Hsp90α Mediates BMI1 Expression in Breast Cancer Stem/Progenitor Cells through Facilitating Nuclear Translocation of c-Myc and EZH2

 ,
,  , , ,
, , ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
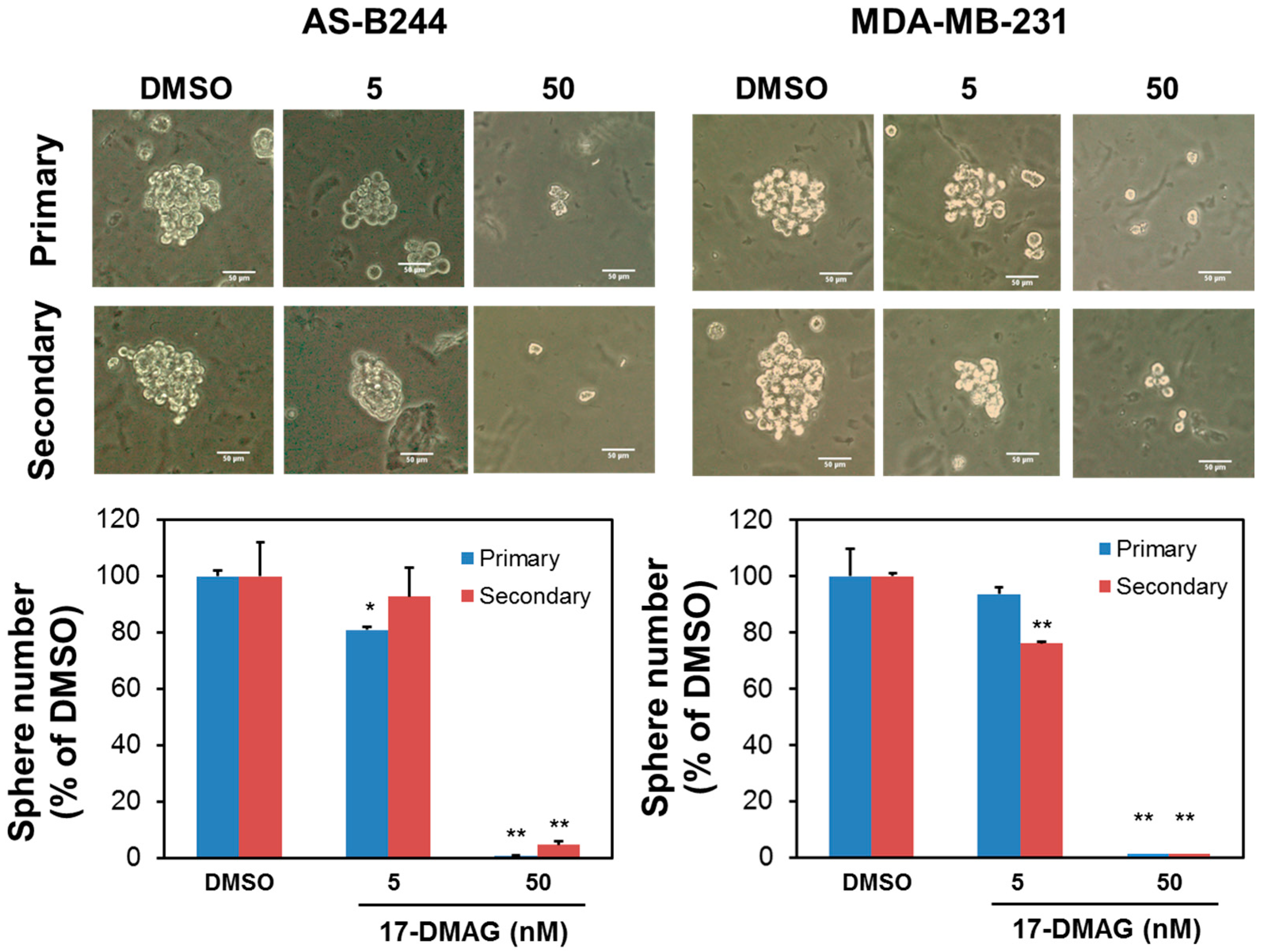
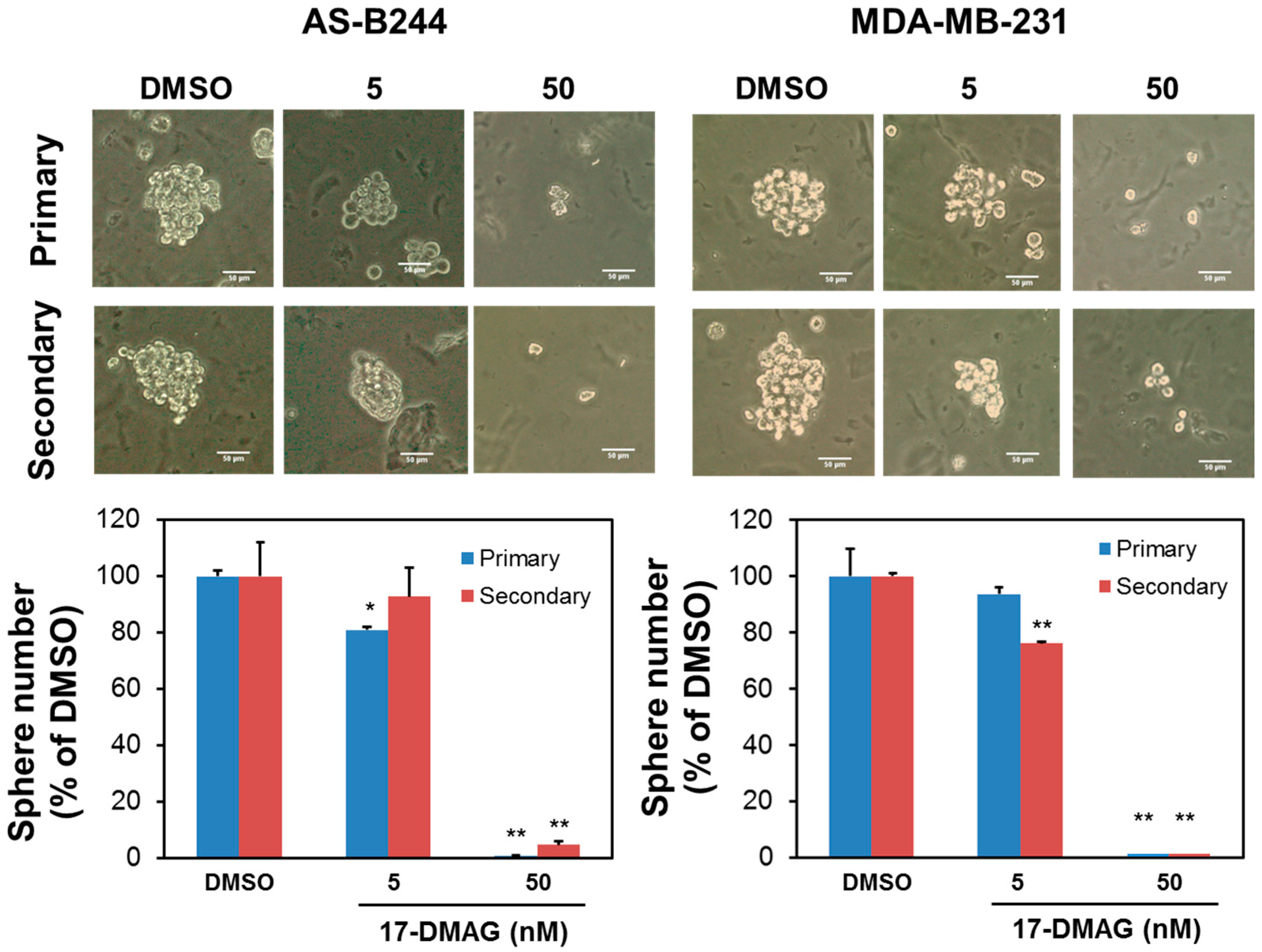
2.1. 17-DMAG Inhibits the Self-Renewal Capability of BCSCs and Downregulates Their Expression of BMI1
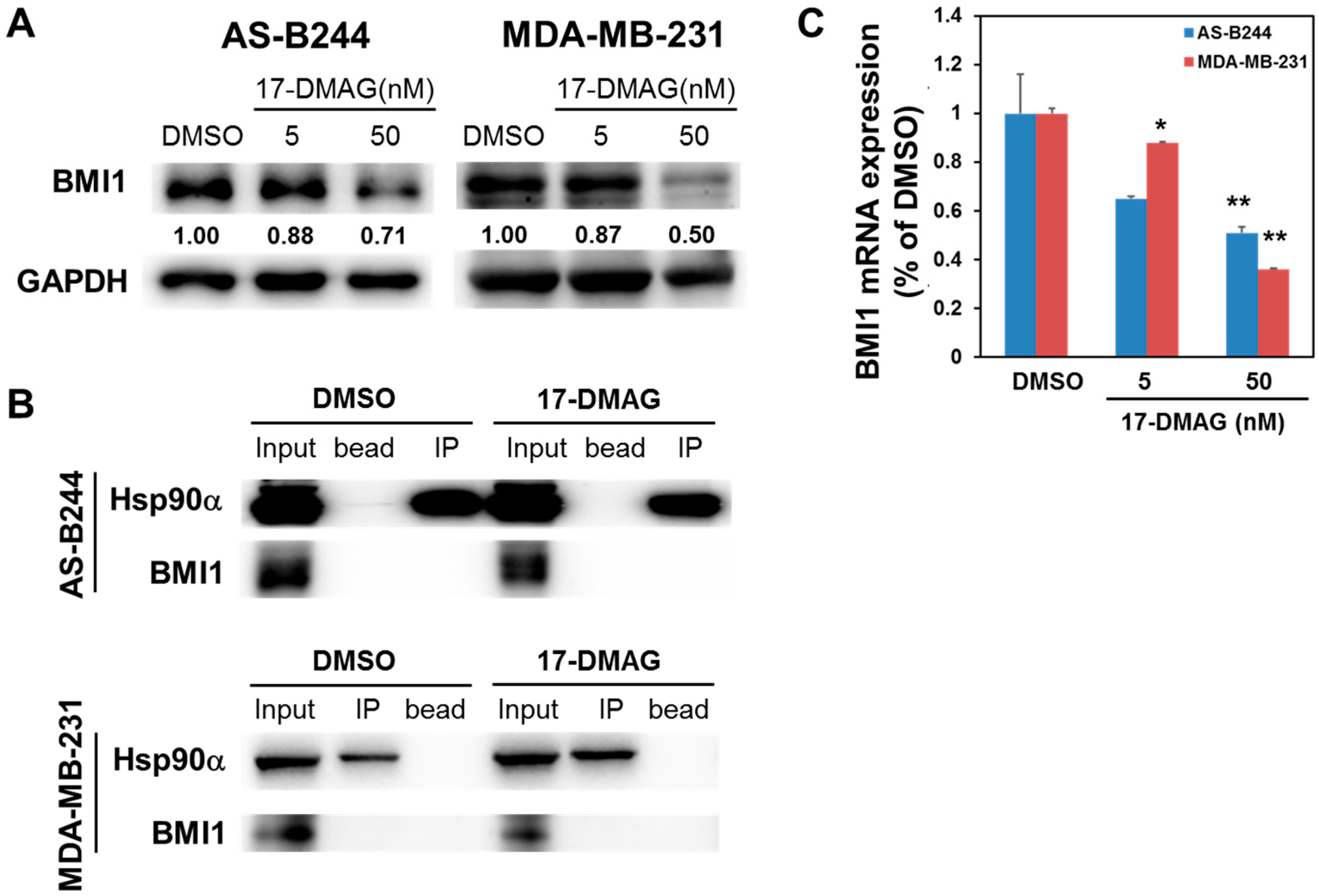
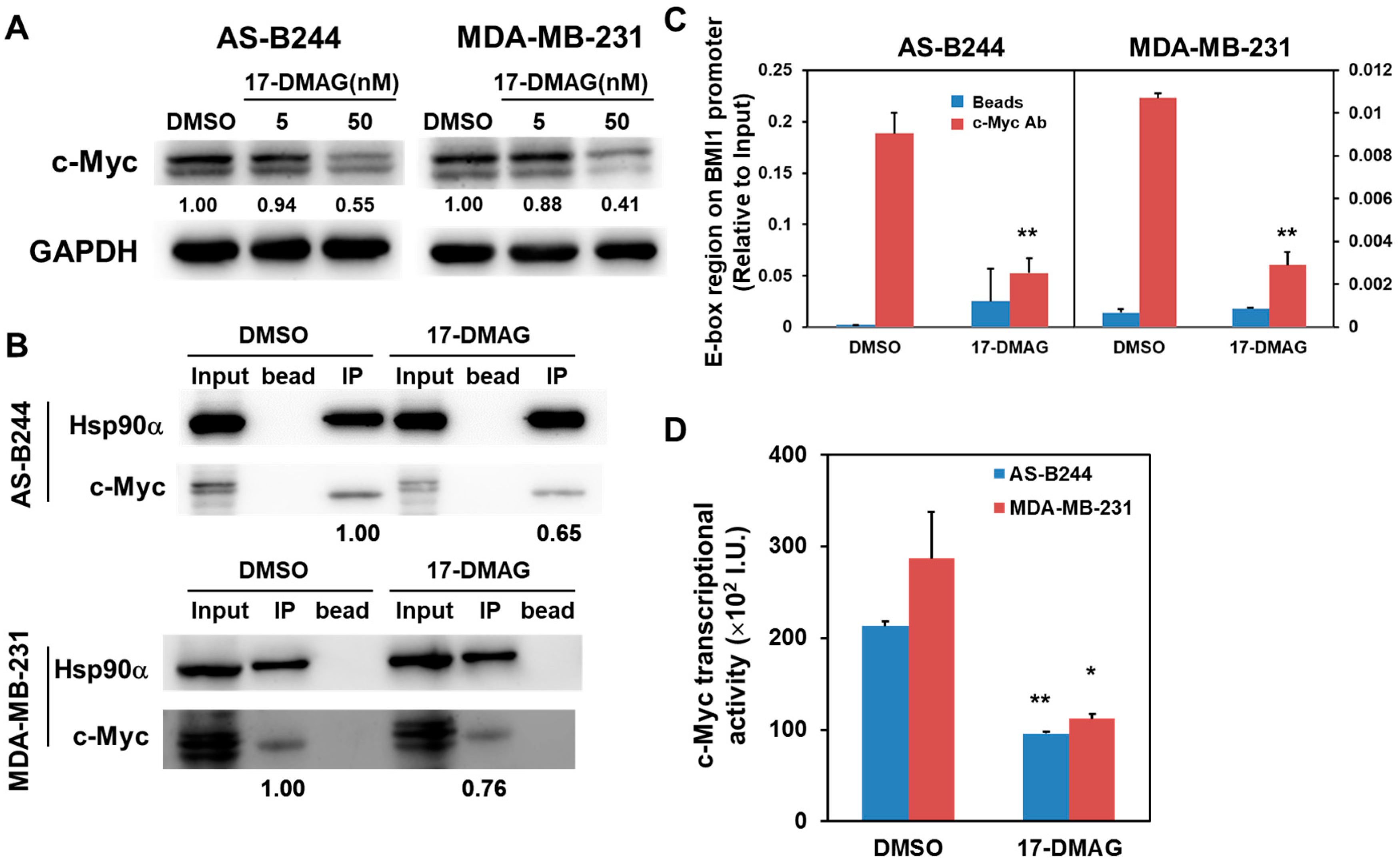
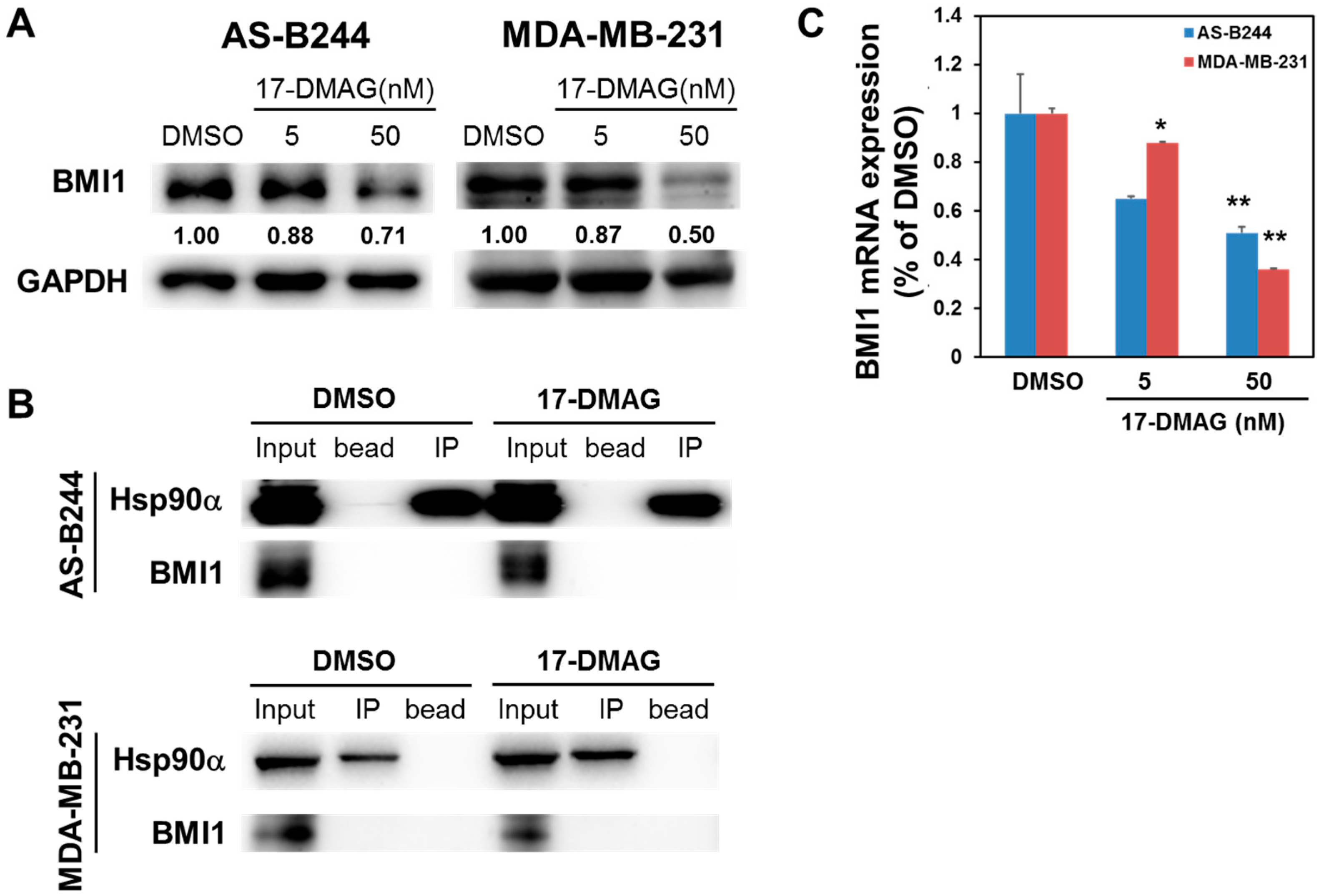
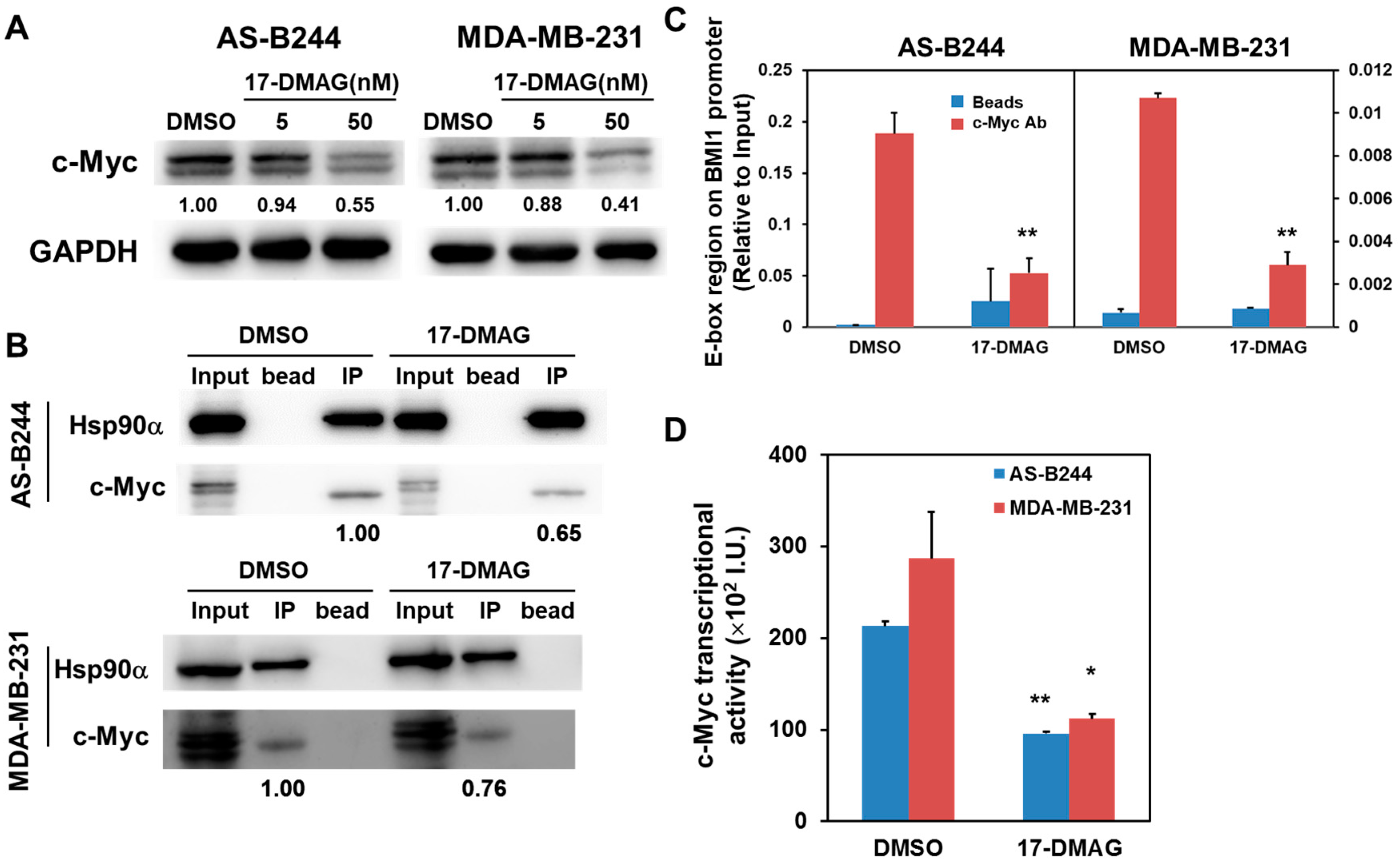
2.2. Inhibitory Effect of 17-DMAG on BMI1 Expression Is Associated with Downregulated c-Myc Transcriptional Activity
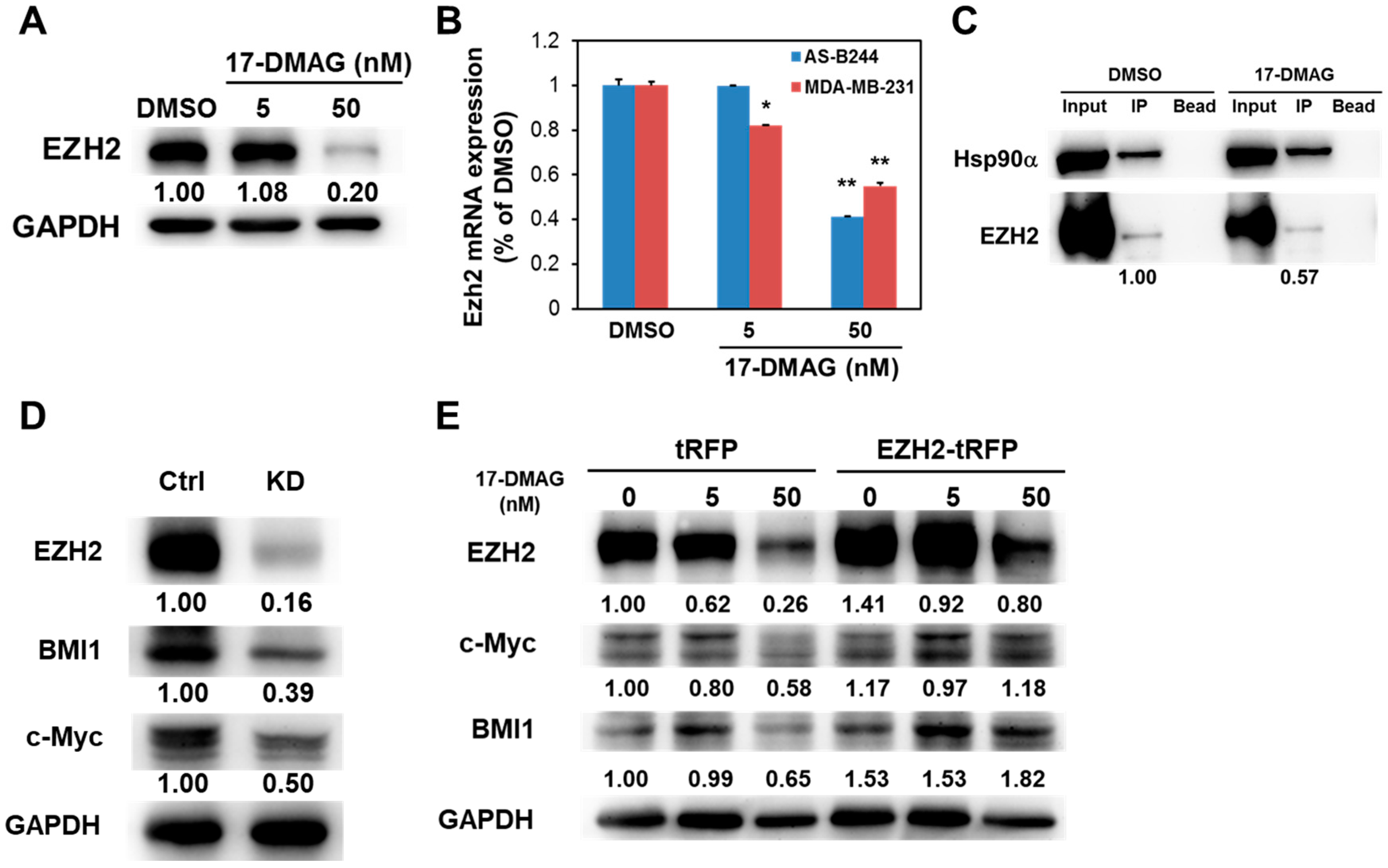
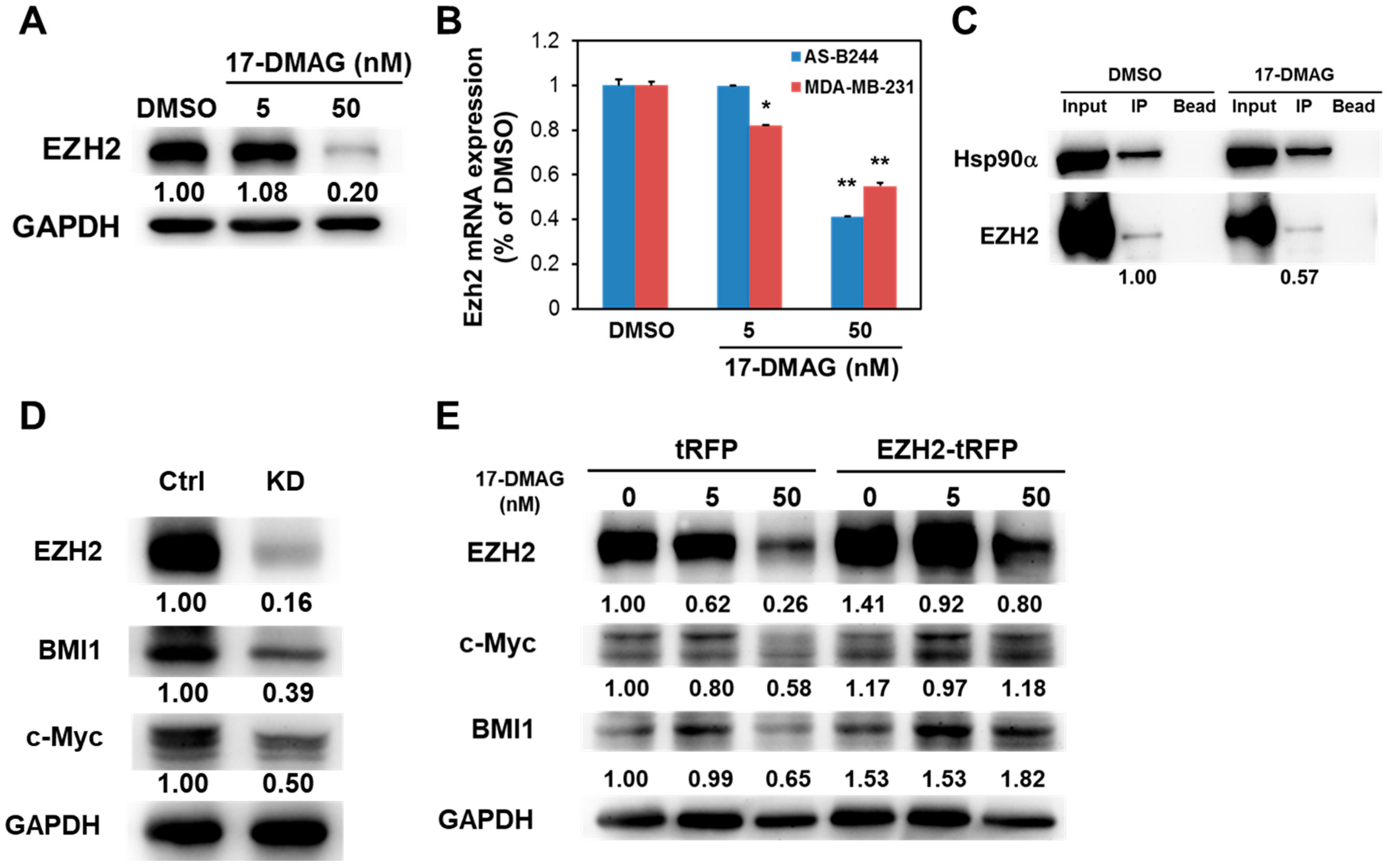
2.3. 17-DMAG-Idnuced Inhibition of BMI1 and c-Myc Expression in BCSCs Is Associated with Downregulated EZH2 Expression
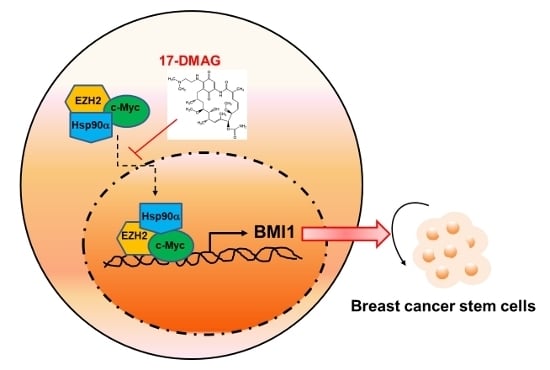
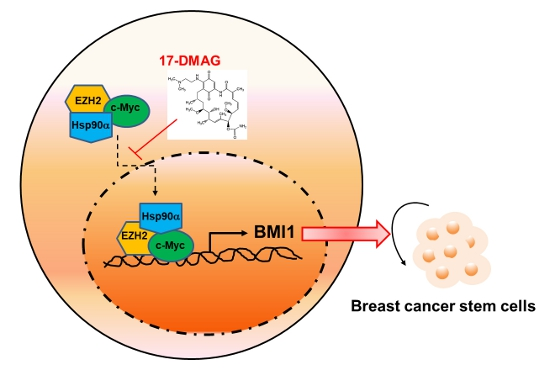
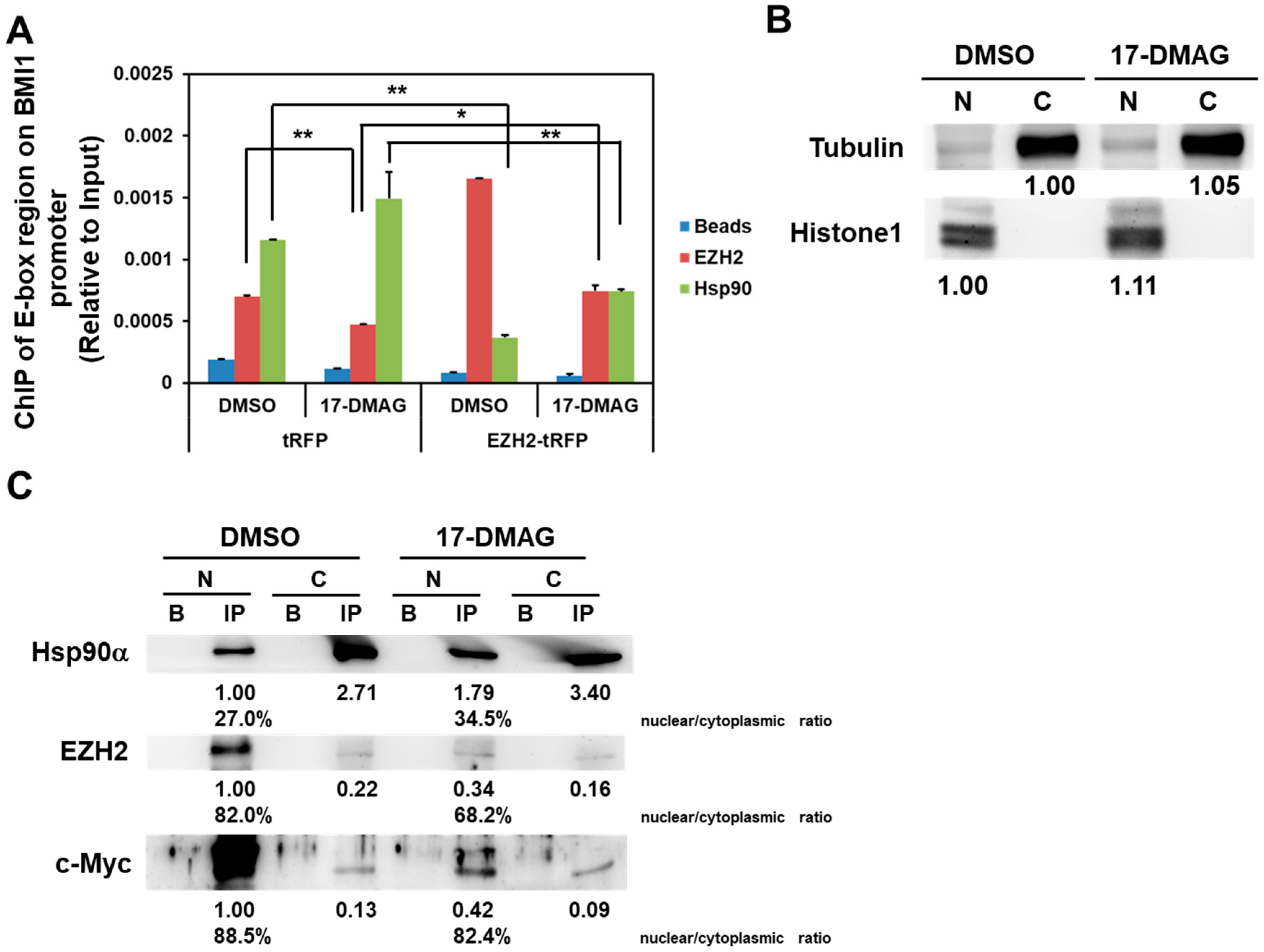
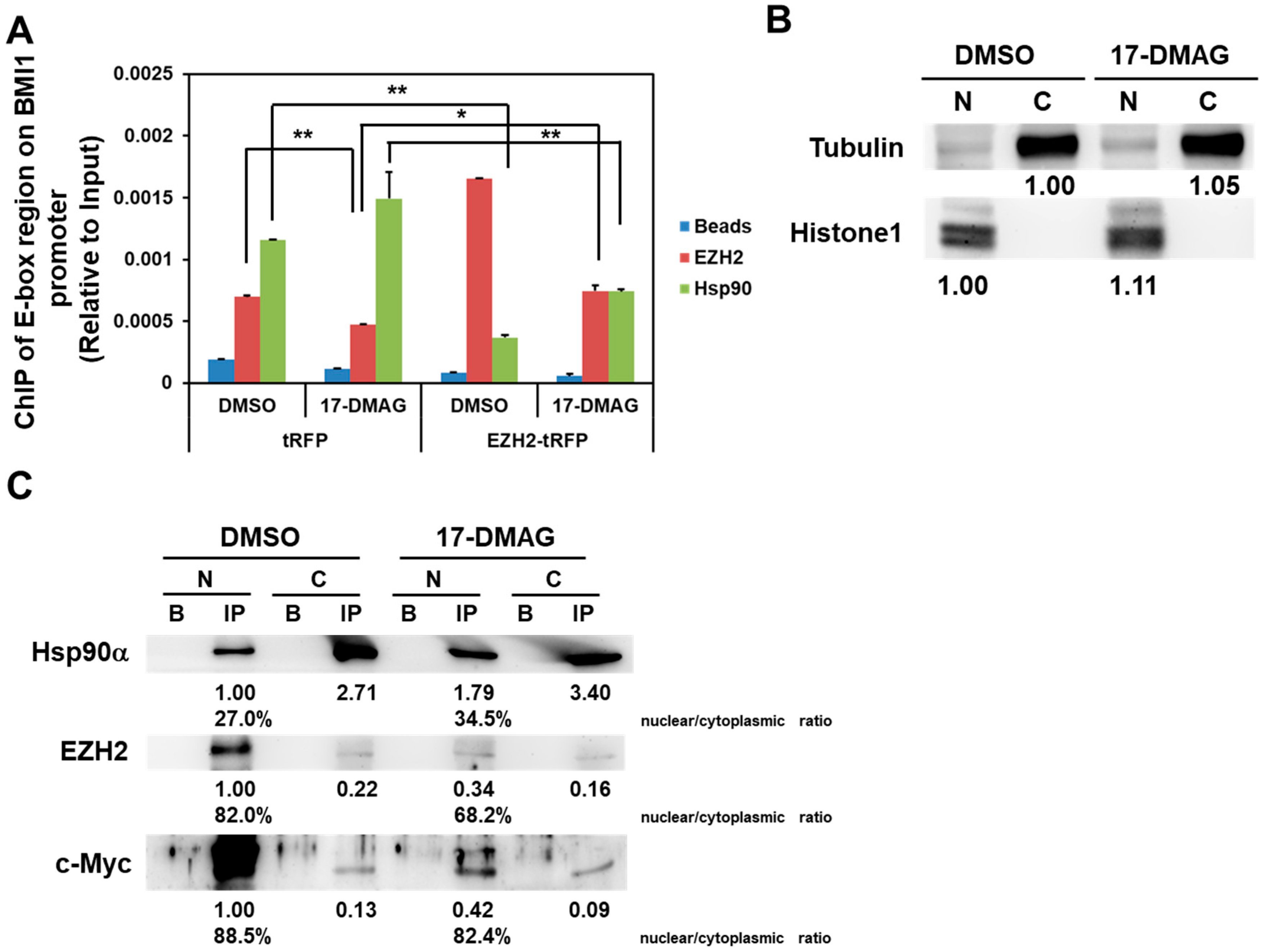
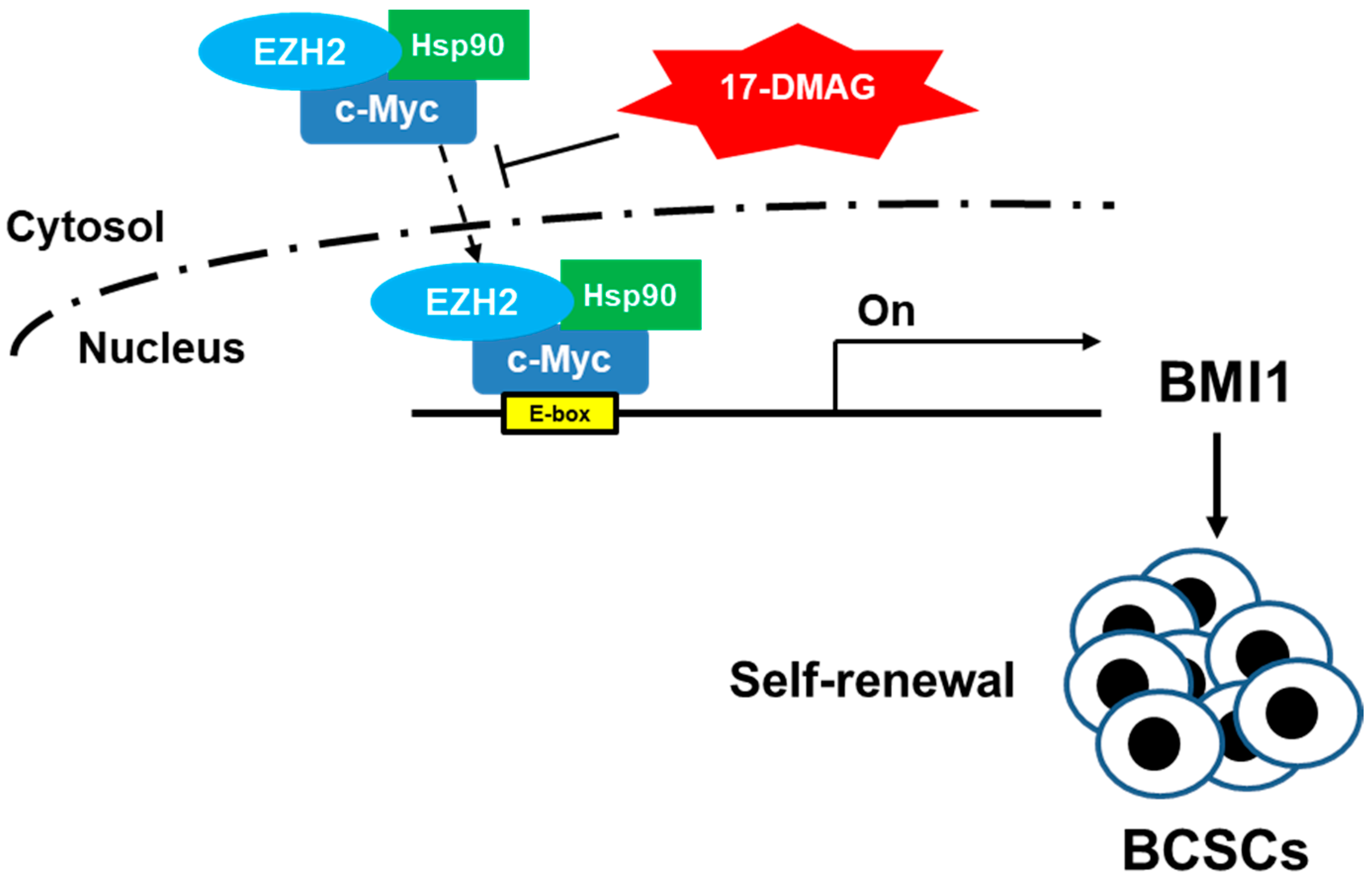
2.4. 17-DMAG Interferes with the Hsp90α-Facilitated Nuclear Translocation of c-Myc and EZH2
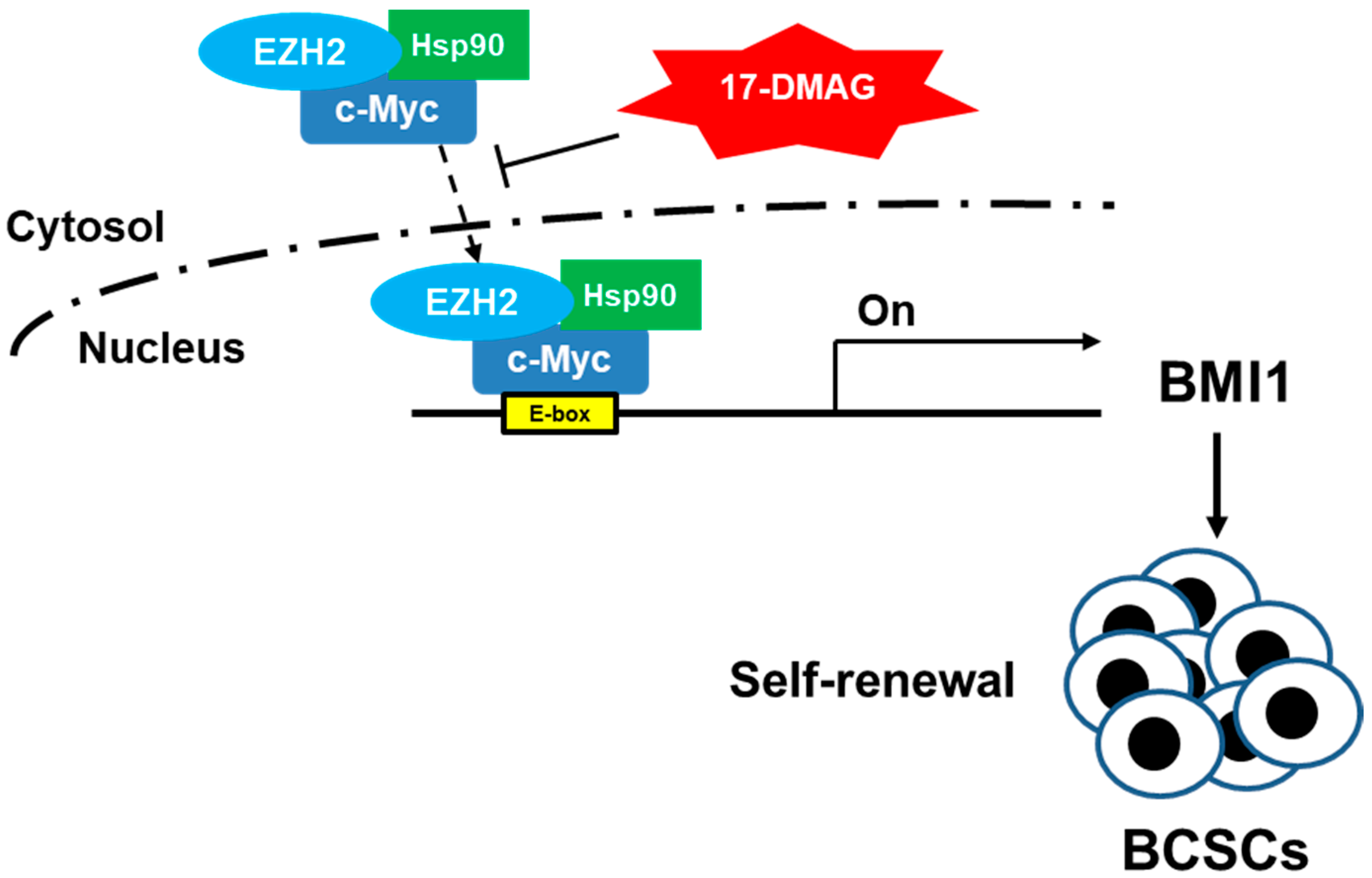
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. Mammosphere Cultivation
4.3. Western Blot
4.4. Quantitative Real-Time RT-PCR
4.5. Immunoprecipitation and ChIP Analysis
4.6. Determination of the Transcriptional Activity of c-Myc
4.7. RNA Interference
4.8. Overexpression of EZH2 with Lentivirus Transduction
4.9. Isolation of Cytosolic and Nuclear Proteins
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| 17-DMAG | 17-Dimethylaminoethylamino-17-demethoxygeldanamycin |
| ALDH | Aldehyde dehydrogenase |
| BCSCs | Breast cancer stem/progenitor cells |
| BMI1 | B lymphoma Mo-MLV insertion region 1 homolog |
| EZH2 | Enhancer of zeste homolog 2 |
| Hsp | Heat shock protein |
| TNBC | Triple-negative breast cancer |
References
- Torre, L.A.; Siegel, R.L.; Ward, E.M.; Jemal, A. Global cancer incidence and mortality rates and trends–An update. Cancer Epidemiol. Biomark. Prev. 2016, 25, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Schnitt, S.J. Classification and prognosis of invasive breast cancer: From morphology to molecular taxonomy. Modern Pathol. 2010, 23, S60–S64. [Google Scholar] [CrossRef] [PubMed]
- Jitariu, A.A.; Cimpean, A.M.; Ribatti, D.; Raica, M. Triple negative breast cancer: The kiss of death. Oncotarget 2017, 8, 46652–46662. [Google Scholar] [CrossRef] [PubMed]
- Brouckaert, O.; Wildiers, H.; Floris, G.; Neven, P. Update on triple-negative breast cancer: Prognosis and management strategies. Int. J. Womens Health 2012, 4, 511–520. [Google Scholar] [PubMed]
- Yi, S.Y.; Hao, Y.B.; Nan, K.J.; Fan, T.L. Cancer stem cells niche: A target for novel cancer therapeutics. Cancer Treat. Rev. 2013, 39, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Hermann, P.C.; Bhaskar, S.; Cioffi, M.; Heeschen, C. Cancer stem cells in solid tumors. Semin. Cancer Biol. 2010, 20, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [PubMed]
- Ginestier, C.; Hur, M.H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.G.; Liu, S.; et al. Aldh1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell. Stem Cell 2007, 1, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Ponti, D.; Costa, A.; Zaffaroni, N.; Pratesi, G.; Petrangolini, G.; Coradini, D.; Pilotti, S.; Pierotti, M.A.; Daidone, M.G. Isolation and in vitro propagation of tumorigenic breast cancer cells with stem/progenitor cell properties. Cancer Res. 2005, 65, 5506–5511. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Yu, C.C.; Wang, B.Y.; Chang, W.W. Tumorsphere as an effective in vitro platform for screening anti-cancer stem cell drugs. Oncotarget 2016, 7, 1215–1226. [Google Scholar] [CrossRef] [PubMed]
- Phillips, T.M.; McBride, W.H.; Pajonk, F. The response of cd24(-/low)/cd44+ breast cancer-initiating cells to radiation. J. Natl. Cancer Inst. 2006, 98, 1777–1785. [Google Scholar] [CrossRef] [PubMed]
- Lagadec, C.; Vlashi, E.; Della Donna, L.; Meng, Y.; Dekmezian, C.; Kim, K.; Pajonk, F. Survival and self-renewing capacity of breast cancer initiating cells during fractionated radiation treatment. Breast Cancer Res. 2010, 12, R13. [Google Scholar] [CrossRef] [PubMed]
- Chuthapisith, S.; Eremin, J.; El-Sheemey, M.; Eremin, O. Breast cancer chemoresistance: Emerging importance of cancer stem cells. Surg. Oncol. 2010, 19, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Calcagno, A.M.; Salcido, C.D.; Gillet, J.P.; Wu, C.P.; Fostel, J.M.; Mumau, M.D.; Gottesman, M.M.; Varticovski, L.; Ambudkar, S.V. Prolonged drug selection of breast cancer cells and enrichment of cancer stem cell characteristics. J. Natl. Cancer Inst. 2010, 102, 1637–1652. [Google Scholar] [CrossRef] [PubMed]
- Siddique, H.R.; Saleem, M. Role of bmi1, a stem cell factor, in cancer recurrence and chemoresistance: Preclinical and clinical evidences. Stem Cells 2012, 30, 372–378. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Bombard, J.; Cintron, K.; Sheedy, J.; Weetall, M.L.; Davis, T.W. Bmi1 as a novel target for drug discovery in cancer. J. Cell. Biochem. 2011, 112, 2729–2741. [Google Scholar] [CrossRef] [PubMed]
- Arnes, J.B.; Collett, K.; Akslen, L.A. Independent prognostic value of the basal-like phenotype of breast cancer and associations with egfr and candidate stem cell marker bmi-1. Histopathology 2008, 52, 370–380. [Google Scholar] [CrossRef] [PubMed]
- Benetatos, L.; Vartholomatos, G.; Hatzimichael, E. Polycomb group proteins and myc: The cancer connection. Cell. Mol. Life Sci. 2014, 71, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.E. Hsp90: Structure and function. Mol. Chaperones 2013, 328, 155–240. [Google Scholar]
- Schopf, F.H.; Biebl, M.M.; Buchner, J. The hsp90 chaperone machinery. Nat. Rev. Mol. Cell Biol. 2017, 18, 345–360. [Google Scholar] [CrossRef] [PubMed]
- Garg, G.; Khandelwal, A.; Blagg, B.S. Anticancer inhibitors of hsp90 function: Beyond the usual suspects. Adv. Cancer Res. 2016, 129, 51–88. [Google Scholar] [PubMed]
- Lee, C.H.; Hong, H.M.; Chang, Y.Y.; Chang, W.W. Inhibition of heat shock protein (hsp) 27 potentiates the suppressive effect of hsp90 inhibitors in targeting breast cancer stem-like cells. Biochimie 2012, 94, 1382–1389. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, M.; Bharali, D.J.; Sudha, T.; Khedr, M.; Guest, I.; Sell, S.; Glinsky, G.V.; Mousa, S.A. Downregulation of bmi1 in breast cancer stem cells suppresses tumor growth and proliferation. Oncotarget 2017, 8, 38731–38742. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.M.; Wang, B.Y.; Lee, C.H.; Lee, H.T.; Li, J.J.; Hong, G.C.; Hung, Y.C.; Chien, P.J.; Chang, C.Y.; Hsu, L.S.; et al. Hinokitiol up-regulates mir-494-3p to suppress bmi1 expression and inhibits self-renewal of breast cancer stem/progenitor cells. Oncotarget 2017. [Google Scholar] [CrossRef]
- Paranjape, A.N.; Balaji, S.A.; Mandal, T.; Krushik, E.V.; Nagaraj, P.; Mukherjee, G.; Rangarajan, A. Bmi1 regulates self-renewal and epithelial to mesenchymal transition in breast cancer cells through nanog. BMC Cancer 2014, 14, 785. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.; Hoenerhoff, M.J.; Bommi, P.; Sainger, R.; Guo, W.J.; Dimri, M.; Band, H.; Band, V.; Green, J.E.; Dimri, G.P. Bmi-1 cooperates with h-ras to transform human mammary epithelial cells via dysregulation of multiple growth-regulatory pathways. Cancer Res. 2007, 67, 10286–10295. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.B.; Liu, G.H.; Zhang, H.; Xing, S.; Hu, L.J.; Zhao, W.F.; Xie, B.; Li, M.Z.; Zeng, B.H.; Li, Y.; et al. Sp1 and c-myc regulate transcription of bmi1 in nasopharyngeal carcinoma. FEBS J. 2013, 280, 2929–2944. [Google Scholar] [CrossRef] [PubMed]
- Carystinos, G.D.; Kandouz, M.; Alaoui-Jamali, M.A.; Batist, G. Unexpected induction of the human connexin 43 promoter by the ras signaling pathway is mediated by a novel putative promoter sequence. Mol. Pharmacol. 2003, 63, 821–831. [Google Scholar] [CrossRef] [PubMed]
- Suva, M.L.; Riggi, N.; Janiszewska, M.; Radovanovic, I.; Provero, P.; Stehle, J.C.; Baumer, K.; Le Bitoux, M.A.; Marino, D.; Cironi, L.; et al. Ezh2 is essential for glioblastoma cancer stem cell maintenance. Cancer Res. 2009, 69, 9211–9218. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; He, S.; Tian, Y.; Gu, Y.; Chen, P.; Li, C.; Huang, J.; Liu, Y.; Yu, H.; Jin, M.; et al. Hsp90 inhibition destabilizes ezh2 protein in alloreactive t cells and reduces graft-versus-host disease in mice. Blood 2017, 129, 2737–2748. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Diaz, I.; Giraldez, T.; Arnau, M.R.; Smits, V.A.; Jaisser, F.; Farman, N.; Alvarez de la Rosa, D. The mineralocorticoid receptor is a constitutive nuclear factor in cardiomyocytes due to hyperactive nuclear localization signals. Endocrinology 2010, 151, 3888–3899. [Google Scholar] [CrossRef] [PubMed]
- Park, I.K.; Morrison, S.J.; Clarke, M.F. Bmi1, stem cells, and senescence regulation. J. Clin. Investig. 2004, 113, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Nishida, Y.; Maeda, A.; Kim, M.J.; Cao, L.; Kubota, Y.; Ishizawa, J.; AlRawi, A.; Kato, Y.; Iwama, A.; Fujisawa, M.; et al. The novel bmi-1 inhibitor ptc596 downregulates mcl-1 and induces p53-independent mitochondrial apoptosis in acute myeloid leukemia progenitor cells. Blood Cancer J. 2017, 7, e527. [Google Scholar] [CrossRef] [PubMed]
- Hollingshead, M.; Alley, M.; Burger, A.M.; Borgel, S.; Pacula-Cox, C.; Fiebig, H.H.; Sausville, E.A. In vivo antitumor efficacy of 17-dmag (17-dimethylaminoethylamino-17-demethoxygeldanamycin hydrochloride), a water-soluble geldanamycin derivative. Cancer Chemother. Pharmacol. 2005, 56, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Eiseman, J.L.; Lan, J.; Lagattuta, T.F.; Hamburger, D.R.; Joseph, E.; Covey, J.M.; Egorin, M.J. Pharmacokinetics and pharmacodynamics of 17-demethoxy 17-(((2-dimethylamino)ethyl)amino)geldanamycin (17dmag, nsc 707545) in c.B-17 scid mice bearing mda-mb-231 human breast cancer xenografts. Cancer Chemother. Pharmacol. 2005, 55, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, R.K.; Egorin, M.J.; Erlichman, C.; Remick, S.C.; Ramalingam, S.S.; Naret, C.; Holleran, J.L.; TenEyck, C.J.; Ivy, S.P.; Belani, C.P. Phase i pharmacokinetic and pharmacodynamic study of 17-dimethylaminoethylamino-17-demethoxygeldanamycin, an inhibitor of heat-shock protein 90, in patients with advanced solid tumors. J. Clin. Oncol. 2010, 28, 1520–1526. [Google Scholar] [CrossRef] [PubMed]
- Duss, S.; Andre, S.; Nicoulaz, A.L.; Fiche, M.; Bonnefoi, H.; Brisken, C.; Iggo, R.D. An oestrogen-dependent model of breast cancer created by transformation of normal human mammary epithelial cells. Breast Cancer Res. 2007, 9, R38. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.H.; Dimri, M.; Dimri, G.P. A positive feedback loop regulates the expression of polycomb group protein bmi1 via wnt signaling pathway. J. Biol. Chem. 2013, 288, 3406–3418. [Google Scholar] [CrossRef] [PubMed]
- Neri, F.; Zippo, A.; Krepelova, A.; Cherubini, A.; Rocchigiani, M.; Oliviero, S. Myc regulates the transcription of the prc2 gene to control the expression of developmental genes in embryonic stem cells. Mol. Cell. Biol. 2012, 32, 840–851. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.C.; Tsai, C.H.; Lai, Y.L.; Yu, C.C.; Chi, W.Y.; Li, J.J.; Chang, W.W. Arecoline-induced myofibroblast transdifferentiation from human buccal mucosal fibroblasts is mediated by zeb1. J. Cell Mol. Med. 2014, 18, 698–708. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, Y.-C.; Chang, W.-W.; Chen, Y.-Y.; Tsai, Y.-H.; Chou, Y.-H.; Tseng, H.-C.; Chen, H.-L.; Wu, C.-C.; Chang-Chien, J.; Lee, H.-T.; et al. Hsp90α Mediates BMI1 Expression in Breast Cancer Stem/Progenitor Cells through Facilitating Nuclear Translocation of c-Myc and EZH2. Int. J. Mol. Sci. 2017, 18, 1986. https://doi.org/10.3390/ijms18091986
Lee Y-C, Chang W-W, Chen Y-Y, Tsai Y-H, Chou Y-H, Tseng H-C, Chen H-L, Wu C-C, Chang-Chien J, Lee H-T, et al. Hsp90α Mediates BMI1 Expression in Breast Cancer Stem/Progenitor Cells through Facilitating Nuclear Translocation of c-Myc and EZH2. International Journal of Molecular Sciences. 2017; 18(9):1986. https://doi.org/10.3390/ijms18091986
Chicago/Turabian StyleLee, Yueh-Chun, Wen-Wei Chang, Yi-Ying Chen, Yu-Hung Tsai, Ying-Hsiang Chou, Hsien-Chun Tseng, Hsin-Lin Chen, Chun-Chieh Wu, Ju Chang-Chien, Hsueh-Te Lee, and et al. 2017. "Hsp90α Mediates BMI1 Expression in Breast Cancer Stem/Progenitor Cells through Facilitating Nuclear Translocation of c-Myc and EZH2" International Journal of Molecular Sciences 18, no. 9: 1986. https://doi.org/10.3390/ijms18091986
APA StyleLee, Y.-C., Chang, W.-W., Chen, Y.-Y., Tsai, Y.-H., Chou, Y.-H., Tseng, H.-C., Chen, H.-L., Wu, C.-C., Chang-Chien, J., Lee, H.-T., Yang, H.-F., & Wang, B.-Y. (2017). Hsp90α Mediates BMI1 Expression in Breast Cancer Stem/Progenitor Cells through Facilitating Nuclear Translocation of c-Myc and EZH2. International Journal of Molecular Sciences, 18(9), 1986. https://doi.org/10.3390/ijms18091986