Clinical Consequences and Molecular Bases of Low Fibrinogen Levels

Abstract
:1. Introduction
2. Diagnosis of Quantitative Fibrinogen Disorders
3. Clinical Features
3.1. Bleeding
3.2. Thrombosis
3.3. Specific Symptoms
3.4. Women’s Health
4. Molecular Bases of Quantitative Fibrinogen Disorders
5. New Models to Study Phenotypic Consequences of Fibrinogen Deficiency
6. Conclusions and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Neerman-Arbez, M.; de Moerloose, P. Hereditary fibrinogen abnormalities. In Williams Hematology, 8th ed.; Kaushansky, K., Lichtman, M., Beutler, E., Kipps, T., Prchal, J., Seligsohn, U., Eds.; McGraw-Hil: New York, NY, USA, 2010; pp. 1–33. [Google Scholar]
- Neerman-Arbez, M.; Honsberger, A.; Antonarakis, S.E.; Morris, M.A. Deletion of the fibrinogen alpha-chain gene (FGA) causes congenital afibrinogenemia. J. Clin. Investig. 1999, 103, 215–218. [Google Scholar] [CrossRef] [PubMed]
- Kattula, S.; Byrnes, J.R.; Wolberg, A.S. Fibrinogen and Fibrin in Hemostasis and Thrombosis. Arterioscler. Thromb. Vasc. Biol. 2017, 37, e13–e21. [Google Scholar] [CrossRef] [PubMed]
- Neerman-Arbez, M.; de Moerloose, P.; Casini, A. Laboratory and Genetic Investigation of Mutations Accounting for Congenital Fibrinogen Disorders. Semin. Thromb. Hemost. 2016, 42, 356–365. [Google Scholar] [PubMed]
- Verhovsek, M.; Moffat, K.A.; Hayward, C.P. Laboratory testing for fibrinogen abnormalities. Am. J. Hematol. 2008, 83, 928–931. [Google Scholar] [CrossRef] [PubMed]
- Mackie, I.J.; Kitchen, S.; Machin, S.J.; Lowe, G.D.; Haemostasis and Thrombosis Task Force of the British Committee for Standards in Haematology. Guidelines on fibrinogen assays. Br. J. Haematol. 2003, 121, 396–404. [Google Scholar] [CrossRef] [PubMed]
- Stanciakova, L.; Kubisz, P.; Dobrotova, M.; Stasko, J. Congenital afibrinogenemia: From etiopathogenesis to challenging clinical management. Expert Rev. Hematol. 2016, 9, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Peyvandi, F. Epidemiology and treatment of congenital fibrinogen deficiency. Thromb. Res. 2012, 130 (Suppl. 2), S7–S11. [Google Scholar] [CrossRef]
- Krammer, B.; Anders, O.; Nagel, H.R.; Burstein, C.; Steiner, M. Screening of dysfibrinogenaemia using the fibrinogen function versus antigen concentration ratio. Thromb. Res. 1994, 76, 577–579. [Google Scholar] [CrossRef]
- Jacquemin, M.; Vanlinthout, I.; Van Horenbeeck, I.; Debasse, M.; Toelen, J.; Schoeters, J.; Lavend’homme, R.; Freson, K.; Peerlinck, K. The amplitude of coagulation curves from thrombin time tests allows dysfibrinogenemia caused by the common mutation FGG-Arg301 to be distinguished from hypofibrinogenemia. Int. J. Lab. Hematol. 2017, 39, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Casini, A.; de Moerloose, P. Can the phenotype of inherited fibrinogen disorders be predicted? Haemophilia 2016, 22, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Kalina, U.; Stohr, H.A.; Bickhard, H.; Knaub, S.; Siboni, S.M.; Mannucci, P.M.; Peyvandi, F. Rotational thromboelastography for monitoring of fibrinogen concentrate therapy in fibrinogen deficiency. Blood Coagul. Fibrinolysis 2008, 19, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Trelinski, J.; Pachniewska, K.; Matczak, J.; Robak, M.; Chojnowski, K. Assessment of Selected ROTEM Parameters, Kinetics of Fibrinogen Polymerization and Plasmin Amidolytic Activity in Patients with Congenital Fibrinogen Defects. Adv. Clin. Exp. Med. 2016, 25, 1255–1263. [Google Scholar] [CrossRef] [PubMed]
- Palla, R.; Peyvandi, F.; Shapiro, A.D. Rare bleeding disorders: Diagnosis and treatment. Blood 2015, 125, 2052–2061. [Google Scholar] [CrossRef] [PubMed]
- Peyvandi, F.; Palla, R.; Menegatti, M.; Siboni, S.M.; Halimeh, S.; Faeser, B.; Pergantou, H.; Platokouki, H.; Giangrande, P.; Peerlinck, K.; et al. Coagulation factor activity and clinical bleeding severity in rare bleeding disorders: Results from the European Network of Rare Bleeding Disorders. J. Thromb. Haemost. 2012, 10, 615–621. [Google Scholar] [CrossRef] [PubMed]
- Peyvandi, F.; Haertel, S.; Knaub, S.; Mannucci, P.M. Incidence of bleeding symptoms in 100 patients with inherited afibrinogenemia or hypofibrinogenemia. J. Thromb. Haemost. 2006, 4, 1634–1637. [Google Scholar] [CrossRef] [PubMed]
- Abolghasemi, H.; Shahverdi, E. Umbilical bleeding: A presenting feature for congenital afibrinogenemia. Blood Coagul. Fibrinolysis 2015, 26, 834–835. [Google Scholar] [CrossRef] [PubMed]
- De Moerloose, P.; Casini, A.; Neerman-Arbez, M. Congenital fibrinogen disorders: An update. Semin. Thromb. Hemost. 2013, 39, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Henselmans, J.M.; Meijer, K.; Haaxma, R.; Hew, J.; van der Meer, J. Recurrent spontaneous intracerebral hemorrhage in a congenitally afibrinogenemic patient: Diagnostic pitfalls and therapeutic options. Stroke 1999, 30, 2479–2482. [Google Scholar] [CrossRef] [PubMed]
- Parameswaran, R.; Dickinson, J.P.; de Lord, S.; Keeling, D.M.; Colvin, B.T. Spontaneous intracranial bleeding in two patients with congenital afibrinogenaemia and the role of replacement therapy. Haemophilia 2000, 6, 705–708. [Google Scholar] [CrossRef] [PubMed]
- Lak, M.; Keihani, M.; Elahi, F.; Peyvandi, F.; Mannucci, P.M. Bleeding and thrombosis in 55 patients with inherited afibrinogenaemia. Br. J. Haematol. 1999, 107, 204–206. [Google Scholar] [CrossRef] [PubMed]
- Reidy, K.; Brand, B.; Jost, B. Severe elbow arthropathy in a patient with congenital afibrinogenemia: A case report. J. Bone Joint Surg. Am. 2010, 92, 456–458. [Google Scholar] [CrossRef] [PubMed]
- Casini, A.; de Moerloose, P.; Neerman-Arbez, M. Clinical Features and Management of Congenital Fibrinogen Deficiencies. Semin. Thromb. Hemost. 2016, 42, 366–374. [Google Scholar] [PubMed]
- Zdziarska, J.; Undas, A.; Basa, J.; Iwaniec, T.; Skotnicki, A.B.; de Moerloose, P.; Neerman-Arbez, M. Severe bleeding and miscarriages in a hypofibrinogenemic woman heterozygous for the gamma Ala82Gly mutation. Blood Coagul. Fibrinolysis 2009, 20, 374–376. [Google Scholar] [CrossRef] [PubMed]
- Casini, A.; Vilar, R.; Beauverd, Y.; Aslan, D.; Devreese, K.; Mondelaers, V.; Alberio, L.; Gubert, C.; de Moerloose, P.; Neerman-Arbez, M. Protein modelling to understand FGB mutations leading to congenital hypofibrinogenaemia. Haemophilia 2017, 23, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Nagler, M.; Kremer Hovinga, J.A.; Alberio, L.; Peter-Salonen, K.; von Tengg-Kobligk, H.; Lottaz, D.; Neerman-Arbez, M.; Lammle, B. Thromboembolism in patients with congenital afibrinogenaemia. Long-term observational data and systematic review. Thromb. Haemost. 2016, 116, 722–732. [Google Scholar] [CrossRef] [PubMed]
- De Moerloose, P.; Boehlen, F.; Neerman-Arbez, M. Fibrinogen and the risk of thrombosis. Semin. Thromb. Hemost. 2010, 36, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Santoro, C.; Massaro, F.; Venosi, S.; Capria, S.; Baldacci, E.; Foa, R.; Mazzucconi, M.G. Severe Thrombotic Complications in Congenital Afibrinogenemia: A Pathophysiological and Management Dilemma. Semin. Thromb. Hemost. 2016, 42, 577–582. [Google Scholar] [PubMed]
- Mosesson, M.W. Update on antithrombin I (fibrin). Thromb. Haemost. 2007, 98, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Dupuy, E.; Soria, C.; Molho, P.; Zini, J.M.; Rosenstingl, S.; Laurian, C.; Bruneval, P.; Tobelem, G. Embolized ischemic lesions of toes in an afibrinogenemic patient: Possible relevance to in vivo circulating thrombin. Thromb. Res. 2001, 102, 211–219. [Google Scholar] [CrossRef]
- Remijn, J.A.; Wu, Y.P.; Ijsseldijk, M.J.; Zwaginga, J.J.; Sixma, J.J.; de Groot, P.G. Absence of fibrinogen in afibrinogenemia results in large but loosely packed thrombi under flow conditions. Thromb. Haemost. 2001, 85, 736–742. [Google Scholar] [PubMed]
- Ni, H.; Denis, C.V.; Subbarao, S.; Degen, J.L.; Sato, T.N.; Hynes, R.O.; Wagner, D.D. Persistence of platelet thrombus formation in arterioles of mice lacking both von Willebrand factor and fibrinogen. J. Clin. Investig. 2000, 106, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Beguin, S.; Hemker, H.C. The influence of fibrinogen and fibrin on thrombin generation—Evidence for feedback activation of the clotting system by clot bound thrombin. Thromb. Haemost. 1994, 72, 713–721. [Google Scholar] [PubMed]
- Korte, W.; Poon, M.C.; Iorio, A.; Makris, M. Thrombosis in Inherited Fibrinogen Disorders. Transfus. Med. Hemother. 2017, 44, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Castaman, G.; Lunardi, M.; Rigo, L.; Mastroeni, V.; Bonoldi, E.; Rodeghiero, F. Severe spontaneous arterial thrombotic manifestations in patients with inherited hypo- and afibrinogenemia. Haemophilia 2009, 15, 533–537. [Google Scholar] [CrossRef] [PubMed]
- Rottenstreich, A.; Lask, A.; Schliamser, L.; Zivelin, A.; Seligsohn, U.; Kalish, Y. Thromboembolic events in patients with severe inherited fibrinogen deficiency. J. Thromb. Thromb. 2015, 42, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Marchi, R.; Walton, B.L.; McGary, C.S.; Lin, F.C.; Ma, A.D.; Pawlinski, R.; Mackman, N.; Campbell, R.A.; Di Paola, J.; Wolberg, A.S. Dysregulated coagulation associated with hypofibrinogenaemia and plasma hypercoagulability: Implications for identifying coagulopathic mechanisms in humans. Thromb. Haemost. 2012, 108, 516–526. [Google Scholar] [CrossRef] [PubMed]
- Miljic, P.; Nedeljkov-Jancic, R.; Zuvela, M.; Subota, V.; Dordevic, V. Coexistence of hypofibrinogenemia and factor V Leiden mutation: Is the balance shifted to thrombosis? Blood Coagul. Fibrinolysis 2014, 25, 628–630. [Google Scholar] [CrossRef] [PubMed]
- Monaldini, L.; Asselta, R.; Duga, S.; Peyvandi, F.; Karimi, M.; Malcovati, M.; Tenchini, M.L. Mutational screening of six afibrinogenemic patients: Identification and characterization of four novel molecular defects. Thromb. Haemost. 2007, 97, 546–551. [Google Scholar] [CrossRef] [PubMed]
- Sumitha, E.; Jayandharan, G.R.; Arora, N.; Abraham, A.; David, S.; Devi, G.S.; Shenbagapriya, P.; Nair, S.C.; George, B.; Mathews, V.; et al. Molecular basis of quantitative fibrinogen disorders in 27 patients from India. Haemophilia 2013, 19, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Asselta, R.; Plate, M.; Robusto, M.; Borhany, M.; Guella, I.; Solda, G.; Afrasiabi, A.; Menegatti, M.; Shamsi, T.; Peyvandi, F.; et al. Clinical and molecular characterisation of 21 patients affected by quantitative fibrinogen deficiency. Thromb. Haemost. 2015, 113, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Lagier, R.; Bouvier, C.A.; Van Strijthem, N. Skeletal changes in congenital fibrinogen abnormalities. Skelet. Radiol. 1980, 5, 233–239. [Google Scholar] [CrossRef]
- Van Meegeren, M.E.; de Rooy, J.W.; Schreuder, H.W.; Brons, P.P. Bone cysts in patients with afibrinogenaemia: A literature review and two new cases. Haemophilia 2014, 20, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Fettah, A.; Gurlek Gokcebay, D.; Culha, V.; Yarali, N.; Tunc, B.; Ozbek, N. A Rare Complication of Congenital Afibrinogenemia: Bone Cysts. Turk. J. Haematol. 2017, 34, 183. [Google Scholar] [CrossRef] [PubMed]
- Akcakus, M.; Patiroglu, T.; Keskin, M.; Koklu, E.; Gozukucuk, A. Nonketotic hyperosmolar coma associated with splenic rupture in congenital afibrinogenemia. J. Pediatr. Hematol. Oncol. 2004, 26, 668–671. [Google Scholar] [CrossRef] [PubMed]
- Arcagok, B.C.; Ozdemir, N.; Tekin, A.; Ozcan, R.; Elicevik, M.; Senyuz, O.F.; Cam, H.; Celkan, T. Spontaneous splenic rupture in a patient with congenital afibrinogenemia. Turk Pediatri Arsivi 2014, 49, 247–249. [Google Scholar] [CrossRef] [PubMed]
- Ehmann, W.C.; Al-Mondhiry, H. Splenic rupture in afibrinogenemia: Conservative versus surgical management. Am. J. Med. 1995, 99, 444. [Google Scholar] [CrossRef]
- Casini, A.; Sokollik, C.; Lukowski, S.W.; Lurz, E.; Rieubland, C.; de Moerloose, P.; Neerman-Arbez, M. Hypofibrinogenemia and liver disease: A new case of Aguadilla fibrinogen and review of the literature. Haemophilia 2015, 21, 820–827. [Google Scholar] [CrossRef] [PubMed]
- Neerman-Arbez, M. To aggregate or not to aggregate. J. Thromb. Haemost. 2007, 5, 1997–1998. [Google Scholar] [CrossRef] [PubMed]
- Puls, F.; Goldschmidt, I.; Bantel, H.; Agne, C.; Brocker, V.; Dammrich, M.; Lehmann, U.; Berrang, J.; Pfister, E.D.; Kreipe, H.H.; et al. Autophagy-enhancing drug carbamazepine diminishes hepatocellular death in fibrinogen storage disease. J. Hepatol. 2013, 59, 626–630. [Google Scholar] [CrossRef] [PubMed]
- Al-Hussaini, A.; Altalhi, A.; El Hag, I.; AlHussaini, H.; Francalanci, P.; Giovannoni, I.; Callea, F. Hepatic fibrinogen storage disease due to the fibrinogen gamma375 Arg → Trp mutation “fibrinogen Aguadilla” is present in Arabs. Saudi J. Gastroenterol. 2014, 20, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Rubbia-Brandt, L.; Neerman-Arbez, M.; Rougemont, A.L.; Male, P.J.; Spahr, L. Fibrinogen gamma375 arg → trp mutation (fibrinogen aguadilla) causes hereditary hypofibrinogenemia, hepatic endoplasmic reticulum storage disease and cirrhosis. Am. J. Surg. Pathol. 2006, 30, 906–911. [Google Scholar] [CrossRef] [PubMed]
- Callea, F.; Brisigotti, M.; Fabbretti, G.; Bonino, F.; Desmet, V.J. Hepatic endoplasmic reticulum storage diseases. Liver 1992, 12, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Castaman, G.; Ruggeri, M.; Rodeghiero, F. Congenital afibrinogenemia: Successful prevention of recurrent hemoperitoneum during ovulation by oral contraceptive. Am. J. Hematol. 1995, 49, 363–364. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Jeong, S.Y.; Cho, D.H. Massive hemoperitoneum due to a ruptured corpus luteum cyst in a patient with congenital hypofibrinogenemia. Obstet. Gynecol. Sci. 2015, 58, 427–430. [Google Scholar] [CrossRef] [PubMed]
- Lebreton, A.; Casini, A.; Alhayek, R.; Kouteich, K.L.; Neerman-Arbez, M.; de Moerloose, P. Successful pregnancy under fibrinogen substitution in a woman with congenital afibrinogenaemia complicated by a postpartum venous thrombosis. Haemophilia 2015, 21, e108–e110. [Google Scholar] [CrossRef] [PubMed]
- Iwaki, T.; Castellino, F.J. Maternal fibrinogen is necessary for embryonic development. Curr. Drug Targets 2005, 6, 535–539. [Google Scholar] [CrossRef] [PubMed]
- Asahina, T.; Kobayashi, T.; Okada, Y.; Itoh, M.; Yamashita, M.; Inamato, Y.; Terao, T. Studies on the role of adhesive proteins in maintaining pregnancy. Horm. Res. 1998, 50 (Suppl. 2), 37–45. [Google Scholar] [CrossRef] [PubMed]
- Oda, T.; Itoh, H.; Kawai, K.; Oda-Kishimoto, A.; Kobayashi, T.; Doi, T.; Uchida, T.; Kanayama, N. Three successful deliveries involving a woman with congenital afibrinogenaemia—Conventional fibrinogen concentrate infusion vs. ‘as required’ fibrinogen concentrate infusion based on changes in fibrinogen clearance. Haemophilia 2016, 22, e478–e481. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Kanayama, N.; Tokunaga, N.; Asahina, T.; Terao, T. Prenatal and peripartum management of congenital afibrinogenaemia. Br. J. Haematol. 2000, 109, 364–366. [Google Scholar] [CrossRef] [PubMed]
- Brennan, S.O.; Wyatt, J.M.; May, S.; De Caigney, S.; George, P.M. Hypofibrinogenemia due to novel 316 Asp → Tyr substitution in the fibrinogen Bbeta chain. Thromb. Haemost. 2001, 85, 450–453. [Google Scholar] [PubMed]
- Frenkel, E.; Duksin, C.; Herman, A.; Sherman, D.J. Congenital hypofibrinogenemia in pregnancy: Report of two cases and review of the literature. Obstet. Gynecol. Surv. 2004, 59, 775–779. [Google Scholar] [CrossRef] [PubMed]
- Ness, P.M.; Budzynski, A.Z.; Olexa, S.A.; Rodvien, R. Congenital hypofibrinogenemia and recurrent placental abruption. Obstet. Gynecol. 1983, 61, 519–523. [Google Scholar] [PubMed]
- Kant, J.A.; Fornace, A.J., Jr.; Saxe, D.; Simon, M.I.; McBride, O.W.; Crabtree, G.R. Evolution and organization of the fibrinogen locus on chromosome 4: Gene duplication accompanied by transposition and inversion. Proc. Natl. Acad. Sci. USA 1985, 82, 2344–2348. [Google Scholar] [CrossRef] [PubMed]
- De Maat, M.P.; Verschuur, M. Fibrinogen heterogeneity: Inherited and noninherited. Curr. Opin. Hematol. 2005, 12, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Blomback, M.; Blomback, B.; Mammen, E.F.; Prasad, A.S. Fibrinogen Detroit—A molecular defect in the N-terminal disulphide knot of human fibrinogen? Nature 1968, 218, 134–137. [Google Scholar] [CrossRef] [PubMed]
- Asselta, R.; Duga, S.; Tenchini, M.L. The molecular basis of quantitative fibrinogen disorders. J. Thromb. Haemost. 2006, 4, 2115–2129. [Google Scholar] [CrossRef] [PubMed]
- Hanss, M.; Biot, F. A database for human fibrinogen variants. Ann. N. Y. Acad. Sci. 2001, 936, 89–90. [Google Scholar] [CrossRef] [PubMed]
- Okumura, N.; Terasawa, F.; Tanaka, H.; Hirota, M.; Ota, H.; Kitano, K.; Kiyosawa, K.; Lord, S.T. Analysis of fibrinogen gamma-chain truncations shows the C-terminus, particularly gammaIle387, is essential for assembly and secretion of this multichain protein. Blood 2002, 99, 3654–3660. [Google Scholar] [CrossRef] [PubMed]
- Nagata, K.; Arai, S.; Taira, C.; Sugano, M.; Honda, T.; Okumura, N. A novel frameshift mutation in the fibrinogen gammaC terminal region, FGG c.1169_1170 del AT, leading to hypofibrinogenemia. Thromb. Res. 2017, 159, 82–85. [Google Scholar] [CrossRef] [PubMed]
- Vu, D.; de Moerloose, P.; Batorova, A.; Lazur, J.; Palumbo, L.; Neerman-Arbez, M. Hypofibrinogenaemia caused by a novel FGG missense mutation (W253C) in the gamma chain globular domain impairing fibrinogen secretion. J. Med. Genet. 2005, 42, e57. [Google Scholar] [CrossRef] [PubMed]
- Vu, D.; Di Sanza, C.; Caille, D.; de Moerloose, P.; Scheib, H.; Meda, P.; Neerman-Arbez, M. Quality control of fibrinogen secretion in the molecular pathogenesis of congenital afibrinogenemia. Hum. Mol. Genet. 2005, 14, 3271–3280. [Google Scholar] [CrossRef] [PubMed]
- Galanakis, D.K.; Neerman-Arbez, M.; Kudryk, B.; Henschen, A. Decreased plasmin resistance by clots of a homophenotypic Aalpha R 16H fibrinogen (Kingsport, slower fibrinopeptide A than fibrinopeptide B release). Blood Coagul. Fibrinolysis 2010, 21, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Brennan, S.O.; Maghzal, G.; Shneider, B.L.; Gordon, R.; Magid, M.S.; George, P.M. Novel fibrinogen gamma375 Arg → Trp mutation (fibrinogen aguadilla) causes hepatic endoplasmic reticulum storage and hypofibrinogenemia. Hepatology 2002, 36, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Brennan, S.O.; Wyatt, J.; Medicina, D.; Callea, F.; George, P.M. Fibrinogen brescia: Hepatic endoplasmic reticulum storage and hypofibrinogenemia because of a gamma284 Gly → Arg mutation. Am. J. Pathol. 2000, 157, 189–196. [Google Scholar] [CrossRef]
- Asselta, R.; Robusto, M.; Braidotti, P.; Peyvandi, F.; Nastasio, S.; D’Antiga, L.; Perisic, V.N.; Maggiore, G.; Caccia, S.; Duga, S. Hepatic fibrinogen storage disease: Identification of two novel mutations (p.Asp316Asn, fibrinogen Pisa and p.Gly366Ser, fibrinogen Beograd) impacting on the fibrinogen gamma-module. J. Thromb. Haemost. 2015, 13, 1459–1467. [Google Scholar] [CrossRef] [PubMed]
- Dib, N.; Quelin, F.; Ternisien, C.; Hanss, M.; Michalak, S.; De Mazancourt, P.; Rousselet, M.C.; Cales, P. Fibrinogen angers with a new deletion (gamma GVYYQ 346-350) causes hypofibrinogenemia with hepatic storage. J. Thromb. Haemost. 2007, 5, 1999–2005. [Google Scholar] [CrossRef] [PubMed]
- Brennan, S.O.; Davis, R.L.; Conard, K.; Savo, A.; Furuya, K.N. Novel fibrinogen mutation gamma314Thr → Pro (fibrinogen AI duPont) associated with hepatic fibrinogen storage disease and hypofibrinogenaemia. Liver Int. 2010, 30, 1541–1547. [Google Scholar] [CrossRef] [PubMed]
- Gut, P.; Reischauer, S.; Stainier, D.Y.R.; Arnaout, R. Little Fish, Big Data: Zebrafish as a Model for Cardiovascular and Metabolic Disease. Physiol. Rev. 2017, 97, 889–938. [Google Scholar] [CrossRef] [PubMed]
- Jagadeeswaran, P.; Gregory, M.; Day, K.; Cykowski, M.; Thattaliyath, B. Zebrafish: A genetic model for hemostasis and thrombosis. J. Thromb. Haemost. 2005, 3, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Jagadeeswaran, P. Zebrafish: A tool to study hemostasis and thrombosis. Curr. Opin. Hematol. 2005, 12, 149–152. [Google Scholar] [CrossRef] [PubMed]
- Fish, R.J.; Vorjohann, S.; Bena, F.; Fort, A.; Neerman-Arbez, M. Developmental expression and organisation of fibrinogen genes in the zebrafish. Thromb. Haemost. 2012, 107, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Weyand, A.C.; Shavit, J.A. Zebrafish as a model system for the study of hemostasis and thrombosis. Curr. Opin. Hematol. 2014, 21, 418–422. [Google Scholar] [CrossRef] [PubMed]
- Day, K.; Krishnegowda, N.; Jagadeeswaran, P. Knockdown of prothrombin in zebrafish. Blood Cells Mol. Dis. 2004, 32, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Khandekar, G.; Jagadeeswaran, P. Role of hepsin in factor VII activation in zebrafish. Blood Cells Mol. Dis. 2014, 52, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Carrillo, M.; Kim, S.; Rajpurohit, S.K.; Kulkarni, V.; Jagadeeswaran, P. Zebrafish von Willebrand factor. Blood Cells Mol. Dis. 2010, 45, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Vo, A.H.; Swaroop, A.; Liu, Y.; Norris, Z.G.; Shavit, J.A. Loss of fibrinogen in zebrafish results in symptoms consistent with human hypofibrinogenemia. PLoS ONE 2013, 8, e74682. [Google Scholar] [CrossRef] [PubMed]
- Fish, R.J.; Di Sanza, C.; Neerman-Arbez, M. Targeted mutation of zebrafish fga models human congenital afibrinogenemia. Blood 2014, 123, 2278–2281. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Kretz, C.A.; Maeder, M.L.; Richter, C.E.; Tsao, P.; Vo, A.H.; Huarng, M.C.; Rode, T.; Hu, Z.; Mehra, R.; et al. Targeted mutagenesis of zebrafish antithrombin III triggers disseminated intravascular coagulation and thrombosis, revealing insight into function. Blood 2014, 124, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Liu, Y.; Huarng, M.C.; Menegatti, M.; Reyon, D.; Rost, M.S.; Norris, Z.G.; Richter, C.E.; Stapleton, A.N.; Chi, N.C.; et al. Genome editing of factor X in zebrafish reveals unexpected tolerance of severe defects in the common pathway. Blood 2017, 130, 666–676. [Google Scholar] [CrossRef] [PubMed]
- Jagadeeswaran, P.; Sheehan, J.P. Analysis of blood coagulation in the zebrafish. Blood Cells Mol. Dis. 1999, 25, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Rost, M.S.; Grzegorski, S.; Shavit, J.A. Quantitative methods for studying hemostasis in zebrafish larvae. In Cellular and Developmental Biology Part B; Detrich, H., Westerfield, M., Zon, L., Eds.; Elsevier: San Diego, CA, USA, 2016; pp. 377–389. [Google Scholar]
- Schurgers, E.; Moorlag, M.; Hemker, C.; Lindhout, T.; Kelchtermans, H.; de Laat, B. Thrombin Generation in Zebrafish Blood. PLoS ONE 2016, 11, e0149135. [Google Scholar] [CrossRef] [PubMed]
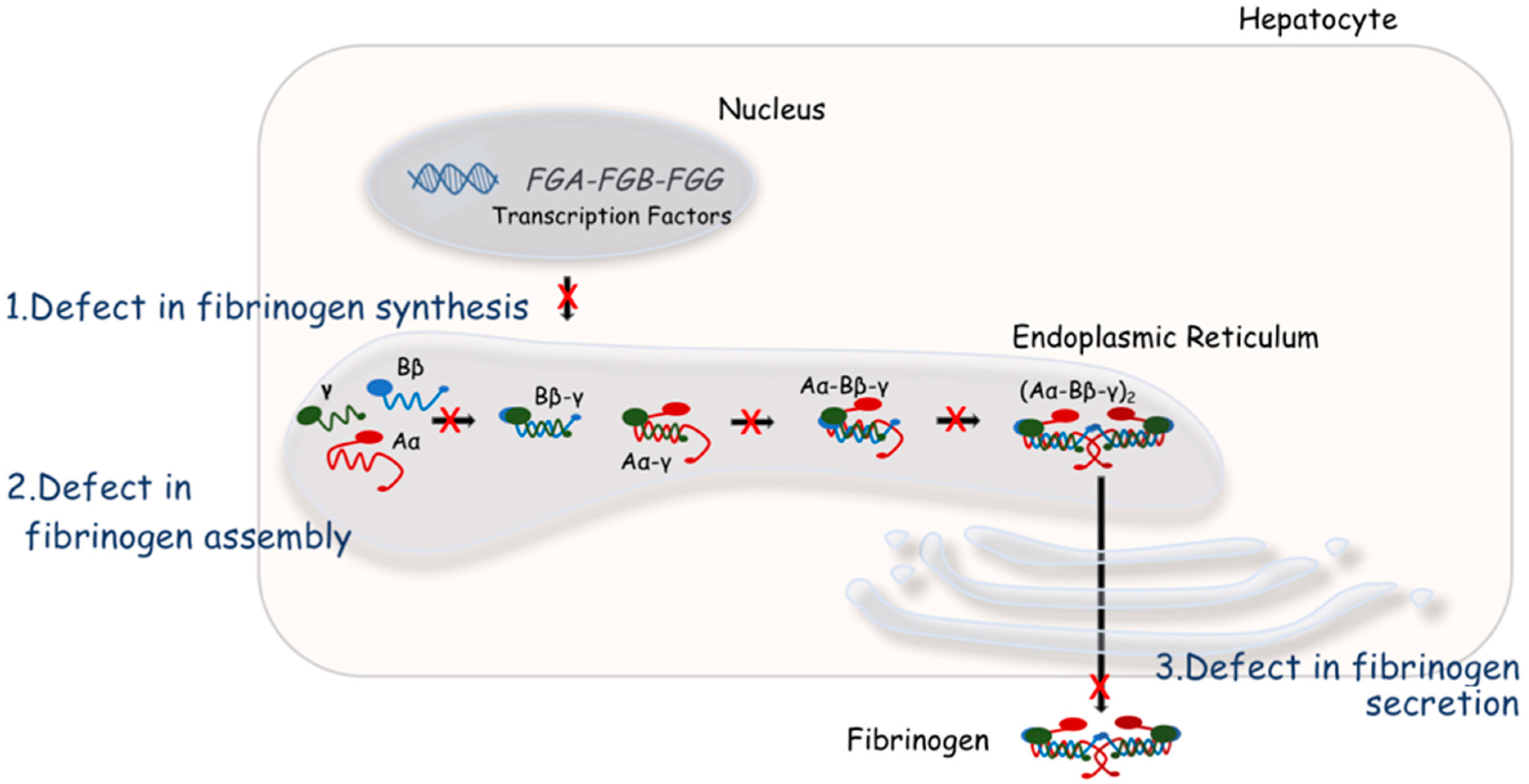

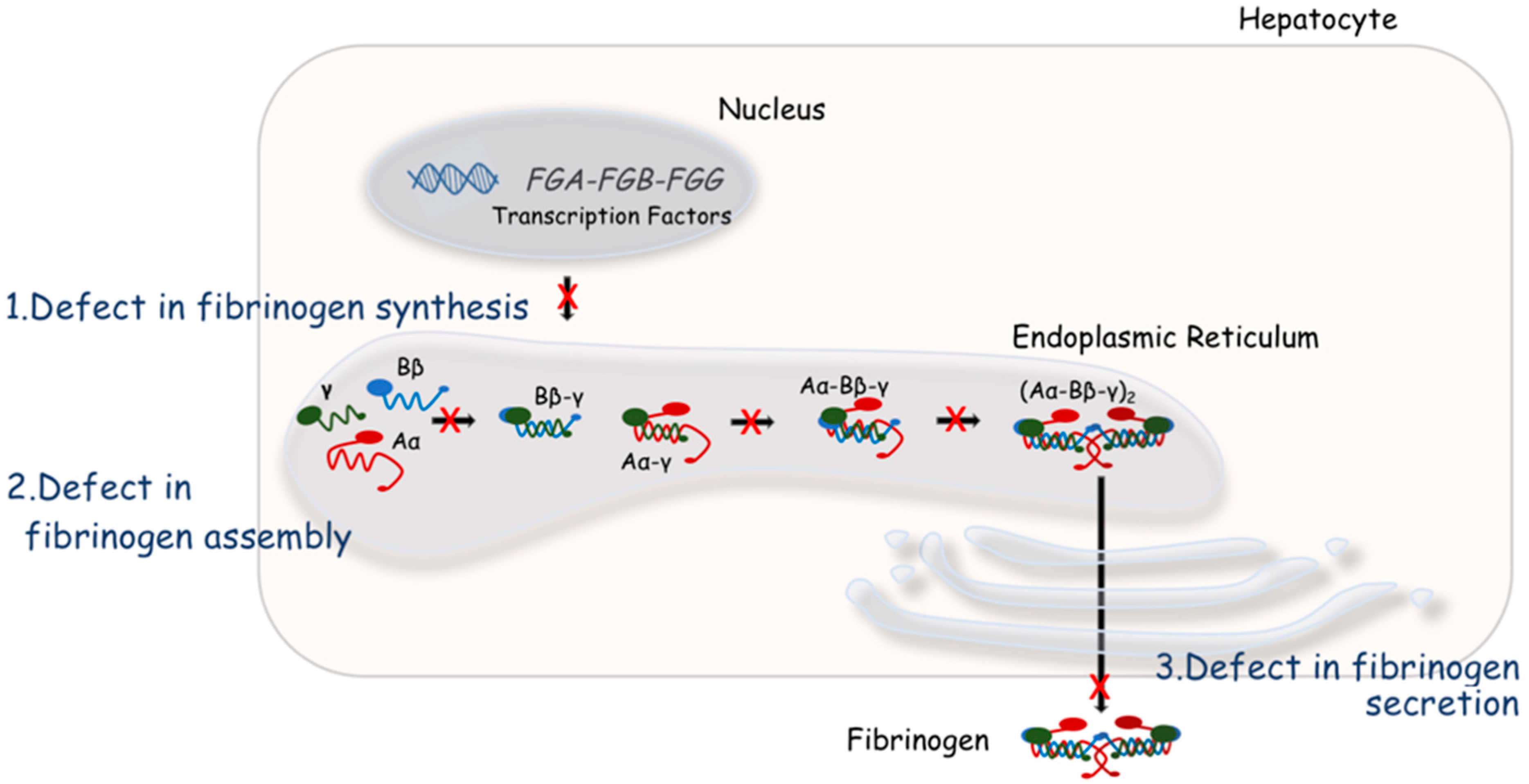{kind=link}
| Author | Patients, n | Males, n (%) | Umbilical Bleeding, n (%) | Muscle Bleeding, n (%) | Joint Bleeding, n (%) | CNS Bleeding, n (%) | Oral Cavity *, n (%) | Menorrhagia, n (% **) | Skin, n (%) | Miscellaneous ***, n (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| Lak [21] | 55 | 27 (49) | 45 (85) | 40 (72) | 30 (55) | 3 (10) | 40 (72) | 14 (50) | NA | 23 (40) |
| Monaldini [39] | 6 | 3 (50) | 0 (0) | 2 (33) | 1 (17) | 1 (17) | 3 (50) | 0 (0) | 1 (17) | 0 (0) |
| de Moerloose [18] | 110 | 45 (41) | 66 (60) | 51 (46) | 33 (30) | 22 (20) | 32 (29) | 36 (55) | 51 (46) | 39 (36) |
| Sumitha [40] | 20 | 12 (60) | 13 (65) | 0 (0) | 1 (5) | 5 (25) | 7 (35) | 3 (38) | 17 (85) | 6 (30) |
| Asselta [41] **** | 13 | 5 (38) | 8 (62) | 0 (0) | 0 (0) | 1 (8) | 8 (62) | 1 (8) | 8 (62) | 6 (46) |
| Nagler [26] | 4 | 3 (75) | 2 (50) | 4 (100) | 4 (100) | 2 (50) | 2 (50) | 1 (100) | 2 (50) | 1 (25) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neerman-Arbez, M.; Casini, A. Clinical Consequences and Molecular Bases of Low Fibrinogen Levels. Int. J. Mol. Sci. 2018, 19, 192. https://doi.org/10.3390/ijms19010192
Neerman-Arbez M, Casini A. Clinical Consequences and Molecular Bases of Low Fibrinogen Levels. International Journal of Molecular Sciences. 2018; 19(1):192. https://doi.org/10.3390/ijms19010192
Chicago/Turabian StyleNeerman-Arbez, Marguerite, and Alessandro Casini. 2018. "Clinical Consequences and Molecular Bases of Low Fibrinogen Levels" International Journal of Molecular Sciences 19, no. 1: 192. https://doi.org/10.3390/ijms19010192
APA StyleNeerman-Arbez, M., & Casini, A. (2018). Clinical Consequences and Molecular Bases of Low Fibrinogen Levels. International Journal of Molecular Sciences, 19(1), 192. https://doi.org/10.3390/ijms19010192