
Chronic Kidney Disease and Exposure to Nephrotoxic Metals



Abstract
1. Introduction
2. Chronic Kidney Disease
3. Chronic Kidney Disease and Exposure to Toxic Metals
4. Environmentally Relevant Toxic Metals
4.1. Arsenic
4.1.1. Renal Handling of Arsenic
4.1.2. Renal Effects of Arsenic Exposure
4.1.3. CKD and the Effects of Arsenic Exposure on the Kidneys
4.2. Cadmium
4.2.1. Renal Handling of Cadmium
4.2.2. Renal Effects of Cadmium Exposure
4.2.3. CKD and the Effects of Cadmium Exposure on the Kidneys
4.3. Lead
4.3.1. Renal Handling of Lead
4.3.2. Renal Effects of Lead Exposure
4.3.3. CKD and the Effects of Lead Exposure on the Kidneys
4.4. Mercury
4.4.1. Renal Handling of Mercury
4.4.2. Renal Effects of Mercury Exposure
4.4.3. CKD and the Effects of Mercury Exposure on the Kidney
5. Summary
Acknowledgments
Conflicts of Interest
References
- Price, R.G. Urinary enzymes, nephrotoxicity and renal disease. Toxicology 1982, 23, 99–134. [Google Scholar] [CrossRef]
- Zalups, R.K.; Diamond, G.L. Mercuric chloride-induced nephrotoxicity in the rat following unilateral nephrectomy and compensatory renal growth. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 1987, 53, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Zalups, R.K.; Cox, C.; Diamond, G.L. Histological and urinalysis assessment of nephrotoxicity induced by mercuric chloride in normal and uninephrectomized rats. In Biological Monitoring of Toxic Metals; Clarkson, T.W., Friberg, L., Nordberg, G.F., Sager, P.R., Eds.; Plenum Publishing Corporation: New York, NY, USA, 1988; pp. 531–545. [Google Scholar]
- Clarkson, T.W.; Magos, L. The effect of sodium maleate on the renal deposition and excretion of mercury. Br. J. Pharmacol. Chemother. 1967, 31, 560–567. [Google Scholar] [CrossRef] [PubMed]
- Magos, L.; Stoytchev, T. Combined effect of sodium maleate and some thiol compounds on mercury excretion and redistribution in rats. Br. J. Pharmacol. 1969, 35, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Trojanowska, B.; Piotrowski, J.K.; Szendzikowski, S. The influence of thioacetamide on the excretion of mercury in rats. Toxicol. Appl. Pharmacol. 1971, 18, 374–386. [Google Scholar] [CrossRef]
- Hall, R.L.; Wilke, W.L.; Fettman, M.J. Renal resistance to mercuric chloride toxicity during prolonged exposure in rats. Vet. Hum. Toxicol. 1986, 28, 305–307. [Google Scholar] [PubMed]
- Eto, K.; Yasutake, A.; Miyamoto, K.; Tokunaga, H.; Otsuka, Y. Chronic effects of methylmercury in rats. II. Pathological aspects. Tohoku J. Exp. Med. 1997, 182, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.; Pallan, S.; Gangji, A.S.; Lukic, D.; Clase, C.M. Mercury-associated nephrotic syndrome: A case report and systematic review of the literature. Am. J. Kidney Dis. 2013, 62, 135–138. [Google Scholar] [CrossRef] [PubMed]
- Vanholder, R.C.; Praet, M.M.; Pattyn, P.A.; Leusen, I.R.; Lameire, N.H. Dissociation of glomerular filtration and renal blood flow in HgCl2-induced acute renal failure. Kidney Int. 1982, 22, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Houser, M.T.; Berndt, W.O. The effect of unilateral nephrectomy on the nephrotoxicity of mercuric chloride in the rat. Toxicol. Appl. Pharmacol. 1986, 83, 506–515. [Google Scholar] [CrossRef]
- Houser, M.T.; Berndt, W.O. Unilateral nephrectomy in the rat: Effects on mercury handling and renal cortical subcellular distribution. Toxicol. Appl. Pharmacol. 1988, 93, 187–194. [Google Scholar] [CrossRef]
- Ramos-Frendo, B.; Perez-Garcia, R.; Lopez-Novoa, J.M.; Hernando-Avendano, L. Increased severity of the acute renal failure induced by HgCl2 on rats with reduced renal mass. Biomedicine 1979, 31, 167–170. [Google Scholar] [PubMed]
- Zalups, R.K. Autometallographic localization of inorganic mercury in the kidneys of rats: Effect of unilateral nephrectomy and compensatory renal growth. Exp. Mol. Pathol. 1991, 54, 10–21. [Google Scholar] [CrossRef]
- Zalups, R.K. Enhanced renal outer medullary uptake of mercury associated with uninephrectomy: Implication of a luminal mechanism. J. Toxicol. Environ. Health 1997, 50, 173–194. [Google Scholar] [CrossRef] [PubMed]
- Zalups, R.K.; Barfuss, D.W.; Kostyniak, P.J. Altered intrarenal accumulation of mercury in uninephrectomized rats treated with methylmercury chloride. Toxicol. Appl. Pharmacol. 1992, 115, 174–182. [Google Scholar] [CrossRef]
- Zalups, R.K.; Klotzbach, J.M.; Diamond, G.L. Enhanced accumulation of injected inorganic mercury in renal outer medulla after unilateral nephrectomy. Toxicol. Appl. Pharmacol. 1987, 89, 226–236. [Google Scholar] [CrossRef]
- Bridges, C.C.; Barfuss, D.W.; Joshee, L.; Zalups, R.K. Compensatory Renal Hypertrophy and the Uptake of Cysteine S-Conjugates of Hg2+ in Isolated S2 Proximal Tubular Segments. Toxicol. Sci. 2016, 154, 278–288. [Google Scholar] [CrossRef] [PubMed]
- Zalups, R.K.; Bridges, C.C. Seventy-five percent nephrectomy and the disposition of inorganic mercury in 2,3-dimercaptopropanesulfonic acid-treated rats lacking functional multidrug-resistance protein 2. J. Pharmacol. Exp. Ther. 2010, 332, 866–875. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention (CDC). Chronic Kidney Disease Surveillance System—United States. Available online: www.cdc.gov/ckd (accessed on 16 September 2016).
- Centers for Disease Control and Prevention (CDC). Summary Health Statistics: National Health Interview Survey; U.S. Department of Human and Heath Services; Centers for Disease Control and Prevention: Atlanta, GA, USA, 2014.
- Jha, V.; Garcia-Garcia, G.; Iseki, K.; Li, Z.; Naicker, S.; Plattner, B.; Saran, R.; Wang, A.Y.; Yang, C.W. Chronic kidney disease: Global dimension and perspectives. Lancet 2013, 382, 260–272. [Google Scholar] [CrossRef]
- Centers of Disease Control and Prevention (CDC). National Chronic Kidney Disease Fact Sheet: General Information and National Estimates on Chronic Kidney Disease in the United States; US Department of Heath and Human Services: Atlanta, GA, USA, 2014. [Google Scholar]
- Fine, L.G.; Norman, J.T.; Kujubu, D.A.; Knecht, A. Renal Hypertrophy. In The Kidney: Physiology and Pathophysiology, 2nd ed.; Seldin, D.W., Giebisch, G., Eds.; Raven Press: New York, NY, USA, 1992; pp. 3113–3133. [Google Scholar]
- Salehmoghaddam, S.; Bradley, T.; Mikhail, N.; Badie-Dezfooly, B.; Nord, E.P.; Trizna, W.; Kheyfets, R.; Fine, L.G. Hypertrophy of basolateral Na-K pump activity in the proximal tubule of the remnant kidney. Lab. Investig. 1985, 53, 443–452. [Google Scholar] [PubMed]
- Toback, F.G.; Smith, P.D.; Lowenstein, L.M. Phospholipid metabolism in the initiation of renal compensatory growth after acute reduction of renal mass. J. Clin. Investig. 1974, 54, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Wolf, G. Cellular mechanisms of tubule hypertrophy and hyperplasia in renal injury. Miner Electrolyte Metab. 1995, 21, 303–316. [Google Scholar] [PubMed]
- Bricker, N.S.; Fine, L.G. The Renal Response to Progressive Nephron Loss. In The Kidney, 2nd ed.; Brenner, B.M., Rector, F.C., Eds.; Saunders: Philadelphia, PA, USA, 1981; Volume 1, pp. 1056–1096. [Google Scholar]
- Klahr, S. Progression of chronic renal disease. Nutrition 1990, 6, 207–212. [Google Scholar] [PubMed]
- Fine, L.G.; Norman, J. Cellular events in renal hypertrophy. Annu. Rev. Physiol. 1989, 51, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Novoa, J.M. The Mechanisms of Age-Associated Glomerular Sclerosis. In The Aging Kidney in Health and Disease; Macias Nunez, J.F., Cameron, J.S., Oreopoulos, D.G., Eds.; Springer: New York, NY, USA, 2008; pp. 113–126. [Google Scholar]
- Roels, H.A.; Lauwerys, R.R.; Bernard, A.M.; Buchet, J.P.; Vos, A.; Oversteyns, M. Assessment of the filtration reserve capacity of the kidney in workers exposed to cadmium. Br. J. Ind. Med. 1991, 48, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Uriu, K.; Kaizu, K.; Qie, Y.L.; Ito, A.; Takagi, I.; Suzuka, K.; Inada, Y.; Hashimoto, O.; Eto, S. Long-term oral intake of low-dose cadmium exacerbates age-related impairment of renal functional reserve in rats. Toxicol. Appl. Pharmacol. 2000, 169, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Zalups, R.K. Reductions in renal mass and the nephropathy induced by mercury. Toxicol. Appl. Pharmacol. 1997, 143, 366–379. [Google Scholar] [CrossRef] [PubMed]
- Agency for Toxic Substances and Disease Registry (ATSDR). Public Health Statement: Arsenic; Centers for Disease Control and Prevention: Atlanta, GA, USA, 2007. [Google Scholar]
- Sattar, A.; Xie, S.; Hafeez, M.A.; Wang, X.; Hussain, H.I.; Iqbal, Z.; Pan, Y.; Iqbal, M.; Shabbir, M.A.; Yuan, Z. Metabolism and toxicity of arsenicals in mammals. Environ. Toxicol. Pharmacol. 2016, 48, 214–224. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.J.; Styblo, M.; Lin, S. The cellular metabolism and systemic toxicity of arsenic. Toxicol. Appl. Pharmacol. 2001, 176, 127–144. [Google Scholar] [CrossRef] [PubMed]
- Calatayud, M.; Barrios, J.A.; Velez, D.; Devesa, V. In vitro study of transporters involved in intestinal absorption of inorganic arsenic. Chem. Res. Toxicol. 2012, 25, 446–453. [Google Scholar] [CrossRef] [PubMed]
- Tseng, W.P.; Chu, H.M.; How, S.W.; Fong, J.M.; Lin, C.S.; Yeh, S. Prevalence of skin cancer in an endemic area of chronic arsenicism in Taiwan. J. Natl. Cancer Inst. 1968, 40, 453–463. [Google Scholar] [PubMed]
- Engel, R.R.; Hopenhayn-Rich, C.; Receveur, O.; Smith, A.H. Vascular effects of chronic arsenic exposure: A review. Epidemiol. Rev. 1994, 16, 184–209. [Google Scholar] [CrossRef] [PubMed]
- Sung, T.C.; Huang, J.W.; Guo, H.R. Association between Arsenic Exposure and Diabetes: A Meta-Analysis. BioMed Res. Int. 2015, 2015, 368087. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.R.; Das, N.K.; Datta, P.K. Pathogenesis, clinical features and pathology of chronic arsenicosis. Indian J. Dermatol. Venereol. Leprol. 2008, 74, 559–570. [Google Scholar] [PubMed]
- Lin, S.; Shi, Q.; Nix, F.B.; Styblo, M.; Beck, M.A.; Herbin-Davis, K.M.; Hall, L.L.; Simeonsson, J.B.; Thomas, D.J. A novel S-adenosyl-l-methionine: Arsenic(III) methyltransferase from rat liver cytosol. J. Biol. Chem. 2002, 277, 10795–10803. [Google Scholar] [CrossRef] [PubMed]
- Healy, S.M.; Casarez, E.A.; Ayala-Fierro, F.; Aposhian, H. Enzymatic methylation of arsenic compounds. V. Arsenite methyltransferase activity in tissues of mice. Toxicol. Appl. Pharmacol. 1998, 148, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.J.; Li, J.; Waters, S.B.; Xing, W.; Adair, B.M.; Drobna, Z.; Devesa, V.; Styblo, M. Arsenic (+3 oxidation state) methyltransferase and the methylation of arsenicals. Exp. Biol. Med. 2007, 232, 3–13. [Google Scholar]
- Petrick, J.S.; Ayala-Fierro, F.; Cullen, W.R.; Carter, D.E.; Vasken Aposhian, H. Monomethylarsonous acid (MMA(III)) is more toxic than arsenite in Chang human hepatocytes. Toxicol. Appl. Pharmacol. 2000, 163, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Styblo, M.; Del Razo, L.M.; Vega, L.; Germolec, D.R.; LeCluyse, E.L.; Hamilton, G.A.; Reed, W.; Wang, C.; Cullen, W.R.; Thomas, D.J. Comparative toxicity of trivalent and pentavalent inorganic and methylated arsenicals in rat and human cells. Arch. Toxicol. 2000, 74, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Mass, M.J.; Tennant, A.; Roop, B.C.; Cullen, W.R.; Styblo, M.; Thomas, D.J.; Kligerman, A.D. Methylated trivalent arsenic species are genotoxic. Chem. Res. Toxicol. 2001, 14, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Styblo, M.; Serves, S.V.; Cullen, W.R.; Thomas, D.J. Comparative inhibition of yeast glutathione reductase by arsenicals and arsenothiols. Chem. Res. Toxicol. 1997, 10, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Chouchane, S.; Snow, E.T. In vitro effect of arsenical compounds on glutathione-related enzymes. Chem. Res. Toxicol. 2001, 14, 517–522. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Del Razo, L.M.; Styblo, M.; Wang, C.; Cullen, W.R.; Thomas, D.J. Arsenicals inhibit thioredoxin reductase in cultured rat hepatocytes. Chem. Res. Toxicol. 2001, 14, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Cullen, W.R.; Thomas, D.J. Methylarsenicals and arsinothiols are potent inhibitors of mouse liver thioredoxin reductase. Chem. Res. Toxicol. 1999, 12, 924–930. [Google Scholar] [CrossRef] [PubMed]
- Petrick, J.S.; Jagadish, B.; Mash, E.A.; Aposhian, H.V. Monomethylarsonous acid (MMA(III)) and arsenite: LD(50) in hamsters and in vitro inhibition of pyruvate dehydrogenase. Chem. Res. Toxicol. 2001, 14, 651–656. [Google Scholar] [CrossRef] [PubMed]
- Fowler, B.A.; Chou, S.J.; Jones, R.L.; Chen, C.J. Arsenic. In Handbook on the Toxicology of Metals, 3rd ed.; Nordberg, G.F., Fowler, B.A., Nordberg, M., Freiberg, L., Eds.; Elsevier: Amsterdam, The Netherlands, 2007; pp. 367–443. [Google Scholar]
- Liu, Z.; Sanchez, M.A.; Jiang, X.; Boles, E.; Landfear, S.M.; Rosen, B.P. Mammalian glucose permease GLUT1 facilitates transport of arsenic trioxide and methylarsonous acid. Biochem. Biophys. Res. Commun. 2006, 351, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Sugawara-Yokoo, M.; Suzuki, T.; Matsuzaki, T.; Naruse, T.; Takata, K. Presence of fructose transporter GLUT5 in the S3 proximal tubules in the rat kidney. Kidney Int. 1999, 56, 1022–1028. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, J.H.; Camp, K.; Maianu, L.; Garvey, W.T. Glucose transporters of rat proximal tubule: Differential expression and subcellular distribution. Am. J. Physiol. 1992, 262, F807–F812. [Google Scholar] [PubMed]
- Lee, T.C.; Ho, I.C.; Lu, W.J.; Huang, J.D. Enhanced expression of multidrug resistance-associated protein 2 and reduced expression of aquaglyceroporin 3 in an arsenic-resistant human cell line. J. Biol. Chem. 2006, 281, 18401–18407. [Google Scholar] [CrossRef] [PubMed]
- Bedford, J.J.; Leader, J.P.; Walker, R.J. Aquaporin expression in normal human kidney and in renal disease. J. Am. Soc. Nephrol. 2003, 14, 2581–2587. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.J.; Tamai, I.; Nezu, J.; Lai, M.L.; Huang, J.D. Organic anion transporting polypeptide-C mediates arsenic uptake in HEK-293 cells. J. Biomed. Sci. 2006, 13, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Aleksunes, L.M.; Augustine, L.M.; Scheffer, G.L.; Cherrington, N.J.; Manautou, J.E. Renal xenobiotic transporters are differentially expressed in mice following cisplatin treatment. Toxicology 2008, 250, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Roggenbeck, B.A.; Banerjee, M.; Leslie, E.M. Cellular arsenic transport pathways in mammals. J. Environ. Sci. 2016, 49, 38–58. [Google Scholar] [CrossRef] [PubMed]
- Kala, S.V.; Kala, G.; Prater, C.I.; Sartorelli, A.C.; Lieberman, M.W. Formation and urinary excretion of arsenic triglutathione and methylarsenic diglutathione. Chem. Res. Toxicol. 2004, 17, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Roggenbeck, B.A.; Carew, M.W.; Charrois, G.J.; Douglas, D.N.; Kneteman, N.M.; Lu, X.; Le, X.C.; Leslie, E.M. Characterization of arsenic hepatobiliary transport using sandwich-cultured human hepatocytes. Toxicol. Sci. 2015, 145, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Yehiayan, L.; Stice, S.; Liu, G.; Matulis, S.; Boise, L.H.; Cai, Y. Dimethylarsinothioyl glutathione as a metabolite in human multiple myeloma cell lines upon exposure to Darinaparsin. Chem. Res. Toxicol. 2014, 27, 754–764. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.J. Unraveling arsenic—Glutathione connections. Toxicol. Sci. 2009, 107, 309–311. [Google Scholar] [CrossRef] [PubMed]
- Scott, N.; Hatlelid, K.M.; MacKenzie, N.E.; Carter, D.E. Reactions of arsenic(III) and arsenic(V) species with glutathione. Chem. Res. Toxicol. 1993, 6, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Delnomdedieu, M.; Basti, M.M.; Styblo, M.; Otvos, J.D.; Thomas, D.J. Complexation of arsenic species in rabbit erythrocytes. Chem. Res. Toxicol. 1994, 7, 621–627. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, H.; Miller, D.S.; Saavedra, J.E.; Keefer, L.K.; Johnson, D.R.; Klaassen, C.D.; Waalkes, M.P. Overexpression of glutathione S-transferase II and multidrug resistance transport proteins is associated with acquired tolerance to inorganic arsenic. Mol. Pharmacol. 2001, 60, 302–309. [Google Scholar] [PubMed]
- Vernhet, L.; Seite, M.P.; Allain, N.; Guillouzo, A.; Fardel, O. Arsenic induces expression of the multidrug resistance-associated protein 2 (MRP2) gene in primary rat and human hepatocytes. J. Pharmacol. Exp. Ther. 2001, 298, 234–239. [Google Scholar] [PubMed]
- Drobna, Z.; Walton, F.S.; Paul, D.S.; Xing, W.; Thomas, D.J.; Styblo, M. Metabolism of arsenic in human liver: The role of membrane transporters. Arch. Toxicol. 2010, 84, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Pei, Q.L.; Li, G.X.; Han, G.; Tian, F.J.; Qin, X.J.; Zhang, R.; Hou, W.S.; Li, X.Y. Effects of MRP2-GSH cotransport system on hepatic arsenic metabolism in rats. Chin. J. Ind. Hyg. Occup. Dis. 2006, 24, 278–280. [Google Scholar]
- Leslie, E.M.; Deeley, R.G.; Cole, S.P. Multidrug resistance proteins: Role of P-glycoprotein, MRP1, MRP2, and BCRP (ABCG2) in tissue defense. Toxicol. Appl. Pharmacol. 2005, 204, 216–237. [Google Scholar] [CrossRef] [PubMed]
- Carew, M.W.; Leslie, E.M. Selenium-dependent and -independent transport of arsenic by the human multidrug resistance protein 2 (MRP2/ABCC2): Implications for the mutual detoxification of arsenic and selenium. Carcinogenesis 2010, 31, 1450–1455. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, M.; Carew, M.W.; Roggenbeck, B.A.; Whitlock, B.D.; Naranmandura, H.; Le, X.C.; Leslie, E.M. A novel pathway for arsenic elimination: Human multidrug resistance protein 4 (MRP4/ABCC4) mediates cellular export of dimethylarsinic acid (DMAV) and the diglutathione conjugate of monomethylarsonous acid (MMAIII). Mol. Pharmacol. 2014, 86, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Kimura, A.; Ishida, Y.; Hayashi, T.; Wada, T.; Yokoyama, H.; Sugaya, T.; Mukaida, N.; Kondo, T. Interferon-gamma plays protective roles in sodium arsenite-induced renal injury by up-regulating intrarenal multidrug resistance-associated protein 1 expression. Am. J. Pathol. 2006, 169, 1118–1128. [Google Scholar] [CrossRef] [PubMed]
- Chin, K.V.; Tanaka, S.; Darlington, G.; Pastan, I.; Gottesman, M.M. Heat shock and arsenite increase expression of the multidrug resistance (MDR1) gene in human renal carcinoma cells. J. Biol. Chem. 1990, 265, 221–226. [Google Scholar] [PubMed]
- Liu, J.; Liu, Y.; Powell, D.A.; Waalkes, M.P.; Klaassen, C.D. Multidrug-resistance mdr1a/1b double knockout mice are more sensitive than wild type mice to acute arsenic toxicity, with higher arsenic accumulation in tissues. Toxicology 2002, 170, 55–62. [Google Scholar] [CrossRef]
- Xie, Y.; Liu, J.; Liu, Y.; Klaassen, C.D.; Waalkes, M.P. Toxicokinetic and genomic analysis of chronic arsenic exposure in multidrug-resistance mdr1a/1b(-/-) double knockout mice. Mol. Cell. Biochem. 2004, 255, 11–18. [Google Scholar] [CrossRef] [PubMed]
- George, B.; You, D.; Joy, M.S.; Aleksunes, L.M. Xenobiotic transporters and kidney injury. Adv. Drug Deliv. Rev. 2017. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Guo, D.; Obianom, O.N.; Su, T.; Polli, J.E.; Shu, Y. Multidrug and toxin extrusion proteins mediate cellular transport of cadmium. Toxicol. Appl. Pharmacol. 2017, 314, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Prasad, G.V.; Rossi, N.F. Arsenic intoxication associated with tubulointerstitial nephritis. Am. J. Kidney Dis. 1995, 26, 373–376. [Google Scholar] [CrossRef]
- Robles-Osorio, M.L.; Sabath-Silva, E.; Sabath, E. Arsenic-mediated nephrotoxicity. Ren. Fail. 2015, 37, 542–547. [Google Scholar] [CrossRef] [PubMed]
- Tsao, D.A.; Tseng, W.C.; Chang, H.R. RKIP expression of liver and kidney after arsenic exposure. Environ. Toxicol. 2017, 32, 1079–1082. [Google Scholar] [CrossRef] [PubMed]
- Yeung, K.; Seitz, T.; Li, S.; Janosch, P.; McFerran, B.; Kaiser, C.; Fee, F.; Katsanakis, K.D.; Rose, D.W.; Mischak, H.; et al. Suppression of Raf-1 kinase activity and MAP kinase signalling by RKIP. Nature 1999, 401, 173–177. [Google Scholar] [PubMed]
- Yeung, K.; Janosch, P.; McFerran, B.; Rose, D.W.; Mischak, H.; Sedivy, J.M.; Kolch, W. Mechanism of suppression of the Raf/MEK/extracellular signal-regulated kinase pathway by the raf kinase inhibitor protein. Mol. Cell. Biol. 2000, 20, 3079–3085. [Google Scholar] [CrossRef] [PubMed]
- Odabaei, G.; Chatterjee, D.; Jazirehi, A.R.; Goodglick, L.; Yeung, K.; Bonavida, B. Raf-1 kinase inhibitor protein: Structure, function, regulation of cell signaling, and pivotal role in apoptosis. Adv. Cancer Res. 2004, 91, 169–200. [Google Scholar] [PubMed]
- Singh, R.D.; Tiwari, R.; Khan, H.; Kumar, A.; Srivastava, V. Arsenic exposure causes epigenetic dysregulation of IL-8 expression leading to proneoplastic changes in kidney cells. Toxicol. Lett. 2015, 237, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Verdugo, M.; Ogra, Y.; Quiroz, W. Mechanisms underlying the toxic effects of antimony species in human embryonic kidney cells (HEK-293) and their comparison with arsenic species. J. Toxicol. Sci. 2016, 41, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Sinha, M.; Manna, P.; Sil, P.C. Arjunolic acid attenuates arsenic-induced nephrotoxicity. Pathophysiology 2008, 15, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Manna, P.; Sil, P.C. Prophylactic role of taurine on arsenic mediated oxidative renal dysfunction via MAPKs/ NF-kappaB and mitochondria dependent pathways. Free Radic. Res. 2009, 43, 995–1007. [Google Scholar] [CrossRef] [PubMed]
- Gong, X.; Ivanov, V.N.; Davidson, M.M.; Hei, T.K. Tetramethylpyrazine (TMP) protects against sodium arsenite-induced nephrotoxicity by suppressing ROS production, mitochondrial dysfunction, pro-inflammatory signaling pathways and programed cell death. Arch. Toxicol. 2015, 89, 1057–1070. [Google Scholar] [CrossRef] [PubMed]
- Orihuela, R.; Kojima, C.; Tokar, E.J.; Person, R.J.; Xu, Y.; Qu, W.; Waalkes, M.P. Oxidative DNA damage after acute exposure to arsenite and monomethylarsonous acid in biomethylation-deficient human cells. Toxicol. Mech. Methods 2013, 23, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, A.; Oshima, Y.; Fujimura, A. An approach to elucidate potential mechanism of renal toxicity of arsenic trioxide. Exp. Hematol. 2007, 35, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Peraza, M.A.; Carter, D.E.; Gandolfi, A.J. Toxicity and metabolism of subcytotoxic inorganic arsenic in human renal proximal tubule epithelial cells (HK-2). Cell Biol. Exp. Toxicol. 2003, 19, 253–264. [Google Scholar] [CrossRef]
- Lee, C.H.; Yu, H.S. Role of mitochondria, ROS, and DNA damage in arsenic induced carcinogenesis. Front. Biosci. 2016, 8, 312–320. [Google Scholar]
- Ganger, R.; Garla, R.; Mohanty, B.P.; Bansal, M.P.; Garg, M.L. Protective Effects of Zinc Against Acute Arsenic Toxicity by Regulating Antioxidant Defense System and Cumulative Metallothionein Expression. Biol. Trace Elem. Res. 2016, 169, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Okayasu, R. Arsenic accumulation, elimination, and interaction with copper, zinc and manganese in liver and kidney of rats. Food Chem. Toxicol. 2008, 46, 3646–3650. [Google Scholar] [CrossRef] [PubMed]
- Kreppel, H.; Bauman, J.W.; Liu, J.; McKim, J.M., Jr.; Klaassen, C.D. Induction of metallothionein by arsenicals in mice. Fundam. Appl. Toxicol. 1993, 20, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Garla, R.; Mohanty, B.P.; Ganger, R.; Sudarshan, M.; Bansal, M.P.; Garg, M.L. Metal stoichiometry of isolated and arsenic substituted metallothionein: PIXE and ESI-MS study. Biometals 2013, 26, 887–896. [Google Scholar] [CrossRef] [PubMed]
- Garla, R.; Kaur, N.; Bansal, M.P.; Garg, M.L.; Mohanty, B.P. Quantum mechanical treatment of As3+-thiol model compounds: Implication for the core structure of As(III)-metallothionein. J. Mol. Model. 2017, 23, 78. [Google Scholar] [CrossRef] [PubMed]
- Qu, W.; Waalkes, M.P. Metallothionein blocks oxidative DNA damage induced by acute inorganic arsenic exposure. Toxicol. Appl. Pharmacol. 2015, 282, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Garla, R.; Ganger, R.; Mohanty, B.P.; Verma, S.; Bansal, M.P.; Garg, M.L. Metallothionein does not sequester arsenic(III) ions in condition of acute arsenic toxicity. Toxicology 2016, 366–367, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Weidemann, D.; Kuo, C.C.; Navas-Acien, A.; Abraham, A.G.; Weaver, V.; Fadrowski, J. Association of arsenic with kidney function in adolescents and young adults: Results from the National Health and Nutrition Examination Survey 2009–2012. Environ. Res. 2015, 140, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Hsueh, Y.M.; Chung, C.J.; Shiue, H.S.; Chen, J.B.; Chiang, S.S.; Yang, M.H.; Tai, C.W.; Su, C.T. Urinary arsenic species and CKD in a Taiwanese population: A case-control study. Am. J. Kidney Dis. 2009, 54, 859–870. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D.; Upham, T.; Barton, C.H. Hemodialysis clearance of arsenic. Clin. Toxicol. 1980, 17, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.Y.; Umans, J.G.; Yeh, F.; Francesconi, K.A.; Goessler, W.; Silbergeld, E.K.; Bandeen-Roche, K.; Guallar, E.; Howard, B.V.; Weaver, V.M.; et al. The association of urine arsenic with prevalent and incident chronic kidney disease: Evidence from the Strong Heart Study. Epidemiology 2015, 26, 601–612. [Google Scholar] [CrossRef] [PubMed]
- Faroon, O.; Ashizawa, A.; Wright, S.; Tucker, P.; Jenkins, K.; Ingerman, L.; Rudisill, C. Toxicological Profile for Cadmium. In U.S. Department of Health and Human Services, Public Health Service; Centers for Disease Control and Prevention; Agency for Toxic Substances and Disease Registry (ATSDR): Atlanta, GA, USA, 2008. [Google Scholar]
- World Health Organization (WHO). Cadmium—Environmental Health Criteria 134; World Health Organization (WHO): Geneva, Switzerland, 1992. [Google Scholar]
- Friberg, L.; Piscator, M.; Nordberg, G.; Kjellstrom, T. Cadmium in the Environmnent, 2nd ed.; CRC Press: Cleveland, OH, USA, 1974. [Google Scholar]
- Elinder, C.G.; Kjellstrom, T.; Lind, B.; Linnman, L.; Piscator, M.; Sundstedt, K. Cadmium exposure from smoking cigarettes: Variations with time and country where purchased. Environ. Res. 1983, 32, 220–227. [Google Scholar] [CrossRef]
- Elinder, C.G.; Lind, B.; Kjellstrom, T.; Linnman, L.; Friberg, L. Cadmium in kidney cortex, liver, and pancreas from Swedish autopsies. Estimation of biological half time in kidney cortex, considering calorie intake and smoking habits. Arch. Environ. Health 1976, 31, 292–302. [Google Scholar] [CrossRef] [PubMed]
- Jarup, L.; Berglund, M.; Elinder, C.G.; Nordberg, G.; Vahter, M. Health effects of cadmium exposure—A review of the literature and a risk estimate. Scand. J. Work Environ. Health 1998, 24 (Suppl. S1), 1–51. [Google Scholar] [PubMed]
- Olsson, I.M.; Bensryd, I.; Lundh, T.; Ottosson, H.; Skerfving, S.; Oskarsson, A. Cadmium in blood and urine—Impact of sex, age, dietary intake, iron status, and former smoking—Association of renal effects. Environ. Health Perspect. 2002, 110, 1185–1190. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control (CDC); Community Futures Development Corporation (CFDC). Fourth National Report on Human Exposure to Environmental Chemicals; Centers for Disease Control (CDC): Atlanta, GA, USA, 2009; pp. 218–226. [Google Scholar]
- Rabenstein, D.L.; Isab, A.A.; Kadima, W.; Mohanakrishnan, P. A proton nuclear magnetic resonance study of the interaction of cadmium with human erythrocytes. Biochim. Biophys. Acta 1983, 762, 531–541. [Google Scholar] [CrossRef]
- Rabenstein, D.L. Metal complexes of glutathione and their biological significance. In Glutathione: Chemical, Biochemical and Medical Aspects, Coenzymes and Cofactors; Dolphin, D., Auromovibic, O., Poulson, R., Eds.; Wiley: New York, NY, USA, 1989; Volume 3, pp. 147–186. [Google Scholar]
- Wang, Y.; Zalups, R.K.; Barfuss, D.W. Potential mechanisms involved in the absorptive transport of cadmium in isolated perfused rabbit renal proximal tubules. Toxicol. Lett. 2010, 193, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Zalups, R.K.; Ahmad, S. Molecular handling of cadmium in transporting epithelia. Toxicol. Appl. Pharmacol. 2003, 186, 163–188. [Google Scholar] [CrossRef]
- Soodvilai, S.; Nantavishit, J.; Muanprasat, C.; Chatsudthipong, V. Renal organic cation transporters mediated cadmium-induced nephrotoxicity. Toxicol. Lett. 2011, 204, 38–42. [Google Scholar] [CrossRef] [PubMed]
- Thevenod, F.; Ciarimboli, G.; Leistner, M.; Wolff, N.A.; Lee, W.K.; Schatz, I.; Keller, T.; Al-Monajjed, R.; Gorboulev, V.; Koepsell, H. Substrate- and cell contact-dependent inhibitor affinity of human organic cation transporter 2: Studies with two classical organic cation substrates and the novel substrate Cd2+. Mol. Pharm. 2013, 10, 3045–3056. [Google Scholar] [CrossRef] [PubMed]
- Sweet, D.H.; Miller, D.S.; Pritchard, J.B. Basolateral localization of organic cation transporter 2 in intact renal proximal tubules. Am. J. Physiol. Ren. Physiol. 2000, 279, F826–F834. [Google Scholar]
- Karbach, U.; Kricke, J.; Meyer-Wentrup, F.; Gorboulev, V.; Volk, C.; Loffing-Cueni, D.; Kaissling, B.; Bachmann, S.; Koepsell, H. Localization of organic cation transporters OCT1 and OCT2 in rat kidney. Am. J. Physiol. Ren. Physiol. 2000, 279, F679–F687. [Google Scholar]
- Felley-Bosco, E.; Diezi, J. Fate of cadmium in rat renal tubules: A micropuncture study. Toxicol. Appl. Pharmacol. 1989, 98, 243–251. [Google Scholar] [CrossRef]
- Barbier, O.; Jacquillet, G.; Tauc, M.; Poujeol, P.; Cougnon, M. Acute study of interaction among cadmium, calcium, and zinc transport along the rat nephron in vivo. Am. J. Physiol. Ren. Physiol. 2004, 287, F1067–F1075. [Google Scholar] [CrossRef] [PubMed]
- Dudley, R.E.; Gammal, L.M.; Klaassen, C.D. Cadmium-induced hepatic and renal injury in chronically exposed rats: Likely role of hepatic cadmium-metallothionein in nephrotoxicity. Toxicol. Appl. Pharmacol. 1985, 77, 414–426. [Google Scholar] [CrossRef]
- Erfurt, C.; Roussa, E.; Thevenod, F. Apoptosis by Cd2+ or CdMT in proximal tubule cells: Different uptake routes and permissive role of endo/lysosomal CdMT uptake. Am. J. Physiol. Cell Physiol. 2003, 285, C1367–C1376. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Sano, K.; Webb, M. The effect of l-cysteine on the portion-selective uptake of cadmium in the renal proximal tubule. Arch. Toxicol. 1987, 60, 365–369. [Google Scholar] [CrossRef] [PubMed]
- Dorian, C.; Gattone, V.H., 2nd; Klaassen, C.D. Accumulation and degradation of the protein moiety of cadmium-metallothionein (CdMT) in the mouse kidney. Toxicol. Appl. Pharmacol. 1992, 117, 242–248. [Google Scholar] [CrossRef]
- Dorian, C.; Gattone, V.H., 2nd; Klaasen, C.D. Renal cadmium deposition and injury as a result of accumulation of cadmium-metallothionein (CdMT) by the proximal convoluted tubules—A light microscopic autoradiography study with 109CdMT. Toxicol. Appl. Pharmacol. 1992, 114, 173–181. [Google Scholar] [CrossRef]
- Foulkes, E.C. Renal tubular transport of cadmium-metallothionein. Toxicol. Appl. Pharmacol. 1978, 45, 505–512. [Google Scholar] [CrossRef]
- Nordberg, M.; Jin, T.; Nordberg, G.F. Cadmium, metallothionein and renal tubular toxicity. IARC Sci. Publ. 1992, 118, 293–297. [Google Scholar]
- Webb, M. Role of metallothionein in cadmium metabolism. In Cadmium; Foulkes, E.C., Ed.; Springer: Berlin, Germany; New York, NY, USA, 1986. [Google Scholar]
- Thevenod, F. Catch me if you can! Novel aspects of cadmium transport in mammalian cells. Biometals 2010, 23, 857–875. [Google Scholar] [CrossRef] [PubMed]
- Thevenod, F. Nephrotoxicity and the proximal tubule. Insights from cadmium. Nephron Physiol. 2003, 93, 87–93. [Google Scholar] [CrossRef]
- Nordberg, G.F.; Goyer, R.; Nordberg, M. Comparative toxicity of cadmium-metallothionein and cadmium chloride on mouse kidney. Arch. Pathol. 1975, 99, 192–197. [Google Scholar] [PubMed]
- Cherian, M.G.; Nordberg, M. Cellular adaptation in metal toxicology and metallothionein. Toxicology 1983, 28, 1–15. [Google Scholar] [PubMed]
- Felley-Bosco, E.; Diezi, J. Fate of cadmium in rat renal tubules: A microinjection study. Toxicol. Appl. Pharmacol. 1987, 91, 204–211. [Google Scholar] [PubMed]
- Murakami, M.; Cain, K.; Webb, M. Cadmium-metallothionein-induced nephropathy: A morphological and autoradiographic study of cadmium distribution, the development of tubular damage and subsequent cell regeneration. J. Appl. Toxicol. 1983, 3, 237–244. [Google Scholar] [PubMed]
- Zalups, R.K.; Gelein, R.M.; Cherian, M.G. Shifts in the dose-effect relationship for the nephropathy induced by cadmium-metallothionein in rats after a reduction in renal mass. J. Pharmacol. Exp. Ther. 1992, 262, 1256–1266. [Google Scholar] [PubMed]
- Abouhamed, M.; Wolff, N.A.; Lee, W.K.; Smith, C.P.; Thevenod, F. Knockdown of endosomal/lysosomal divalent metal transporter 1 by RNA interference prevents cadmium-metallothionein-1 cytotoxicity in renal proximal tubule cells. Am. J. Physiol. Ren. Physiol. 2007, 293, F705–F712. [Google Scholar]
- Ferguson, C.J.; Wareing, M.; Ward, D.T.; Green, R.; Smith, C.P.; Riccardi, D. Cellular localization of divalent metal transporter DMT-1 in rat kidney. Am. J. Physiol. Ren. Physiol. 2001, 280, F803–F814. [Google Scholar]
- Scheuhammer, A.M.; Cherian, M.G. Quantification of metallothioneins by a silver-saturation method. Toxicol. Appl. Pharmacol. 1986, 82, 417–425. [Google Scholar] [PubMed]
- Wang, B.; Schneider, S.N.; Dragin, N.; Girijashanker, K.; Dalton, T.P.; He, L.; Miller, M.L.; Stringer, K.F.; Soleimani, M.; Richardson, D.D.; et al. Enhanced cadmium-induced testicular necrosis and renal proximal tubule damage caused by gene-dose increase in a Slc39a8-transgenic mouse line. Am. J. Physiol. Cell Physiol. 2007, 292, C1523–C1535. [Google Scholar] [PubMed]
- Girijashanker, K.; He, L.; Soleimani, M.; Reed, J.M.; Li, H.; Liu, Z.; Wang, B.; Dalton, T.P.; Nebert, D.W. Slc39a14 gene encodes ZIP14, a metal/bicarbonate symporter: Similarities to the ZIP8 transporter. Mol. Pharmacol. 2008, 73, 1413–1423. [Google Scholar] [PubMed]
- Fujishiro, H.; Okugaki, S.; Kubota, K.; Fujiyama, T.; Miyataka, H.; Himeno, S. The role of ZIP8 down-regulation in cadmium-resistant metallothionein-null cells. J. Appl. Toxicol. 2009, 29, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Fujishiro, H.; Yano, Y.; Takada, Y.; Tanihara, M.; Himeno, S. Roles of ZIP8, ZIP14, and DMT1 in transport of cadmium and manganese in mouse kidney proximal tubule cells. Metallomics 2012, 4, 700–708. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Wang, B.; Hay, E.B.; Nebert, D.W. Discovery of ZIP transporters that participate in cadmium damage to testis and kidney. Toxicol. Appl. Pharmacol. 2009, 238, 250–257. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Witkowska, K.; Afonso Guerra-Assuncao, J.; Ren, M.; Ng, F.L.; Mauro, C.; Tucker, A.T.; Caulfield, M.J.; Ye, S. A blood pressure-associated variant of the SLC39A8 gene influences cellular cadmium accumulation and toxicity. Hum. Mol. Genet. 2016, 25, 4117–4126. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, M.; Matsumoto, T.; Morimoto, R.; Arioka, S.; Omote, H.; Moriyama, Y. A human transporter protein that mediates the final excretion step for toxic organic cations. Proc. Natl. Acad. Sci. USA 2005, 102, 17923–17928. [Google Scholar] [CrossRef] [PubMed]
- Masuda, S.; Terada, T.; Yonezawa, A.; Tanihara, Y.; Kishimoto, K.; Katsura, T.; Ogawa, O.; Inui, K. Identification and functional characterization of a new human kidney-specific H+/organic cation antiporter, kidney-specific multidrug and toxin extrusion 2. J. Am. Soc. Nephrol. 2006, 17, 2127–2135. [Google Scholar] [CrossRef] [PubMed]
- Carriere, P.; Mantha, M.; Champagne-Paradis, S.; Jumarie, C. Characterization of basolateral-to-apical transepithelial transport of cadmium in intestinal TC7 cell monolayers. Biometals 2011, 24, 857–874. [Google Scholar] [CrossRef] [PubMed]
- Prozialeck, W.C.; Edwards, J.R. Early biomarkers of cadmium exposure and nephrotoxicity. Biometals 2010, 23, 793–809. [Google Scholar] [CrossRef] [PubMed]
- Elinder, C.G.; Gerhardsson, L.; Oberdorster, G. Biological monitoring of toxic metals: Overview. In Biological Monitoring of Toxic Metals; Clarkson, T.W., Friberg, L., Nordberg, G.F., Sager, P.R., Eds.; Plenum Press: London, UK, 1988; pp. 1–71. [Google Scholar]
- Nordberg, G.F.; Nogawa, K.; Nordberg, M.; Friedmann, J.M. Cadmium. In Handbook on the Toxicology of Metals; Nordberg, G.F., Fowler, B.A., Nordberg, M., Friberg, L., Eds.; Elsevier: Amsterdam, The Netherlands, 2007; pp. 445–486. [Google Scholar]
- Nordberg, G.F.; Nordberg, M. Biological monitoring of cadmium. In Biological Monitoring of Toxic Metals; Clarkson, T.W., Friberg, L., Nordberg, G.F., Sager, P.R., Eds.; Plenum Press: New York, NY, USA, 2001; pp. 151–168. [Google Scholar]
- Johri, N.; Jacquillet, G.; Unwin, R. Heavy metal poisoning: The effects of cadmium on the kidney. Biometals 2010, 23, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Prozialeck, W.C.; VanDreel, A.; Ackerman, C.D.; Stock, I.; Papaeliou, A.; Yasmine, C.; Wilson, K.; Lamar, P.C.; Sears, V.L.; Gasiorowski, J.Z.; et al. Evaluation of cystatin C as an early biomarker of cadmium nephrotoxicity in the rat. Biometals 2016, 29, 131–146. [Google Scholar] [CrossRef] [PubMed]
- Akesson, A.; Lundh, T.; Vahter, M.; Bjellerup, P.; Lidfeldt, J.; Nerbrand, C.; Samsioe, G.; Stromberg, U.; Skerfving, S. Tubular and glomerular kidney effects in Swedish women with low environmental cadmium exposure. Environ. Health Perspect. 2005, 113, 1627–1631. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, K.; Iwao, S.; Sugita, M.; Sakurai, H. Increased urinary beta 2-microglobulin in cadmium exposure: Dose-effect relationship and biological significance of beta 2-microglobulin. Environ. Health Perspect. 1979, 28, 147–153. [Google Scholar] [PubMed]
- Jin, T.; Nordberg, G.; Wu, X.; Ye, T.; Kong, Q.; Wang, Z.; Zhuang, F.; Cai, S. Urinary N-acetyl-beta-d-glucosaminidase isoenzymes as biomarker of renal dysfunction caused by cadmium in a general population. Environ. Res. 1999, 81, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Prozialeck, W.C.; Vaidya, V.S.; Liu, J.; Waalkes, M.P.; Edwards, J.R.; Lamar, P.C.; Bernard, A.M.; Dumont, X.; Bonventre, J.V. Kidney injury molecule-1 is an early biomarker of cadmium nephrotoxicity. Kidney Int. 2007, 72, 985–993. [Google Scholar] [CrossRef] [PubMed]
- Jarup, L.; Alfven, T. Low level cadmium exposure, renal and bone effects—The OSCAR study. Biometals 2004, 17, 505–509. [Google Scholar] [CrossRef] [PubMed]
- Jarup, L.; Hellstrom, L.; Alfven, T.; Carlsson, M.D.; Grubb, A.; Persson, B.; Pettersson, C.; Spang, G.; Schutz, A.; Elinder, C.G. Low level exposure to cadmium and early kidney damage: The OSCAR study. Occup. Environ. Med. 2000, 57, 668–672. [Google Scholar] [CrossRef] [PubMed]
- Noonan, C.W.; Sarasua, S.M.; Campagna, D.; Kathman, S.J.; Lybarger, J.A.; Mueller, P.W. Effects of exposure to low levels of environmental cadmium on renal biomarkers. Environ. Health Perspect. 2002, 110, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, M.E.; Wong, L.Y.; Osterloh, J.D. Smoking status and urine cadmium above levels associated with subclinical renal effects in U.S. adults without chronic kidney disease. Int. J. Hyg. Environ. Health 2011, 214, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Choi, S.J.; Kim, D.W.; Kim, N.Y.; Park, C.H.; Yu, S.D.; Kim, D.S.; Park, K.S.; Song, J.S.; Kim, H.; et al. Risk assessment of low-level cadmium and arsenic on the kidney. J. Toxicol. Environ. Health A 2009, 72, 1493–1498. [Google Scholar] [CrossRef] [PubMed]
- Sabolic, I.; Ljubojevic, M.; Herak-Kramberger, C.M.; Brown, D. Cd-MT causes endocytosis of brush-border transporters in rat renal proximal tubules. Am. J. Physiol. Ren. Physiol. 2002, 283, F1389–F1402. [Google Scholar] [CrossRef]
- Herak-Kramberger, C.M.; Brown, D.; Sabolic, I. Cadmium inhibits vacuolar H(+)-ATPase and endocytosis in rat kidney cortex. Kidney Int. 1998, 53, 1713–1726. [Google Scholar] [CrossRef] [PubMed]
- Wolff, N.A.; Abouhamed, M.; Verroust, P.J.; Thevenod, F. Megalin-dependent internalization of cadmium-metallothionein and cytotoxicity in cultured renal proximal tubule cells. J. Pharmacol. Exp. Ther. 2006, 318, 782–791. [Google Scholar] [CrossRef] [PubMed]
- Santoyo-Sanchez, M.P.; Pedraza-Chaverri, J.; Molina-Jijon, E.; Arreola-Mendoza, L.; Rodriguez-Munoz, R.; Barbier, O.C. Impaired endocytosis in proximal tubule from subchronic exposure to cadmium involves angiotensin II type 1 and cubilin receptors. BMC Nephrol. 2013, 14, 211. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.Y.; Wu, K.H.; Chiu, W.T.; Wang, S.H.; Shih, C.M. The cadmium-induced death of mesangial cells results in nephrotoxicity. Autophagy 2009, 5, 571–572. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Templeton, D.M. Cadmium activates CaMK-II and initiates CaMK-II-dependent apoptosis in mesangial cells. FEBS Lett. 2007, 581, 1481–1486. [Google Scholar] [CrossRef] [PubMed]
- Jarup, L.; Persson, B.; Elinder, C.G. Decreased glomerular filtration rate in solderers exposed to cadmium. Occup. Environ. Med. 1995, 52, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Brzoska, M.M.; Kaminski, M.; Dziki, M.; Moniuszko-Jakoniuk, J. Changes in the structure and function of the kidney of rats chronically exposed to cadmium. II. Histoenzymatic studies. Arch. Toxicol. 2004, 78, 226–231. [Google Scholar] [PubMed]
- Jarup, L.; Elinder, C.G. Incidence of renal stones among cadmium exposed battery workers. Br. J. Ind. Med. 1993, 50, 598–602. [Google Scholar] [CrossRef] [PubMed]
- Aoshima, K.; Kasuya, M. Preliminary study on serum levels of 1,25-dihydroxyvitamin D and 25-hydroxyvitamin D in cadmium-induced renal tubular dysfunction. Toxicol. Lett. 1991, 57, 91–99. [Google Scholar] [PubMed]
- Tsuritani, I.; Honda, R.; Ishizaki, M.; Yamada, Y.; Kido, T.; Nogawa, K. Impairment of vitamin D metabolism due to environmental cadmium exposure, and possible relevance to sex-related differences in vulnerability to the bone damage. J. Toxicol. Environ. Health 1992, 37, 519–533. [Google Scholar] [CrossRef] [PubMed]
- Galazyn-Sidorczuk, M.; Brzoska, M.M.; Jurczuk, M.; Moniuszko-Jakoniuk, J. Oxidative damage to proteins and DNA in rats exposed to cadmium and/or ethanol. Chem. Biol. Interact. 2009, 180, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Matovic, V.; Buha, A.; Ethukic-Cosic, D.; Bulat, Z. Insight into the oxidative stress induced by lead and/or cadmium in blood, liver and kidneys. Food Chem. Toxicol. 2015, 78, 130–140. [Google Scholar] [CrossRef] [PubMed]
- So, K.Y.; Oh, S.H. Cadmium-induced heme-oxygenase-1 expression plays dual roles in autophagy and apoptosis and is regulated by both PKC-delta and PKB/Akt activation in NRK52E kidney cells. Toxicology 2016, 370, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Gu, D.; Zhou, M.; Shi, H.; Yan, S.; Cai, Y. Regulatory role of miR-125a/b in the suppression by selenium of cadmium-induced apoptosis via the mitochondrial pathway in LLC-PK1 cells. Chem. Biol. Interact. 2016, 243, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Babaknejad, N.; Moshtaghie, A.A.; Nayeri, H.; Hani, M.; Bahrami, S. Protective Role of Zinc and Magnesium against Cadmium Nephrotoxicity in Male Wistar Rats. Biol. Trace Elem. Res. 2016, 174, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.S.; Ho, W.C.; Caffrey, J.L.; Sonawane, B. Low serum zinc is associated with elevated risk of cadmium nephrotoxicity. Environ. Res. 2014, 134, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Jihen el, H.; Imed, M.; Fatima, H.; Abdelhamid, K. Protective effects of selenium (Se) and zinc (Zn) on cadmium (Cd) toxicity in the liver and kidney of the rat: Histology and Cd accumulation. Food Chem. Toxicol. 2008, 46, 3522–3527. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, Y.; Habeebu, S.S.; Klaassen, C.D. Metallothionein-null mice are highly susceptible to the hematotoxic and immunotoxic effects of chronic CdCl2 exposure. Toxicol. Appl. Pharmacol. 1999, 159, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Brzoska, M.M.; Roszczenko, A.; Galazyn-Sidorczuk, M.; Majewska, K. Zinc supplementation can protect from enhanced risk of femoral neck fracture in male rats chronically exposed to cadmium. Exp. Toxicol. Pathol. 2011, 63, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Jihen el, H.; Fatima, H.; Nouha, A.; Baati, T.; Imed, M.; Abdelhamid, K. Cadmium retention increase: A probable key mechanism of the protective effect of zinc on cadmium-induced toxicity in the kidney. Toxicol. Lett. 2010, 196, 104–109. [Google Scholar] [CrossRef] [PubMed]
- Jacquillet, G.; Barbier, O.; Cougnon, M.; Tauc, M.; Namorado, M.C.; Martin, D.; Reyes, J.L.; Poujeol, P. Zinc protects renal function during cadmium intoxication in the rat. Am. J. Physiol. Ren. Physiol. 2006, 290, F127–F137. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Liu, J.; Gao, J.; Shahzad, M.; Han, Z.; Wang, Z.; Li, J.; Sjolinder, H. Zinc supplementation protects against cadmium accumulation and cytotoxicity in Madin-Darby bovine kidney cells. PLoS ONE 2014, 9, e103427. [Google Scholar] [CrossRef] [PubMed]
- Matovic, V.; Buha, A.; Bulat, Z.; Dukic-Cosic, D. Cadmium toxicity revisited: Focus on oxidative stress induction and interactions with zinc and magnesium. Arhiv Hig. Rada Toksikol. 2011, 62, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Matovic, V.; Plamenac Bulat, Z.; Djukic-Cosic, D.; Soldatovic, D. Antagonism between cadmium and magnesium: A possible role of magnesium in therapy of cadmium intoxication. Magnes. Res. 2010, 23, 19–26. [Google Scholar] [PubMed]
- Quamme, G.A. Free cadmium activity in renal epithelial cells is enhanced by Mg2+ depletion. Kidney Int. 1992, 41, 1237–1244. [Google Scholar] [CrossRef] [PubMed]
- Djukic-Cosic, D.; Ninkovic, M.; Malicevic, Z.; Matovic, V.; Soldatovic, D. Effect of magnesium pretreatment on reduced glutathione levels in tissues of mice exposed to acute and subacute cadmium intoxication: A time course study. Magnes. Res. 2007, 20, 177–186. [Google Scholar] [PubMed]
- Huang, M.; Choi, S.J.; Kim, D.W.; Kim, N.Y.; Bae, H.S.; Yu, S.D.; Kim, D.S.; Kim, H.; Choi, B.S.; Yu, I.J.; et al. Evaluation of factors associated with cadmium exposure and kidney function in the general population. Environ. Toxicol. 2011, 28, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Ferraro, P.M.; Costanzi, S.; Naticchia, A.; Sturniolo, A.; Gambaro, G. Low level exposure to cadmium increases the risk of chronic kidney disease: Analysis of the NHANES 1999–2006. BMC Public Health 2010, 10, 304. [Google Scholar] [CrossRef] [PubMed]
- Navas-Acien, A.; Tellez-Plaza, M.; Guallar, E.; Muntner, P.; Silbergeld, E.; Jaar, B.; Weaver, V. Blood cadmium and lead and chronic kidney disease in US adults: A joint analysis. Am. J. Epidemiol. 2009, 170, 1156–1164. [Google Scholar] [CrossRef] [PubMed]
- Hellstrom, L.; Elinder, C.G.; Dahlberg, B.; Lundberg, M.; Jarup, L.; Persson, B.; Axelson, O. Cadmium exposure and end-stage renal disease. Am. J. Kidney Dis. 2001, 38, 1001–1008. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Lamberts, L.V.; Behets, G.J.; Zhao, T.; Zhou, M.; Liu, G.; Hou, X.; Guan, G.; D’Haese, P.C. Selenium, lead, and cadmium levels in renal failure patients in China. Biol. Trace Elem. Res. 2009, 131, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Gonick, H.C. Nephrotoxicity of cadmium & lead. Ind. J. Med. Res. 2008, 128, 335–352. [Google Scholar]
- Lauwerys, R.; Bernard, A.; Cardenas, A. Monitoring of early nephrotoxic effects of industrial chemicals. Toxicol. Lett. 1992, 64–65, 33–42. [Google Scholar] [CrossRef]
- Ginsberg, G.L. Cadmium risk assessment in relation to background risk of chronic kidney disease. J. Toxicol. Environ. Health A 2012, 75, 374–390. [Google Scholar] [CrossRef] [PubMed]
- Hwangbo, Y.; Weaver, V.M.; Tellez-Plaza, M.; Guallar, E.; Lee, B.K.; Navas-Acien, A. Blood cadmium and estimated glomerular filtration rate in Korean adults. Environ. Health Perspect. 2011, 119, 1800–1805. [Google Scholar] [CrossRef] [PubMed]
- Porter, G.A. Risk factors for toxic nephropathies. Toxicol. Lett. 1989, 46, 269–279. [Google Scholar] [CrossRef]
- Roels, H.A.; Hoet, P.; Lison, D. Usefulness of biomarkers of exposure to inorganic mercury, lead, or cadmium in controlling occupational and environmental risks of nephrotoxicity. Ren. Fail. 1999, 21, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Mueller, P.W.; Price, R.G.; Finn, W.F. New approaches for detecting thresholds of human nephrotoxicity using cadmium as an example. Environ. Health Perspect. 1998, 106, 227–230. [Google Scholar] [CrossRef] [PubMed]
- Jayatilake, N.; Mendis, S.; Maheepala, P.; Mehta, F.R. Chronic kidney disease of uncertain aetiology: Prevalence and causative factors in a developing country. BMC Nephrol. 2013, 14, 180. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.H.; Hyun, Y.Y.; Lee, K.B.; Chang, Y.; Ryu, S.; Oh, K.H.; Ahn, C. Environmental heavy metal exposure and chronic kidney disease in the general population. J. Korean Med. Sci. 2015, 30, 272–277. [Google Scholar] [CrossRef] [PubMed]
- Thomas, L.D.; Elinder, C.G.; Wolk, A.; Akesson, A. Dietary cadmium exposure and chronic kidney disease: A population-based prospective cohort study of men and women. Int. J. Hyg. Environ. Health 2014, 217, 720–725. [Google Scholar] [CrossRef] [PubMed]
- Satarug, S.; Vesey, D.A.; Gobe, G.C. Kidney Cadmium Toxicity, Diabetes and High Blood Pressure: The Perfect Storm. Tohoku J. Exp. Med. 2017, 241, 65–87. [Google Scholar] [CrossRef] [PubMed]
- Hayslett, J.P. Functional adaptation to reduction in renal mass. Physiol. Rev. 1979, 59, 137–164. [Google Scholar] [PubMed]
- Zalups, R.K.; Fraser, J.; Koropatnick, J. Enhanced transcription of metallothionein genes in rat kidney: Effect of uninephrectomy and compensatory renal growth. Am. J. Physiol. 1995, 268, F643–F650. [Google Scholar] [PubMed]
- Edwards, J.R.; Prozialeck, W.C. Cadmium, diabetes and chronic kidney disease. Toxicol. Appl. Pharmacol. 2009, 238, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Nordberg, G.F.; Jin, T.; Wu, X.; Lu, J.; Chen, L.; Lei, L.; Hong, F.; Nordberg, M. Prevalence of kidney dysfunction in humans-relationship to cadmium dose, metallothionein, immunological and metabolic factors. Biochimie 2009, 91, 1282–1285. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, Y.; Wang, W.; Wu, Y. Association of urinary cadmium with risk of diabetes: A meta-analysis. Environ. Sci. Pollut. Res. Int. 2017, 24, 10083–10090. [Google Scholar] [CrossRef] [PubMed]
- Schrijvers, B.F.; De Vriese, A.S.; Flyvbjerg, A. From hyperglycemia to diabetic kidney disease: The role of metabolic, hemodynamic, intracellular factors and growth factors/cytokines. Endocr. Rev. 2004, 25, 971–1010. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.R.; Early, J.L.; Nonavinakere, V.K.; Mallory, Z. Effect of cadmium on blood glucose level in the rat. Toxicol. Lett. 1990, 54, 199–205. [Google Scholar] [CrossRef]
- Chapatwala, K.D.; Boykin, M.; Butts, A.; Rajanna, B. Effect of intraperitoneally injected cadmium on renal and hepatic gluconeogenic enzymes in rats. Drug Chem. Toxicol. 1982, 5, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Lei, L.J.; Jin, T.Y.; Zhou, Y.F. Insulin expression in rats exposed to cadmium. Biomed. Environ. Sci. 2007, 20, 295–301. [Google Scholar] [PubMed]
- Merali, Z.; Singhal, R.L. Diabetogenic effects of chronic oral cadmium adminstration to neonatal rats. Br. J. Pharmacol. 1980, 69, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Lei, L.J.; Jin, T.Y.; Zhou, Y.F. Effects of cadmium on levels of insulin in rats. J. Hyg. Res. 2005, 34, 394–396. [Google Scholar]
- Jin, T.; Nordberg, G.; Sehlin, J.; Wallin, H.; Sandberg, S. The susceptibility to nephrotoxicity of streptozotocin-induced diabetic rats subchronically exposed to cadmium chloride in drinking water. Toxicology 1999, 142, 69–75. [Google Scholar] [CrossRef]
- Bernard, A.; Schadeck, C.; Cardenas, A.; Buchet, J.P.; Lauwerys, R. Potentiation of diabetic glomerulopathy in uninephrectomized rats subchronically exposed to cadmium. Toxicol. Lett. 1991, 58, 51–57. [Google Scholar] [CrossRef]
- Jin, T.; Nordberg, G.F.; Sehlin, J.; Leffler, P.; Wu, J. The susceptibility of spontaneously diabetic mice to cadmium-metallothionein nephrotoxicity. Toxicology 1994, 89, 81–90. [Google Scholar] [CrossRef]
- Haswell-Elkins, M.; Satarug, S.; O'Rourke, P.; Moore, M.; Ng, J.; McGrath, V.; Walmby, M. Striking association between urinary cadmium level and albuminuria among Torres Strait Islander people with diabetes. Environ. Res. 2008, 106, 379–383. [Google Scholar] [CrossRef] [PubMed]
- Buchet, J.P.; Lauwerys, R.; Roels, H.; Bernard, A.; Bruaux, P.; Claeys, F.; Ducoffre, G.; de Plaen, P.; Staessen, J.; Amery, A.; et al. Renal effects of cadmium body burden of the general population. Lancet 1990, 336, 699–702. [Google Scholar] [CrossRef]
- Satarug, S.; Ujjin, P.; Vanavanitkun, Y.; Baker, J.R.; Moore, M.R. Influence of body iron store status and cigarette smoking on cadmium body burden of healthy Thai women and men. Toxicol. Lett. 2004, 148, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Berglund, M.; Akesson, A.; Nermell, B.; Vahter, M. Intestinal absorption of dietary cadmium in women depends on body iron stores and fiber intake. Environ. Health Perspect. 1994, 102, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Akesson, A.; Berglund, M.; Schutz, A.; Bjellerup, P.; Bremme, K.; Vahter, M. Cadmium exposure in pregnancy and lactation in relation to iron status. Am. J. Public Health 2002, 92, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.W.; Kim, K.Y.; Choi, B.S.; Youn, P.; Ryu, D.Y.; Klaassen, C.D.; Park, J.D. Regulation of metal transporters by dietary iron, and the relationship between body iron levels and cadmium uptake. Arch. Toxicol. 2007, 81, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Vesey, D.A. Transport pathways for cadmium in the intestine and kidney proximal tubule: Focus on the interaction with essential metals. Toxicol. Lett. 2010, 198, 13–19. [Google Scholar] [CrossRef] [PubMed]
- DeWitt, R.D. Pediatric lead exposure and the water crisis in Flint, Michigan. J. Am. Acad. Phys. Assist. 2017, 30, 43–46. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, C.; Yard, E.; Dignam, T.; Buchanan, S.; Condon, S.; Brown, M.J.; Raymond, J.; Rogers, H.S.; Sarisky, J.; de Castro, R.; et al. Blood Lead Levels Among Children Aged. MMWR 2016, 65, 650–654. [Google Scholar] [PubMed]
- Shah, K.K.; Oleske, J.M.; Gomez, H.F.; Davidow, A.L.; Bogden, J.D. Blood Lead Concentrations of Children in the United States: A Comparison of States Using Two Very Large Databases. J. Pediatr. 2017. [Google Scholar] [CrossRef] [PubMed]
- Centers of Disease Control and Prevention (CDC). Very high blood lead levels among adults—United States, 2002–2011. MMWR 2013, 62, 967–971. [Google Scholar]
- Raymond, J.; Brown, M.J. Childhood Blood Lead Levels in Children Aged. MMWR 2017, 66, 1–10. [Google Scholar] [PubMed]
- Alarcon, W.A. Elevated Blood Lead Levels Among Employed Adults—United States, 1994–2013. MMWR 2016, 63, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Goyer, R.A. Lead toxicity: Current concerns. Environ. Health Perspect. 1993, 100, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Dapul, H.; Laraque, D. Lead poisoning in children. Adv. Pediatr. 2014, 61, 313–333. [Google Scholar] [CrossRef] [PubMed]
- Fowler, B.A. Mechanisms of Kidney Cell Injury from Metals. Environ. Health Perspect. 1992, 100, 57–63. [Google Scholar] [CrossRef]
- Smith, D.R.; Kahng, M.W.; Quintanilla-Vega, B.; Fowler, B.A. High-affinity renal lead-binding proteins in environmentally-exposed humans. Chem. Biol. Interact. 1998, 115, 39–52. [Google Scholar] [CrossRef]
- Oskarsson, A.; Squibb, K.S.; Fowler, B.A. Intracellular binding of lead in the kidney: The partial isolation and characterization of postmitochondrial lead binding components. Biochem. Biophys. Res. Commun. 1982, 104, 290–298. [Google Scholar] [CrossRef]
- DuVal, G.; Fowler, B.A. Preliminary purification and characterization studies of a low molecular weight, high affinity cytosolic lead-binding protein in rat brain. Biochem. Biophys. Res. Commun. 1989, 159, 177–184. [Google Scholar] [CrossRef]
- Fowler, B.A.; DuVal, G. Effects of lead on the kidney: Roles of high-affinity lead-binding proteins. Environ. Health Perspect. 1991, 91, 77–80. [Google Scholar] [CrossRef] [PubMed]
- Swenberg, J.A.; Short, B.; Borghoff, S.; Strasser, J.; Charbonneau, M. The comparative pathobiology of alpha 2u-globulin nephropathy. Toxicol. Appl. Pharmacol. 1989, 97, 35–46. [Google Scholar] [CrossRef]
- Goyer, R.A. Mechanisms of lead and cadmium nephrotoxicity. Toxicol. Lett. 1989, 46, 153–162. [Google Scholar] [CrossRef]
- Marchetti, C. Role of calcium channels in heavy metal toxicity. ISRN Toxicol. 2013, 2013, 184360. [Google Scholar] [CrossRef] [PubMed]
- Goyer, R.A. Toxic and essential metal interactions. Annu. Rev. Nutr. 1997, 17, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, E.E.; Edwards, B.B.; Jensen, R.L.; Mahaffey, K.R.; Fomon, S.J. Absorption and retention of lead by infants. Pediatr. Res. 1978, 12, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, M.; Lv, Q.; Chen, G.; Li, Y.; Li, S.; Mo, Y.; Ou, S.; Yuan, Z.; Huang, M.; et al. Blood lead level and its relationship to essential elements in preschool children from Nanning, China. J. Trace Elem. Med. Biol. 2015, 30, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Six, K.M.; Goyer, R.A. Experimental enhancement of lead toxicity by low dietary calcium. J. Lab. Clin. Med. 1970, 76, 933–942. [Google Scholar] [PubMed]
- Bogden, J.D.; Gertner, S.B.; Christakos, S.; Kemp, F.W.; Yang, Z.; Katz, S.R.; Chu, C. Dietary calcium modifies concentrations of lead and other metals and renal calbindin in rats. J. Nutr. 1992, 122, 1351–1360. [Google Scholar] [PubMed]
- Mahaffey, K.R.; Gartside, P.S.; Glueck, C.J. Blood lead levels and dietary calcium intake in 1- to 11-year-old children: The Second National Health and Nutrition Examination Survey, 1976 to 1980. Pediatrics 1986, 78, 257–262. [Google Scholar] [PubMed]
- Blake, K.C.; Mann, M. Effect of calcium and phosphorus on the gastrointestinal absorption of 203Pb in man. Environ. Res. 1983, 30, 188–194. [Google Scholar] [CrossRef]
- Barton, J.C. Active transport of lead-210 by everted segments of rat duodenum. Am. J. Physiol. 1984, 247, G193–G198. [Google Scholar] [PubMed]
- Giebisch, G.; Windhager, E. Transport of urea, glucose, phosphate, calcium, magnesium, and organic solutes. In Medical Physiology, 2nd ed.; Boron, W.F., Boulpaep, E.L., Eds.; Elsevier: Philadelphia, PA, USA, 2012; pp. 797–820. [Google Scholar]
- Kerper, L.E.; Hinkle, P.M. Cellular uptake of lead is activated by depletion of intracellular calcium stores. J. Biol. Chem. 1997, 272, 8346–8352. [Google Scholar] [CrossRef] [PubMed]
- Simons, T.J. Lead transport and binding by human erythrocytes in vitro. Pflugers Arch. 1993, 423, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Simons, T.J. Lead-calcium interactions in cellular lead toxicity. Neurotoxicology 1993, 14, 77–85. [Google Scholar] [PubMed]
- Simons, T.J.; Pocock, G. Lead enters bovine adrenal medullary cells through calcium channels. J. Neurochem. 1987, 48, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.; Hajela, R.K.; Atchison, W.D. Characteristics of block by Pb2+ of function of human neuronal L-, N-, and R-type Ca2+ channels transiently expressed in human embryonic kidney 293 cells. Mol. Pharmacol. 2002, 62, 1418–1430. [Google Scholar] [CrossRef] [PubMed]
- Chiu, T.Y.; Teng, H.C.; Huang, P.C.; Kao, F.J.; Yang, D.M. Dominant role of Orai1 with STIM1 on the cytosolic entry and cytotoxicity of lead ions. Toxicol. Sci. 2009, 110, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Pfleger, H.; Wolf, H.U. Activation of membrane-bound high-affinity calcium ion-sensitive adenosine triphosphatase of human erythrocytes by bivalent metal ions. Biochem. J. 1975, 147, 359–361. [Google Scholar] [CrossRef] [PubMed]
- Alexander, J.; Aaseth, J.; Mikalsen, A. Excretion of lead in rat bile—The role of glutathione. Acta Pharmacol. Toxicol. 1986, 59 (Suppl. S7), 486–489. [Google Scholar] [CrossRef]
- Patrick, L. Lead toxicity part II: The role of free radical damage and the use of antioxidants in the pathology and treatment of lead toxicity. Altern. Med. Rev. 2006, 11, 114–127. [Google Scholar] [PubMed]
- Basgen, J.M.; Sobin, C. Early chronic low-level lead exposure produces glomerular hypertrophy in young C57BL/6J mice. Toxicol. Lett. 2014, 225, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Ritz, E.; Mann, J.; Stoeppler, M. Lead and the kidney. Adv. Nephrol. Necker Hosp. 1988, 17, 241–274. [Google Scholar] [PubMed]
- Navarro-Moreno, L.G.; Quintanar-Escorza, M.A.; Gonzalez, S.; Mondragon, R.; Cerbon-Solorzano, J.; Valdes, J.; Calderon-Salinas, J.V. Effects of lead intoxication on intercellular junctions and biochemical alterations of the renal proximal tubule cells. Toxicol. In Vitro 2009, 23, 1298–1304. [Google Scholar] [CrossRef] [PubMed]
- Cardenas, A.; Roels, H.; Bernard, A.M.; Barbon, R.; Buchet, J.P.; Lauwerys, R.R.; Rosello, J.; Ramis, I.; Mutti, A.; Franchini, I.; et al. Markers of early renal changes induced by industrial pollutants. II. Application to workers exposed to lead. Br. J. Ind. Med. 1993, 50, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Soliman, M.M.; Baiomy, A.A.; Yassin, M.H. Molecular and Histopathological Study on the Ameliorative Effects of Curcumin Against Lead Acetate-Induced Hepatotoxicity and Nephrototoxicity in Wistar Rats. Biol. Trace Elem. Res. 2015, 167, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Xu, Y.; Shen, J.; Han, L.; Chen, X.; Feng, X.; Kuang, X. Urinary KIM-1: A novel biomarker for evaluation of occupational exposure to lead. Sci. Rep. 2016, 6, 38930. [Google Scholar] [CrossRef] [PubMed]
- Garcon, G.; Leleu, B.; Marez, T.; Zerimech, F.; Haguenoer, J.M.; Furon, D.; Shirali, P. Biomonitoring of the adverse effects induced by the chronic exposure to lead and cadmium on kidney function: Usefulness of alpha-glutathione S-transferase. Sci. Total Environ. 2007, 377, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Gao, X.; Guo, M.; Jiang, H.; Cao, Y.; Zhang, N. The Protective Effect of Baicalin Against Lead-Induced Renal Oxidative Damage in Mice. Biol. Trace Elem. Res. 2017, 175, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, H.; Hu, M.; Cao, J.; Chen, D.; Liu, Z. Oxidative stress and apoptotic changes in primary cultures of rat proximal tubular cells exposed to lead. Arch. Toxicol. 2009, 83, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Dewanjee, S.; Sahu, R.; Karmakar, S.; Gangopadhyay, M. Toxic effects of lead exposure in Wistar rats: Involvement of oxidative stress and the beneficial role of edible jute (Corchorus olitorius) leaves. Food Chem. Toxicol. 2013, 55, 78–91. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Wang, Z.K.; Wang, Z.Y.; Yang, D.B.; Liu, Z.P.; Wang, L. Mitochondrial permeability transition and its regulatory components are implicated in apoptosis of primary cultures of rat proximal tubular cells exposed to lead. Arch. Toxicol. 2016, 90, 1193–1209. [Google Scholar] [CrossRef] [PubMed]
- Bonora, M.; Morganti, C.; Morciano, G.; Giorgi, C.; Wieckowski, M.R.; Pinton, P. Comprehensive analysis of mitochondrial permeability transition pore activity in living cells using fluorescence-imaging-based techniques. Nat. Protoc. 2016, 11, 1067–1080. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Cao, J.; Chen, D.; Liu, X.; Lu, H.; Liu, Z. Role of oxidative stress, apoptosis, and intracellular homeostasis in primary cultures of rat proximal tubular cells exposed to cadmium. Biol. Trace Elem. Res. 2009, 127, 53–68. [Google Scholar] [CrossRef] [PubMed]
- Jacotot, E.; Deniaud, A.; Borgne-Sanchez, A.; Touat, Z.; Briand, J.P.; Le Bras, M.; Brenner, C. Therapeutic peptides: Targeting the mitochondrion to modulate apoptosis. Biochim. Biophys. Acta 2006, 1757, 1312–1323. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, G.W.; Ar, D. Lead activates calmodulin sensitive processes. Life Sci. 1983, 33, 1001–1006. [Google Scholar] [CrossRef]
- Habermann, E.; Crowell, K.; Janicki, P. Lead and other metals can substitute for Ca2+ in calmodulin. Arch. Toxicol. 1983, 54, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Stoclet, J.C.; Gerard, D.; Kilhoffer, M.C.; Lugnier, C.; Miller, R.; Schaeffer, P. Calmodulin and its role in intracellular calcium regulation. Prog. Neurobiol. 1987, 29, 321–364. [Google Scholar] [CrossRef]
- Wang, H.; Wang, Z.K.; Jiao, P.; Zhou, X.P.; Yang, D.B.; Wang, Z.Y.; Wang, L. Redistribution of subcellular calcium and its effect on apoptosis in primary cultures of rat proximal tubular cells exposed to lead. Toxicology 2015, 333, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Cao, H.; Zhang, Y.; Ma, J.; Wang, J.; Gao, Y.; Zhang, X.; Zhang, F.; Chu, L. Nephroprotective effect of calcium channel blockers against toxicity of lead exposure in mice. Toxicol. Lett. 2013, 218, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Staessen, J.; Yeoman, W.B.; Fletcher, A.E.; Markowe, H.L.; Marmot, M.G.; Rose, G.; Semmence, A.; Shipley, M.J.; Bulpitt, C.J. Blood lead concentration, renal function, and blood pressure in London civil servants. Br. J. Ind. Med. 1990, 47, 442–447. [Google Scholar] [CrossRef] [PubMed]
- Ekong, E.B.; Jaar, B.G.; Weaver, V.M. Lead-related nephrotoxicity: A review of the epidemiologic evidence. Kidney Int. 2006, 70, 2074–2084. [Google Scholar] [CrossRef] [PubMed]
- Fadrowski, J.J.; Navas-Acien, A.; Tellez-Plaza, M.; Guallar, E.; Weaver, V.M.; Furth, S.L. Blood lead level and kidney function in US adolescents: The Third National Health and Nutrition Examination Survey. Arch. Int. Med. 2010, 170, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Spector, J.T.; Navas-Acien, A.; Fadrowski, J.; Guallar, E.; Jaar, B.; Weaver, V.M. Associations of blood lead with estimated glomerular filtration rate using MDRD, CKD-EPI and serum cystatin C-based equations. Nephrol. Dial. Transplant. 2011, 26, 2786–2792. [Google Scholar] [CrossRef] [PubMed]
- Landrigan, P.J. Current issues in the epidemiology and toxicology of occupational exposure to lead. Environ. Health Perspect. 1990, 89, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Serrato, M.I.; Fortoul, T.I.; Rojas-Martinez, R.; Mendoza-Alvarado, L.R.; Canales-Trevino, L.; Bochichio-Riccardelli, T.; Avila-Costa, M.R.; Olaiz-Fernandez, G. Lead blood concentrations and renal function evaluation: Study in an exposed Mexican population. Environ. Res. 2006, 100, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.; Chung, J.H.; Kim, S.J.; Koh, E.S.; Yoon, H.E.; Park, C.W.; Chang, Y.S.; Shin, S.J. Blood lead and cadmium levels and renal function in Korean adults. Clin. Exp. Nephrol. 2014, 18, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Sommar, J.N.; Svensson, M.K.; Bjor, B.M.; Elmstahl, S.I.; Hallmans, G.; Lundh, T.; Schon, S.M.; Skerfving, S.; Bergdahl, I.A. End-stage renal disease and low level exposure to lead, cadmium and mercury; a population-based, prospective nested case-referent study in Sweden. Environ. Health 2013, 12, 9. [Google Scholar] [CrossRef] [PubMed]
- Agency for Toxic Substances and Disease Registry (ATSDR). Toxicological Profile for Mercury. In ATSDR’s Toxicological Profiles; U.S. Department of Health and Human Services, Public Health Service (PHS); Centers for Disease Control and Prevention: Atlanta, GA, USA, 2008. [Google Scholar]
- Clarkson, T.W.; Magos, L. The toxicology of mercury and its chemical compounds. Crit. Rev. Toxicol. 2006, 36, 609–662. [Google Scholar] [CrossRef] [PubMed]
- Rooney, J.P. The role of thiols, dithiols, nutritional factors and interacting ligands in the toxicology of mercury. Toxicology 2007, 234, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Zalups, R.K. Molecular interactions with mercury in the kidney. Pharmacol. Rev. 2000, 52, 113–143. [Google Scholar] [PubMed]
- Risher, J.F.; De Rosa, C.T. Inorganic: The other mercury. J. Environ. Health 2007, 70, 9–16. [Google Scholar] [PubMed]
- Gage, J.C. Distribution and Excretion of Methyl and Phenyl Mercury Salts. Br. J. Ind. Med. 1964, 21, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Norseth, T.; Clarkson, T.W. Studies on the biotransformation of 203Hg-labeled methyl mercury chloride in rats. Arch. Environ. Health 1970, 21, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Norseth, T.; Clarkson, T.W. Biotransformation of methylmercury salts in the rat studied by specific determination of inorganic mercury. Biochem. Pharmacol. 1970, 19, 2775–2783. [Google Scholar] [CrossRef]
- Omata, S.; Sato, M.; Sakimura, K.; Sugano, H. Time-dependent accumulation of inorganic mercury in subcellular fractions of kidney, liver, and brain of rats exposed to methylmercury. Arch. Toxicol. 1980, 44, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, J.B. Toxicokinetics of mercuric chloride and methylmercuric chloride in mice. J. Toxicol. Environ. Health 1992, 37, 85–122. [Google Scholar] [CrossRef] [PubMed]
- Zalups, R.K. Early aspects of the intrarenal distribution of mercury after the intravenous administration of mercuric chloride. Toxicology 1993, 79, 215–228. [Google Scholar] [CrossRef]
- Murphy, M.J.; Culliford, E.J.; Parsons, V. A case of poisoning with mercuric chloride. Resuscitation 1979, 7, 35–44. [Google Scholar] [CrossRef]
- Rowens, B.; Guerrero-Betancourt, D.; Gottlieb, C.A.; Boyes, R.J.; Eichenhorn, M.S. Respiratory failure and death following acute inhalation of mercury vapor. A clinical and histologic perspective. Chest 1991, 99, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Samuels, E.R.; Heick, H.M.; McLaine, P.N.; Farant, J.P. A case of accidental inorganic mercury poisoning. J. Anal. Toxicol. 1982, 6, 120–122. [Google Scholar] [CrossRef] [PubMed]
- Yasutake, A.; Hirayama, K.; Inoue, M. Mechanism of urinary excretion of methylmercury in mice. Arch. Toxicol. 1989, 63, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Hughes, W.L. A physicochemical rationale for the biological activity of mercury and its compounds. Ann. N. Y. Acad. Sci. 1957, 65, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Fuhr, B.J.; Rabenstein, D.L. Nuclear magnetic resonance studies of the solution chemistry of metal complexes. IX. The binding of cadmium, zinc, lead, and mercury by glutathione. J. Am. Chem. Soc. 1973, 95, 6944–6950. [Google Scholar] [CrossRef] [PubMed]
- Rubino, F.M.; Verduci, C.; Giampiccolo, R.; Pulvirenti, S.; Brambilla, G.; Colombi, A. Molecular characterization of homo- and heterodimeric mercury(II)-bis-thiolates of some biologically relevant thiols by electrospray ionization and triple quadrupole tandem mass spectrometry. J. Am. Soc. Mass Spectrom. 2004, 15, 288–300. [Google Scholar] [CrossRef] [PubMed]
- Cannon, V.T.; Barfuss, D.W.; Zalups, R.K. Molecular homology and the luminal transport of Hg2+ in the renal proximal tubule. J. Am. Soc. Nephrol. 2000, 11, 394–402. [Google Scholar] [PubMed]
- Cannon, V.T.; Zalups, R.K.; Barfuss, D.W. Amino acid transporters involved in luminal transport of mercuric conjugates of cysteine in rabbit proximal tubule. J. Pharmacol. Exp. Ther. 2001, 298, 780–789. [Google Scholar] [PubMed]
- Zalups, R.K. Basolateral uptake of inorganic mercury in the kidney. Toxicol. Appl. Pharmacol. 1998, 151, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Zalups, R.K.; Lash, L.H. Binding of mercury in renal brush-border and basolateral membrane-vesicles. Biochem. Pharmacol. 1997, 53, 1889–1900. [Google Scholar] [CrossRef]
- Zalups, R.K.; Minor, K.H. Luminal and basolateral mechanisms involved in the renal tubular uptake of inorganic mercury. J. Toxicol. Environ. Health 1995, 46, 73–100. [Google Scholar] [CrossRef] [PubMed]
- Zalups, R.K.; Barfuss, D.W. Accumulation and handling of inorganic mercury in the kidney after coadministration with glutathione. J. Toxicol. Environ. Health 1995, 44, 385–399. [Google Scholar] [CrossRef] [PubMed]
- Zalups, R.K.; Barfuss, D.W. Nephrotoxicity of inorganic mercury co-administrated with l-cysteine. Toxicology 1996, 109, 15–29. [Google Scholar] [CrossRef]
- Zalups, R.K.; Barfuss, D.W. Participation of mercuric conjugates of cysteine, homocysteine, and N-acetylcysteine in mechanisms involved in the renal tubular uptake of inorganic mercury. J. Am. Soc. Nephrol. 1998, 9, 551–561. [Google Scholar] [PubMed]
- Bridges, C.C.; Bauch, C.; Verrey, F.; Zalups, R.K. Mercuric conjugates of cysteine are transported by the amino acid transporter system b(0,+): Implications of molecular mimicry. J. Am. Soc. Nephrol. 2004, 15, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Bridges, C.C.; Zalups, R.K. Homocysteine, system b0,+ and the renal epithelial transport and toxicity of inorganic mercury. Am. J. Pathol. 2004, 165, 1385–1394. [Google Scholar] [CrossRef]
- Bridges, C.C.; Zalups, R.K. System B0,+ and the transport of thiol-S-conjugates of methylmercury. J. Pharmacol. Exp. Ther. 2006, 319, 948–956. [Google Scholar] [CrossRef] [PubMed]
- Zalups, R.K.; Barfuss, D.W. Pretreatment with p-aminohippurate inhibits the renal uptake and accumulation of injected inorganic mercury in the rat. Toxicology 1995, 103, 23–35. [Google Scholar] [CrossRef]
- Zalups, R.K.; Barfuss, D.W. Small aliphatic dicarboxylic acids inhibit renal uptake of administered mercury. Toxicol. Appl. Pharmacol. 1998, 148, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Zalups, R.K. Organic anion transport and action of gamma-glutamyl transpeptidase in kidney linked mechanistically to renal tubular uptake of inorganic mercury. Toxicol. Appl. Pharmacol. 1995, 132, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Zalups, R.K. Basolateral uptake of mercuric conjugates of N-acetylcysteine and cysteine in the kidney involves the organic anion transport system. J. Toxicol. Environ. Health A 1998, 55, 13–29. [Google Scholar] [CrossRef] [PubMed]
- Ferrier, B.; Martin, M.; Roch-Ramel, F. Effects of p-aminohippurate and pyrazinoate on the renal excretion of salicylate in the rat: A micropuncture study. J. Pharmacol. Exp. Ther. 1983, 224, 451–458. [Google Scholar] [PubMed]
- Koh, A.S.; Simmons-Willis, T.A.; Pritchard, J.B.; Grassl, S.M.; Ballatori, N. Identification of a mechanism by which the methylmercury antidotes N-acetylcysteine and dimercaptopropanesulfonate enhance urinary metal excretion: Transport by the renal organic anion transporter-1. Mol. Pharmacol. 2002, 62, 921–926. [Google Scholar] [CrossRef] [PubMed]
- Kojima, R.; Sekine, T.; Kawachi, M.; Cha, S.H.; Suzuki, Y.; Endou, H. Immunolocalization of multispecific organic anion transporters, OAT1, OAT2, and OAT3, in rat kidney. J. Am. Soc. Nephrol. 2002, 13, 848–857. [Google Scholar] [PubMed]
- Motohashi, H.; Sakurai, Y.; Saito, H.; Masuda, S.; Urakami, Y.; Goto, M.; Fukatsu, A.; Ogawa, O.; Inui, K. Gene expression levels and immunolocalization of organic ion transporters in the human kidney. J. Am. Soc. Nephrol. 2002, 13, 866–874. [Google Scholar] [PubMed]
- Pritchard, J.B. Coupled transport of p-aminohippurate by rat kidney basolateral membrane vesicles. Am. J. Physiol. 1988, 255, F597–F604. [Google Scholar] [PubMed]
- Shimomura, A.; Chonko, A.M.; Grantham, J.J. Basis for heterogeneity of para-aminohippurate secretion in rabbit proximal tubules. Am. J. Physiol. 1981, 240, F430–F436. [Google Scholar] [PubMed]
- Tanaka, T.; Naganuma, A.; Miura, N.; Imura, N. Role of testosterone in gamma-glutamyltranspeptidase-dependent renal methylmercury uptake in mice. Toxicol. Appl. Pharmacol. 1992, 112, 58–63. [Google Scholar] [CrossRef]
- Ullrich, K.J.; Rumrich, G.; Fritzsch, G.; Kloss, S. Contraluminal para-aminohippurate (PAH) transport in the proximal tubule of the rat kidney. II. Specificity: Aliphatic dicarboxylic acids. Pflugers Arch. 1987, 408, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Zalups, R.K.; Ahmad, S. Handling of cysteine S-conjugates of methylmercury in MDCK cells expressing human OAT1. Kidney Int. 2005, 68, 1684–1699. [Google Scholar] [CrossRef] [PubMed]
- Zalups, R.K.; Ahmad, S. Handling of the homocysteine S-conjugate of methylmercury by renal epithelial cells: Role of organic anion transporter 1 and amino acid transporters. J. Pharmacol. Exp. Ther. 2005, 315, 896–904. [Google Scholar] [CrossRef] [PubMed]
- Zalups, R.K.; Ahmad, S. Transport of N-acetylcysteine S-conjugates of methylmercury in Madin-Darby canine kidney cells stably transfected with human isoform of organic anion transporter 1. J. Pharmacol. Exp. Ther. 2005, 314, 1158–1168. [Google Scholar] [CrossRef] [PubMed]
- Aslamkhan, A.G.; Han, Y.H.; Yang, X.P.; Zalups, R.K.; Pritchard, J.B. Human renal organic anion transporter 1-dependent uptake and toxicity of mercuric-thiol conjugates in Madin-Darby canine kidney cells. Mol. Pharmacol. 2003, 63, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Zalups, R.K.; Ahmad, S. Homocysteine and the renal epithelial transport and toxicity of inorganic mercury: Role of basolateral transporter organic anion transporter 1. J. Am. Soc. Nephrol. 2004, 15, 2023–2031. [Google Scholar] [CrossRef] [PubMed]
- Zalups, R.K.; Aslamkhan, A.G.; Ahmad, S. Human organic anion transporter 1 mediates cellular uptake of cysteine-S conjugates of inorganic mercury. Kidney Int. 2004, 66, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Cherian, M.G.; Clarkson, T.W. Biochemical changes in rat kidney on exposure to elemental mercury vapor: Effect on biosynthesis of metallothionein. Chem. Biol. Interact. 1976, 12, 109–120. [Google Scholar] [CrossRef]
- Zalups, R.K.; Koropatnick, J. Temporal changes in metallothionein gene transcription in rat kidney and liver: Relationship to content of mercury and metallothionein protein. J. Pharmacol. Exp. Ther. 2000, 295, 74–82. [Google Scholar] [PubMed]
- Ruprecht, J. Scientific Monograph for Dimaval; Heyltex Corporation: Houston, TX, USA, 2008. [Google Scholar]
- Aposhian, H.V. DMSA and DMPS—Water soluble antidotes for heavy metal poisoning. Annu. Rev. Pharmacol. Toxicol. 1983, 23, 193–215. [Google Scholar] [CrossRef] [PubMed]
- Aposhian, H.V.; Maiorino, R.M.; Gonzalez-Ramirez, D.; Zuniga-Charles, M.; Xu, Z.; Hurlbut, K.M.; Junco-Munoz, P.; Dart, R.C.; Aposhian, M.M. Mobilization of heavy metals by newer, therapeutically useful chelating agents. Toxicology 1995, 97, 23–38. [Google Scholar] [CrossRef]
- Aposhian, H.V.; Maiorino, R.M.; Rivera, M.; Bruce, D.C.; Dart, R.C.; Hurlbut, K.M.; Levine, D.J.; Zheng, W.; Fernando, Q.; Carter, D.; et al. Human studies with the chelating agents, DMPS and DMSA. J. Toxicol. Clin. Toxicol. 1992, 30, 505–528. [Google Scholar] [CrossRef] [PubMed]
- Planas-Bohne, F. The effect of 2,3-dimercaptorpropane-1-sulfonate and dimercaptosuccinic acid on the distribution and excretion of mercuric chloride in rats. Toxicology 1981, 19, 275–278. [Google Scholar] [CrossRef]
- Zalups, R.K. Influence of 2,3-dimercaptopropane-1-sulfonate (DMPS) and meso-2,3-dimercaptosuccinic acid (DMSA) on the renal disposition of mercury in normal and uninephrectomized rats exposed to inorganic mercury. J. Pharmacol. Exp. Ther. 1993, 267, 791–800. [Google Scholar] [PubMed]
- Zalups, R.K.; Bridges, C.C. Relationships between the renal handling of DMPS and DMSA and the renal handling of mercury. Chem. Res. Toxicol. 2012, 25, 1825–1838. [Google Scholar] [CrossRef] [PubMed]
- Rodiger, M.; Zhang, X.; Ugele, B.; Gersdorff, N.; Wright, S.H.; Burckhardt, G.; Bahn, A. Organic anion transporter 3 (OAT3) and renal transport of the metal chelator 2,3-dimercapto-1-propanesulfonic acid (DMPS). Can. J. Physiol. Pharmacol. 2010, 88, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Bahn, A.; Knabe, M.; Hagos, Y.; Rodiger, M.; Godehardt, S.; Graber-Neufeld, D.S.; Evans, K.K.; Burckhardt, G.; Wright, S.H. Interaction of the metal chelator 2,3-dimercapto-1-propanesulfonate with the rabbit multispecific organic anion transporter 1 (rbOAT1). Mol. Pharmacol. 2002, 62, 1128–1136. [Google Scholar] [CrossRef] [PubMed]
- Burckhardt, B.C.; Drinkuth, B.; Menzel, C.; Konig, A.; Steffgen, J.; Wright, S.H.; Burckhardt, G. The renal Na(+)-dependent dicarboxylate transporter, NaDC-3, translocates dimethyl- and disulfhydryl-compounds and contributes to renal heavy metal detoxification. J. Am. Soc. Nephrol. 2002, 13, 2628–2638. [Google Scholar] [CrossRef] [PubMed]
- Islinger, F.; Gekle, M.; Wright, S.H. Interaction of 2,3-dimercapto-1-propane sulfonate with the human organic anion transporter hOAT1. J. Pharmacol. Exp. Ther. 2001, 299, 741–747. [Google Scholar] [PubMed]
- Bridges, C.C.; Joshee, L.; Zalups, R.K. MRP2 and the DMPS- and DMSA-mediated elimination of mercury in TR(-) and control rats exposed to thiol S-conjugates of inorganic mercury. Toxicol. Sci. 2008, 105, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Bridges, C.C.; Joshee, L.; Zalups, R.K. Multidrug resistance proteins and the renal elimination of inorganic mercury mediated by 2,3-dimercaptopropane-1-sulfonic acid and meso-2,3-dimercaptosuccinic acid. J. Pharmacol. Exp. Ther. 2008, 324, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Bridges, C.C.; Joshee, L.; Zalups, R.K. Effect of DMPS and DMSA on the Placental and Fetal Disposition of Methylmercury. Placenta 2009, 30, 800–805. [Google Scholar] [CrossRef] [PubMed]
- Bridges, C.C.; Zalups, R.K.; Joshee, L. Toxicological significance of renal Bcrp: Another potential transporter in the elimination of mercuric ions from proximal tubular cells. Toxicol. Appl. Pharmacol. 2015, 285, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Aremu, D.A.; Madejczyk, M.S.; Ballatori, N. N-acetylcysteine as a potential antidote and biomonitoring agent of methylmercury exposure. Environ. Health Perspect. 2008, 116, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Madejczyk, M.S.; Aremu, D.A.; Simmons-Willis, T.A.; Clarkson, T.W.; Ballatori, N. Accelerated urinary excretion of methylmercury following administration of its antidote N-acetylcysteine requires Mrp2/Abcc2, the apical multidrug resistance-associated protein. J. Pharmacol. Exp. Ther. 2007, 322, 378–384. [Google Scholar] [CrossRef] [PubMed]
- McDowell, E.M.; Nagle, R.B.; Zalme, R.C.; McNeil, J.S.; Flamenbaum, W.; Trump, B.F. Studies on the pathophysiology of acute renal failure. I. Correlation of ultrastructure and function in the proximal tubule of the rat following administration of mercuric chloride. Virchows Arch. B Cell Pathol. 1976, 22, 173–196. [Google Scholar] [PubMed]
- Zalme, R.C.; McDowell, E.M.; Nagle, R.B.; McNeil, J.S.; Flamenbaum, W.; Trump, B.F. Studies on the pathophysiology of acute renal failure. II. A histochemical study of the proximal tubule of the rat following administration of mercuric chloride. Virchows Arch. B Cell Pathol. 1976, 22, 197–216. [Google Scholar] [PubMed]
- Rodin, A.E.; Crowson, C.N. Mercury nephrotoxicity in the rat. 2. Investigation of the intracellular site of mercury nephrotoxicity by correlated serial time histologic and histoenzymatic studies. Am. J. Pathol. 1962, 41, 485–499. [Google Scholar] [PubMed]
- Gritzka, T.L.; Trump, B.F. Renal tubular lesions caused by mercuric chloride. Electron microscopic observations: Degeneration of the pars recta. Am. J. Pathol. 1968, 52, 1225–1277. [Google Scholar] [PubMed]
- Zalups, R.K. Evidence for basolateral uptake of cadmium in the kidneys of rats. Toxicol. Appl. Pharmacol. 2000, 164, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Gotelli, C.A.; Astolfi, E.; Cox, C.; Cernichiari, E.; Clarkson, T.W. Early biochemical effects of an organic mercury fungicide on infants: “dose makes the poison”. Science 1985, 227, 638–640. [Google Scholar] [CrossRef] [PubMed]




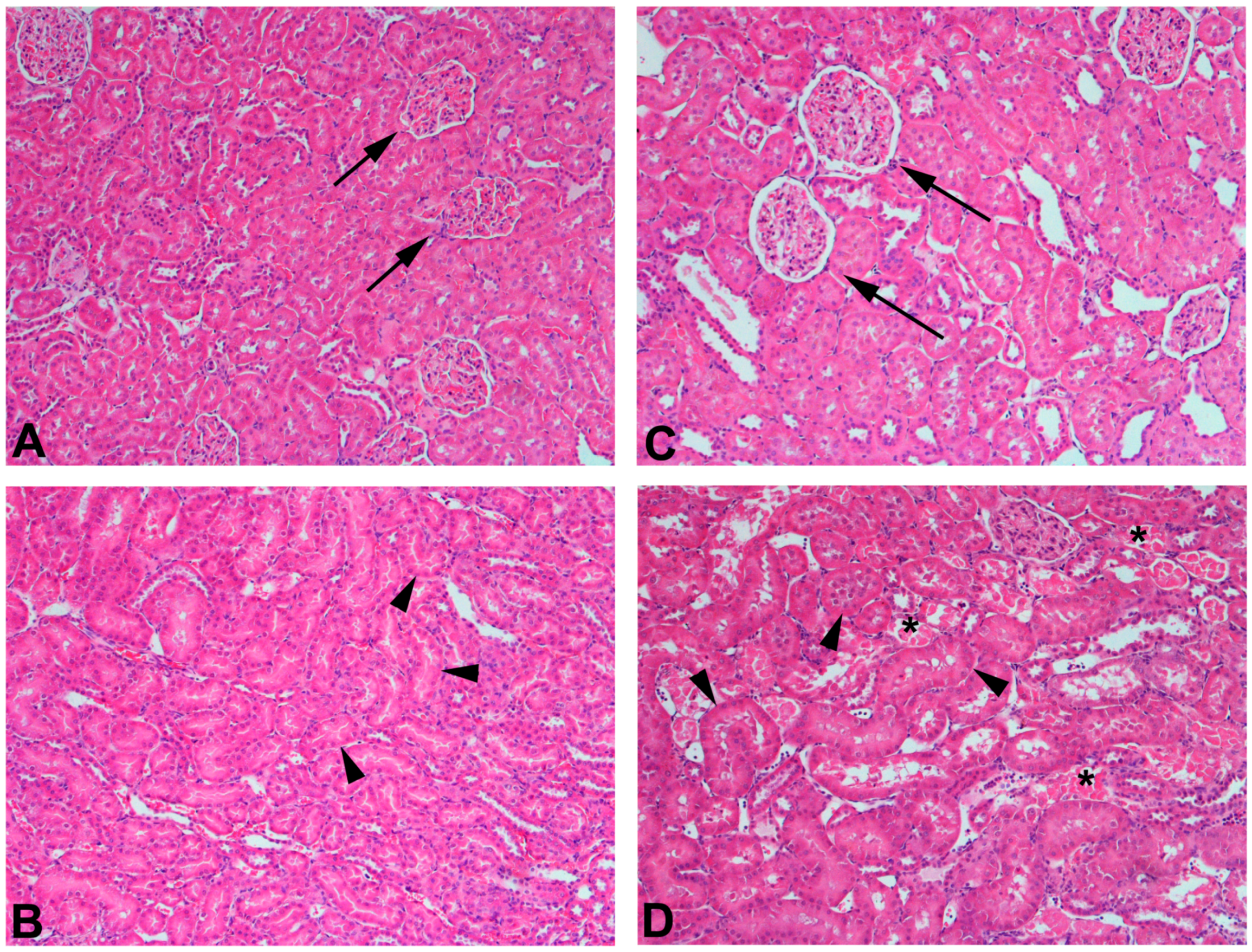{kind=link}
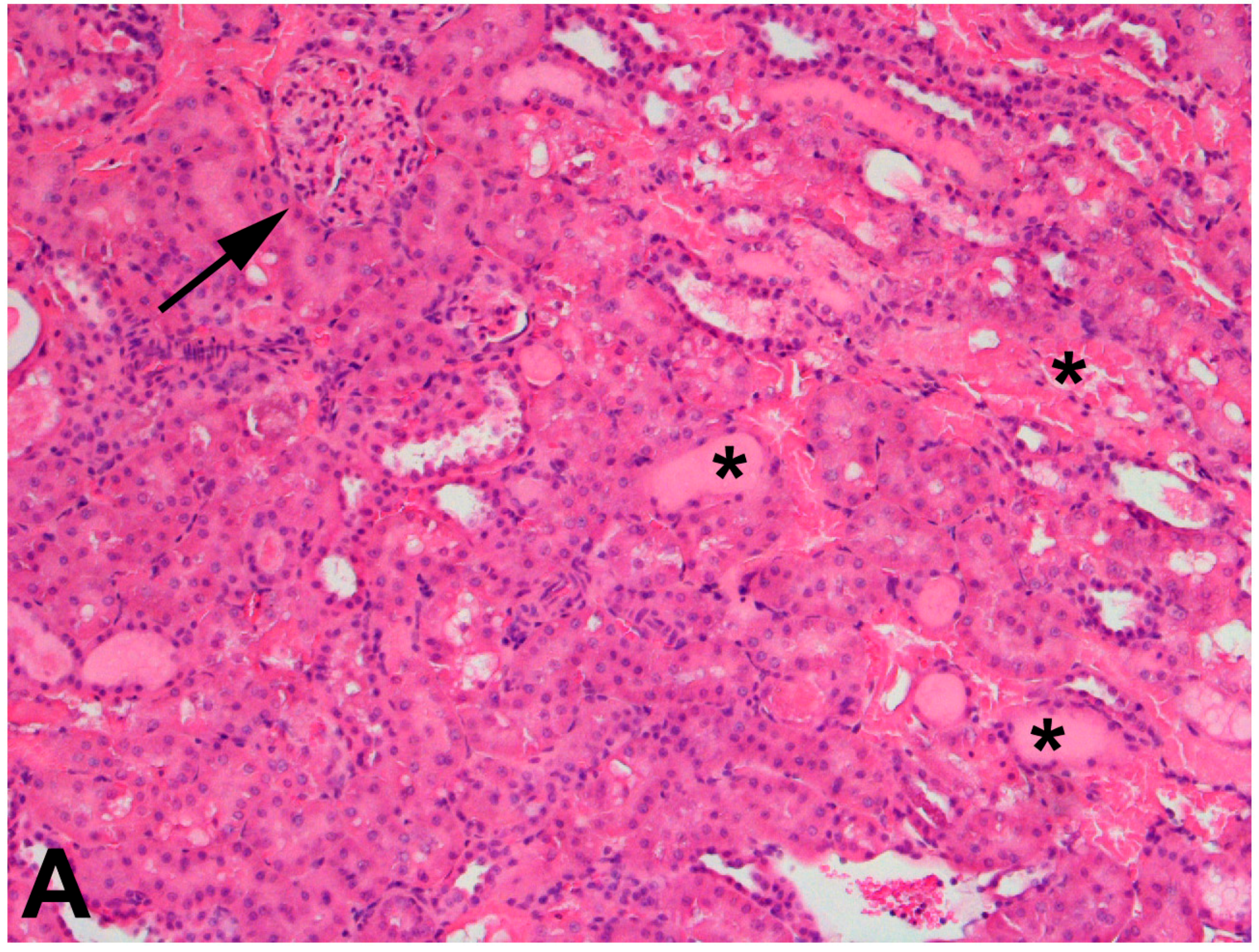{kind=link}
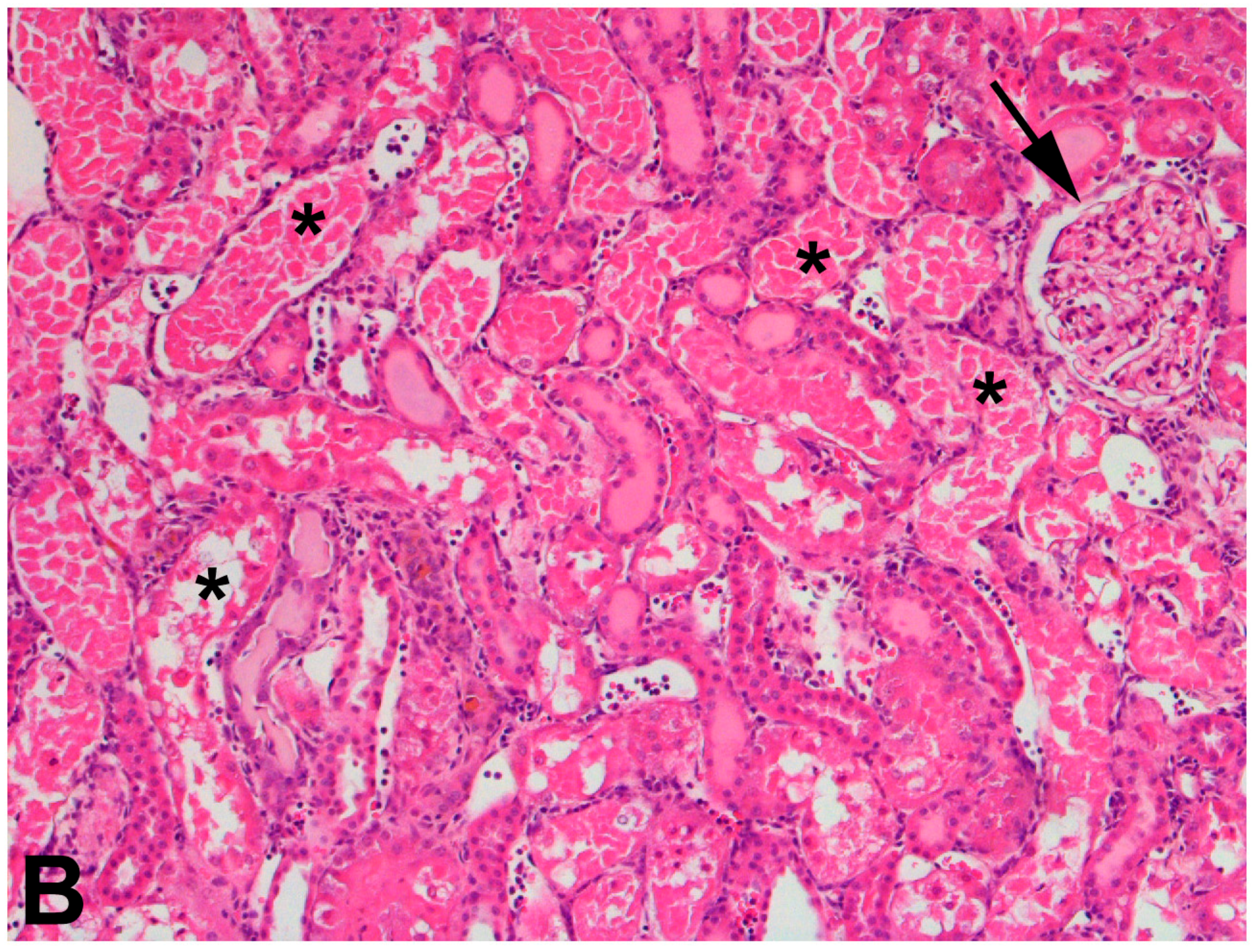{kind=link}
| Transport Mechanism | Species | |
|---|---|---|
| Apical uptake | ? | ? |
| Basolateral uptake | GLUT1 | AsIII and MAsIII |
| GLUT5 | AsIII and MAsIII | |
| Aquaporin 3 | AsIII | |
| OATP2B1 | ? | |
| Apical export | MRP2 | As(GS)3, MAs(GS)2 |
| P-glycoprotein | iAs, As(GS)3, MAs(GS)2 | |
| MRP4 | iAsIII, iAsV, MAsIII, MAsV, DMAV | |
| MATE | ? | |
| Basolateral export | GLUT1 | AsIII and MAsIII |
| GLUT5 | AsIII and MAsIII | |
| OATP2B1 | AsIII and MAsIII |
| Transport Mechanism | Species | |
|---|---|---|
| Apical uptake | Receptor-mediated endocytosis | CdMT |
| DMT1 | Cd2+ | |
| ZIP8 | Cd2+ | |
| ZIP14 | Cd2+ | |
| Basolateral uptake | OCT2 | Thiol S-conjugate of Cd |
| Apical export | MATE1 | Cd2+ |
| MATE2-K | Cd2+ | |
| MRP2 | Thiol S-conjugate of Cd | |
| P-glycoprotein | Thiol S-conjugate of Cd | |
| MRP4 | ? | |
| BCRP | ? | |
| Basolateral export | ? | ? |
| Transport Mechanism | Species | |
|---|---|---|
| Apical uptake | Receptor-mediated endocytosis | Pb-protein complexes |
| Ca2+ channels | Pb2+ | |
| Basolateral uptake | ? | ? |
| Apical export | MRP2 | GSH-Pb |
| BCRP | GSH-Pb | |
| Basolateral export | Ca2+-ATPase | Pb2+ |
| Transport Mechanism | Species | |
|---|---|---|
| Apical uptake | System b0,+ | Cys- and Hcy-S-conjugates of Hg2+ |
| System B0,+ | Cys-S-conjugates of MeHg | |
| Basolateral uptake | OAT1 | Cys-, Hcy-, NAC-S-conjugates of Hg2+ and MeHg |
| OAT3 | Cys-S-conjugates of Hg2+ | |
| Apical export | MRP2 | DMPS-, DMSA-, NAC-S-conjugates of Hg2+ DMPS-, DMSA-S-conjugates of MeHg |
| BCRP | DMPS- and Cys-S-conjugates of Hg2+ | |
| Basolateral export | ? | ? |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Orr, S.E.; Bridges, C.C. Chronic Kidney Disease and Exposure to Nephrotoxic Metals. Int. J. Mol. Sci. 2017, 18, 1039. https://doi.org/10.3390/ijms18051039
Orr SE, Bridges CC. Chronic Kidney Disease and Exposure to Nephrotoxic Metals. International Journal of Molecular Sciences. 2017; 18(5):1039. https://doi.org/10.3390/ijms18051039
Chicago/Turabian StyleOrr, Sarah E., and Christy C. Bridges. 2017. "Chronic Kidney Disease and Exposure to Nephrotoxic Metals" International Journal of Molecular Sciences 18, no. 5: 1039. https://doi.org/10.3390/ijms18051039
APA StyleOrr, S. E., & Bridges, C. C. (2017). Chronic Kidney Disease and Exposure to Nephrotoxic Metals. International Journal of Molecular Sciences, 18(5), 1039. https://doi.org/10.3390/ijms18051039