The Endocannabinoid System and Autism Spectrum Disorders: Insights from Animal Models


Abstract
1. Introduction
2. EC-modulation of ASD-Like Behaviors
2.1. EC System in Genetic-Based Models
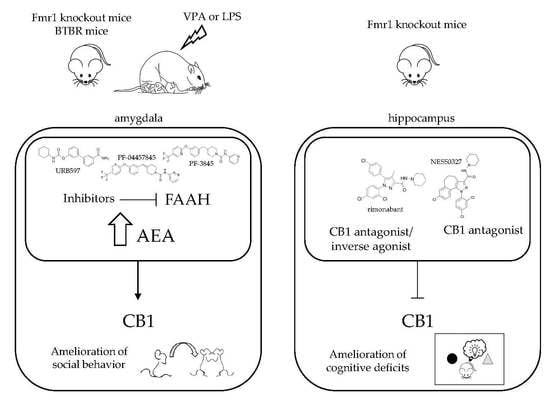
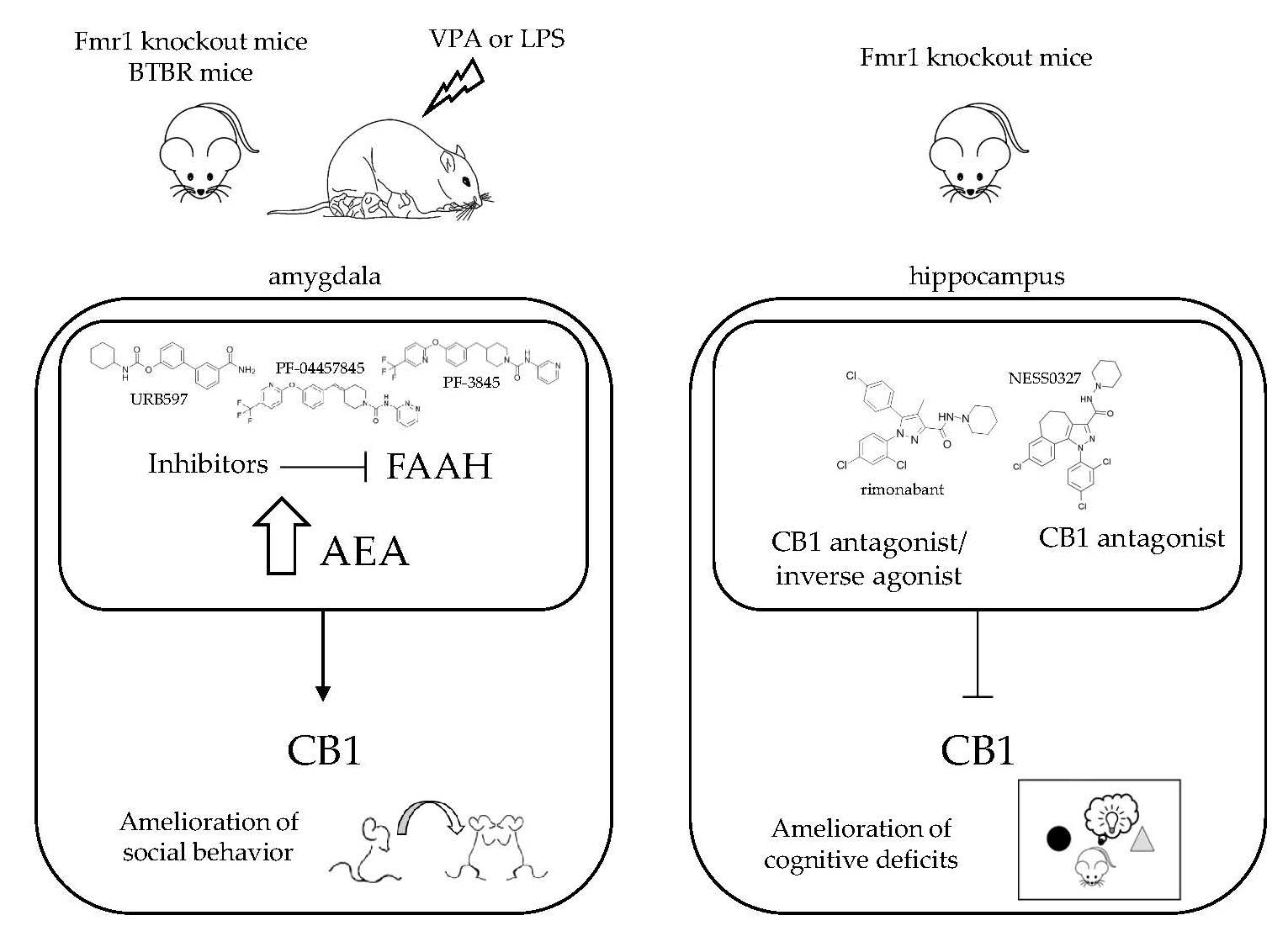
2.1.1. Fragile X Mental Retardation (Fmr1) Knockout Mice
2.1.2. Neuroligin-3 (NLGN3) Mouse Models
2.1.3. BTBR Mouse Model
2.2. The EC System in Environmental-Based Models
2.2.1. Prenatal VPA Exposure
2.2.2. Postnatal LPS Injection
3. Possible Mechanisms
4. Conclusions
Conflicts of Interest
References
- Di Marzo, V.; Piscitelli, F. The endocannabinoid system and its modulation by phytocannabinoids. Neurotherapeutics 2015, 12, 692–698. [Google Scholar] [CrossRef] [PubMed]
- Di Marzo, V.; de Petrocellis, L. Endocannabinoids as regulators of transient receptor potential (TRP) channels: A further opportunity to develop new endocannabinoid-based therapeutic drugs. Curr. Med. Chem. 2010, 17, 1430–1449. [Google Scholar] [CrossRef] [PubMed]
- Pistis, M.; Melis, M. From surface to nuclear receptors: The endocannabinoid family extends its assets. Curr. Med. Chem. 2010, 17, 1450–1467. [Google Scholar] [CrossRef] [PubMed]
- Moriconi, A.; Cerbara, I.; Maccarrone, M.; Topai, A. GPR55: Current knowledge and future perspectives of a purported “Type-3” cannabinoid receptor. Curr. Med. Chem. 2010, 17, 1411–1429. [Google Scholar] [CrossRef] [PubMed]
- Iannotti, F.A.; di Marzo, V.; Petrosino, S. Endocannabinoids and endocannabinoid-related mediators: Targets, metabolism and role in neurological disorders. Prog. Lipid Res. 2016, 62, 107–128. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Kunos, G. Modulating the endocannabinoid system in human health and disease—successes and failures. FEBS J. 2013, 280, 1918–1943. [Google Scholar] [CrossRef] [PubMed]
- Rubino, T.; Zamberletti, E.; Parolaro, D. Endocannabinoids and mental disorders. Handb. Exp. Pharmacol. 2015, 231, 261–283. [Google Scholar] [PubMed]
- Marsicano, G.; Lutz, B. Neuromodulatory functions of the endocannabinoid system. J. Endocrinol. Investig. 2006, 29, 27–46. [Google Scholar]
- Marco, E.M.; Laviola, G. The endocannabinoid system in the regulation of emotions throughout lifespan: A discussion on therapeutic perspectives. J. Psychopharmacol. 2012, 26, 150–163. [Google Scholar] [CrossRef] [PubMed]
- Trezza, V.; Damsteegt, R.; Manduca, A.; Petrosino, S.; van Kerkhof, L.W.; Pasterkamp, R.J.; Zhou, Y.; Campolongo, P.; Cuomo, V.; di Marzo, V.; et al. Endocannabinoids in amygdala and nucleus accumbens mediate social play reward in adolescent rats. J. Neurosci. 2012, 32, 14899–14908. [Google Scholar] [CrossRef] [PubMed]
- Trezza, V.; Vanderschuren, L.J. Divergent effects of anandamide transporter inhibitors with different target selectivity on social play behavior in adolescent rats. J. Pharmacol. Exp. Ther. 2009, 328, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Trezza, V.; Vanderschuren, L.J. Bidirectional cannabinoid modulation of social behavior in adolescent rats. Psychopharmacology 2008, 197, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Rubino, T.; Realini, N.; Castiglioni, C.; Guidali, C.; Vigano, D.; Marras, E.; Petrosino, S.; Perletti, G.; Maccarrone, M.; di Marzo, V.; et al. Role in anxiety behavior of the endocannabinoid system in the prefrontal cortex. Cereb. Cortex 2008, 18, 1292–1301. [Google Scholar] [CrossRef] [PubMed]
- Katona, I.; Freund, T.F. Endocannabinoid signaling as a synaptic circuit breaker in neurological disease. Nat. Med. 2008, 14, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, H.C.; Leggett, J.D.; Wood, S.A.; Castrique, E.S.; Kershaw, Y.M.; Lightman, S.L. Regulation of the hypothalamic-pituitary-adrenal axis circadian rhythm by endocannabinoids is sexually diergic. Endocrinology 2010, 151, 3720–3727. [Google Scholar] [CrossRef] [PubMed]
- Vaughn, L.K.; Denning, G.; Stuhr, K.L.; de Wit, H.; Hill, M.N.; Hillard, C.J. Endocannabinoid signalling: Has it got rhythm? Br. J. Pharmacol. 2010, 160, 530–543. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, B.; Kent, L.; Suckling, J.; Bullmore, E.; Baron-Cohen, S. Variations in the human cannabinoid receptor (CNR1) gene modulate striatal responses to happy faces. Eur. J. Neurosci. 2006, 23, 1944–1948. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, B.; Baron-Cohen, S. Variation in the human cannabinoid receptor CNR1 gene modulates gaze duration for happy faces. Mol. Autism 2011, 2, 10. [Google Scholar] [CrossRef] [PubMed]
- Domschke, K.; Dannlowski, U.; Ohrmann, P.; Lawford, B.; Bauer, J.; Kugel, H.; Heindel, W.; Young, R.; Morris, P.; Arolt, V.; et al. Cannabinoid receptor 1 (CNR1) gene: Impact on antidepressant treatment response and emotion processing in major depression. Eur. Neuropsychopharmacol. 2008, 18, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Baron-Cohen, S. Autism: Research into causes and intervention. Pediatr. Rehabil. 2004, 7, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Purcell, A.E.; Jeon, O.H.; Zimmerman, A.W.; Blue, M.E.; Pevsner, J. Postmortem brain abnormalities of the glutamate neurotransmitter system in autism. Neurology 2001, 57, 1618–1628. [Google Scholar] [CrossRef] [PubMed]
- Lai, M.C.; Lombardo, M.V.; Baron-Cohen, S. Autism. Lancet 2014, 383, 896–910. [Google Scholar] [CrossRef]
- Pasciuto, E.; Borrie, S.C.; Kanellopoulos, A.K.; Santos, A.R.; Cappuyns, E.; D’Andrea, L.; Pacini, L.; Bagni, C. Autism Spectrum Disorders: Translating human deficits into mouse behavior. Neurobiol. Learn. Mem. 2015, 124, 71–87. [Google Scholar] [CrossRef] [PubMed]
- Crawley, J.N. Translational animal models of autism and neurodevelopmental disorders. Dialogues Clin. Neurosci. 2012, 14, 293–305. [Google Scholar] [PubMed]
- Ergaz, Z.; Weinstein-Fudim, L.; Ornoy, A. Genetic and non-genetic animal models for autism spectrum disorders (ASD). Reprod. Toxicol. 2016, 64, 116–140. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.C.; Gonzales, E.L.; Lazaro, M.T.; Choi, C.S.; Bahn, G.H.; Yoo, H.J.; Shin, C.Y. Clinical and neurobiological relevance of current animal models of autism spectrum disorders. Biomol. Ther. 2016, 24, 207–243. [Google Scholar] [CrossRef] [PubMed]
- Servadio, M.; Vanderschuren, L.J.; Trezza, V. Modeling autism-relevant behavioral phenotypes in rats and mice: Do ‘autistic’ rodents exist? Behav. Pharmacol. 2015, 26, 522–540. [Google Scholar] [CrossRef] [PubMed]
- Garber, K.B.; Visootsak, J.; Warren, S.T. Fragile X syndrome. Eur. J. Hum. Genet. 2008, 16, 666–672. [Google Scholar] [CrossRef] [PubMed]
- Hagerman, R.; Hoem, G.; Hagerman, P. Fragile X and autism: Intertwined at the molecular level leading to targeted treatments. Mol. Autism 2010, 1, 12. [Google Scholar] [CrossRef] [PubMed]
- Kazdoba, T.M.; Leach, P.T.; Silverman, J.L.; Crawley, J.N. Modeling fragile X syndrome in the Fmr1 knockout mouse. Intractable Rare Dis. Res. 2014, 3, 118–133. [Google Scholar] [CrossRef] [PubMed]
- Comery, T.A.; Harris, J.B.; Willems, P.J.; Oostra, B.A.; Irwin, S.A.; Weiler, I.J.; Greenough, W.T. Abnormal dendritic spines in fragile X knockout mice: Maturation and pruning deficits. Proc. Natl. Acad. Sci. USA 1997, 94, 5401–5404. [Google Scholar] [CrossRef] [PubMed]
- Irwin, S.A.; Galvez, R.; Greenough, W.T. Dendritic spine structural anomalies in fragile-X mental retardation syndrome. Cereb. Cortex 2000, 10, 1038–1044. [Google Scholar] [CrossRef] [PubMed]
- Huber, K.M.; Gallagher, S.M.; Warren, S.T.; Bear, M.F. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc. Natl. Acad. Sci. USA 2002, 99, 7746–7750. [Google Scholar] [CrossRef] [PubMed]
- Nakamoto, M.; Nalavadi, V.; Epstein, M.P.; Narayanan, U.; Bassell, G.J.; Warren, S.T. Fragile X mental retardation protein deficiency leads to excessive mGluR5-dependent internalization of AMPA receptors. Proc. Natl. Acad. Sci. USA 2007, 104, 15537–15542. [Google Scholar] [CrossRef] [PubMed]
- Nosyreva, E.D.; Huber, K.M. Metabotropic receptor-dependent long-term depression persists in the absence of protein synthesis in the mouse model of fragile X syndrome. J. Neurophysiol. 2006, 95, 3291–3295. [Google Scholar] [CrossRef] [PubMed]
- Centonze, D.; Rossi, S.; Mercaldo, V.; Napoli, I.; Ciotti, M.T.; de Chiara, V.; Musella, A.; Prosperetti, C.; Calabresi, P.; Bernardi, G.; et al. Abnormal striatal GABA transmission in the mouse model for the fragile X syndrome. Biol. Psychiatry 2008, 63, 963–973. [Google Scholar] [CrossRef] [PubMed]
- Curia, G.; Papouin, T.; Seguela, P.; Avoli, M. Down regulation of tonic GABAergic inhibition in a mouse model of fragile X syndrome. Cereb. Cortex 2009, 19, 1515–1520. [Google Scholar] [CrossRef] [PubMed]
- Olmos-Serrano, J.L.; Paluszkiewicz, S.M.; Martin, B.S.; Kaufmann, W.E.; Corbin, J.G.; Huntsman, M.M. Defective GABAergic neurotransmission and pharmacological rescue of neuronal hyperexcitability in the amygdala in a mouse model of fragile X syndrome. J. Neurosci. 2010, 30, 9929–9938. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Alger, B.E. Enhanced endocannabinoid signaling elevates neuronal excitability in fragile X syndrome. J. Neurosci. 2010, 30, 5724–5729. [Google Scholar] [CrossRef] [PubMed]
- Maccarrone, M.; Rossi, S.; Bari, M.; de Chiara, V.; Rapino, C.; Musella, A.; Bernardi, G.; Bagni, C.; Centonze, D. Abnormal mGlu 5 receptor/endocannabinoid coupling in mice lacking FMRP and BC1 RNA. Neuropsychopharmacology 2010, 35, 1500–1509. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.M.; Sepers, M.; Henstridge, C.M.; Lassalle, O.; Neuhofer, D.; Martin, H.; Ginger, M.; Frick, A.; DiPatrizio, N.V.; Mackie, K.; et al. Uncoupling of the endocannabinoid signalling complex in a mouse model of fragile X syndrome. Nat. Commun. 2012, 3, 1080. [Google Scholar] [CrossRef] [PubMed]
- Busquets-Garcia, A.; Gomis-Gonzalez, M.; Guegan, T.; Agustin-Pavon, C.; Pastor, A.; Mato, S.; Perez-Samartin, A.; Matute, C.; de la Torre, R.; Dierssen, M.; et al. Targeting the endocannabinoid system in the treatment of fragile X syndrome. Nat. Med. 2013, 19, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Gomis-Gonzalez, M.; Busquets-Garcia, A.; Matute, C.; Maldonado, R.; Mato, S.; Ozaita, A. Possible therapeutic doses of cannabinoid type 1 receptor antagonist reverses key alterations in fragile X syndrome mouse model. Genes 2016, 7, 56. [Google Scholar] [CrossRef] [PubMed]
- Qin, M.; Zeidler, Z.; Moulton, K.; Krych, L.; Xia, Z.; Smith, C.B. Endocannabinoid-mediated improvement on a test of aversive memory in a mouse model of fragile X syndrome. Behav. Brain Res. 2015, 291, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Wei, D.; Dinh, D.; Lee, D.; Li, D.; Anguren, A.; MorenoSanz, G.; Gall, C.M.; Piomelli, D. Enhancement of anandamide-mediated endocannabinoid signaling corrects autism-related social impairment. Cannabis Cannabinoid Res. 2016, 1, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Jamain, S.; Radyushkin, K.; Hammerschmidt, K.; Granon, S.; Boretius, S.; Varoqueaux, F.; Ramanantsoa, N.; Gallego, J.; Ronnenberg, A.; Winter, D.; et al. Reduced social interaction and ultrasonic communication in a mouse model of monogenic heritable autism. Proc. Natl. Acad. Sci. USA 2008, 105, 1710–1715. [Google Scholar] [CrossRef] [PubMed]
- Sudhof, T.C. Neuroligins and neurexins link synaptic function to cognitive disease. Nature 2008, 455, 903–911. [Google Scholar] [CrossRef] [PubMed]
- Jamain, S.; Quach, H.; Betancur, C.; Rastam, M.; Colineaux, C.; Gillberg, I.C.; Soderstrom, H.; Giros, B.; Leboyer, M.; Gillberg, C.; et al. Paris autism research international sibpair, S., mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat. Genet. 2003, 34, 27–29. [Google Scholar] [CrossRef] [PubMed]
- Comoletti, D.; de Jaco, A.; Jennings, L.L.; Flynn, R.E.; Gaietta, G.; Tsigelny, I.; Ellisman, M.H.; Taylor, P. The Arg451Cys-neuroligin-3 mutation associated with autism reveals a defect in protein processing. J. Neurosci. 2004, 24, 4889–4893. [Google Scholar] [CrossRef] [PubMed]
- Etherton, M.; Foldy, C.; Sharma, M.; Tabuchi, K.; Liu, X.; Shamloo, M.; Malenka, R.C.; Sudhof, T.C. Autism-linked neuroligin-3 R451C mutation differentially alters hippocampal and cortical synaptic function. Proc. Natl. Acad. Sci. USA 2011, 108, 13764–13769. [Google Scholar] [CrossRef] [PubMed]
- Radyushkin, K.; Hammerschmidt, K.; Boretius, S.; Varoqueaux, F.; El-Kordi, A.; Ronnenberg, A.; Winter, D.; Frahm, J.; Fischer, J.; Brose, N.; et al. Neuroligin-3-deficient mice: Model of a monogenic heritable form of autism with an olfactory deficit. Genes Brain Behav. 2009, 8, 416–425. [Google Scholar] [CrossRef] [PubMed]
- Foldy, C.; Malenka, R.C.; Sudhof, T.C. Autism-associated neuroligin-3 mutations commonly disrupt tonic endocannabinoid signaling. Neuron 2013, 78, 498–509. [Google Scholar] [CrossRef] [PubMed]
- Speed, H.E.; Masiulis, I.; Gibson, J.R.; Powell, C.M. Increased cortical inhibition in autism-linked neuroligin-3R451C mice is due in part to loss of endocannabinoid signaling. PLoS ONE 2015, 10, e0140638. [Google Scholar] [CrossRef] [PubMed]
- McFarlane, H.G.; Kusek, G.K.; Yang, M.; Phoenix, J.L.; Bolivar, V.J.; Crawley, J.N. Autism-like behavioral phenotypes in BTBR T + tf/J mice. Genes Brain Behav. 2008, 7, 152–163. [Google Scholar] [CrossRef] [PubMed]
- Silverman, J.L.; Tolu, S.S.; Barkan, C.L.; Crawley, J.N. Repetitive self-grooming behavior in the BTBR mouse model of autism is blocked by the mGluR5 antagonist MPEP. Neuropsychopharmacology 2010, 35, 976–989. [Google Scholar] [CrossRef] [PubMed]
- Onaivi, E.S.; Benno, R.; Halpern, T.; Mehanovic, M.; Schanz, N.; Sanders, C.; Yan, X.; Ishiguro, H.; Liu, Q.R.; Berzal, A.L.; et al. Consequences of cannabinoid and monoaminergic system disruption in a mouse model of autism spectrum disorders. Curr. Neuropharmacol. 2011, 9, 209–214. [Google Scholar] [CrossRef] [PubMed]
- McTighe, S.M.; Neal, S.J.; Lin, Q.; Hughes, Z.A.; Smith, D.G. The BTBR mouse model of autism spectrum disorders has learning and attentional impairments and alterations in acetylcholine and kynurenic acid in prefrontal cortex. PLoS ONE 2013, 8, e62189. [Google Scholar] [CrossRef] [PubMed]
- Benno, R.; Smirnova, Y.; Vera, S.; Liggett, A.; Schanz, N. Exaggerated responses to stress in the BTBR T + tf/J mouse: An unusual behavioral phenotype. Behav. Brain Res. 2009, 197, 462–465. [Google Scholar] [CrossRef] [PubMed]
- Amaral, D.G.; Schumann, C.M.; Nordahl, C.W. Neuroanatomy of autism. Trends Neurosci. 2008, 31, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Hallmayer, J.; Cleveland, S.; Torres, A.; Phillips, J.; Cohen, B.; Torigoe, T.; Miller, J.; Fedele, A.; Collins, J.; Smith, K.; et al. Genetic heritability and shared environmental factors among twin pairs with autism. Arch. Gen. Psychiatry 2011, 68, 1095–1102. [Google Scholar] [CrossRef] [PubMed]
- Sandin, S.; Lichtenstein, P.; Kuja-Halkola, R.; Larsson, H.; Hultman, C.M.; Reichenberg, A. The familial risk of autism. JAMA 2014, 311, 1770–1777. [Google Scholar] [CrossRef] [PubMed]
- Mabunga, D.F.; Gonzales, E.L.; Kim, J.W.; Kim, K.C.; Shin, C.Y. Exploring the validity of valproic acid animal model of autism. Exp. Neurobiol. 2015, 24, 285–300. [Google Scholar] [CrossRef] [PubMed]
- Roullet, F.I.; Lai, J.K.; Foster, J.A. In utero exposure to valproic acid and autism—a current review of clinical and animal studies. Neurotoxicol. Teratol. 2013, 36, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.; King, J.; Cunningham, M.; Stephan, M.; Kerr, B.; Hersh, J.H. Fetal valproate syndrome and autism: Additional evidence of an association. Dev. Med. Child. Neurol. 2001, 43, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Ornoy, A. Valproic acid in pregnancy: How much are we endangering the embryo and fetus? Reprod. Toxicol. 2009, 28, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Moore, S.J.; Turnpenny, P.; Quinn, A.; Glover, S.; Lloyd, D.J.; Montgomery, T.; Dean, J.C. A clinical study of 57 children with fetal anticonvulsant syndromes. J. Med. Genet. 2000, 37, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Schneider, T.; Przewlocki, R. Behavioral alterations in rats prenatally exposed to valproic acid: Animal model of autism. Neuropsychopharmacology 2005, 30, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Schneider, T.; Roman, A.; Basta-Kaim, A.; Kubera, M.; Budziszewska, B.; Schneider, K.; Przewlocki, R. Gender-specific behavioral and immunological alterations in an animal model of autism induced by prenatal exposure to valproic acid. Psychoneuroendocrinology 2008, 33, 728–740. [Google Scholar] [CrossRef] [PubMed]
- Halladay, A.K.; Bishop, S.; Constantino, J.N.; Daniels, A.M.; Koenig, K.; Palmer, K.; Messinger, D.; Pelphrey, K.; Sanders, S.J.; Singer, A.T.; et al. Sex and gender differences in autism spectrum disorder: Summarizing evidence gaps and identifying emerging areas of priority. Mol. Autism 2015, 6, 36. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, T.; Kulangara, K.; Antoniello, K.; Markram, H. Elevated NMDA receptor levels and enhanced postsynaptic long-term potentiation induced by prenatal exposure to valproic acid. Proc. Natl. Acad. Sci. USA 2007, 104, 13501–13506. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, T.; Silberberg, G.; Markram, H. Hyperconnectivity of local neocortical microcircuitry induced by prenatal exposure to valproic acid. Cereb. Cortex 2008, 18, 763–770. [Google Scholar] [CrossRef] [PubMed]
- Martin, H.G.; Manzoni, O.J. Late onset deficits in synaptic plasticity in the valproic acid rat model of autism. Front. Cell Neurosci. 2014, 8, 23. [Google Scholar] [CrossRef] [PubMed]
- Kerr, D.M.; Downey, L.; Conboy, M.; Finn, D.P.; Roche, M. Alterations in the endocannabinoid system in the rat valproic acid model of autism. Behav. Brain Res. 2013, 249, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Servadio, M.; Melancia, F.; Manduca, A.; di Masi, A.; Schiavi, S.; Cartocci, V.; Pallottini, V.; Campolongo, P.; Ascenzi, P.; Trezza, V. Targeting anandamide metabolism rescues core and associated autistic-like symptoms in rats prenatally exposed to valproic acid. Transl. Psychiatry 2016, 6, e902. [Google Scholar] [CrossRef] [PubMed]
- Kerr, D.M.; Gilmartin, A.; Roche, M. Pharmacological inhibition of fatty acid amide hydrolase attenuates social behavioural deficits in male rats prenatally exposed to valproic acid. Pharmacol. Res. 2016, 113, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Ornoy, A.; Weinstein-Fudim, L.; Ergaz, Z. Prenatal factors associated with autism spectrum disorder (ASD). Reprod. Toxicol. 2015, 56, 155–169. [Google Scholar] [CrossRef] [PubMed]
- Patterson, P.H. Modeling autistic features in animals. Pediatr. Res. 2011, 69, 34R–40R. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.E.; Li, J.; Garbett, K.; Mirnics, K.; Patterson, P.H. Maternal immune activation alters fetal brain development through interleukin-6. J. Neurosci. 2007, 27, 10695–10702. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, E.Y.; Patterson, P.H. Activation of the maternal immune system induces endocrine changes in the placenta via IL-6. Brain Behav. Immun. 2011, 25, 604–615. [Google Scholar] [CrossRef] [PubMed]
- Malkova, N.V.; Yu, C.Z.; Hsiao, E.Y.; Moore, M.J.; Patterson, P.H. Maternal immune activation yields offspring displaying mouse versions of the three core symptoms of autism. Brain Behav. Immun. 2012, 26, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Hava, G.; Vered, L.; Yael, M.; Mordechai, H.; Mahoud, H. Alterations in behavior in adult offspring mice following maternal inflammation during pregnancy. Dev. Psychobiol. 2006, 48, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Golan, H.M.; Lev, V.; Hallak, M.; Sorokin, Y.; Huleihel, M. Specific neurodevelopmental damage in mice offspring following maternal inflammation during pregnancy. Neuropharmacology 2005, 48, 903–917. [Google Scholar] [CrossRef] [PubMed]
- Romero, E.; Ali, C.; Molina-Holgado, E.; Castellano, B.; Guaza, C.; Borrell, J. Neurobehavioral and immunological consequences of prenatal immune activation in rats: Influence of antipsychotics. Neuropsychopharmacology 2007, 32, 1791–1804. [Google Scholar] [CrossRef] [PubMed]
- Patterson, P.H. Immune involvement in schizophrenia and autism: Etiology, pathology and animal models. Behav. Brain Res. 2009, 204, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Doenni, V.M.; Gray, J.M.; Song, C.M.; Patel, S.; Hill, M.N.; Pittman, Q.J. Deficient adolescent social behavior following early-life inflammation is ameliorated by augmentation of anandamide signaling. Brain Behav. Immun. 2016, 58, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Castillo, P.E.; Younts, T.J.; Chavez, A.E.; Hashimotodani, Y. Endocannabinoid signaling and synaptic function. Neuron 2012, 76, 70–81. [Google Scholar] [CrossRef] [PubMed]
- Keimpema, E.; Hokfelt, T.; Harkany, T.; Doherty, P. The molecular interplay between endocannabinoid and neurotrophin signals in the nervous system and beyond. Eur. J. Neurosci. 2014, 39, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Lipina, C.; Irving, A.J.; Hundal, H.S. Mitochondria: A possible nexus for the regulation of energy homeostasis by the endocannabinoid system? Am. J. Physiol. Endocrinol. Metab. 2014, 307, E1–E13. [Google Scholar] [CrossRef] [PubMed]
- Turcotte, C.; Blanchet, M.R.; Laviolette, M.; Flamand, N. The CB2 receptor and its role as a regulator of inflammation. Cell Mol. Life Sci. 2016, 73, 4449–4470. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.Y.; Chen, C. Endocannabinoids in synaptic plasticity and neuroprotection. Neuroscientist 2015, 21, 152–168. [Google Scholar] [CrossRef] [PubMed]
- Wei, D.; Lee, D.; Cox, C.D.; Karsten, C.A.; Penagarikano, O.; Geschwind, D.H.; Gall, C.M.; Piomelli, D. Endocannabinoid signaling mediates oxytocin-driven social reward. Proc. Natl. Acad. Sci. USA 2015, 112, 14084–14089. [Google Scholar] [CrossRef] [PubMed]
- Richter, J.D.; Klann, E. Making synaptic plasticity and memory last: Mechanisms of translational regulation. Genes Dev. 2009, 23, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, A.; Kaphzan, H.; Alvarez-Dieppa, A.C.; Murphy, J.P.; Pierre, P.; Klann, E. Genetic removal of p70 S6 kinase 1 corrects molecular, synaptic, and behavioral phenotypes in fragile X syndrome mice. Neuron 2012, 76, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Huber, K.M.; Klann, E.; Costa-Mattioli, M.; Zukin, R.S. Dysregulation of mammalian target of rapamycin signaling in mouse models of autism. J. Neurosci. 2015, 35, 13836–13842. [Google Scholar] [CrossRef] [PubMed]
- Siniscalco, D.; Sapone, A.; Giordano, C.; Cirillo, A.; de Magistris, L.; Rossi, F.; Fasano, A.; Bradstreet, J.J.; Maione, S.; Antonucci, N. Cannabinoid receptor type 2, but not type 1, is up-regulated in peripheral blood mononuclear cells of children affected by autistic disorders. J. Autism Dev. Disord. 2013, 43, 2686–2695. [Google Scholar] [CrossRef] [PubMed]
- Siniscalco, D.; Bradstreet, J.J.; Cirillo, A.; Antonucci, N. The in vitro GcMAF effects on endocannabinoid system transcriptionomics, receptor formation, and cell activity of autism-derived macrophages. J. Neuroinflam. 2014, 11, 78. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Lee, J.; Kim, E. Excitation/inhibition imbalance in animal models of autism spectrum disorders. Biol. Psychiatry 2017, 81, 838–847. [Google Scholar] [CrossRef] [PubMed]
- Cellot, G.; Cherubini, E. GABAergic signaling as therapeutic target for autism spectrum disorders. Front. Pediatr. 2014, 2, 70. [Google Scholar] [CrossRef] [PubMed]
- Hashimotodani, Y.; Ohno-Shosaku, T.; Kano, M. Endocannabinoids and synaptic function in the CNS. Neuroscientist 2007, 13, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Galve-Roperh, I.; Palazuelos, J.; Aguado, T.; Guzman, M. The endocannabinoid system and the regulation of neural development: Potential implications in psychiatric disorders. Eur. Arch. Psychiatry Clin. Neurosci. 2009, 259, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Ruiz, J.J.; Berrendero, F.; Hernandez, M.L.; Romero, J.; Ramos, J.A. Role of endocannabinoids in brain development. Life Sci. 1999, 65, 725–736. [Google Scholar] [CrossRef]

{kind=link}
| Drug | Mechanism of Action |
|---|---|
| URB597 PF-3845 PF-04457845 | fatty acid amide hydrolase (FAAH) inhibitors |
| JZL184 | monoacylglycerol lipase (MAGL) inhibitor |
| Rimonabant | cannabinoid type 1 (CB1) receptor antagonist/inverse agonist |
| NESS0327 | neutral CB1 receptor antagonist |
| AM630 | cannabinoid type 2 (CB2) receptor antagonist/inverse agonist |
| Drug | Dose | Model | Outcome | Reference | |
|---|---|---|---|---|---|
| Anandamide (AEA) | URB597 | Acute 0.3 mg/kg | Fragile X Mental Retardation (Fmr1) knockout mice C57BL/6J background | improvement of aversive memory and anxiety-like behaviors | [44] |
| URB597 | Acute 0.3 mg/kg | Fmr1 knockout mice FVB background | amelioration of social impairments | [45] | |
| URB597 | Acute 0.3 and 1 mg/kg | BTBR mice | amelioration of social impairments | [45] | |
| URB597 | Acute | valproic acid (VPA) exposure in Wistar rats | normalization of communication abnormalities and reversal of social deficits | [74] | |
| PF-3845 | Acute 10 mg/kg | VPA exposure in Sprague-Dawley rats | reversal of social deficits | [75] | |
| PF-04457845 | Acute 1 mg/kg | lipopolysaccharide (LPS) administration in Sprague-Dawley rats | reversal of social deficits | [85] | |
| 2-arachidonoyl glycerol (2-AG) | JZL184 | Acute 16 mg/kg | Fmr1 knockout mice C57BL/6J background | normalization of locomotion and anxiety-like responses | [41] |
| Cannabinoid type 1 (CB1) receptor | Rimonabant | Acute 1 mg/kg | Fmr1 knockout mice FVB background | amelioration of cognitive deficits, seizure susceptibility and nociceptive desensitization | [42] |
| Rimonabant | Chronic 1 mg/kg | Fmr1 knockout mice FVB background | amelioration of cognitive deficits | [42] | |
| Rimonabant | Acute 0.3–1 mg/kg | Fmr1 knockout mice FVB background | amelioration of cognitive deficits | [43] | |
| Rimonabant | Chronic 0.03–1 mg/kg | Fmr1 knockout mice FVB background | amelioration of cognitive deficits | [43] | |
| NESS0327 | Chronic 0.1 mg/kg | Fmr1 knockout mice FVB background | amelioration of cognitive deficits | [43] | |
| Cannabinoid type 2 (CB2) receptor | AM630 | Acute 1 mg/kg | Fmr1 knockout mice FVB background | normalization of anxiety-like behavior and audiogenic seizure susceptibility | [42] |
| Model | Alteration | Brain Area | Reference |
|---|---|---|---|
| Fragile X Mental Retardation (Fmr1) knockout mice | ↓ EC-mediated long-term depression (LTD) at excitatory synapses | forebrain | [41] |
| ↓ EC-mediated LTD at inhibitory synapses | dorsal striatum, hippocampus | [39,40] | |
| Neuroligin (NLGN)3R451C knockin and NLGN3 knockout mice | ↓ tonic EC signaling at cholecystokinin-expressing (CCK) basket cell synapses | hippocampus | [52] |
| Valproic acid (VPA) exposure in rats | ↓ tonic EC signaling at interneurons other than parvalbumin (PV)- or somatostatin (SOM)-positive | somatosensory cortex | [53] |
| ↓ diacylglycerol lipase (DAGL)-α mRNA | cerebellum | [73] | |
| ↑ monoacylglycerol lipase (MAGL) activity and ↓ peroxisome proliferator-activated receptor (PPAR)-γ and G-protein coupled receptor (GPR)55 | hippocampus | [73] | |
| ↓ PPARα and GPR55 | frontal cortex | [73] | |
| altered phosphorylation of cannabinoid type 1 (CB1) receptor | amygdala, hippocampus, dorsal striatum | [74] | |
| ↓ N-acyl-phosphatidylethanolamine phospholipase D (NAPE PLD) and ↑ fatty acid amide hydrolase (FAAH) | whole brain | [74] | |
| lipopolysaccharide (LPS) administration in rats | ↓ CB1 receptor, ↑ anandamide (AEA) levels and ↑ FAAH activity | amygdala | [85] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zamberletti, E.; Gabaglio, M.; Parolaro, D. The Endocannabinoid System and Autism Spectrum Disorders: Insights from Animal Models. Int. J. Mol. Sci. 2017, 18, 1916. https://doi.org/10.3390/ijms18091916
Zamberletti E, Gabaglio M, Parolaro D. The Endocannabinoid System and Autism Spectrum Disorders: Insights from Animal Models. International Journal of Molecular Sciences. 2017; 18(9):1916. https://doi.org/10.3390/ijms18091916
Chicago/Turabian StyleZamberletti, Erica, Marina Gabaglio, and Daniela Parolaro. 2017. "The Endocannabinoid System and Autism Spectrum Disorders: Insights from Animal Models" International Journal of Molecular Sciences 18, no. 9: 1916. https://doi.org/10.3390/ijms18091916
APA StyleZamberletti, E., Gabaglio, M., & Parolaro, D. (2017). The Endocannabinoid System and Autism Spectrum Disorders: Insights from Animal Models. International Journal of Molecular Sciences, 18(9), 1916. https://doi.org/10.3390/ijms18091916