Mitochondrial Liver Toxicity of Valproic Acid and Its Acid Derivatives Is Related to Inhibition of α-Lipoamide Dehydrogenase



Abstract
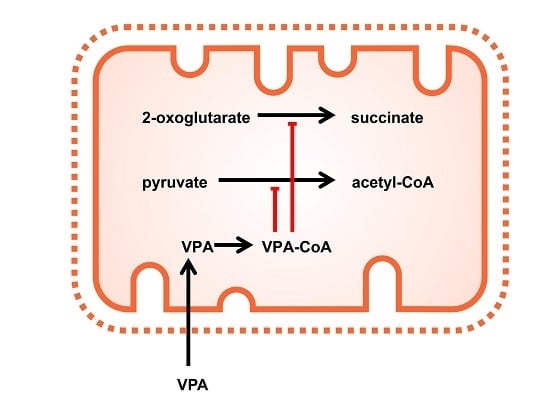
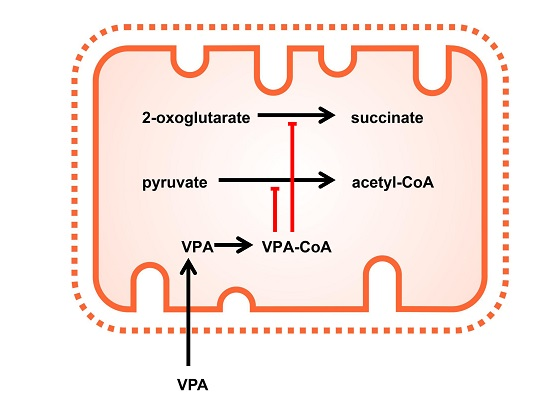
1. Introduction
2. Results
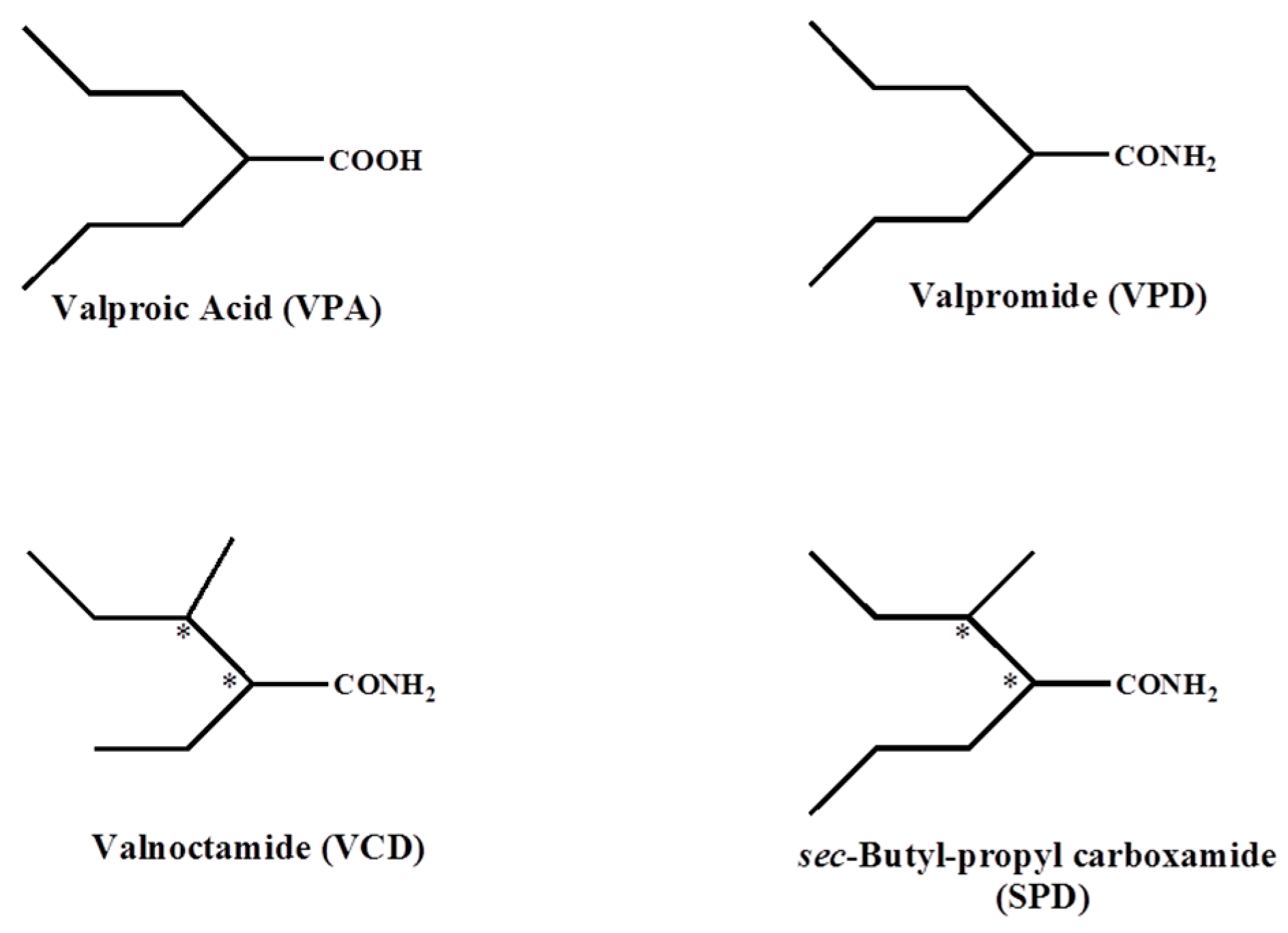
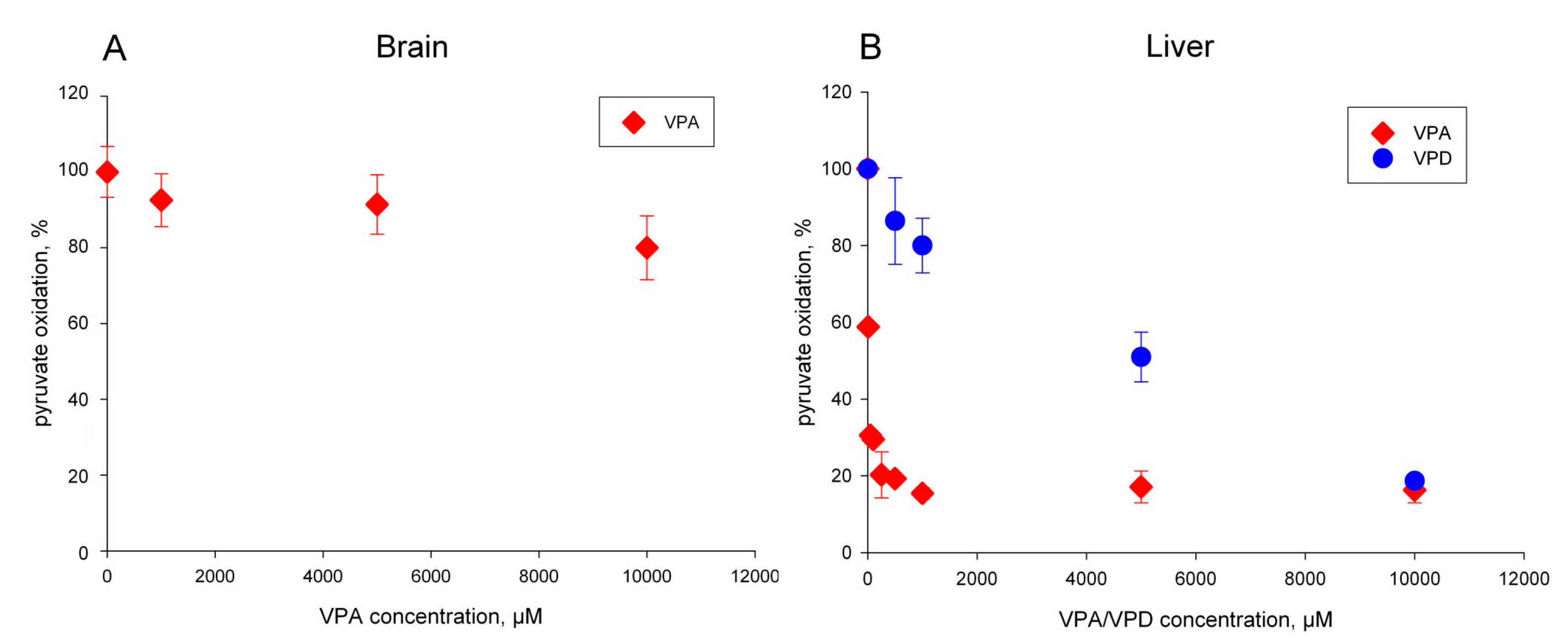
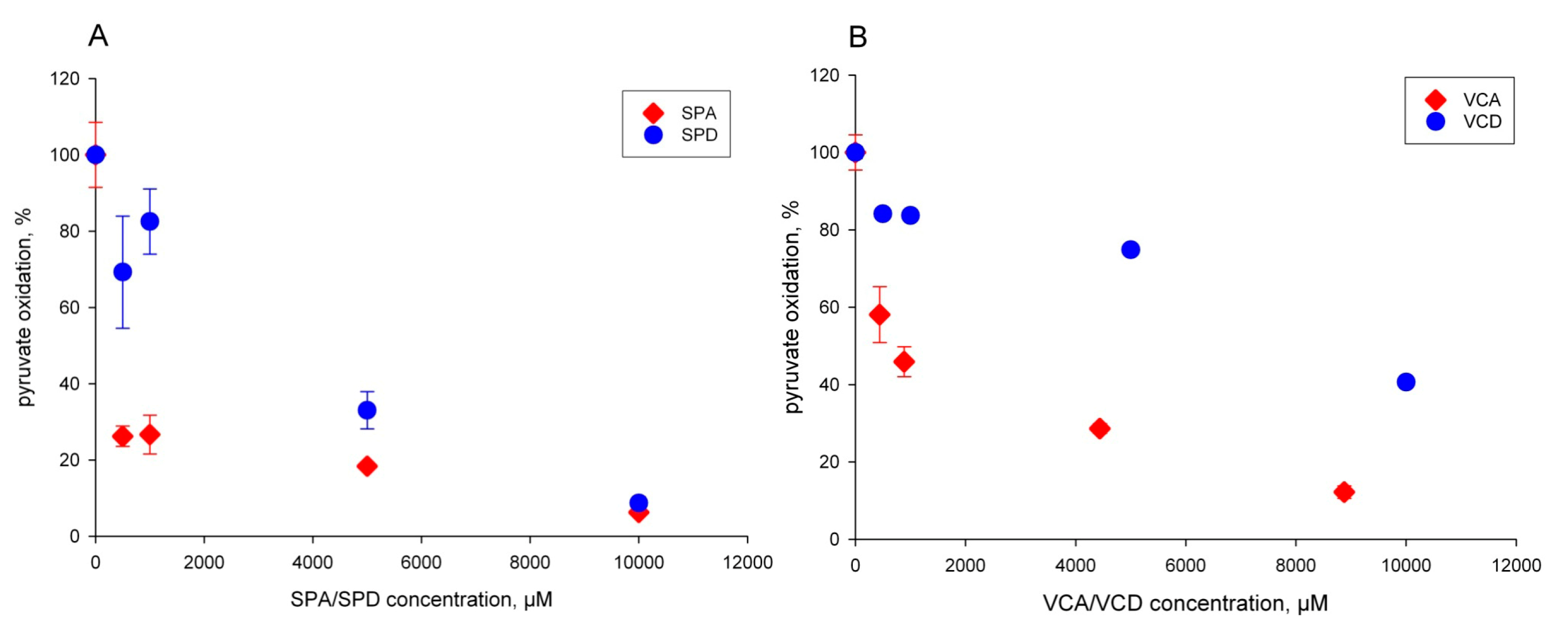
2.1. The Pyruvate Oxidation of Rat Liver Mitochondria Is Selectively Inhibited by Valproic Acid (VPA) and Its Acid Derivatives
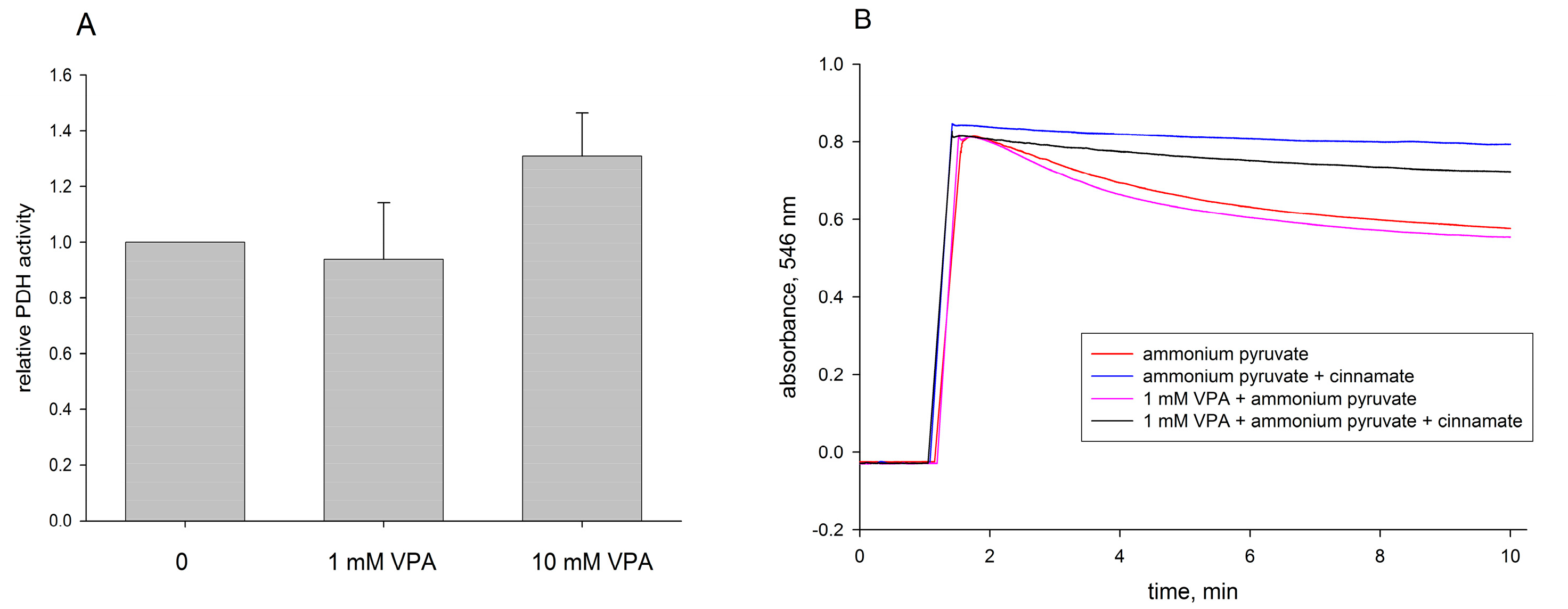
2.2. Pyruvate Dehydrogenase and Pyruvate Transport of Rat Liver Mitochondria Are Not Affected by VPA
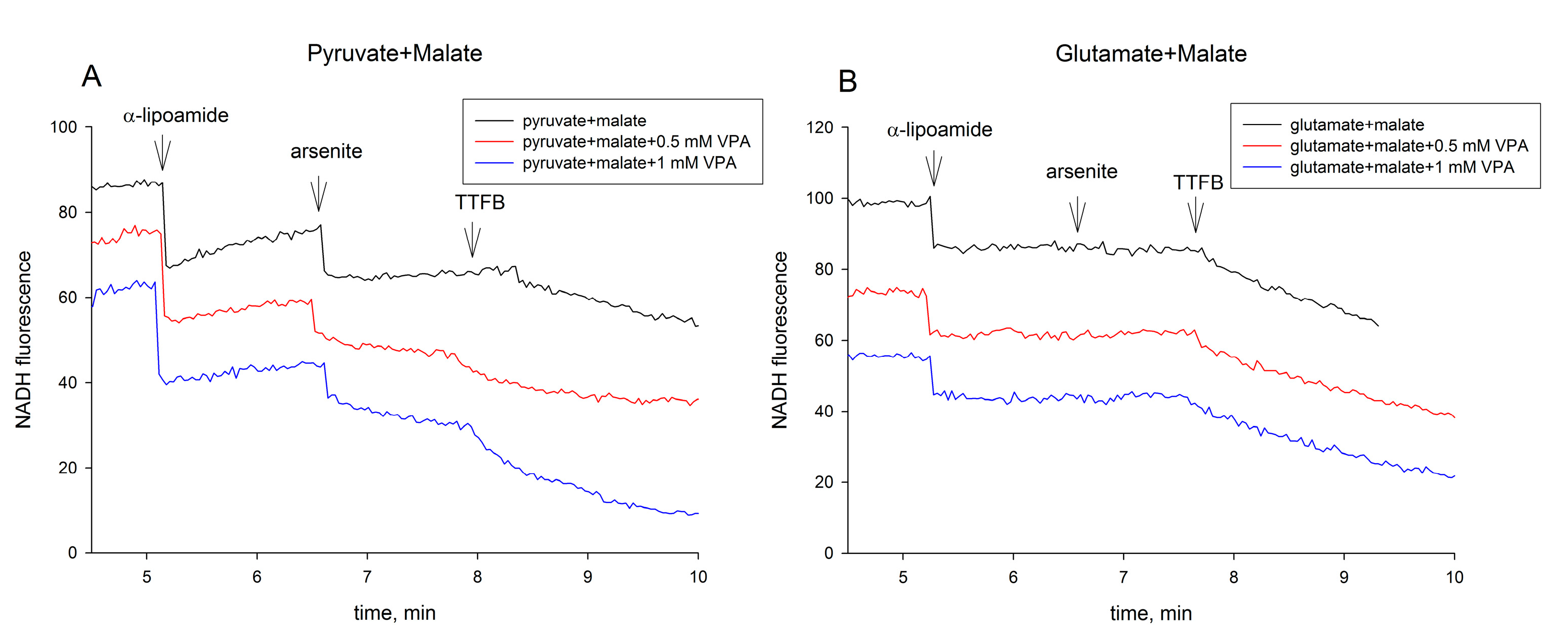
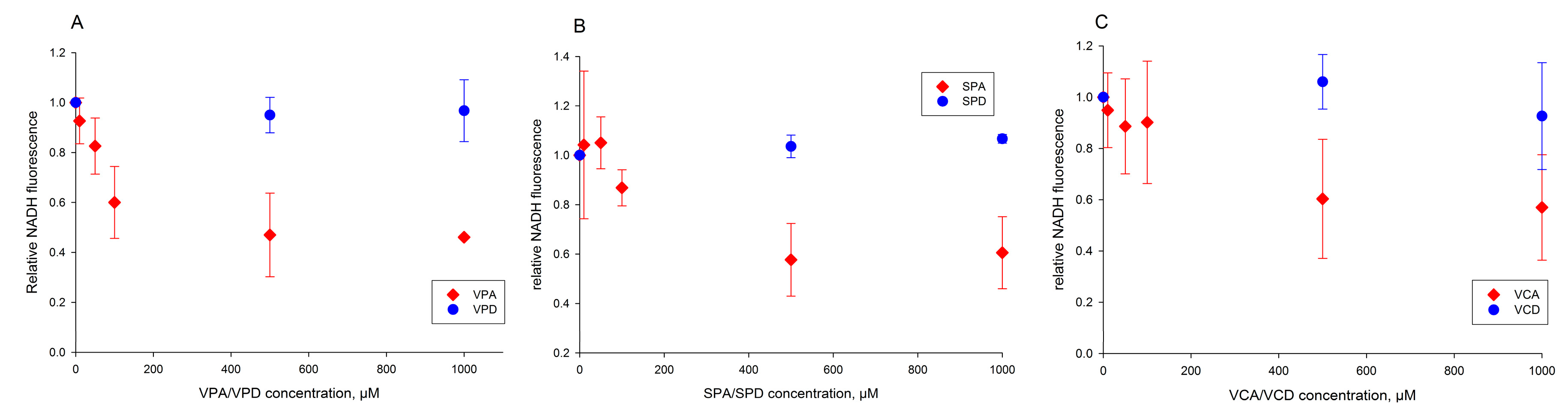
2.3. Valproic Acid and Their Acid Derivatives Inhibit α-Lipoamide Dehydrogenase
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Isolation of Rat Brain and Rat Liver Mitochondria
4.3. Determination of Substrate Oxidation Rates
4.4. Determination of Pyruvate Dehydrogenase (PDH) Activity and Measurement of Mitochondrial Pyruvate Uptake
4.5. Determination of α-Lipoamide Dehydrogenase Activity in Intact Mitochondria
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| VPA | Valproic acid |
| VPD | Valpromide |
| VCA | Valnoctic acid |
| VCD | Valnoctamide |
| SPA | sec-Butylpropylacetic acid |
| SPD | sec-Butylpropylacetamide |
| CNS | Central nervous system |
| PDH | Pyruvate dehydrogenase |
| SE | Status epilepticus |
References
- Stewart, J.D.; Horvath, R.; Baruffini, E.; Ferrero, I.; Bulst, S.; Watkins, P.B.; Fontana, R.J.; Day, C.P.; Chinnery, P.F. Polymerase γ gene POLG determines the risk of sodium valproate-induced liver toxicity. Hepatology 2010, 52, 1791–1796. [Google Scholar] [CrossRef] [PubMed]
- Saneto, R.P.; Lee, I.C.; Koenig, M.K.; Bao, X.; Weng, S.W.; Naviaux, R.K.; Wong, L.J. POLG DNA testing as an emerging standard of care before instituting valproic acid therapy for pediatric seizure disorders. Seizure 2010, 19, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.F.; Ruiter, J.P.; Illst, L.; Jakobs, C.; Duran, M.; de Almeida, I.T.; Wanders, R.J. Valproate inhibits the mitochondrial pyruvate-driven oxidative phosphorylation in vitro. J. Inherit. Metab. Dis. 1997, 20, 397–400. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.F.; Ruiter, J.P.; IJlst, L.; Allers, P.; ten Brink, H.J.; Jakobs, C.; Duran, M.; de Almeida, I.T.; Wanders, R.J. Synthesis and intramitochondrial levels of valproyl-coenzyme A metabolites. Anal. Biochem. 2001, 290, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Becker, C.M.; Harris, R.A. Influence of valproic acid on hepatic carbohydrate and lipid metabolism. Arch. Biochem. Biophys. 1983, 223, 381–392. [Google Scholar] [CrossRef]
- Swartzentruber, M.S.; Harris, R.A. Inhibition of metabolic processes by coenzyme-A-sequestering aromatic acids. Prevention by para-chloro- and para-nitrobenzoic acids. Biochem. Pharmacol. 1987, 36, 3147–3153. [Google Scholar] [CrossRef]
- Ponchaut, S.; van Hoof, F.; Veitch, K. In vitro effects of valproate and valproate metabolites on mitochondrial oxidations: Relevance of CoA sequestration to the observed inhibitions. Biochem. Pharmacol. 1992, 43, 2435–2442. [Google Scholar] [CrossRef]
- Aires, C.C.; Soveral, G.; Luís, P.B.; ten Brink, H.J.; de Almeida, I.T.; Duran, M.; Wanders, R.J.; Silva, M.F. Pyruvate uptake is inhibited by valproic acid and metabolites in mitochondrial membranes. FEBS Lett. 2008, 582, 3359–3366. [Google Scholar] [CrossRef] [PubMed]
- Luís, P.B.; Ruiter, J.; IJlst, L.; de Almeida, I.T.; Duran, M.; Wanders, R.J.; Silva, M.F. Valproyl-CoA inhibits the activity of ATP- and GTP-dependent succinate:CoA ligases. J. Inherit. Metab. Dis. 2014, 37, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Aires, C.C.; van Cruchten, A.; Ijlst, L.; de Almeida, I.T.; Duran, M.; Wanders, R.J.; Silva, M.F. New insights on the mechanisms of valproate-induced hyperammonemianhibition of hepatic N-acetylglutamate synthase activity by valproyl-CoA. J. Hepatol. 2011, 55, 426–434. [Google Scholar] [CrossRef] [PubMed]
- Luís, P.B.; Ruiter, J.P.; Aires, C.C.; Soveral, G.; de Almeida, I.T.; Duran, M.; Wanders, R.J.; Silva, M.F. Valproic acid metabolites inhibit dihydrolipoyl dehydrogenase activity leading to impaired 2-oxoglutarate-driven oxidative phosphorylation. Biochim. Biophys. Acta 2007, 1767, 1126–1133. [Google Scholar] [CrossRef] [PubMed]
- Barel, S.; Yagen, B.; Schurig, V.; Soback, S.; Pisani, F.; Perucca, E.; Bialer, M. Stereoselective pharmacokinetic analysis of valnoctamide in healthy subjects and in patients with epilepsy. Clin. Pharmacol. Ther. 1997, 61, 442–449. [Google Scholar] [CrossRef]
- Bialer, M.; Yagen, B. Valproic acid—Second generation. Neurotherapeutics 2007, 4, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Bialer, M. Chemical properties of antiepileptic drugs (AEDs). Adv. Drug Deliv. Rev. 2007, 64, 887–895. [Google Scholar] [CrossRef] [PubMed]
- Bialer, M.; Haj-Yehia, A.; Barzaghi, N.; Pisani, F.; Perucca, E. Pharmacokinetics of a valpromide isomer, valnoctamide, in healthy subjects. Eur. J. Clin. Pharmacol. 1990, 38, 289–291. [Google Scholar] [CrossRef] [PubMed]
- Bialer, M. Clinical pharmacology of valpromide. Clin. Pharmacokinet. 1991, 20, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Bialer, M.; White, H.S. Key factors in the discovery and development of new antiepileptic drugs (AEDs). Nat. Rev. Drug Discov. 2010, 9, 68–83. [Google Scholar] [CrossRef] [PubMed]
- Curia, G.; Longo, D.; Biagini, G.; Jones, R.S.; Avoli, M. The pilocarpine model of temporal lobe epilepsy. J. Neurosci. Methods 2008, 172, 143–157. [Google Scholar] [CrossRef] [PubMed]
- White, H.S.; Alex, A.B.; Pollock, A.; Hen, N.; Shekh-Ahmad, T.; Wilcox, K.S.; McDonough, J.H.; Stables, J.P.; Kaufmann, D.; Yagen, B.; et al. A new derivative of valproic acid amide possesses a broad-spectrum antiseizure profile and unique activity against status epilepticus and organophosphate neuronal damage. Epilepsia 2012, 53, 134–146. [Google Scholar] [CrossRef] [PubMed]
- Pouliot, W.; Bialer, M.; Hen, N.; Shekh-Ahmad, T.; Kaufmann, D.; Yagen, B.; Ricks, K.; Roach, B.; Nelson, C.; Dudek, F.E. Electrographic analysis of the effect of sec-butyl-propylacetamide on pharmacoresistant status epilepticus. Neuroscience 2013, 231, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, D.; Yagen, B.; Minert, A.; Wlodarczyk, B.; Finnell, R.H.; Schurig, V.; Devor, M.; Bialer, M. Evaluation of the antiallodynic, teratogenic and pharmacokinetic profile of stereoisomers of valnoctamide, an amide derivative of a chiral isomer of valproic acid. Neuropharmacology 2010, 52, 1228–1236. [Google Scholar] [CrossRef] [PubMed]
- Shekh-Ahmad, T.; Hen, N.; Yagen, B.; McDonough, J.H.; Finnell, R.H.; Wlodarczyk, B.J.; Bialer, M. Stereoselective anticonvuslant and pharmacokinetic analysis of valnoctamide, a CNS-active derivative of valproic acid with low teratogenic potential. Epilepsia 2014, 55, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Finnell, R.H.; Bennett, G.D.; Karras, S.B.; Mohl, V.K. Common hierarchies of susceptibility to the induction of neural tube defects in mouse embryos by valproic acid and its 4-propyl-4-pentenoic acid metabolite. Teratology 1988, 38, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Bialer, M.; Johannessen, S.I.; Levy, R.H.; Perucca, E.; Tomson, T.; White, H.S. Progress report on new antiepileptic drugs: A summary of the Eleventh Eilat Conference (EILAT XI). Epilepsy Res. 2013, 103, 2–30. [Google Scholar] [CrossRef] [PubMed]
- Bar-Klein, G.; Swissa, E.; Kamintsky, L.; Shekh-Ahmad, T.; Saar-Ashkenazy, R.; Hubary, Y.; Shrot, S.; Stetlander, L.; Eisenkraft, A.; Friedman, A.; et al. sec-Butyl-propylacetamide (SPD) and two of its stereoisomers rapidly terminate paraoxon-induced status epilepticus in rats. Epilepsia 2014, 55, 1953–1958. [Google Scholar] [CrossRef] [PubMed]
- Luís, P.B.; Ruiter, J.P.; Ijlst, L.; de Almeida, I.T.; Duran, M.; Mohsen, A.W.; Vockley, J.; Wanders, R.J.; Silva, M.F. Role of isovaleryl-CoA dehydrogenase and short branched-chain acyl-CoA dehydrogenase in the metabolism of valproic acidmplications for the branched-chain amino acid oxidation pathway. Drug Metab. Dispos. 2011, 39, 1155–1160. [Google Scholar] [CrossRef] [PubMed]
- Turnbull, D.M.; Bone, A.J.; Bartlett, K.; Koundakjian, P.P.; Sherratt, H.S. The effect of valproate on intermediary metabolism in isolated rat hepatocytes and intact rats. Biochem. Pharmacol. 1983, 32, 1887–1892. [Google Scholar] [CrossRef]
- Zsurka, G.; Baron, M.; Stewart, J.D.; Kornblum, C.; Bös, M.; Sassen, R.; Taylor, R.W.; Elger, C.E.; Chinnery, P.F.; Kunz, W.S. Clonally expanded mitochondrial DNA mutations in epileptic individuals with mutated DNA polymerase γ. J. Neuropathol. Exp. Neurol. 2008, 67, 857–866. [Google Scholar] [CrossRef] [PubMed]
- Zsurka, G.; Kunz, W.S. Mitochondrial dysfunction and seizures: The neuronal energy crisis. Lancet Neurol. 2015, 14, 956–966. [Google Scholar] [CrossRef]
- Levy, R.H.; Shen, D.D.; Abbott, F.S.; Riggs, K.W.; Hachad, H. Valproic acidchemistry, biotransformaton and pharmacokinetics. In Antiepileptic Drugs, 5th ed.; Levy, R.H., Mattson, R.H., Meldrum, B.S., Perucca, W., Eds.; Lippincott, Williams & Wilkins: Philadelphia, PA, USA, 2002; pp. 780–800. [Google Scholar]
- Bersudsky, Y.; Applebaum, J.; Gaiduk, Y.; Sharony, L.; Mishory, A.; Podberezsky, A.; Agam, G.; Belmaker, R.H. Valnoctamide as a valproate substitute with low teratogenic potential in mania: A double-blind, controlled, add-on clinical trial. Bipolar Disord. 2010, 12, 376–382. [Google Scholar] [CrossRef] [PubMed]
- Wlodarczyk, B.J.; Ogle, K.; Lin, L.Y.; Bialer, M.; Finnell, R.H. Comparative teratogenicity analysis of valnoctamide, risperidone, and olanzapine in mice. Bipolar Disord. 2015, 17, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, R.E.; Hamud, F.; Fiskum, G.; Varghese, P.J.; Sharpe, S. Cerebral ischemia and reperfusionrevention of brain mitochondrial injury by lidoflazine. J. Cereb. Blood Flow Metab. 1987, 7, 752–758. [Google Scholar] [CrossRef] [PubMed]
- Kudin, A.P.; Bimpong-Buta, N.Y.; Vielhaber, S.; Elger, C.E.; Kunz, W.S. Characterization of superoxide-producing sites in isolated brain mitochondria. J. Biol. Chem. 2004, 279, 4127–4135. [Google Scholar] [CrossRef] [PubMed]
- Steinbrecht, I.; Kunz, W. Use of “cycling” technic for random quantitative determination of the degree of reduction of NAD and NADP system in rat liver mitochondria with continuous recording of the measurements. Acta Biol. Med. Ger. 1970, 25, 731–747. [Google Scholar] [PubMed]
- Wisniewski, E.; Kunz, W.S.; Gellerich, F.N. Phosphate affects the distribution of flux control among the enzymes of oxidative phosphorylation in rat skeletal muscle mitochondria. J. Biol. Chem. 1993, 268, 9343–9346. [Google Scholar] [PubMed]
- Scislowski, P.W.D.; Davis, E.J. A sensitive spectrophotometric assay of pyruvate dehydrogenase activity. Anal. Biochem. 1986, 155, 400–404. [Google Scholar] [CrossRef]
- Eboli, M.L.; Paradies, G.; Galeotti, T.; Papa, S. Pyruvate transport in tumour-cell mitochondria. Biochim. Biophys. Acta 1977, 460, 183–187. [Google Scholar] [CrossRef]
- Patel, M.S.; Hong, Y.S. Lipoic acid as an antioxidant. The role of dihydrolipoamide dehydrogenase. Methods Mol. Biol. 1998, 108, 337–346. [Google Scholar] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| VPA | VPD | VCA | VCD | SPA | SPD | |
|---|---|---|---|---|---|---|
| pyruvate oxidation | 50 µM | 5 mM | 900 µM | 8 mM | 50 µM | 3 mM |
| 2-oxoglutarate oxidation | 50 µM | 8 mM | 200 µM | 5 mM | 50 µM | 1 mM |
| glutamate oxidation | 10 mM | 3 mM | 2 mM | 4.5 mM | 2 mM | 2 mM |
| α-lipoamide dehydrogenase | 80 µM | >1 mM 1 | 300 µM | >1 mM 1 | 100 µM | >1 mM 1 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kudin, A.P.; Mawasi, H.; Eisenkraft, A.; Elger, C.E.; Bialer, M.; Kunz, W.S. Mitochondrial Liver Toxicity of Valproic Acid and Its Acid Derivatives Is Related to Inhibition of α-Lipoamide Dehydrogenase. Int. J. Mol. Sci. 2017, 18, 1912. https://doi.org/10.3390/ijms18091912
Kudin AP, Mawasi H, Eisenkraft A, Elger CE, Bialer M, Kunz WS. Mitochondrial Liver Toxicity of Valproic Acid and Its Acid Derivatives Is Related to Inhibition of α-Lipoamide Dehydrogenase. International Journal of Molecular Sciences. 2017; 18(9):1912. https://doi.org/10.3390/ijms18091912
Chicago/Turabian StyleKudin, Alexei P., Hafiz Mawasi, Arik Eisenkraft, Christian E. Elger, Meir Bialer, and Wolfram S. Kunz. 2017. "Mitochondrial Liver Toxicity of Valproic Acid and Its Acid Derivatives Is Related to Inhibition of α-Lipoamide Dehydrogenase" International Journal of Molecular Sciences 18, no. 9: 1912. https://doi.org/10.3390/ijms18091912
APA StyleKudin, A. P., Mawasi, H., Eisenkraft, A., Elger, C. E., Bialer, M., & Kunz, W. S. (2017). Mitochondrial Liver Toxicity of Valproic Acid and Its Acid Derivatives Is Related to Inhibition of α-Lipoamide Dehydrogenase. International Journal of Molecular Sciences, 18(9), 1912. https://doi.org/10.3390/ijms18091912