Abstract
Reactive oxygen species (ROS) act as signaling molecules that control physiological processes, including cell adaptation to stress. Redox signaling via ROS has quite recently become the focus of much attention in numerous pathological contexts, including neurodegenerative diseases, kidney and cardiovascular disease. Imbalance in ROS formation and degradation has also been implicated in essential hypertension. Essential hypertension is characterized by multiple genetic and environmental factors which do not completely explain its associated risk factors. Thereby, even if advances in therapy have led to a significant reduction in hypertension-associated complications, to interfere with the unbalance of redox signals might represent an additional therapeutic challenge. The decrease of nitric oxide (NO) levels, the antioxidant activity commonly found in preclinical models of hypertension and the ability of antioxidant approaches to reduce ROS levels have spurred clinicians to investigate the contribution of ROS in humans. Indeed, particular effort has recently been devoted to understanding how redox signaling may contribute to vascular pathobiology in human hypertension. However, although biomarkers of oxidative stress have been found to positively correlate with blood pressure in preclinical model of hypertension, human data are less convincing. We herein provide an overview of the most relevant mechanisms via which oxidative stress might contribute to the pathophysiology of essential hypertension. Moreover, alternative approaches, which are directed towards improving antioxidant machinery and/or interfering with ROS production, are also discussed.
1. Introduction
Hypertension, a common chronic condition, is a public health problem the world over [1,2]. Interestingly, more than 90% of cases do not have a known cause and current therapies simply aim to control the major symptom of hypertension: elevated arterial blood pressure. Despite extensive research, the intricate and multifactorial nature of the disease means that its pathophysiology is still unclear [3]. The pathophysiological mechanisms that contribute to high blood pressure are complex and include inflammation, remodeling, stiffness, calcification and associated atherosclerosis [4,5]. In this regard, the loss of appropriate endothelial functions, such as reduced vasodilation, increased vasoconstriction and loss of endothelial integrity, seem to play a major role. Oxidative stress, caused by excess ROS generation, decreased nitric oxide (NO) levels and reduced antioxidant capability in the cardiovascular, renal and central nervous systems, is a common feature. ROS regulate cellular processes, such as differentiation, proliferation, apoptosis, cell cycles and migration [6,7], under physiological conditions. ROS control endothelial function and vascular tone in the vascular system meaning that increased ROS production and/or weakened antioxidant defense mechanisms contribute to endothelium and smooth muscle cell (VSMC) dysfunction, which ultimately results in progressive organ failure [8,9,10,11]. The loss of redox homeostasis, characterized by diminished NO bioavailability and increased ROS production, also induces endothelial dysfunction, arterial remodeling and vascular inflammation [8,9,10,11]. ROS production can therefore lead to increased contractility, VSMC growth and apoptosis, monocyte migration, lipid peroxidation, inflammation and increased deposition of extracellular matrix proteins [7,11,12,13,14,15]. As originally reported by Redon and colleagues [16], increased levels of oxidative stress byproducts and decreased endogenous antioxidant enzyme action in peripheral mononuclear cells are common in patients with hypertension. Similar results have been obtained in differing experimental models of hypertension, including nephro-vascular hypertension [17,18,19] and Angiotensin II (Angio-II)-induced hypertension [20,21]. In addition, patients suffering from hypertension are connoted by a significantly higher level of plasma H2O2 than normotensive subjects [22,23]. Since these initial reports were published, intense efforts have been devoted to identifying the specific role that ROS play in these effects [24,25,26,27,28,29]. A “EU-ROS” position paper recently revised all these issues [30]. This concise review will discuss ROS action in vascular biology and its contribution to the development of endothelial dysfunction and vascular remodeling that leads to vascular tree fibrotic changes and hypertension.
2. Redox Signaling
Blood pressure homeostasis is regulated by a dynamic equilibrium of varying mechanisms [31]. In fact, a variety of molecules that belong to the “ROS group” and have different effects on cellular function are involved in the molecular events that control blood pressure [7,32,33]. The uncontrolled generation of ROS promotes oxidative stress and consequent DNA, protein and lipid damage, leading to cell injury and cytotoxicity [7,34]. Although ROS are generated during the reduction of molecular oxygen, the different chemical properties of individual ROS molecules have important implications and functions in cellular redox signaling. ROS usually include unstable free radicals, such as superoxide (O2−), and non-free radicals, such as hydrogen peroxide (H2O2), which are produced as intermediates in reduction–oxidation (redox) processes [7]. The life spans of O2− and •OH mean that these different ROS species can activate signaling pathways that sometimes have conflicting consequences. For example, it has been shown that, unlike H2O2, which has vasodilatory effects in a number of vascular diseases [35,36], O2− acts as a vasoconstrictor and leads to endothelial dysfunction [8]. In addition, ROS byproducts are also derived from a number of metabolic processes, which include xanthine oxidoreductase activity, uncoupled NO synthase (NOS), nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (Nox) and mitochondrial respiratory enzymes [31].
2.1. Xanthine Oxidase
Xanthine oxidase (XO) is a hepatic enzyme that catalyzes the production of uric acid, NO and ROS [37,38]. XO exists in two different forms: xanthine dehydrogenase (XDH) and XO [39]. Compelling evidence suggests that the cellular ratio of XO to XDH is critical to the development of atherosclerosis, endothelial dysfunction, nephro-vascular hypertension and cardiovascular disease in general [40]. Spontaneously hypertensive rats are characterized by the presence of elevated levels of endothelial XO and increased ROS production, which is associated with increases in XO activity and arteriolar tone [41]. Laakso et al. [42] have shown that xanthine oxidoreductase activity is associated with the development of hypertension-associated end-organ failure. Clinical data have demonstrated that the enzyme’s activity is increased both in hypertensive patients [40] and in patients with Angio-II-associated coronary disease [43]. Moreover, both endothelial XO [44] and plasma XO [45] activity are increased in human atherosclerotic plaques, suggesting XO-derived superoxide contributes to the development of hypertension-induced atherosclerosis [46,47]. Based on these data, circulating uric acid has been indicated as a potential oxidative stress biomarker [48,49].
2.2. Nitric Oxide Synthase and Arginase
Nitric oxide synthases (NOS) are a family of enzymes that catalyze the production of NO and citrulline from oxygen and l-arginine substrates [50]. In mammals, three isoforms of NOS (NOS1–3) encoded by distinct genes have been identified [50,51]. Two of them, NOS1 or neuronal NOS (nNOS), and NOS3 or endothelial NOS (eNOS) are constitutively expressed and synthetize NO in a calcium-dependent manner [52], while the latter enzyme, NOS2 or inducible NOS (iNOS), is a calcium-independent enzyme, which expression is regulated by inflammatory cytokines and bacterial toxins such as LPS [53] Unlike nNOS and eNOS, the expression of iNOS has been shown to induce detrimental effects by means of peroxynitrite-mediated apoptosis [54]. In fact, treatment with peroxynitrite scavenger prevented such damaging signals [54]. Moreover, data obtained in myocardial iNOS-overexpressing mice suggested a role of the enzyme in cardiac remodeling processes [55]. However, since these data have been questioned by a number of studies [56,57,58,59], the biological effects of iNOS are still controversial.
In the vascular system, NO production by eNOS is a fundamental mechanism of vascular homeostasis regulation [51]. Arginine is the foremost source for NO via NOS [60]. Indeed, four sets of enzymes are involved in arginine metabolism: arginase (ARG), NOS, arginine:glycine amidinotransferase (AGAT), and arginine decarboxylase (ADC). In particular, in the cytosol NOS metabolizes arginine to NO and l-citrulline; in the mitochondria ADC and AGAT metabolize Arginine into agmatine and creatine respectively; while both in the cytosol and in the mitochondria, ARG metabolizes arginine to urea and l-ornithine, the precursor of polyamines [60]. ARG consist of two distinct isoforms [61]. In particular, the isoform II of ARG is widely expressed [61]. In all expressing tissues, including the vascular system, ARG plays an important role in regulating the synthesis of proline and polyamines (putrescine, spermidine, and spermine) as well as NO [61,62,63]. This implies that, being arginine a limiting substrate for eNOS, ARG represents a crucial mediator of endothelial dysfunction. In this regard, an increased ARG activity has been linked to the development of high blood pressure in salt-loaded salt-sensitive Dahl rats [64]. Similar results were described in deoxycorticosterone acetate (DOCA)-salt hypertensive rats [65] and in endothelial cells of coronary arterioles isolated from renovascular hypertensive pigs [66]. Likewise, in spontaneously hypertensive rats, the increased expression and activity of aortic ARG has been associated with the attenuated relaxation responses to acetylcholine [67]. In fact, the non-selective arginase inhibitor, α-difluoromethylornithine, prevents the development of hypertension [67]. Finally, it has been shown that hemodynamic changes going along with the development of hypertension represent potent inducers of ARG mRNA expression in vascular cells [68].
All these data sustain the notion that reduced NO bioactivity, due to different mechanisms, is a common mechanism of endothelial dysfunction and the development of hypertension [69]. Indeed, the loss of NO bioavailability, due to a reduced synthesis or a defective anti-oxidant response contributes to endothelial dysfunction mainly because ROS production exceeds available antioxidant defense mechanisms. Abnormalities in its production and/or bioavailability go along with, and even precede, hypertension [69]. NO rapidly reacts with anion superoxide (O2−) to form peroxynitrite (ONOO−), which itself can cause vasoconstriction and lead to NOS uncoupling, lipid peroxidation and vascular damage [51,69,70]. Consistently, limited L-arginine availability promotes uncoupling of eNOS resulting in oxygen free radical formation [71].
Increased oxidative stress therefore contributes to the activation of the renin-angiotensin system, changes in glucose metabolism and renal impairment in hypertensive subjects. These effects lead to prolonged redox signaling and reduced NO bioavailability in renal microvasculature, resulting in increased afferent arteriolar tone and hypertension [72]. In this regard, there is some evidence to suggest that the mechanisms involved in reduced NO bioavailability can be attributed to decreased NO production and/or increased NO degradation [72]. Moreover, it has been reported that NO-mediated relaxation, in response to acetylcholine, is blunted in patients with hypertension or pre-hypertensive conditions [73]. The impairment of NO synthesis has also been reported in hypertensive patients with dysfunctional endothelia [74], diabetes mellitus [75], hypercholesterolemia [76] and cigarette smoke addiction [77].
Decreased NO production has been originally detected in pre-clinical models of hypertension [78,79] and diabetes [80,81]. In fact, marked renal and endothelial dysfunction, as well as myocardial infarction and dyslipidemia, have been described in knockout mice for n/i/eNOS isoforms [81].
Of particular interest, recent evidence sustains a role of NO in the maintenance of sodium balance and normotension [82,83]. This is consistent with the original observations that NO is synthesized by the macula densa (MD) and acts to suppress the tubuloglomerular feedback (TGF) [84,85,86]. An intriguing study by Wang et al. [87] demonstrated that a selective inhibition of NOS1β, a primary splice variant of nNOS expressed in the macula densa, in NOS1αKO and WT mice was associated with blunted glomerular filtration rate, diuretic and natriuretic responses after a high-salt (HS) diet [87]. Consistently, Lu and colleagues [88] demonstrated that deletion of NOS1β enhances TGF response and promotes the development of salt-sensitive hypertension. All together, these data suggest a specific role of NOS1β in renal hemodynamic auto-regulation and its potential dysregulation in salt sensitivity hypertension.
2.3. NADPH Oxidases
The group of mammalian NADPH oxidases is made up of seven members, Nox1 to Nox5 and Duox1 and -2, which are structurally similar; all contain FAD and NADPH binding sites, six transmembrane domains and bear two heme groups [89,90]. Depending on the amount of ROS produced in the vascular system, Nox proteins can have both beneficial and detrimental effects. While Nox proteins play an important role in the control of vascular tone and the formation of new blood vessels in physiological conditions [89,90], in hypertensive settings, it has been postulated that Nox expression and activity may lead to inflammation and fibrosis, as well as vascular remodeling [90,91,92]. Superoxide production via membrane NADH/NADPH oxidase activation in Angio-II-induced hypertension sustains the detrimental role of Nox in the pathogenesis of hypertension [21,93]. Rey and colleagues [94] have demonstrated that Nox inhibition attenuates blood pressure elevation in C57Bl/6Tac mice. In Angio-II–induced hypertension, a role for Nox proteins has also been reported in mice lacking p47phox. Indeed, it has been demonstrated that Nox proteins are a major source of O2− and thus contribute to Angio-II-mediated hypertension [95]. In an intriguing work by Matsuno et al. [96], Nox1-null mice exhibited a reduction in ROS activity and a hypertensive response to Angio-II after seven days of treatment, demonstrating that the Nox1/NADPH oxidase-derived ROS in pressor response to Angio-II relies on reduced NO bioavailability. Accordingly, the smooth muscle-specific over-expression of Nox1 potentiates the hypertensive response to Angio-II [97], and similar effects were found in endothelial specific Nox2 overexpression [98]. Finally, ROS also regulate prostanoids resulting in vasoconstriction and reduced endothelium-dependent vasodilation [99,100]. Cyclooxygenase (COX), responsible for the formation of prostanoids, can produce ROS by itself via NADPH [99,100] and COX-derived prostanoids act as autocrine ROS inducers. This implies that ROS can act both upstream and downstream of the COX-prostanoid system [99,100,101] and strengthen the role that Nox proteins play in the development of hypertension through multiple mechanisms.
A fine investigation of NADPH oxidase-dependent ROS generation has been obtained by animal models of salt-sensitive hypertension. In knock-out mice lacking the gp91PHOX gene, which encodes for a NADPH oxidase subunit, Ang II-induced hypertension was markedly attenuated [102]. Blunted arterial pressure was also found in salt-resistant SSp67phox null rats subjected to HS diet [103]. Moreover, NADPH oxidase upregulation and increase arterial pressure and urinary albumin excretion were found in MnSOD−/−, but not wildtype mice, subjected to HS diet [104]. These recent data support the original results in DOCA-salt hypertensive rats, where long term administration of the NADPH oxidase inhibitor, apocynin, significantly decreased systolic blood pressure and aortic O2− production [105].
2.4. Mitochondria
Mitochondria mediate a variety of cellular processes, such as the maintaining of cellular redox status, cell survival and death, calcium homeostasis, while also participating in thermogenesis [106,107]. In physiological conditions, the electron transport chain (ETC) of mitochondria is in charge of efficiently carrying electrons from NADH through complexes I to V, the site of ATP generation. However, it has been demonstrated that hypertension may mediate mitochondria dysfunction [28,108]. Indeed, the mitochondria become dysfunctional under hypertension and mitochondrial superoxide radicals can have a significant impact on this disease. Furthermore, ROS from NADPH oxidase have been shown to enter the mitochondria under hypertension and promote electron loss and ROS production from the ETC [109]. A reduction in antioxidant enzymatic activity in patients with hypertension has also been reported [110]. There is therefore increasing interest in the role that antioxidants play in restoring mitochondrial function in this clinical setting [111]. Itani and colleagues [112] have recently shown that the inhibition of cyclophilin D (CypD), a regulatory subunit of the mitochondrial permeability transition pore (mPTP), which plays a critical role in mitochondrial ROS (mtROS) production, prevents Angio-II-induced hypertension in mice. Moreover, an interesting study by Widder [113] has shown that mitochondrial ROS contribute to Angio-II-induced myocardial hypertrophy, sustained vascular dysfunction, ROS generation and the development of hypertension. This would appear to suggest that mtROS contribute to the deleterious effects of Angio-II, which may be mediated via the direct interaction of Angio-II with mitochondrial components.
3. Oxidative Stress and Hypertension
While the rate of ROS production/clearance is balanced in physiological conditions, in oxidative stress conditions, the unbalance rate translates into increased ROS bioavailability and oxidative stress-mediated cellular damage [110,111,112,113,114]. Indeed, oxidative stress is a common feature of many vascular pathological conditions, such as atherosclerosis, hypercholesterolemia, hypertension, diabetes and heart failure. In the early 1990s, Nakazono et al. [115] demonstrated the existence of a relationship between ROS and hypertension in hypertensive rats by administering the anti-oxidant superoxide dismutase (SOD) mimetics. More recently, it has been shown that mice that lack ROS-producing enzymes display lower blood pressure than wild-type mice and that Angio-II infusion was unable to induce hypertension. Additionally, the ROS production-mediated expression of pro-inflammatory gene products [116] and the modulation of the early signal transduction pathways that are involved in the control of cell growth and death have been reported to contribute to hypertension-induced tissue damage [116,117]. Clinical studies have also demonstrated that patients with hypertension produced excessive amounts of ROS, exhibited higher levels of plasma H2O2 than normotensive subjects [16,118,119,120] and are characterized by unbalanced levels of antioxidant defense mechanisms [121]. This implies that oxidative signaling pathways are attractive targets with which to prevent the development of hypertension and possibly its complications [122,123]. However, the mechanisms involved in oxidative stress processes in hypertension are not yet fully understood. Decreases in NO bioavailability and the oxidation of biological active molecules, such as LDL, were the first well-characterized processes. Indeed, lipid abnormalities and oxidative stress also indirectly contribute to the development of vascular injury and the early phases of renal disease by stimulating mesangial cell (MC) proliferation [124]. In this regard, our laboratory demonstrated that adhesion molecules, such as β4 integrin, can exert stringent control on Rac-1/ROS-mediated MC cycle progression when exposed to ox-LDL [125]. These data point to the crucial role that Nox-activity-generated ROS play in the progression of kidney injuries that lead to hypertension. The oxidation of membrane fatty acids can lead to the formation of F2-isoprostanes, which are present in the blood of patients with diabetes and/or hypercholesterolemia. It is worth noting that plasma F2-isoprostanes are increased in animals with experimental hypertension and in humans with nephron-vascular hypertension [126,127]. Intriguing studies have recently demonstrated the presence of increased ROS generation in pulmonary arterial hypertension (PAH) patients [128,129], suggesting that ROS contribute to abnormal endothelial proliferation and PAH development. Moreover, it has been reported that Nox activity and expression are upregulated in animals and humans with PAH, suggesting that Nox-derived ROS may contribute to the development of PAH [130,131]. All the above data support the notion that the development of hypertension is associated with and, more importantly, could be caused by oxidative stress. This concept has been proven using a genetic approach, which reduces the generation of ROS, and using the overexpression of antioxidant enzymes [132]. The targeting of ROS and the increasing of NO bioavailability using specific compounds may well be a means of treating hypertension and a challenge for the future.
4. Antioxidant Therapies in Hypertension
Antioxidant defense systems in biological systems adapt themselves to the changing levels of oxidants in order to maintain an oxidant-antioxidant balance. Natural defenses against ROS include enzymatic and non-enzymatic systems. Non-enzymatic antioxidants include ascorbic acid (vitamin C) and α-tocopherol (vitamin E), and histidine dipeptides. Vitamin C and E were found to inhibit LDL oxidation, via ROS scavenging and by improving NO bioavailability [133,134]. Enzymatic antioxidant systems include enzymes such as superoxide dismutase, glutathione peroxidase and catalase. Compelling evidences have shown inverse correlations between the various circulating antioxidants and hypertension [135,136,137]. In fact, hypertensive subjects have reduced activity and/or decreased antioxidant enzyme content, including that of SOD, glutathione peroxidase and catalase [137,138,139,140]. Reducing oxidative damage by scavenging ROS with antioxidants should therefore ameliorate vascular injury and prevent the increase of blood pressure.
4.1. Clinical Studies
Antioxidants are molecules that can donate electrons and/or hydrogen atoms to oxidants, prevent the initiation of ROS chain reactions, remove free radical intermediates and inhibit other oxidative reactions [7]. The ability of antioxidants to trap ROS makes them capable of reducing oxidative damage and might possibly control blood pressure [141,142]. Vitamins C and E have been used as a therapeutic approach to decreasing oxidative stress and reducing blood pressure by acting on NADPH oxidase and NO bioavailability [143]. The oral administration of vitamin C has been shown to improve endothelium-dependent vasodilatory responsiveness by enhancing antioxidant status and reducing blood pressure in patients with hypertension [143,144]. XO inhibitors have also been used and allopurinol has, in fact, been investigated in clinical studies involving patients with hypertension [145]. Allopurinol administration was associated with improved cardiovascular outcomes when compared to matched non-exposed controls, suggesting that allopurinol has a positive effect on cardiovascular events [145]. Unfortunately, interventional clinical trials with antioxidants have not been as effective as expected [146,147,148,149,150].
Such disappointing outcomes can, however, be partially explained by insufficient dosage and the timing of the delivery of active compounds to the sites where they are needed. It has also been reported that high doses of antioxidants can increase oxidative stress [151].
4.2. Future Therapeutic Challenges
Progress in the search for the best Nox inhibitors has been made [152]. In fact, rather than scavenging ROS with non-specific antioxidant drugs, targeting compounds that inhibit specific NOX actions have been suggested as being more effective in modulating ROS production and could become future antihypertensive therapy development options (see Table 1). One of the first NADPH inhibitors used in preclinical studies was diphenyliodonium, which is very potent but lacks specificity [153]. A naturally occurring NADPH oxidase inhibitor, apocynin, was found to blunt the development of hypertension and to prevent endothelial dysfunction in hypertensive rats when orally administered [154]. However, similar to diphenyliodonium, apocynin is aspecific and lacks selectivity [155]. The second-generation of NOX inhibitors are more specific and selective [94,156,157,158]. Several NADPH potential oxidase inhibiting compounds are currently under investigation, however, none of the available Nox inhibitors is ready for clinical application and preclinical studies are not yet completed for many of them [159,160,161,162,163]. Further studies are needed to better understand the relationship between hypertension and the role of NADPH oxidase, because Nox inhibitors’ ability to prevent the formation of ROS means they present considerable advantages over antioxidants, which act only to moderate the effects of ROS that have been already produced.

Table 1.
Data on novel XO and NOX inhibitors as therapeutics.
Moreover, we must also keep in mind the fact that the role of oxidative stress in priming hypertension in humans is still debated, even if oxidative stress biomarkers positively correlate with blood pressure in essential hypertension [32,41,78,108,119] and circulating SOD was recently found to be a marker of cardiovascular alterations in hypertensive and diabetic patients [164]. Our laboratory has recently found that the naturally occurring unacylated form of ghrelin (UnAG) is a potent antioxidant compound, which acts on both progenitors and mature endothelial cells. It interferes with Rac1 activation in progenitor cells, even in humans [165], and induces SOD2 expression in preclinical models of acute ROS generation in vascular cells [166,167]. We have no data on its efficacy in hypertension; however, the potent anti-oxidant effects of UnAG might be tested in essential hypertension for its clinical benefits or as an oxidative stress proof of concept.
5. Conclusions
Compelling evidence from experimental and clinical studies indicates that oxidative stress might not only be the etiological cause of hypertension, but might also amplify the development of the disease. Despite the great enthusiasm for the efficacy of antioxidant compounds in preclinical studies, their translation into clinical benefits has failed due to possible confounding factors in patients with co-morbidity. Basic/clinical trials are therefore required to elucidate the role of antioxidants as novel anti-hypertensive therapeutics. One constant point that emerges from this review is that antioxidants would be more efficient in the early stages of the disease when deleterious ROS effects can be reversed. It is worth noting that recently identified inhibitors, which are directed against specific NOX isoenzymes, may be a novel approach to the treatment and prevention of hypertension-associated oxidative damage (Figure 1). However, the lack of evidence from large-scale randomized, controlled and multi-centric trials suggests that further studies are needed if we are to understand the specific contribution of NOX inhibition. To conclude, selective and specific ROS scavengers, rather than non-selective ROS inhibitors, should be developed to evaluate their long-term benefits in the clinic. However, it is always worth remembering that a suitable lifestyle should be recommended above all else since environmental factors are the main triggers of oxidative stress.
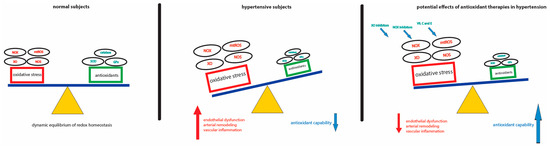
Figure 1.
Schematic representation of ROS-mediated damage in hypertension and potential therapeutic approaches. The dynamic equilibrium of ROS production/clearance is balanced in physiological conditions (left). In hypertensive setting, the loss of redox homeostasis, characterized by a reduced antioxidant capability and increased ROS production, drives endothelial dysfunction, arterial remodeling and vascular inflammation (middle). Selective and specific ROS scavengers may be a novel approach to the treatment of hypertension-associated oxidative damage (right).
Acknowledgments
This work has been supported by grants obtained by Maria Felice Brizzi from Ministero dell’Istruzione, Università e Ricerca (MIUR) ex 60%.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| GTx | Glutathione peroxidase |
| mtROS | Mitochondrial ROS |
| NOS | Nitric oxide synthases |
| NOX | NADPH oxidase |
| SOD | Superoxide dismutase |
| Vit. C and E | Vitamin C and E |
| XO | Xanthine oxidase |
References
- Sliwa, K.; Stewart, S.; Gersh, B.J. Hypertension: A global perspective. Circulation 2011, 123, 2892–2896. [Google Scholar] [CrossRef] [PubMed]
- Bauer, U.E.; Briss, P.A.; Goodman, R.A.; Bowman, B.A. Prevention of chronic disease in the 21st century: Elimination of the leading preventable causes of premature death and disability in the USA. Lancet 2014, 384, 45–52. [Google Scholar] [CrossRef]
- Montezano, A.C.; Touyz, R.M. Molecular mechanisms of hypertension—Reactive oxygen species and antioxidants: A basic science update for the clinician. Can. J. Cardiol. 2012, 28, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Mulvany, M.J.; Baumbach, G.L.; Aalkjaer, C.; Heagerty, A.M.; Korsgaard, N.; Schiffrin, E.L.; Heistad, D.D. Vascular remodeling. Hypertension 1996, 28, 505–506. [Google Scholar] [PubMed]
- Hayashi, K.; Naiki, T. Adaptation and remodeling of vascular wall; biomechanical response to hypertension. J. Mech. Behav. Biomed. Mater. 2009, 2, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Lushchak, V.I. Free radicals, reactive oxygen species, oxidative stress and its classification. Chem. Biol. Interact. 2014, 224, 164–175. [Google Scholar] [CrossRef] [PubMed]
- Droge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Harrison, D.G. Endothelial dysfunction in cardiovascular diseases: The role of oxidant stress. Circ. Res. 2000, 87, 840–844. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.R.; Witting, P.K.; Drummond, G.R. Redox control of endothelial function and dysfunction: Molecular mechanisms and therapeutic opportunities. Antioxid. Redox Signal. 2008, 10, 1713–1765. [Google Scholar] [CrossRef] [PubMed]
- Schulz, E.; Anter, E.; Keaney, J.F., Jr. Oxidative stress, antioxidants, and endothelial function. Curr. Med. Chem. 2004, 11, 1093–1104. [Google Scholar] [CrossRef] [PubMed]
- Taniyama, Y.; Griendling, K.K. Reactive oxygen species in the vasculature: Molecular and cellular mechanisms. Hypertension 2003, 42, 1075–1081. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.P. Radical-free biology of oxidative stress. Am. J. Physiol. Cell Physiol. 2008, 295, C849–C868. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.D.; Ridnour, L.A.; Isenberg, J.S.; Flores-Santana, W.; Switzer, C.H.; Donzelli, S.; Hussain, P.; Vecoli, C.; Paolocci, N.; Ambs, S.; et al. The chemical biology of nitric oxide: Implications in cellular signaling. Free Radic. Biol. Med. 2008, 45, 18–31. [Google Scholar] [CrossRef] [PubMed]
- Lambeth, J.D. NOX enzymes and the biology of reactive oxygen. Nat. Rev. Immunol. 2004, 4, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Ray, P.D.; Huang, B.W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef] [PubMed]
- Redón, J.; Oliva, M.R.; Tormos, C.; Giner, V.; Chaves, J.; Iradi, A.; Sáez, G.T. Antioxidant activities and oxidative stress byproducts in human hypertension. Hypertension 2003, 41, 1096–1101. [Google Scholar] [CrossRef] [PubMed]
- Higashi, Y.; Sasaki, S.; Nakagawa, K.; Matsuura, H.; Oshima, T.; Chayama, K. Endothelial function and oxidative stress in renovascular hypertension. N. Engl. J. Med. 2002, 346, 1954–1962. [Google Scholar] [CrossRef] [PubMed]
- Heitzer, T.; Wenzel, U.; Hink, U.; Krollner, D.; Skatchkov, M.; Stahl, R.A.; MacHarzina, R.; Bräsen, J.H.; Meinertz, T.; Münzel, T. Increased NAD(P)H oxidase-mediated superoxide production in renovascular hypertension: Evidence for an involvement of protein kinase C. Kidney Int. 1999, 55, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Jung, O.; Schreiber, J.G.; Geiger, H.; Pedrazzini, T.; Busse, R.; Brandes, R.P. gp91phox-containing NADPH oxidase mediates endothelial dysfunction in renovascular hypertension. Circulation 2004, 109, 1795–1801. [Google Scholar] [CrossRef] [PubMed]
- Laursen, J.B.; Rajagopalan, S.; Galis, Z.; Tarpey, M.; Freeman, B.A.; Harrison, D.G. Role of superoxide in angiotensin II-induced but not catecholamine-induced hypertension. Circulation 1997, 95, 588–593. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, S.; Kurz, S.; Münzel, T.; Tarpey, M.; Freeman, B.A.; Griendling, K.K.; Harrison, D.G. Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. Contribution to alterations of vasomotor tone. J. Clin. Investig. 1996, 97, 1916–1923. [Google Scholar] [CrossRef] [PubMed]
- Lacy, F.; O’Connor, D.T.; Schmid-Schönbein, G.W. Plasma hydrogen peroxide production in hypertensives and normotensive subjects at genetic risk of hypertension. J. Hypertens. 1998, 16, 291–303. [Google Scholar] [CrossRef] [PubMed]
- Lacy, F.; Kailasam, M.T.; O’Connor, D.T.; Schmid-Schönbein, G.W.; Parmer, R.J. Plasma hydrogen peroxide production in human essential hypertension: Role of heredity, gender, and ethnicity. Hypertension 2000, 36, 878–884. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.Y.; Wong, W.T.; Leung, F.P.; Zhang, Y.; Wang, Y.X.; Lee, H.K.; Ng, C.F.; Chen, Z.Y.; Yao, X.; Au, C.L.; et al. Oxidative stress-dependent cyclooxygenase-2-derived prostaglandin f(2α) impairs endothelial function in renovascular hypertensive rats. Antioxid. Redox Signal. 2012, 16, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Ungvari, Z.; Csiszar, A.; Kaminski, P.M.; Wolin, M.S.; Koller, A. Chronic high pressure-induced arterial oxidative stress: Involvement of protein kinase C-dependent NAD(P)H oxidase and local renin-angiotensin system. Am. J. Pathol. 2004, 165, 219–226. [Google Scholar] [CrossRef]
- Callera, G.E.; Tostes, R.C.; Yogi, A.; Montezano, A.C.; Touyz, R.M. Endothelin-1-induced oxidative stress in DOCA-salt hypertension involves NADPH-oxidase-independent mechanisms. Clin. Sci. Lond. 2006, 110, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Nguyen Dinh Cat, A.; Montezano, A.C.; Burger, D.; Touyz, R.M. Angiotensin II, NADPH oxidase, and redox signaling in the vasculature. Antioxid. Redox Signal. 2013, 19, 1110–1120. [Google Scholar] [CrossRef] [PubMed]
- Dikalov, S.I.; Ungvari, Z. Role of mitochondrial oxidative stress in hypertension. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, 1417–1427. [Google Scholar] [CrossRef] [PubMed]
- Araujo, M.; Wilcox, C.S. Oxidative stress in hypertension: Role of the kidney. Antioxid. Redox Signal. 2014, 20, 74–101. [Google Scholar] [CrossRef] [PubMed]
- Egea, J.; Fabregat, I.; Frapart, Y.M.; Ghezzi, P.; Görlach, A.; Kietzmann, T.; Kubaichuk, K.; Knaus, U.G.; Lopez, M.G.; Olaso-Gonzalez, G.; et al. European contribution to the study of ROS: A summary of the findings and prospects for the future from the COST action BM1203 (EU-ROS). Redox Biol. 2017, 13, 94–162. [Google Scholar] [CrossRef] [PubMed]
- Mancia, G.; De Backer, G.; Dominiczak, A.; Cifkova, R.; Fagard, R.; Germano, G.; Grassi, G.; Heagerty, A.M.; Kjeldsen, S.E.; Laurent, S.; et al. Management of Arterial Hypertension of the European Society of Hypertension; European Society of Cardiology. 2007 Guidelines for the Management of Arterial Hypertension: The Task Force for the Management of Arterial Hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). J. Hypertens. 2007, 25, 1105–1187. [Google Scholar] [CrossRef] [PubMed]
- Touyz, R.M.; Schiffrin, E.L. Reactive oxygen species in vascular biology: Implications in hypertension. Histochem. Cell Biol. 2004, 122, 339–352. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D.; Rodriguez-Iturbe, B. Mechanisms of disease: Oxidative stress and inflammation in the pathogenesis of hypertension. Nat. Clin. Pract. Nephrol. 2006, 2, 582–593. [Google Scholar] [CrossRef] [PubMed]
- D’Autréaux, B.; Toledano, M.B. ROS as signalling molecules: Mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol. 2007, 8, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhao, H.; Li, H.; Kalyanaraman, B.; Nicolosi, A.C.; Gutterman, D.D. Mitochondrial sources of H2O2 generation play a key role in flow-mediated dilation in human coronary resistance arteries. Circ. Res. 2003, 93, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Matoba, T.; Shimokawa, H.; Nakashima, M.; Hirakawa, Y.; Mukai, Y.; Hirano, K.; Kanaide, H.; Takeshita, A. Hydrogen peroxide is an endothelium-derived hyperpolarizing factor in mice. J. Clin. Investig. 2000, 106, 1521–1530. [Google Scholar] [CrossRef] [PubMed]
- Nagler, L.G.; Vartanyan, L.S. Subunit structure of bovine milk xanthine oxidase. Biochim. Biophys. Acta 1976, 427, 78–90. [Google Scholar] [CrossRef]
- McCord, J.M. Oxygen-derived free radicals in postischemic tissue injury. N. Engl. J. Med. 1985, 312, 159–163. [Google Scholar] [PubMed]
- Harrison, R. Structure and function of xanthine oxidoreductase: Where are we now? Free Radic. Biol. Med. 2002, 33, 774–797. [Google Scholar] [CrossRef]
- Berry, C.E.; Hare, J.M. Xanthine oxidoreductase and cardiovascular disease: Molecular mechanisms and pathophysiological implications. J. Physiol. 2004, 555, 589–606. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; DeLano, F.A.; Parks, D.A.; Jamshidi, N.; Granger, D.N.; Ishii, H.; Suematsu, M.; Zweifach, B.W.; Schmid-Schönbein, G.W. Xanthine oxidase activity associated with arterial blood pressure in spontaneously hypertensive rats. Proc. Natl. Acad. Sci. USA 1998, 95, 4754–4759. [Google Scholar] [CrossRef] [PubMed]
- Laakso, J.T.; Teräväinen, T.L.; Martelin, E.; Vaskonen, T.; Lapatto, R. Renal xanthine oxidoreductase activity during development of hypertension in spontaneously hypertensive rats. J. Hypertens. 2004, 22, 1333–1340. [Google Scholar] [CrossRef] [PubMed]
- Landmesser, U.; Spiekermann, S.; Preuss, C.; Sorrentino, S.; Fischer, D.; Manes, C.; Mueller, M.; Drexler, H. Angiotensin II induces endothelial xanthine oxidase activation: Role for endothelial dysfunction in patients with coronary disease. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 943–948. [Google Scholar] [CrossRef] [PubMed]
- Ohara, Y.; Peterson, T.E.; Harrison, D.G. Hypercholesterolemia increases endothelial superoxide anion production. J. Clin. Investig. 1993, 91, 2546–2551. [Google Scholar] [CrossRef] [PubMed]
- White, C.R.; Darley-Usmar, V.; Berrington, W.R.; McAdams, M.; Gore, J.Z.; Thompson, J.A.; Parks, D.A.; Tarpey, M.M.; Freeman, B.A. Circulating plasma xanthine oxidase contributes to vascular dysfunction in hypercholesterolemic rabbits. Proc. Natl. Acad. Sci. USA 1996, 93, 8745–8749. [Google Scholar] [CrossRef] [PubMed]
- Guzik, T.J.; Sadowski, J.; Guzik, B.; Jopek, A.; Kapelak, B.; Przybylowski, P.; Wierzbicki, K.; Korbut, R.; Harrison, D.G.; Channon, K.M. Coronary artery superoxide production and nox isoform expression in human coronary artery disease. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Patetsios, P.; Song, M.; Shutze, W.P.; Pappas, C.; Rodino, W.; Ramirez, J.A.; Panetta, T.F. Identification of uric acid and xanthine oxidase in atherosclerotic plaque. Am. J. Cardiol. 2001, 88, 188–191. [Google Scholar] [CrossRef]
- Riegersperger, M.; Covic, A.; Goldsmith, D. Allopurinol, uric acid, and oxidative stress in cardiorenal disease. Int. Urol. Nephrol. 2011, 43, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Beattie, C.J.; Fulton, R.L.; Higgins, P.; Padmanabhan, S.; McCallum, L.; Walters, M.R.; Dominiczak, A.F.; Touyz, R.M.; Dawson, J. Allopurinol initiation and change in blood pressure in older adults with hypertension. Hypertension 2014, 64, 1102–1107. [Google Scholar] [CrossRef] [PubMed]
- Alderton, W.K.; Cooper, C.E.; Knowles, R.G. Nitric oxide synthases: Structure, function and inhibition. Biochem. J. 2001, 357, 593–615. [Google Scholar] [CrossRef] [PubMed]
- Balligand, J.L.; Feron, O.; Dessy, C. eNOS activation by physical forces: From short-term regulation of contraction to chronic remodeling of cardiovascular tissues. Physiol. Rev. 2009, 89, 481–534. [Google Scholar] [CrossRef] [PubMed]
- Fleming, I.; Busse, R. Signal transduction of eNOS activation. Cardiovasc. Res. 1999, 43, 532–541. [Google Scholar] [CrossRef]
- Nathan, C. Inducible nitric oxide synthase: What difference does it make? J. Clin. Investig. 1997, 100, 2417–2423. [Google Scholar] [CrossRef] [PubMed]
- Arstall, M.A.; Sawyer, D.B.; Fukazawa, R.; Kelly, R.A. Cytokine-mediated apoptosis in cardiac myocytes: The role of inducible nitric oxide synthase induction and peroxynitrite generation. Circ. Res. 1999, 85, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Mungrue, I.N.; Gros, R.; You, X.; Pirani, A.; Azad, A.; Csont, T.; Schulz, R.; Butany, J.; Stewart, D.J.; Husain, M. Cardiomyocyte overexpression of iNOS in mice results in peroxynitrite generation, heart block, and sudden death. J. Clin. Investig. 2002, 109, 735–743. [Google Scholar] [CrossRef] [PubMed]
- Heger, J.; Gödecke, A.; Flögel, U.; Merx, M.W.; Molojavyi, A.; Kühn-Velten, W.N.; Schrader, J. Cardiac-specific overexpression of inducible nitric oxide synthase does not result in severe cardiac dysfunction. Circ. Res. 2002, 90, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Flögel, U.; Merx, M.W.; Godecke, A.; Decking, U.K.; Schrader, J. Myoglobin: A scavenger of bioactive NO. Proc. Natl. Acad. Sci. USA 2001, 98, 735–740. [Google Scholar] [CrossRef] [PubMed]
- Gödecke, A.; Molojavyi, A.; Heger, J.; Flögel, U.; Ding, Z.; Jacoby, C.; Schrader, J. Myoglobin protects the heart from inducible nitric-oxide synthase (iNOS)-mediated nitrosative stress. J. Biol. Chem. 2003, 278, 21761–21766. [Google Scholar] [CrossRef] [PubMed]
- Funakoshi, H.; Kubota, T.; Kawamura, N.; Machida, Y.; Feldman, A.M.; Tsutsui, H.; Shimokawa, H.; Takeshita, A. Disruption of inducible nitric oxide synthase improves beta-adrenergic inotropic responsiveness but not the survival of mice with cytokine-induced cardiomyopathy. Circ. Res. 2002, 90, 959–965. [Google Scholar] [CrossRef] [PubMed]
- Forstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Meininger, C.J.; Hawker, J.R., Jr.; Haynes, T.E.; Kepka-Lenhart, D.; Mistry, S.K.; Morris, S.M., Jr.; Wu, G. Regulatory role of arginase I and II in nitric oxide, polyamine, and proline syntheses in endothelial cells. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E75–E82. [Google Scholar] [PubMed]
- Odenlund, M.; Holmqvist, B.; Baldetorp, B.; Hellstrand, P.; Nilsson, B.O. Polyamine synthesis inhibition induces S phase cell cycle arrest in vascular smooth muscle cells. Amino Acids 2009, 36, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Orlando, G.F.; Wolf, G.; Engelmann, M. Role of neuronal nitric oxide synthase in the regulation of the neuroendocrine stress response in rodents: Insights from mutant mice. Amino Acids 2008, 35, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Johnson, F.K.; Johnson, R.A.; Peyton, K.J.; Durante, W. Arginase inhibition restores arteriolar endothelial function in Dahl rats with salt-induced hypertension. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 288, R1057–R1062. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, S.; Richert, L.; Berthelot, A. Increased arginase activity in aorta of mineralocorticoid-salt hypertensive rats. Clin. Exp. Hypertens. 2000, 22, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Hein, T.W.; Wang, W.; Miller, M.W.; Fossum, T.W.; McDonald, M.M.; Humphrey, J.D.; Kuo, L. Upregulation of vascular arginase in hypertension decreases nitric oxide-mediated dilation of coronary arterioles. Hypertension 2004, 44, 935–943. [Google Scholar] [CrossRef] [PubMed]
- Demougeot, C.; Prigent-Tesssier, A.; Marie, C.; Berthelot, A. Arginase inhibition reduced endothelial dysfunction and blood pressure rising in spontaneously hypertensive rats. J. Hypertens. 2005, 23, 971–978. [Google Scholar] [CrossRef] [PubMed]
- Durante, W.; Liao, L.; Reyna, S.V.; Peyton, K.J.; Schafer, A.I. Physiologic cyclic stretch directs L-arginine transport and metabolism to collagen synthesis in vascular smooth muscle cells. FASEB J. 2000, 14, 1775–1783. [Google Scholar] [CrossRef] [PubMed]
- Taddei, S.; Virdis, A.; Ghiadoni, L.; Sudano, I.; Salvetti, A. Endothelial dysfunction in hypertension. J. Cardiovasc. Pharmacol. 2001, 38, S11–S14. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef] [PubMed]
- Berka, V.; Wu, G.; Yeh, H.C.; Palmer, G.; Tsai, A.L. Three different oxygen-induced radical species in endothelial nitric oxide synthase oxygenase domain under regulation by l-arginine and tetrahydrobiopterin. J. Biol. Chem. 2004, 279, 32243–32251. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, C.S. Oxidative stress and nitric oxide deficiency in the kidney: A critical link to hypertension? Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 289, R913–R935. [Google Scholar] [CrossRef] [PubMed]
- Taddei, S.; Virdis, A.; Mattei, P.; Salvetti, A. Vasodilation to acetylcholine in primary and secondary forms of human hypertension. Hypertension 1993, 21, 929–933. [Google Scholar] [CrossRef] [PubMed]
- Higashi, Y.; Sasaki, S.; Nakagawa, K.; Fukuda, Y.; Matsuura, H.; Oshima, T.; Chayama, K. Tetrahydrobiopterin enhances forearm vascular response to acetylcholine in both normotensive and hypertensive individuals. Am. J. Hypertens. 2002, 15, 326–332. [Google Scholar] [CrossRef]
- Heitzer, T.; Krohn, K.; Albers, S.; Meinertz, T. Tetrahydrobiopterin improves endothelium-dependent vasodilation by increasing nitric oxide activity in patients with Type II diabetes mellitus. Diabetologia 2000, 43, 1435–1438. [Google Scholar] [CrossRef] [PubMed]
- Stroes, E.; Kastelein, J.; Cosentino, F.; Erkelens, W.; Wever, R.; Koomans, H.; Lüscher, T.; Rabelink, T. Tetrahydrobiopterin restores endothelial function in hypercholesterolemia. J. Clin. Investig. 1997, 99, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Heitzer, T.; Brockhoff, C.; Mayer, B.; Warnholtz, A.; Mollnau, H.; Henne, S.; Meinertz, T.; Münzel, T. Tetrahydrobiopterin improves endothelium-dependent vasodilation in chronic smokers: Evidence for a dysfunctional nitric oxide synthase. Circ. Res. 2000, 86, E36–E41. [Google Scholar] [CrossRef] [PubMed]
- Kerr, S.; Brosnan, M.J.; McIntyre, M.; Reid, J.L.; Dominiczak, A.F.; Hamilton, C.A. Superoxide anion production is increased in a model of genetic hypertension: Role of the endothelium. Hypertension 1999, 33, 1353–1358. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Witte, K.; August, M.; Brausch, I.; Gödtel-Armbrust, U.; Habermeier, A.; Closs, E.I.; Oelze, M.; Münzel, T.; Förstermann, U. Reversal of endothelial nitric oxide synthase uncoupling and up-regulation of endothelial nitric oxide synthase expression lowers blood pressure in hypertensive rats. J. Am. Coll. Cardiol. 2006, 47, 2536–2544. [Google Scholar] [CrossRef] [PubMed]
- Hink, U.; Li, H.; Mollnau, H.; Oelze, M.; Matheis, E.; Hartmann, M.; Skatchkov, M.; Thaiss, F.; Stahl, R.A.; Warnholtz, A.; et al. Mechanisms underlying endothelial dysfunction in diabetes mellitus. Circ. Res. 2001, 88, E14–E22. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, M.; Shimokawa, H.; Otsuji, Y.; Yanagihara, N. Pathophysiological relevance of NO signaling in the cardiovascular system: Novel insight from mice lacking all NO synthases. Pharmacol. Ther. 2010, 128, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Hyndman, K.A.; Boesen, E.I.; Elmarakby, A.A.; Brands, M.W.; Huang, P.; Kohan, D.E.; Pollock, D.M.; Pollock, J.S. Renal collecting duct NOS1 maintains fluid-electrolyte homeostasis and blood pressure. Hypertension 2013, 62, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Cowley, A.W., Jr.; Yang, C.; Zheleznova, N.N.; Staruschenko, A.; Kurth, T.; Rein, L.; Kumar, V.; Sadovnikov, K.; Dayton, A.; Hoffman, M.; et al. Evidence of the Importance of Nox4 in Production of Hypertension in Dahl Salt-Sensitive Rats. Hypertension 2016, 67, 440–450. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.L.; Garvin, J.L.; Carretero, O.A. Role of macula densa nitric oxide and cGMP in the regulation of tubuloglomerular feedback. Kidney Int. 2000, 58, 2053–2060. [Google Scholar] [CrossRef] [PubMed]
- Welch, W.J.; Tojo, A.; Lee, J.U.; Kang, D.G.; Schnackenberg, C.G.; Wilcox, C.S. Nitric oxide synthase in the JGA of the SHR: Expression and role in tubuloglomerular feedback. Am. J. Physiol. 1999, 277, F130–F138. [Google Scholar] [PubMed]
- Liu, R.; Carretero, O.A.; Ren, Y.; Garvin, J.L. Increased intracellular pH at the macula densa activates nNOS during tubuloglomerular feedback. Kidney Int. 2005, 67, 1837–1843. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chandrashekar, K.; Wang, L.; Lai, E.Y.; Wei, J.; Zhang, G.; Wang, S.; Zhang, J.; Juncos, L.A.; Liu, R. Inhibition of Nitric Oxide Synthase 1 Induces Salt-Sensitive Hypertension in Nitric Oxide Synthase 1α Knockout and Wild-Type Mice. Hypertension 2016, 67, 792–799. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Wei, J.; Stec, D.E.; Roman, R.J.; Ge, Y.; Cheng, L.; Liu, E.Y.; Zhang, J.; Hansen, P.B.; Fan, F.; et al. Macula Densa Nitric Oxide Synthase 1β Protects against Salt-Sensitive Hypertension. Am. Soc. Nephrol. 2016, 27, 2346–2356. [Google Scholar] [CrossRef] [PubMed]
- Brandes, R.P.; Kreuzer, J. Vascular NADPH oxidases: Molecular mechanisms of activation. Cardiovasc. Res. 2005, 65, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Lassegue, B.; Griendling, K.K. NADPH oxidases: Functions and pathologies in the vasculature. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Ago, T.; Kuroda, J.; Kamouchi, M.; Sadoshima, J.; Kitazono, T. Pathophysiological roles of NADPH oxidase/Nox family proteins in the vascular system review and perspective. Circ. J. 2011, 75, 1791–1800. [Google Scholar] [CrossRef] [PubMed]
- Gray, S.P.; Di Marco, E.; Okabe, J.; Szyndralewiez, C.; Heitz, F.; Montezano, A.C.; de Haan, J.B.; Koulis, C.; El-Osta, A.; Andrews, K.L.; et al. NADPH oxidase 1 plays a key role in diabetes mellitus-accelerated atherosclerosis. Circulation 2013, 127, 1888–1902. [Google Scholar] [CrossRef] [PubMed]
- Fukui, T.; Ishizaka, N.; Rajagopalan, S.; Laursen, J.B.; Capers, Q., 4th; Taylor, W.R.; Harrison, D.G.; de Leon, H.; Wilcox, J.N.; Griendling, K.K. p22phox mRNA expression and NADPH oxidase activity are increased in aortas from hypertensive rats. Circ. Res. 1997, 80, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Rey, F.E.; Cifuentes, M.E.; Kiarash, A.; Quinn, M.T.; Pagano, P.J. Novel competitive inhibitor of NAD(P)H oxidase assembly attenuates vascular O2− and systolic blood pressure in mice. Circ. Res. 2001, 89, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Landmesser, U.; Cai, H.; Dikalov, S.; McCann, L.; Hwang, J.; Jo, H.; Holland, S.M.; Harrison, D.G. Role of p47(phox) in vascular oxidative stress and hypertension caused by angiotensin II. Hypertension 2002, 40, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Matsuno, K.; Yamada, H.; Iwata, K.; Jin, D.; Katsuyama, M.; Matsuki, M.; Takai, S.; Yamanishi, K.; Miyazaki, M.; Matsubara, H.; et al. Nox1 is involved in angiotensin II-mediated hypertension: A study in Nox1-deficient mice. Circulation 2005, 112, 2677–2685. [Google Scholar] [CrossRef] [PubMed]
- Dikalova, A.; Clempus, R.; Lassègue, B.; Cheng, G.; McCoy, J.; Dikalov, S.; San Martin, A.; Lyle, A.; Weber, D.S.; Weiss, D.; et al. Nox1 overexpression potentiates angiotensin II-induced hypertension and vascular smooth muscle hypertrophy in transgenic mice. Circulation 2005, 112, 2668–2676. [Google Scholar] [CrossRef] [PubMed]
- Bendall, J.K.; Rinze, R.; Adlam, D.; Tatham, A.L.; de Bono, J.; Wilson, N.; Volpi, E.; Channon, K.M. Endothelial Nox2 overexpression potentiates vascular oxidative stress and hemodynamic response to angiotensin II: Studies in endothelial-targeted Nox2 transgenic mice. Circ. Res. 2007, 100, 1016–1025. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.T.; Tian, X.Y.; Huang, Y. Endothelial dysfunction in diabetes and hypertension: Cross talk in RAS, BMP4, and ROS-dependent COX-2-derived prostanoids. J. Cardiovasc. Pharmacol. 2013, 61, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Hernanz, R.; Briones, A.M.; Salaices, M.; Alonso, M.J. New roles for old pathways? A circuitous relationship between reactive oxygen species and cyclo-oxygenase in hypertension. Clin. Sci. Lond. 2014, 126, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Revelles, S.; Avendaño, M.S.; García-Redondo, A.B.; Alvarez, Y.; Aguado, A.; Pérez-Girón, J.V.; García-Redondo, L.; Esteban, V.; Redondo, J.M.; Alonso, M.J.; et al. Reciprocal relationship between reactive oxygen species and cyclooxygenase-2 and vascular dysfunction in hypertension. Antioxid. Redox Signal. 2013, 18, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Haque, M.Z.; Majid, D.S. High salt intake delayed angiotensin II-induced hypertension in mice with a genetic variant of NADPH oxidase. Am. J. Hypertens. 2011, 24, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Evans, L.C.; Ryan, R.P.; Broadway, E.; Skelton, M.M.; Kurth, T.; Cowley, A.W., Jr. Null mutation of the nicotinamide adenine dinucleotide phosphate-oxidase subunit p67phox protects the Dahl-S rat from salt-induced reductions in medullary blood flow and glomerular filtration rate. Hypertension 2015, 65, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Jin, K.; Vaziri, N.D. Salt-sensitive hypertension in mitochondrial superoxide dismutase deficiency is associated with intra-renal oxidative stress and inflammation. Clin. Exp. Nephrol. 2014, 18, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Beswick, R.A.; Dorrance, A.M.; Leite, R.; Webb, R.C. NADH/NADPH oxidase and enhanced superoxide production in the mineralocorticoid hypertensive rat. Hypertension 2001, 38, 1107–1111. [Google Scholar] [CrossRef] [PubMed]
- McBride, H.M.; Neuspiel, M.; Wasiak, S. Mitochondria: More than just a powerhouse. Curr. Biol. 2006, 16, R551–R560. [Google Scholar] [CrossRef] [PubMed]
- Duchen, M.R. Mitochondria in health and disease: Perspectives on a new mitochondrial biology. Mol. Asp. Med. 2004, 25, 365–451. [Google Scholar] [CrossRef] [PubMed]
- De Cavanagh, E.M.; Toblli, J.E.; Ferder, L.; Piotrkowski, B.; Stella, I.; Fraga, C.G.; Inserra, F. Angiotensin II blockade improves mitochondrial function in spontaneously hypertensive rats. Cell. Mol. Biol. 2005, 51, 573–578. [Google Scholar] [PubMed]
- Doughan, A.K.; Harrison, D.G.; Dikalov, S.I. Molecular mechanisms of angiotensin II-mediated mitochondrial dysfunction: Linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ. Res. 2008, 102, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Xiang, W.; Potts, J.; Floyd, M.; Sharan, C.; Yang, H.; Ross, J.; Nyanda, A.M.; Guo, Z. Reduction in extracellular superoxide dismutase activity in African-American patients with hypertension. Free Radic. Biol. Med. 2006, 41, 1384–1391. [Google Scholar] [CrossRef] [PubMed]
- Dikalova, A.E.; Bikineyeva, A.T.; Budzyn, K.; Nazarewicz, R.R.; McCann, L.; Lewis, W.; Harrison, D.G.; Dikalov, S.I. Therapeutic targeting of mitochondrial superoxide in hypertension. Circ. Res. 2010, 107, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Itani, H.A.; Dikalova, A.E.; McMaster, W.G.; Nazarewicz, R.R.; Bikineyeva, A.T.; Harrison, D.G.; Dikalov, S.I. Mitochondrial Cyclophilin D in Vascular Oxidative Stress and Hypertension. Hypertension 2016, 67, 1218–1227. [Google Scholar] [CrossRef] [PubMed]
- Widder, J.D.; Fraccarollo, D.; Galuppo, P.; Hansen, J.M.; Jones, D.P.; Ertl, G.; Bauersachs, J. Attenuation of angiotensin II-induced vascular dysfunction and hypertension by overexpression of Thioredoxin 2. Hypertension 2009, 54, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Wang, Q.; Zhu, J.; Xiao, Q.; Zhang, L. Reactive Oxygen Species: Key Regulators in Vascular Health and Diseases. Br. J. Pharmacol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Nakazono, K.; Watanabe, N.; Matsuno, K.; Sasaki, J.; Sato, T.; Inoue, M. Does superoxide underlie the pathogenesis of hypertension? Proc. Natl. Acad. Sci. USA 1991, 88, 10045–10048. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Ma, X.; Xie, X.; Shen, G.X. Involvement of NADPH oxidase in oxidized LDL-induced upregulation of heat shock factor-1 and plasminogen activator inhibitor-1 in vascular endothelial cells. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E104–E111. [Google Scholar] [CrossRef] [PubMed]
- Gulati, P.; Klöhn, P.C.; Krug, H.; Göttlicher, M.; Markova, B.; Böhmer, F.D.; Herrlich, P. Redox regulation in mammalian signal transduction. IUBMB Life 2001, 52, 25–28. [Google Scholar] [CrossRef] [PubMed]
- Stojiljkovic, M.P.; Lopes, H.F.; Zhang, D.; Morrow, J.D.; Goodfriend, T.L.; Egan, B.M. Increasing plasma fatty acids elevates F2-isoprostanes in humans: Implications for the cardiovascular risk factor cluster. J. Hypertens. 2002, 20, 1215–1221. [Google Scholar] [CrossRef] [PubMed]
- San José, G.; Fortuño, A.; Moreno, M.U.; Robador, P.A.; Bidegain, J.; Varo, N.; Beloqui, O.; Díez, J.; Zalba, G. The angiotensin-converting enzyme insertion/deletion polymorphism is associated with phagocytic NADPH oxidase-dependent superoxide generation: Potential implication in hypertension. Clin. Sci. Lond. 2009, 116, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, T.; Dohi, Y.; Yamashita, S.; Hirowatari, Y.; Fujii, S.; Ohte, N. Serotonin in peripheral blood reflects oxidative stress and plays a crucial role in atherosclerosis: Novel insights toward holistic anti-atherothrombotic strategy. Atherosclerosis 2016, 246, 157–160. [Google Scholar] [CrossRef] [PubMed]
- Touyz, R.M.; Yao, G.; Viel, E.; Amiri, F.; Schiffrin, E.L. Angiotensin II and endothelin-1 regulate MAP kinases through different redox-dependent mechanisms in human vascular smooth muscle cells. J. Hypertens. 2004, 22, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Basset, O.; Deffert, C.; Foti, M.; Bedard, K.; Jaquet, V.; Ogier-Denis, E.; Krause, K.H. NADPH oxidase 1 deficiency alters caveolin phosphorylation and angiotensin II-receptor localization in vascular smooth muscle. Antioxid. Redox Signal. 2009, 11, 2371–2384. [Google Scholar] [CrossRef] [PubMed]
- Lai, E.Y.; Solis, G.; Luo, Z.; Carlstrom, M.; Sandberg, K.; Holland, S.; Wellstein, A.; Welch, W.J.; Wilcox, C.S. p47(phox) is required for afferent arteriolar contractile responses to angiotensin II and perfusion pressure in mice. Hypertension 2012, 59, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Ha, H.; Lee, H.B. Oxidative stress in diabetic nephropathy: Basic and clinical information. Curr. Diabetes Rep. 2001, 1, 282–287. [Google Scholar] [CrossRef]
- Dentelli, P.; Rosso, A.; Zeoli, A.; Gambino, R.; Pegoraro, L.; Pagano, G.; Falcioni, R.; Brizzi, M.F. Oxidative stress-mediated mesangial cell proliferation requires RAC-1/reactive oxygen species production and beta4 integrin expression. J. Biol. Chem. 2007, 282, 26101–26110. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, M.C.; Sanabria, E.; Manriquez, M.C.; Romero, J.C.; Juncos, L.A. Role of endothelin and isoprostanes in slow pressor responses to angiotensin II. Hypertension 2001, 37, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Minuz, P.; Patrignani, P.; Gaino, S.; Degan, M.; Menapace, L.; Tommasoli, R.; Seta, F.; Capone, M.L.; Tacconelli, S.; Palatresi, S.; et al. Increased oxidative stress and platelet activation in patients with hypertension and renovascular disease. Circulation 2002, 106, 2800–2805. [Google Scholar] [CrossRef] [PubMed]
- Rawat, D.K.; Alzoubi, A.; Gupte, R.; Chettimada, S.; Watanabe, M.; Kahn, A.G.; Okada, T.; McMurtry, I.F.; Gupte, S.A. Increased reactive oxygen species, metabolic maladaptation, and autophagy contribute to pulmonary arterial hypertension-induced ventricular hypertrophy and diastolic heart failure. Hypertension 2014, 64, 1266–1274. [Google Scholar] [CrossRef] [PubMed]
- Al Ghouleh, I.; Sahoo, S.; Meijles, D.N.; Amaral, J.H.; de Jesus, D.S.; Sembrat, J.; Rojas, M.; Goncharov, D.A.; Goncharova, E.A.; Pagano, P.J. Endothelial Nox1 Oxidase Assembly in Human Pulmonary Arterial Hypertension; Driver of Gremlin1-Mediated Proliferation. Clin. Sci. Lond. 2017, 131, 2019–2035. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Luo, X.J.; Yang, Z.B.; Zhang, J.J.; Li, T.B.; Zhang, X.J.; Ma, Q.L.; Zhang, G.G.; Hu, C.P.; Peng, J. Inhibition of NOX/VPO1 pathway and inflammatory reaction by trimethoxystilbene in prevention of cardiovascular remodeling in hypoxia-induced pulmonary hypertensive rats. J. Cardiovasc. Pharmacol. 2014, 63, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Mittal, M.; Roth, M.; Konig, P.; Hofmann, S.; Dony, E.; Goyal, P.; Selbitz, A.C.; Schermuly, R.T.; Ghofrani, H.A.; Kwapiszewska, G.; et al. Hypoxia-dependent regulation of nonphagocytic NADPH oxidase subunit NOX4 in the pulmonary vasculature. Circ. Res. 2007, 101, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.E.; Hong, K.W.; Jin, H.S.; Oh, B. Association analysis of reactive oxygen species-hypertension genes discovered by literature mining. Genom. Inform. 2012, 10, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Jialal, I.; Grundy, S.M. Preservation of the endogenous antioxidants in low density lipoprotein by ascorbate but not probucol during oxidative modification. J. Clin. Investig. 1991, 87, 597–601. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.R.; Stocker, R. Molecular action of vitamin E in lipoprotein oxidation: Implications for atherosclerosis. Free Radic. Biol. Med. 2000, 28, 1795–1805. [Google Scholar] [CrossRef]
- Fraga, C.G.; Oteiza, P.I.; Galleano, M. In vitro measurements and interpretation of total antioxidant capacity. Biochim. Biophys. Acta 2014, 1840, 931–934. [Google Scholar] [CrossRef] [PubMed]
- Pinchuk, I.; Shoval, H.; Dotan, Y.; Lichtenberg, D. Evaluation of antioxidants: Scope, limitations and relevance of assays. Chem. Phys. Lipids 2012, 165, 638–647. [Google Scholar] [CrossRef] [PubMed]
- González, J.; Valls, N.; Brito, R.; Rodrigo, R. Essential hypertension and oxidative stress: New insights. World J. Cardiol. 2014, 6, 353–366. [Google Scholar] [CrossRef] [PubMed]
- Simic, D.V.; Mimic-Oka, J.; Pljesa-Ercegovac, M.; Savic-Radojevic, A.; Opacic, M.; Matic, D.; Ivanovic, B.; Simic, T. Byproducts of oxidative protein damage and antioxidant enzyme activities in plasma of patients with different degrees of essential hypertension. J. Hum. Hypertens. 2006, 20, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Mullan, B.A.; Young, I.S.; Fee, H.; McCance, D.R. Ascorbic acid reduces blood pressure and arterial stiffness in type 2 diabetes. Hypertension 2002, 40, 804–809. [Google Scholar] [CrossRef] [PubMed]
- Takac, I.; Schröder, K.; Brandes, R.P. The Nox family of NADPH oxidases: Friend or foe of the vascular system? Curr. Hypertens. Rep. 2012, 14, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Lassègue, B.; Clempus, R.E. Vascular NAD(P)H oxidases: Specific features, expression, and regulation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003, 285, R277–R297. [Google Scholar] [CrossRef] [PubMed]
- Briones, A.M.; Touyz, R.M. Oxidative stress and hypertension: Current concepts. Curr. Hypertens. Rep. 2010, 12, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Rodrigo, R.; Prat, H.; Passalacqua, W.; Araya, J.; Bächler, J.P. Decrease in oxidative stress through supplementation of vitamins C and E is associated with a reduction in blood pressure in patients with essential hypertension. Clin. Sci. Lond. 2008, 114, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Newberry, S.J. What is the evidence that vitamin C supplements lower blood pressure? Am. J. Clin. Nutr. 2012, 95, 997–998. [Google Scholar] [CrossRef] [PubMed]
- MacIsaac, R.L.; Salatzki, J.; Higgins, P.; Walters, M.R.; Padmanabhan, S.; Dominiczak, A.F.; Touyz, R.M.; Dawson, J. Allopurinol and cardiovascular outcomes in adults with hypertension. Hypertension 2016, 67, 535–540. [Google Scholar] [CrossRef] [PubMed]
- Vivekananthan, D.P.; Penn, M.S.; Sapp, S.K.; Hsu, A.; Topol, E.J. Use of antioxidant vitamins for the prevention of cardiovascular disease: Meta-analysis of randomised trials. Lancet 2003, 361, 2017–2023. [Google Scholar] [CrossRef]
- Montezano, A.C.; Touyz, R.M. Oxidative stress, Noxs, and hypertension: Experimental evidence and clinical controversies. Ann. Med. 2012, 44, S2–S16. [Google Scholar] [CrossRef] [PubMed]
- Collins, A.R.; Lyon, C.J.; Xia, X.; Liu, J.Z.; Tangirala, R.K.; Yin, F.; Boyadjian, R.; Bikineyeva, A.; Praticò, D.; Harrison, D.G.; et al. Age-accelerated atherosclerosis correlates with failure to upregulate antioxidant genes. Circ. Res. 2009, 104, e42–e54. [Google Scholar] [CrossRef] [PubMed]
- Czernichow, S.; Bertrais, S.; Blacher, J.; Galan, P.; Briançon, S.; Favier, A.; Safar, M.; Hercberg, S. Effect of supplementation with antioxidants upon long-term risk of hypertension in the SU.VI.MAX study: Association with plasma antioxidant levels. J. Hypertens. 2005, 23, 2013–2018. [Google Scholar] [CrossRef] [PubMed]
- Sesso, H.D.; Buring, J.E.; Christen, W.G.; Kurth, T.; Belanger, C.; MacFadyen, J.; Bubes, V.; Manson, J.E.; Glynn, R.J.; Gaziano, J.M. Vitamins E and C in the prevention of cardiovascular disease in men: The Physicians’ Health Study II randomized controlled trial. JAMA 2008, 300, 2123–2133. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.R.; Pastor-Barriuso, R.; Dalal, D.; Riemersma, R.A.; Appel, L.J.; Guallar, E. Meta-analysis: High-dosage vitamin E supplementation may increase all-cause mortality. Ann. Intern. Med. 2005, 142, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Drummond, G.R.; Selemidis, S.; Griendling, K.K.; Sobey, C.G. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat. Rev. Drug Discov. 2011, 10, 453–471. [Google Scholar] [CrossRef] [PubMed]
- Wind, S.; Beuerlein, K.; Eucker, T.; Müller, H.; Scheurer, P.; Armitage, M.E.; Ho, H.; Schmidt, H.H.; Wingler, K. Comparative pharmacology of chemically distinct NADPH oxidase inhibitors. Br. J. Pharmacol. 2010, 161, 885–898. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M.; Wang, H.D.; McNeill, J.R. Role of oxidative stress and nitric oxide in regulation of spontaneous tone in aorta of DOCA-salt hypertensive rats. Br. J. Pharmacol. 2004, 141, 562–573. [Google Scholar] [CrossRef] [PubMed]
- Jaquet, V.; Scapozza, L.; Clark, R.A.; Krause, K.H.; Lambeth, J.D. Small-molecule NOX inhibitors: ROS-generating NADPH oxidases as therapeutic targets. Antioxid. Redox Signal. 2009, 11, 2535–2552. [Google Scholar] [CrossRef] [PubMed]
- Spychalowicz, A.; Wilk, G.; Śliwa, T.; Ludew, D.; Guzik, T.J. Novel therapeutic approaches in limiting oxidative stress and inflammation. Curr. Pharm. Biotechnol. 2012, 13, 2456–2466. [Google Scholar] [CrossRef] [PubMed]
- Streeter, J.; Thiel, W.; Brieger, K.; Miller, F.J., Jr. Opportunity nox: The future of NADPH oxidases as therapeutic targets in cardiovascular disease. Cardiovasc. Ther. 2013, 31, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Gianni, D.; Taulet, N.; Zhang, H.; DerMardirossian, C.; Kister, J.; Martinez, L.; Roush, W.R.; Brown, S.J.; Bokoch, G.M.; Rosen, H. A novel and specific NADPH oxidase-1 (Nox1) small-molecule inhibitor blocks the formation of functional invadopodia in human colon cancer cells. ACS Chem. Biol. 2010, 5, 981–993. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.A.; Neupane, G.P.; Lee, E.S.; Jeong, B.S.; Park, B.C.; Thapa, P. NADPH oxidase inhibitors: A patent review. Expert Opin. Ther. Pat. 2011, 21, 1147–1158. [Google Scholar] [CrossRef] [PubMed]
- Altenhöfer, S.; Radermacher, K.A.; Kleikers, P.W.; Wingler, K.; Schmidt, H.H. Evolution of NADPH Oxidase Inhibitors: Selectivity and Mechanisms for Target Engagement. Antioxid. Redox Signal. 2015, 23, 406–427. [Google Scholar] [CrossRef] [PubMed]
- Ranayhossaini, D.J.; Rodriguez, A.I.; Sahoo, S.; Chen, B.B.; Mallampalli, R.K.; Kelley, E.E.; Csanyi, G.; Gladwin, M.T.; Romero, G.; Pagano, P.J. Selective recapitulation of conserved and nonconserved regions of putative NOXA1 protein activation domain confers isoform-specific inhibition of Nox1 oxidase and attenuation of endothelial cell migration. J. Biol. Chem. 2013, 288, 36437–36450. [Google Scholar] [CrossRef] [PubMed]
- Somanna, N.K.; Valente, A.J.; Krenz, M.; Fay, W.P.; Delafontaine, P.; Chandrasekar, B. The Nox1/4 Dual Inhibitor GKT137831 or Nox4 Knockdown Inhibits Angiotensin-II-Induced Adult Mouse Cardiac Fibroblast Proliferation and Migration. AT1 Physically Associates With Nox4. J. Cell. Physiol. 2016, 231, 1130–1141. [Google Scholar] [CrossRef] [PubMed]
- Gray, S.P.; Jha, J.C.; Kennedy, K.; van Bommel, E.; Chew, P.; Szyndralewiez, C.; Touyz, R.M.; Schmidt, H.H.; Cooper, M.E.; Jandeleit-Dahm, K.A. Combined NOX1/4 inhibition with GKT137831 in mice provides dose-dependent reno- and atheroprotection even in established micro- and macrovascular disease. Diabetologia 2017, 60, 927–937. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Marcos, M.A.; Blázquez-Medela, A.M.; Gamella-Pozuelo, L.; Recio-Rodriguez, J.I.; García-Ortiz, L.; Martínez-Salgado, C. Serum Superoxide Dismutase Is Associated with Vascular Structure and Function in Hypertensive and Diabetic Patients. Oxid. Med. Cell. Longev. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Togliatto, G.; Trombetta, A.; Dentelli, P.; Baragli, A.; Rosso, A.; Granata, R.; Ghigo, D.; Pegoraro, L.; Ghigo, E.; Brizzi, M.F. Unacylated ghrelin rescues endothelial progenitor cell function in individuals with type 2 diabetes. Diabetes 2010, 59, 1016–1025. [Google Scholar] [CrossRef] [PubMed]
- Togliatto, G.; Trombetta, A.; Dentelli, P.; Cotogni, P.; Rosso, A.; Tschöp, M.H.; Granata, R.; Ghigo, E.; Brizzi, M.F. Unacylated ghrelin promotes skeletal muscle regeneration following hindlimb ischemia via SOD-2-mediated miR-221/222 expression. J. Am. Heart Assoc. 2013, 2, e000376. [Google Scholar] [CrossRef] [PubMed]
- Togliatto, G.; Trombetta, A.; Dentelli, P.; Gallo, S.; Rosso, A.; Cotogni, P.; Granata, R.; Falcioni, R.; Delale, T.; Ghigo, E.; et al. Unacylated ghrelin induces oxidative stress resistance in a glucose intolerance and peripheral artery disease mouse model by restoring endothelial cell miR-126 expression. Diabetes 2015, 64, 1370–1382. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).