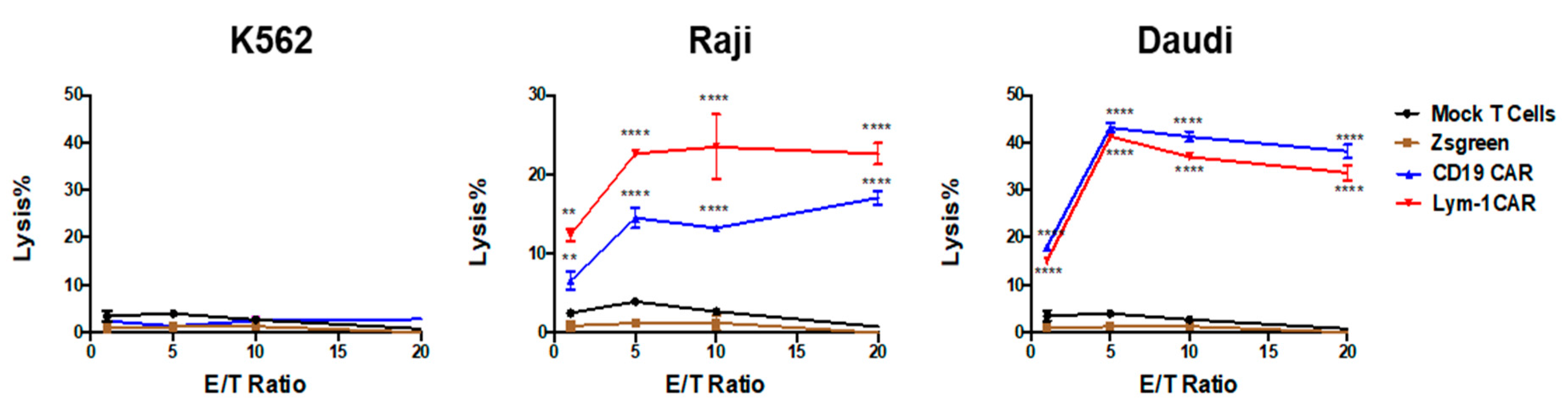
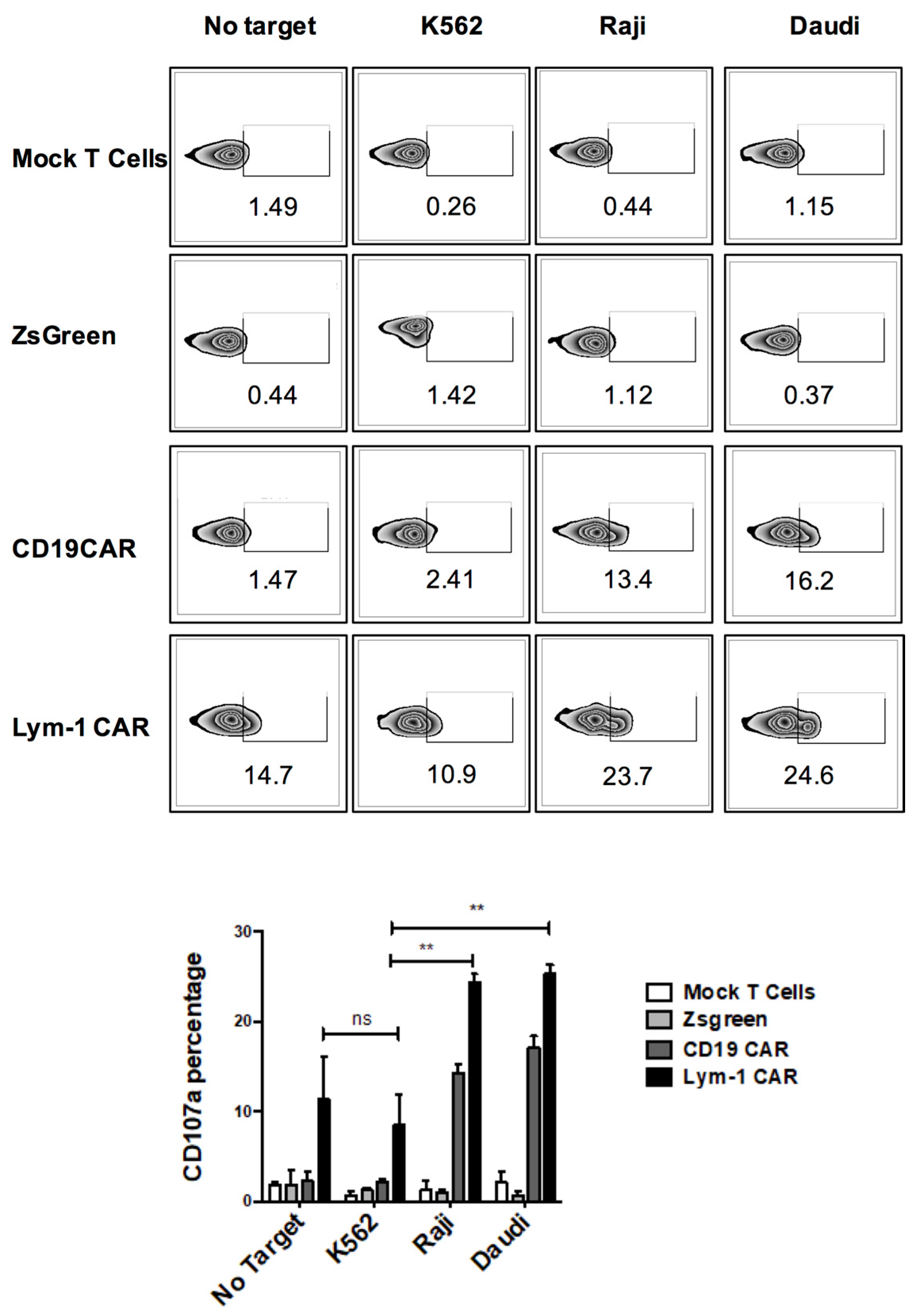
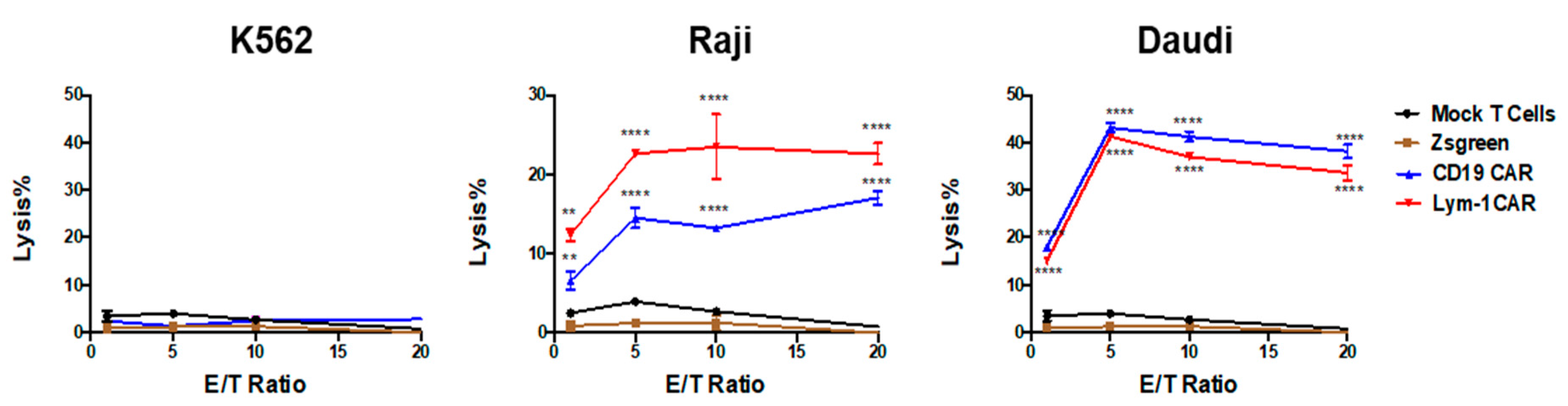
2.2. Epitope-Driven Upregulation of CD107a and Epitope-Dependent Cytotoxicity of Lym-1 and CD19 CAR T Cells
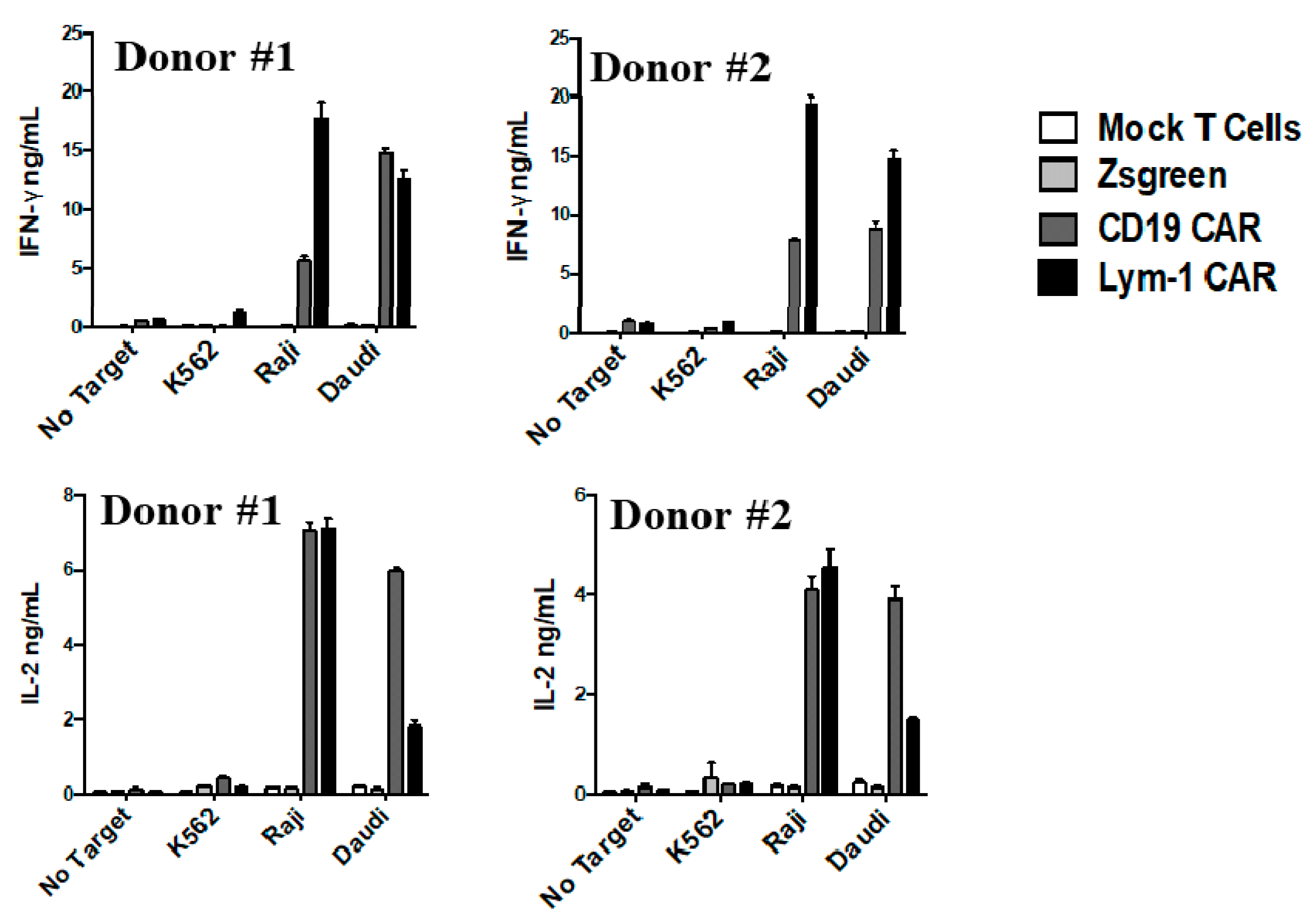
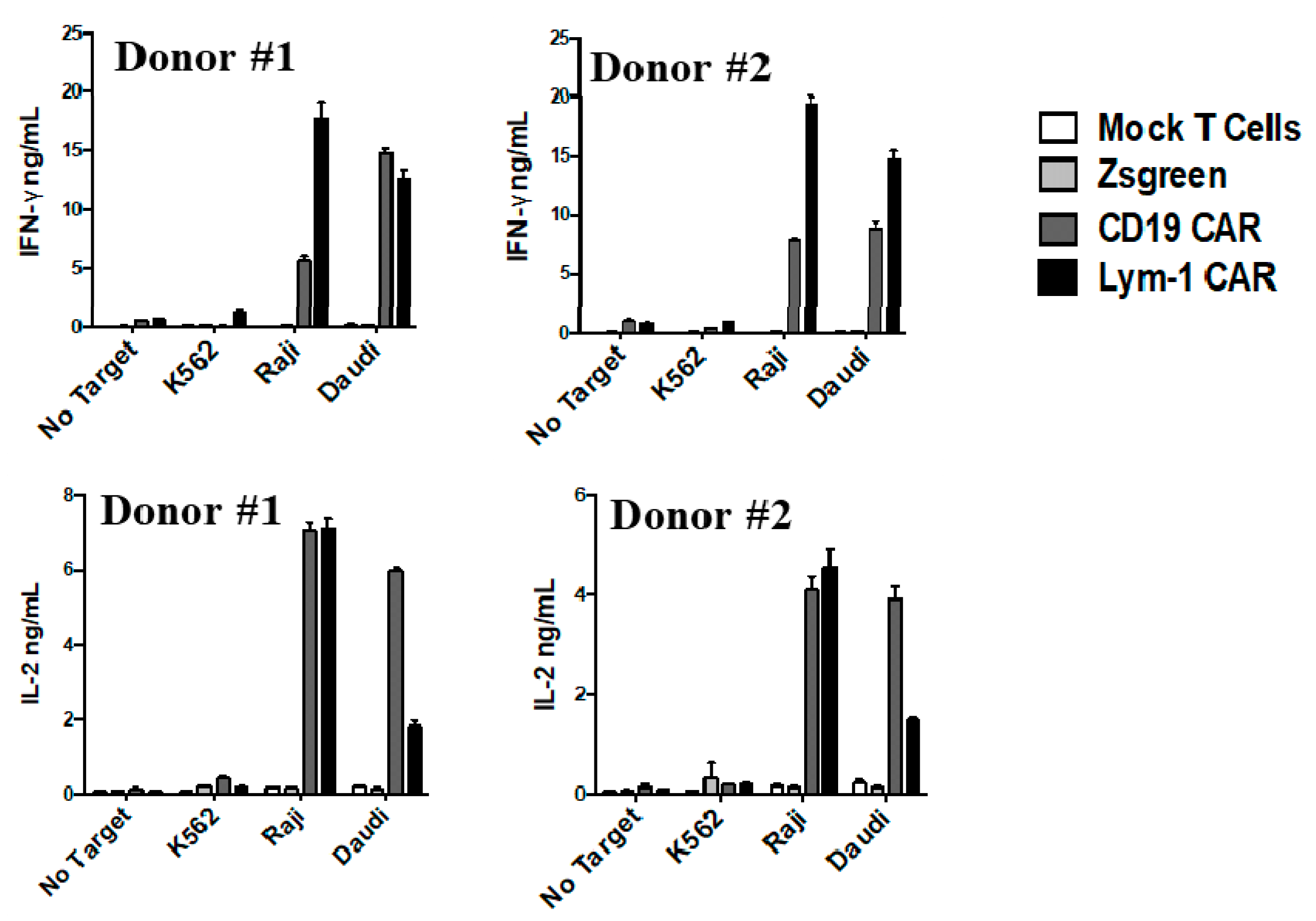
2.3. Epitope-Driven Release of Cytokines from Lym-1 and CD19 CAR T Cells
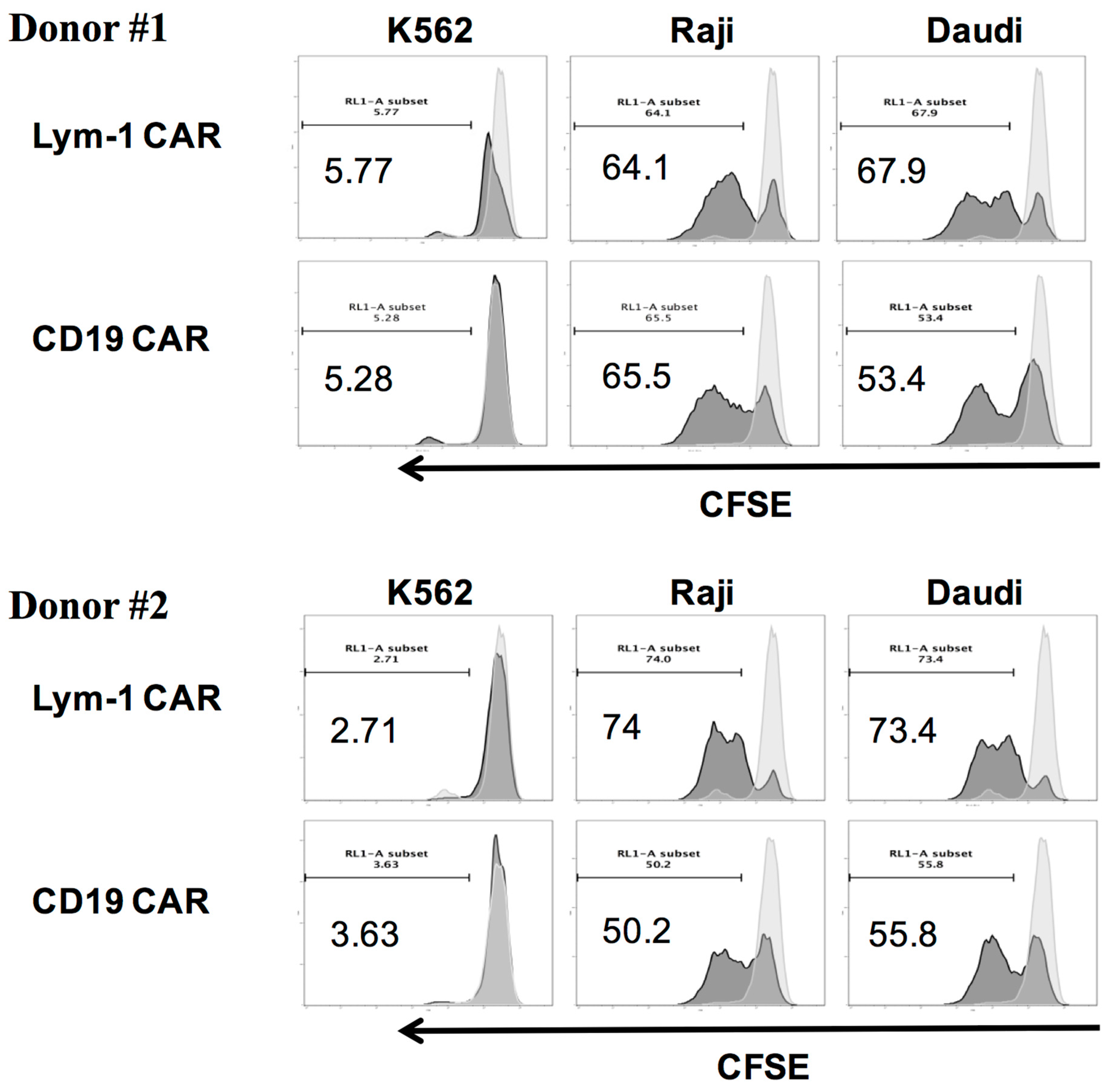
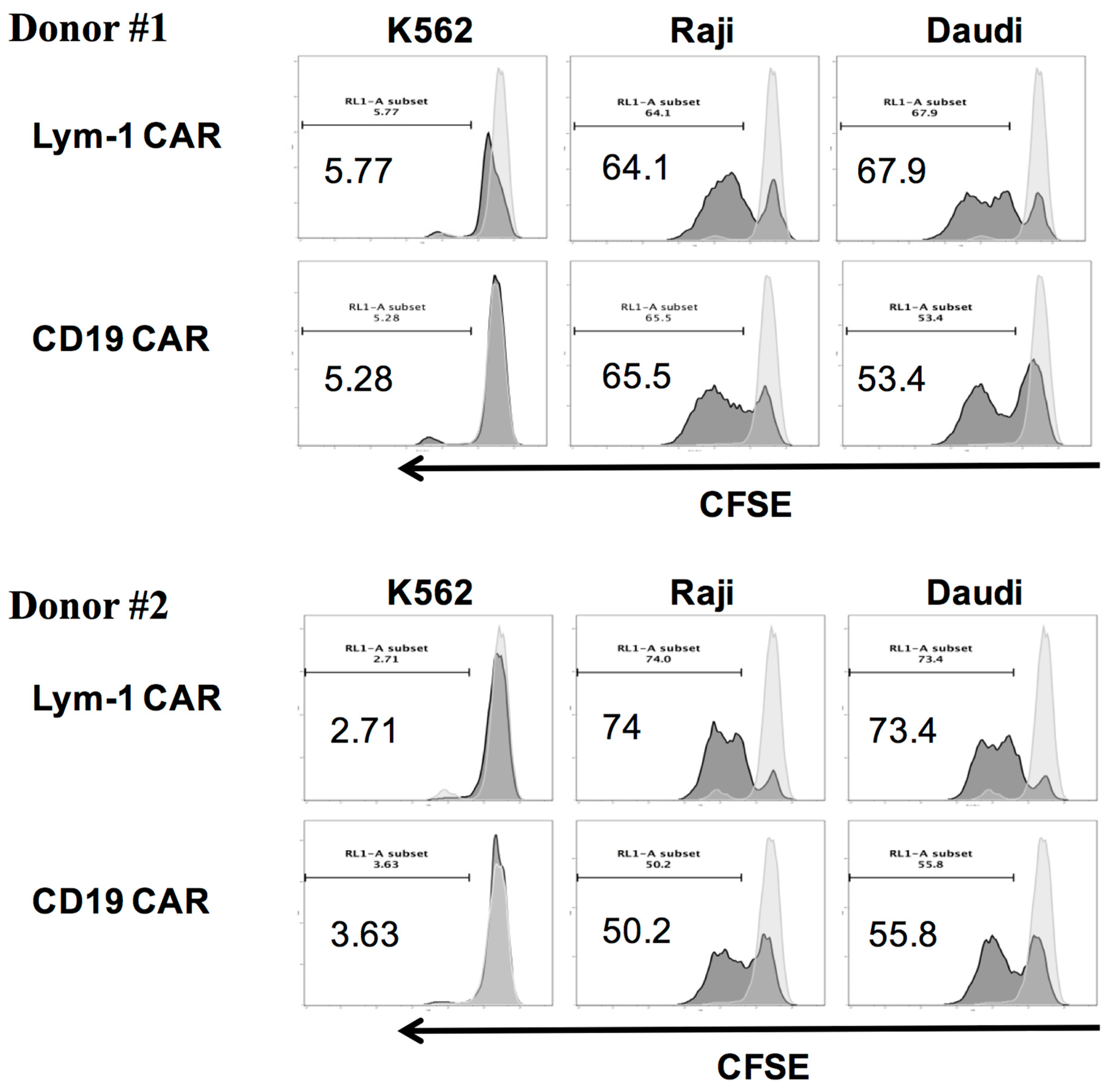
2.4. Epitope-Driven Proliferation of Lym-1 and CD19 CAR T Cells
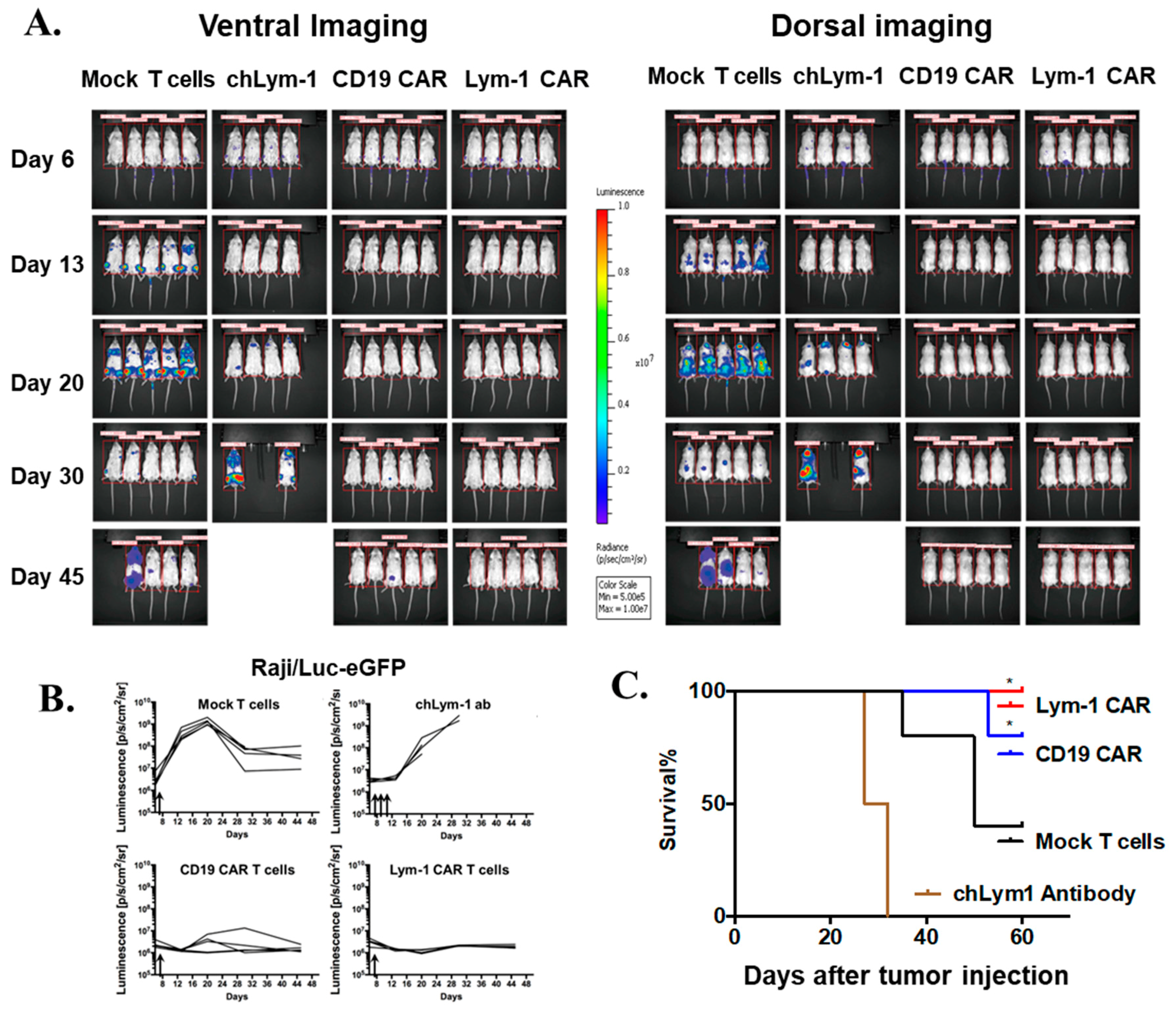
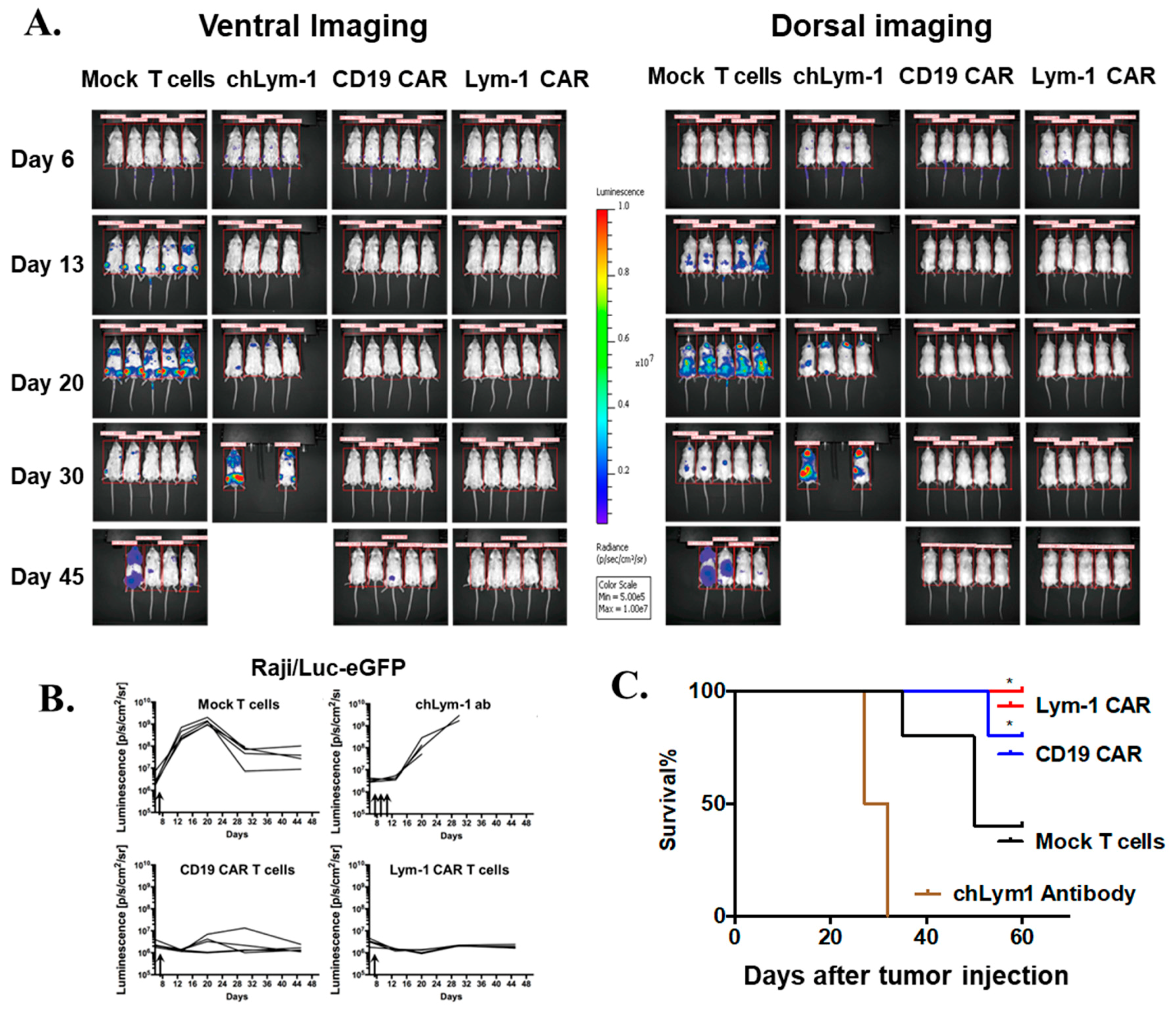
2.5. Lym-1 and CD19 CAR T Cells Eradicate Raji/Luc-GFP Xenograft Tumors in NSG Mice
3. Discussion
4. Materials and Methods
4.1. Antibodies
4.2. Cell Lines
4.3. Vector Construction and Preparation of Lentivirus
4.4. Primary T Cell Isolation and Transduction
4.5. Cytotoxicity
4.6. Cytokine Production and Degranulation Assays
4.7. CFSE Proliferation Assay
4.8. Raji/Luc-GFP Xenograft Studies in NOD Scid-IL2Rgammanull (NSG) Mice
4.9. Statistical Analysis
5. Patents
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ALL | acute lymphoblastic leukemia |
| ASCT | allogenic stem cell transplantation |
| B-NHL | B-cell non-Hodgkin lymphomas |
| CAR | chimeric antigen receptor |
| CLL | chronic lymphocytic leukemia |
| ORR | objective response rates |
| OS | overall survival |
| R/R | relapsed or Refractory |
| NHL | non-Hodgkin lymphoma |
| NSG | NOD scid IL2Rgammanull |
References
- Shankland, K.R.; Armitage, J.O.; Hancock, B.W. Non-Hodgkin lymphoma. Lancet 2012, 380, 848–857. [Google Scholar] [CrossRef]
- Armitage, J.O.; Gascoyne, R.D.; Lunning, M.A.; Cavalli, F. Non-Hodgkin lymphoma. Lancet 2017, 390, 298–310. [Google Scholar] [CrossRef]
- Rovira, J.; Valera, A.; Colomo, L.; Setoain, X.; Rodriguez, S.; Martinez-Trillos, A.; Gine, E.; Dlouhy, I.; Magnano, L.; Gaya, A.; et al. Prognosis of patients with diffuse large B cell lymphoma not reaching complete response or relapsing after frontline chemotherapy or immunochemotherapy. Ann. Hematol. 2015, 94, 803–812. [Google Scholar] [CrossRef] [PubMed]
- Nagle, S.J.; Woo, K.; Schuster, S.J.; Nasta, S.D.; Stadtmauer, E.; Mick, R.; Svoboda, J. Outcomes of patients with relapsed/refractory diffuse large B-cell lymphoma with progression of lymphoma after autologous stem cell transplantation in the rituximab era. Am. J. Hematol. 2013, 88, 890–894. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.C.; Riddell, S.R. Designing chimeric antigen receptors to effectively and safely target tumors. Curr. Opin. Immunol. 2015, 33, 9–15. [Google Scholar] [CrossRef]
- Ramos, C.A.; Dotti, G. Chimeric antigen receptor (CAR)-engineered lymphocytes for cancer therapy. Expert Opin. Biol. Ther. 2011, 11, 855–873. [Google Scholar] [CrossRef] [PubMed]
- Ruella, M.; Barrett, D.M.; Kenderian, S.S.; Shestova, O.; Hofmann, T.J.; Perazzelli, J.; Klichinsky, M.; Aikawa, V.; Nazimuddin, F.; Kozlowski, M.; et al. Dual CD19 and CD123 targeting prevents antigen-loss relapses after CD19-directed immunotherapies. J. Clin. Investig. 2016, 126, 3814–3826. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Teachey, D.T.; Porter, D.L.; Grupp, S.A. CD19-targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Blood 2015, 125, 4017–4023. [Google Scholar] [CrossRef] [PubMed]
- Grupp, S.A.; Maude, S.L.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Callahan, C.; Lacey, S.F.; Levine, B.L.; Melenhorst, J.J.; Motley, L.; et al. Durable Remissions in Children with Relapsed/Refractory ALL Treated with T Cells Engineered with a CD19-Targeted Chimeric Antigen Receptor (CTL019). Blood 2015, 126, 681. [Google Scholar]
- Crump, M.; Neelapu, S.S.; Farooq, U.; Neste, E.V.D.; Kuruvilla, J.; Ahmed, M.A.; Link, B.K.; Hay, A.E.; Cerhan, J.R.; Zhu, L.; et al. Outcomes in refractory aggressive diffuse large b-cell lymphoma (DLBCL): Results from the international SCHOLAR-1 study. J. Clin. Oncol. 2016, 34, 7516. [Google Scholar] [CrossRef]
- Locke, F.L.; Neelapu, S.S.; Bartlett, N.L.; Siddiqi, T.; Chavez, J.C.; Hosing, C.M.; Ghobadi, A.; Budde, L.E.; Bot, A.; Rossi, J.M.; et al. Phase 1 Results of ZUMA-1: A Multicenter Study of KTE-C19 Anti-CD19 CAR T Cell Therapy in Refractory Aggressive Lymphoma. Mol. Ther. 2017, 25, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Kochenderfer, J.N.; Somerville, R.; Lu, L.; Iwamoto, A.; Yang, J.C.; Klebanoff, C.; Kammula, U.; Sherry, R.M.; Victoria, S.; Yuan, C.; et al. Anti-CD19 CAR T Cells Administered after Low-Dose Chemotherapy Can Induce Remissions of Chemotherapy-Refractory Diffuse Large B-Cell Lymphoma. Blood 2014, 124, 550. [Google Scholar]
- Kochenderfer, J.N.; Dudley, M.E.; Kassim, S.H.; Somerville, R.P.; Carpenter, R.O.; Stetler-Stevenson, M.; Yang, J.C.; Phan, G.Q.; Hughes, M.S.; Sherry, R.M.; et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J. Clin. Oncol. 2015, 33, 540–549. [Google Scholar] [CrossRef] [PubMed]
- Kochenderfer, J.N.; Somerville, R.P.T.; Lu, T.; Shi, V.; Bot, A.; Rossi, J.; Xue, A.; Goff, S.L.; Yang, J.C.; Sherry, R.M.; et al. Lymphoma Remissions Caused by Anti-CD19 Chimeric Antigen Receptor T Cells Are Associated With High Serum Interleukin-15 Levels. J. Clin. Oncol. 2017, 35, 1803–1813. [Google Scholar] [CrossRef] [PubMed]
- Epstein, A.L.; Marder, R.J.; Winter, J.N.; Stathopoulos, E.; Chen, F.M.; Parker, J.W.; Taylor, C.R. Two new monoclonal antibodies, Lym-1 and Lym-2, reactive with human B-lymphocytes and derived tumors, with immunodiagnostic and immunotherapeutic potential. Cancer Res. 1987, 47, 830–840. [Google Scholar] [PubMed]
- Rose, L.M.; Deng, C.T.; Scott, S.L.; Xiong, C.Y.; Lamborn, K.R.; Gumerlock, P.H.; DeNardo, G.L.; Meares, C.F. Critical Lym-1 binding residues on polymorphic HLA-DR molecules. Mol. Immunol. 1999, 36, 789–797. [Google Scholar] [CrossRef]
- DeNardo, G.L.; DeNardo, S.J.; Lamborn, K.R.; Goldstein, D.S.; Levy, N.B.; Lewis, J.P.; O’Grady, L.F.; Raventos, A.; Kroger, L.A.; Macey, D.J.; et al. Low-dose, fractionated radioimmunotherapy for B-cell malignancies using 131I-Lym-1 antibody. Cancer Biother. Radiopharm. 1998, 13, 239–254. [Google Scholar] [CrossRef] [PubMed]
- Hu, E.; Epstein, A.L.; Naeve, G.S.; Gill, I.; Martin, S.; Sherrod, A.; Nichols, P.; Chen, D.; Mazumder, A.; Levine, A.M. A phase 1a clinical trial of LYM-1 monoclonal antibody serotherapy in patients with refractory B cell malignancies. Hematol. Oncol. 1989, 7, 155–166. [Google Scholar] [CrossRef] [PubMed]
- DeNardo, S.J.; DeNardo, G.L.; O’Grady, L.F.; Hu, E.; Sytsma, V.M.; Mills, S.L.; Levy, N.B.; Macey, D.J.; Miller, C.H.; Epstein, A.L. Treatment of B cell malignancies with 131I Lym-1 monoclonal antibodies. Int. J. Cancer Suppl. 1988, 3, 96–101. [Google Scholar] [CrossRef] [PubMed]
- DeNardo, S.J.; DeNardo, G.L.; Kukis, D.L.; Shen, S.; Kroger, L.A.; DeNardo, D.A.; Goldstein, D.S.; Mirick, G.R.; Salako, Q.; Mausner, L.F.; et al. 67Cu-2IT-BAT-Lym-1 pharmacokinetics, radiation dosimetry, toxicity and tumor regression in patients with lymphoma. J. Nucl. Med. 1999, 40, 302–310. [Google Scholar] [PubMed]
- Zhang, N.; Khawli, L.A.; Hu, P.; Epstein, A.L. Lym-1-induced apoptosis of non-Hodgkin’s lymphomas produces regression of transplanted tumors. Cancer Biother. Radiopharm. 2007, 22, 342–356. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Zeng, X.; Li, Y.; Wang, S.; Wang, Z.; Sun, Y.; Gao, H.; Zhang, G.; Feng, M.; Ju, D. Autophagy plays a critical role in ChLym-1-induced cytotoxicity of non-hodgkin’s lymphoma cells. PLoS ONE 2013, 8, e72478. [Google Scholar] [CrossRef] [PubMed]
- Alcantar-Orozco, E.M.; Gornall, H.; Baldan, V.; Hawkins, R.E.; Gilham, D.E. Potential limitations of the NSG humanized mouse as a model system to optimize engineered human T cell therapy for cancer. Hum. Gene Ther. Methods 2013, 24, 310–320. [Google Scholar] [CrossRef] [PubMed]
- Denardo, G.L.; Denardo, S.J.; Kukis, D.L.; O’Donnell, R.T.; Shen, S.; Goldstein, D.S.; Kroger, L.A.; Salako, Q.; Denardo, D.A.; Mirick, G.R.; et al. Maximum tolerated dose of 67Cu-2IT-BAT-LYM-1 for fractionated radioimmunotherapy of non-Hodgkin’s lymphoma: A pilot study. Anticancer Res. 1998, 18, 2779–2788. [Google Scholar] [PubMed]
- Long, A.H.; Haso, W.M.; Shern, J.F.; Wanhainen, K.M.; Murgai, M.; Ingaramo, M.; Smith, J.P.; Walker, A.J.; Kohler, M.E.; Venkateshwara, V.R.; et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat. Med. 2015, 21, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.S.; Matsushita, M.; Plotkin, J.; Riviere, I.; Sadelain, M. Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell-mediated tumor eradication. Mol. Ther. 2010, 18, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Glasky, M.S.; Yun, A.; Alauddin, M.M.; Hornick, J.L.; Khawli, L.A.; Epstein, A.L. A human-mouse chimeric Lym-1 monoclonal antibody with specificity for human lymphomas expressed in a baculovirus system. Hum. Antibodies Hybridomas 1995, 6, 57–67. [Google Scholar] [PubMed]
- Zah, E.; Lin, M.Y.; Silva-Benedict, A.; Jensen, M.C.; Chen, Y.Y. T Cells Expressing CD19/CD20 Bispecific Chimeric Antigen Receptors Prevent Antigen Escape by Malignant B Cells. Cancer Immunol. Res. 2016, 4, 498–508. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, L.; Hu, P.; Wolfe, B.; Gonsalves, C.; Ren, L.; Khawli, L.A.; Kaslow, H.R.; Epstein, A.L. Lym-1 Chimeric Antigen Receptor T Cells Exhibit Potent Anti-Tumor Effects against B-Cell Lymphoma. Int. J. Mol. Sci. 2017, 18, 2773. https://doi.org/10.3390/ijms18122773
Zheng L, Hu P, Wolfe B, Gonsalves C, Ren L, Khawli LA, Kaslow HR, Epstein AL. Lym-1 Chimeric Antigen Receptor T Cells Exhibit Potent Anti-Tumor Effects against B-Cell Lymphoma. International Journal of Molecular Sciences. 2017; 18(12):2773. https://doi.org/10.3390/ijms18122773
Chicago/Turabian StyleZheng, Long, Peisheng Hu, Brandon Wolfe, Caryn Gonsalves, Luqing Ren, Leslie A. Khawli, Harvey R. Kaslow, and Alan L. Epstein. 2017. "Lym-1 Chimeric Antigen Receptor T Cells Exhibit Potent Anti-Tumor Effects against B-Cell Lymphoma" International Journal of Molecular Sciences 18, no. 12: 2773. https://doi.org/10.3390/ijms18122773
APA StyleZheng, L., Hu, P., Wolfe, B., Gonsalves, C., Ren, L., Khawli, L. A., Kaslow, H. R., & Epstein, A. L. (2017). Lym-1 Chimeric Antigen Receptor T Cells Exhibit Potent Anti-Tumor Effects against B-Cell Lymphoma. International Journal of Molecular Sciences, 18(12), 2773. https://doi.org/10.3390/ijms18122773