
Melatonin, a Full Service Anti-Cancer Agent: Inhibition of Initiation, Progression and Metastasis



 ,
,  and
and
Abstract
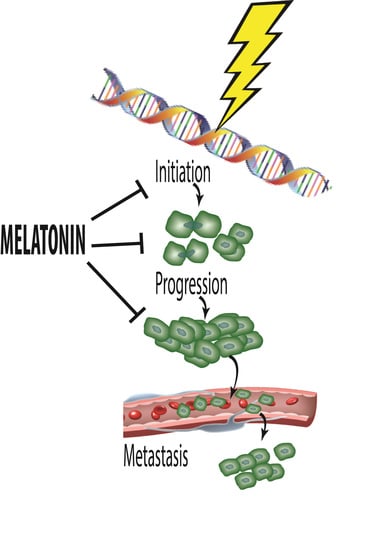
:
1. Introduction
2. Cancer Initiation: Genomic Damage and Instability
2.1. Ionizing Radiation-Induced DNA Damage
2.2. Melatonin as a RadioProtector
2.3. Other DNA Damaging Agents/Processes
2.4. Transposable Elements and DNA Damage
2.5. Melatonin and DNA Repair
3. Melatonin and Cancer Progression
3.1. Breast Cancer
3.2. Synergistic Actions of Melatonin in Breast Cancer
3.3. Ovarian Cancer
3.4. Leiomyosarcoma
3.5. Pancreatic Cancer
3.6. Hepatic Cancer
3.7. Colorectal Cancer
3.8. Lung Cancer
3.9. Bone Cancer
3.10. Glioblastoma
3.11. Leukemia
3.12. Prostate Cancer
4. Melatonin: Causing Treatment Resistant Cancer to Become Sensitive to Treatment
5. Role of Melatonin in Cancer Metastasis
6. Concluding Remarks and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Reiter, R.J. Mechanisms of cancer inhibition by melatonin. J. Pineal Res. 2004, 37, 213–214. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.X.; Chen, L.D.; Poeggeler, B.; Manchester, L.C.; Reiter, R.J. Melatonin: A potent, endogenous hydroxyl radical scavenger. J. Pineal Res. 1993, 1, 57–60. [Google Scholar]
- Poeggeler, B.; Saarela, S.; Reiter, R.J.; Tan, D.X.; Chen, L.D.; Manchester, L.C.; Barlow-Walden, L. Melatonin—A highly potent endogenous radical scavenger and electron donor: New aspects of the oxidation chemistry of the indole assessed in vitro. Ann. N. Y. Acad. Sci. 1994, 738, 419–420. [Google Scholar] [CrossRef] [PubMed]
- Allegra, M.; Reiter, R.J.; Tan, D.X.; Gentile, C.; Tesoriere, L.; Livrea, M.A. The chemistry of melatonin’s interactions with reactive species. J. Pineal Res. 2003, 34, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zavodnik, I.B.; Domanski, A.V.; Lapshina, E.A.; Bryszewska, M.; Reiter, R.J. Melatonin directly scavenges free radicals generated in red blood cells and in a cell-free system: Chemiluminescence measurements and theoretical calculations. Life Sci. 2006, 79, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Velkov, Z.A.; Velkov, Y.Z.; Galunska, B.T.; Paskalev, D.N.; Tadjer, A.V. Melatonin: Quantum-chemical and biochemical investigation of antioxidant activity. Eur. J. Med. Chem. 2009, 44, 2834–2839. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Tan, D.X.; Kim, S.J.; Qi, W. Melatonin as a pharmacological agent against oxidative damage to lipids and DNA. Proc. West. Pharmacol. Soc. 1998, 41, 229–236. [Google Scholar] [PubMed]
- Reiter, R.J. Oxidative damage to nuclear DNA: Amelioration by melatonin. Neuro Endocrinol. Lett. 1999, 20, 145–150. [Google Scholar] [PubMed]
- Karbownik, M.; Tan, D.X.; Reiter, R.J. Melatonin reduces the oxidation of nuclear DNA and membrane lipids induced by the carcinogen δ-aminolevulinic acid. Int. J. Cancer 2000, 88, 7–11. [Google Scholar] [CrossRef]
- Qi, W.; Reiter, R.J.; Tan, D.X.; Manchester, L.C.; Siu, A.W.; Garcia, J.J. Increased levels of oxidatively damaged DNA induced by chromium (III) and H2O2: Protection by melatonin and related molecules. J. Pineal Res. 2000, 29, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J. Melatonin: Lowering the high price of free radicals. News Physiol. Sci. 2000, 15, 246–250. [Google Scholar] [PubMed]
- Karbownik, M.; Lewinski, A.; Reiter, R.J. Anticarcinogenic actions of melatonin which involve antioxidative processes: Comparison with other antioxidants. Int. J. Biochem. Cell Biol. 2001, 33, 735–753. [Google Scholar] [CrossRef]
- Karbownik, M.; Reiter, R.J. Melatonin protects against oxidative stress caused by δ-aminolevulinic acid: Implications for cancer. Cancer Investig. 2002, 20, 276–286. [Google Scholar] [CrossRef]
- Lane, D.P.; Benchimol, S. p53: Oncogene or anti-oncogene? Genes Dev. 1990, 4, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Aschauer, L.; Muller, P.A. Novel targets and interaction partners of mutant p53 gain-of-function. Biochem. Soc. Trans. 2016, 44, 460–466. [Google Scholar] [CrossRef] [PubMed]
- Cerutti, P.; Ghosh, R.; Oya, Y.; Amstad, P. The role of cellular antioxidant defense in oxidant carcinogenesis. Environ. Health Perspect. 1994, 102 (Suppl. S10), 123–129. [Google Scholar] [CrossRef] [PubMed]
- Blask, D.E.; Brainard, G.C.; Dauchy, R.T.; Hanifin, J.P.; Davidson, L.K.; Krause, J.A.; Sauer, L.A.; Rivera-Bermudez, M.A.; Dubocovich, M.L.; Jasser, S.A.; et al. Melatonin-depleted blood from premenopausal women exposed to light at night stimulates growth of human breast cancer xenografts in nude rats. Cancer Res. 2005, 65, 11174–11184. [Google Scholar] [CrossRef] [PubMed]
- Dubocovich, M.L.; Rivera-Bermudez, M.A.; Gerdin, M.J.; Masana, M.I. Molecular pharmacology, regulation and function of mammalian melatonin receptors. Front. Biosci. 2003, 8, d1093–d1108. [Google Scholar] [CrossRef] [PubMed]
- Dubocovich, M.L.; Delagrange, P.; Krause, D.N.; Sugden, D.; Cardinali, D.P.; Olcese, J. International Union of Basic and Clinical Pharmacology. LXXV. Nomenclature, classification, and pharmacology of G-protein-coupled melatonin receptors. Pharmacol. Rev. 2010, 62, 343–380. [Google Scholar] [CrossRef] [PubMed]
- Sauer, L.A.; Dauchy, R.T.; Blask, D.E.; Armstrong, B.J.; Scalici, S. 13-Hydroxyoctadecadienoic acid is a mitogenic signal for linoleic acid-dependent growth of hepatoma 7288CTL in vivo. Cancer Res. 1999, 59, 4688–4692. [Google Scholar] [PubMed]
- Burdge, G. α-Linoleic metabolism in men and women: Nutritional and biological implications. Curr. Opin. Clin. Nutr. Metab. Care 2004, 7, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Blask, D.E.; Dauchy, R.T.; Sauer, L.A.; Krause, J.A. Melatonin uptake and growth prevention in rat hepatoma 7288CTC in response to dietary melatonin: Melatonin receptor-mediated inhibition of tumor linoleic and metabolism to the growth signaling molecule 13-hydroxyoctadecadienic acid and the potential role of phytomelatonin. Carcinogenesis 2004, 25, 951–960. [Google Scholar] [PubMed]
- Dubbels, R.; Reiter, R.J.; Klenke, E.; Goebel, A.; Schnakenberg, E.; Ehlers, C.; Schiwara, H.W.; Schloot, W. Melatonin in edible plants identified by radioimmunoassay and high performance liquid chromatography-mass spectrometry. J. Pineal Res. 1995, 18, 28–31. [Google Scholar] [CrossRef] [PubMed]
- Hattori, A.; Migitaka, H.; Iigo, M.; Itoh, M.; Yamamato, K.; Ohtani-Kaneko, R.; Hara, M.; Suzuki, T.; Reiter, R.J. Identification of melatonin in plants and its effects on plasma melatonin levels and binding to melatonin receptors in vertebrates. Biochem. Mol. Biol. Int. 1995, 35, 627–634. [Google Scholar] [PubMed]
- Leon-Blanco, M.M.; Guerrero, J.M.; Reiter, R.J.; Calvo, J.R.; Pozo, D. Melatonin inhibits telomerase activity in the MCF-7 tumor cell line both in vivo and in vitro. J. Pineal Res. 2003, 35, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Manuelidis, L. Genomic stability and instability in different neuroepithelial tumors: A role for chromosome structure? J. Neurooncol. 1994, 18, 225–239. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.R.; Backburn, E.H. Telomeres and telomerase. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2004, 359, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Masutomi, K.; Hahn, W.C. Telomerase and tumorigenesis. Cancer Lett. 2003, 194, 163–172. [Google Scholar] [CrossRef]
- Ishikawa, N.; Nakamura, K.; Izumiyama-Shimomura, N.; Aida, J.; Matsuda, Y.; Arai, T.; Takubo, K. Changes in telomere status with aging: An update. Geriatr. Gerontol. Int. 2016, 16, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Girgert, R.; Bartsch, C.; Hill, S.M.; Kreienberg, R.; Hanf, V. Tracking the elusive antiestrogenic effect of melatonin: A new methodological approach. Neuroendocrinol. Lett. 2003, 24, 440–444. [Google Scholar] [PubMed]
- Sanchez-Barcelo, E.J.; Cos, S.; Mediavilla, D.; Martinez-Campa, C.; Gonzalez, A.; Alonso-Gonzalez, C. Melatonin-estrogen interactions in breast cancer. J. Pineal Res. 2005, 38, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Kilic, E.; Kilic, U.; Reiter, R.J.; Bassetti, C.L.; Hermann, D.M. Prophylactic use of melatonin protects against focal cerebral ischemia in mice: Role of endothelin converting enzyme-1. J. Pineal Res. 2004, 37, 247–251. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.; Rghigh, A. Polymorphisms of endothelin-1 gene: An overview. Curr. Clin. Pharmacol. 2016, 11, 191–210. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.B.; Peng, T.C.; Huang, D. Correlations between plasma endothelin-1 and breakthrough pain in patients with cancer. OncoTargets Ther. 2015, 8, 3703–3706. [Google Scholar]
- Lissoni, P.; Ardizzoia, A.; Barni, S.; Brivio, F.; Tsi, E.; Rovelli, F.; Tancini, G.; Maestroni, G.J.; Fumagalli, L. Efficacy and tolerability of cancer neuroimmunotherapy with subcutaneous low-dose interleukin-2 and the pineal hormone melatonin—A progress report of 200 patients with advanced solid neoplasm. Oncol. Rep. 1995, 2, 1063–1068. [Google Scholar] [CrossRef] [PubMed]
- Lissoni, P.; Rovelli, F.; Malugani, F.; Bucovec, R.; Conti, A.; Maestroni, G.J. Anti-angiogenic activity of melatonin in advanced cancer patients. Neuroendocrinol. Lett. 2001, 22, 45–47. [Google Scholar] [PubMed]
- Lissoni, P.; Chilelli, M.; Villa, S.; Cerizza, L.; Tancini, G. Five years survival of metastatic non-small cell lung cancer patients treated with chemotherapy alone or chemotherapy and melatonin: A randomized trial. J. Pineal Res. 2003, 35, 12–15. [Google Scholar] [CrossRef] [PubMed]
- Jung-Hynes, B.; Reiter, R.J.; Ahmad, N. Sirtuins, melatonin and circadian rhythms: Building a bridge between aging and cancer. J. Pineal Res. 2010, 48, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Mediavilla, M.D.; Sanchez-Barcelo, E.J.; Tan, D.X.; Manchester, L.C.; Reiter, R.J. Basic mechanisms involved in the anti-cancer effects of melatonin. Curr. Med. Chem. 2010, 17, 4462–4481. [Google Scholar] [CrossRef] [PubMed]
- Hill, S.M.; Belancio, V.P.; Dauchy, R.T.; Xiang, S.; Brimer, S.; Mao, L.; Hauch, A.; Lundberg, P.W.; Summers, W.; Yuan, L.; et al. Melatonin: An inhibitor of breast cancer. Endocr. Relat. Cancer 2015, 22, R183–R204. [Google Scholar] [CrossRef] [PubMed]
- Foss, E.J.; Lao, U.; Dalrymple, E.; Adrianse, R.L.; Loe, T.; Bedalov, A. SIR2 suppresses replication gaps and genome instability by balancing replication between repetitive and unique sequences. Proc. Natl. Acad. Sci. USA 2017, 114, 552–557. [Google Scholar] [CrossRef] [PubMed]
- Sholl, L.M.; Barletta, J.A.; Hornick, J.L. Radiation-associated neoplasia: Clinical, pathological and genomic correlates. Histopathology 2017, 70, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Sage, E.; Skikazono, N. Radiation-induced clustered DNA lesions: Repair and mutagenesis. Free Radic. Biol. Med. 2016. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Oxidative stress and cancers: Have we moved forward? Biochem. J. 2007, 401, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kamran, M.Z.; Ranjan, A.; Kaur, N.; Sur, S.; Tandon, Z. Radioprotective agents: Strategies and translational advances. Med. Res. Rev. 2016, 36, 461–493. [Google Scholar] [CrossRef] [PubMed]
- Rosen, E.M.; Day, R.; Singh, V.K. New approaches to radiation protection. Font. Oncol. 2014, 4, 381. [Google Scholar] [CrossRef] [PubMed]
- Blickenstaff, R.T.; Brandstadter, S.M.; Reddy, S.; Witt, R. Potential radioprotective agents: 1. Homologs of melatonin. J. Pharm. Sci. 1994, 83, 216–218. [Google Scholar] [CrossRef] [PubMed]
- Nakano, T.; Xu, X.; Salem, A.M.H.; Shoulkamy, M.L.; Ide, H. Radiation-induced DNA-protein cross links: Mechanisms and biological significance. Free Radic. Biol. Med. 2017. [Google Scholar] [CrossRef] [PubMed]
- Poeggeler, B.; Reiter, R.J.; Tan, D.X.; Chen, L.D.; Manchester, L.C. Melatonin, hydroxyl radical-mediated oxidative damage and aging: A hypothesis. J. Pineal Res. 1993, 14, 151–168. [Google Scholar] [CrossRef] [PubMed]
- Vijayalaxmi; Meltz, M.L.; Reiter, R.J.; Herman, T.S.; Kumar, S. Melatonin and protection from whole body irradiation: Survival studies in mice. Mutat. Res. 1999, 425, 21–27. [Google Scholar] [CrossRef]
- Vijayalazmi; Meltz, M.L.; Reiter, R.J.; Herman, T.S. Melatonin and protection from genetic damage in blood and bone marrow: Whole-body radiation studies in mice. J. Pineal Res. 1999, 27, 221–225. [Google Scholar] [CrossRef]
- Malaker, K. Clinical experience with radioprotectors. In Radioprotectors: Chemical, Biological, and Clinical Perspectives; Bump, E.A., Malaker, K., Eds.; CRC Press: Boca Raton, FL, USA, 1998; pp. 373–409. [Google Scholar]
- Vijayalaxmi; Reiter, R.J.; Meltz, M.L. Melatonin protects human blood lymphocytes from radiation-induced chromosome damage. Mutat. Res. 1995, 346, 23–31. [Google Scholar] [CrossRef]
- Vijayalaxmi; Reiter, R.J.; Leal, B.Z.; Meltz, M.L. Effect of melatonin mitotic and proliferative indices and sister chromatid exchanges in human blood lymphocytes. Mutat. Res. 1996, 351, 187–192. [Google Scholar] [CrossRef]
- Vijayalaxmi; Reiter, R.J.; Herman, T.S.; Meltz, M.L. Melatonin and radioprotection from genetic damage: In vivo/in vitro studies with human volunteers. Mutat. Res. 1996, 371, 221–228. [Google Scholar] [CrossRef]
- Neville, S.; Arendt, J.; Ioannides, G. A study of the mutagenicity of melatonin and 6-hydroxymelatonin. J. Pineal Res. 1989, 6, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Jahnke, G.; Marr, M.; Myers, C.; Wilson, R.; Travlos, G.; Price, C. Maternal and developmental toxicity evaluation of melatonin administered orally to pregnant Sprague-Dawley rats. Toxicol. Sci. 1999, 50, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Andersen, L.P.; Gogenur, I.; Rosenberg, J.; Reiter, R.J. The safety of melatonin in humans. Clin. Drug Investig. 2016, 36, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Karbownik, M.; Reiter, R.J. Antioxidative effects of melatonin in protection against cellular damage caused by ionizing radiation. Proc. Soc. Exp. Biol. Med. 2000, 225, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Sainz, R.M.; Mayo, J.C.; Rodriguez, C.; Tan, D.X.; Lopez-Burillo, S.; Reiter, R.J. Melatonin and cell death: Differential actions on apoptosis in normal and cancer cells. Cell. Mol. Life Sci. 2003, 60, 1407–1426. [Google Scholar] [CrossRef] [PubMed]
- Vijayalaxmi; Thomas, C.R.; Reiter, R.J.; Herman, T.S. Melatonin: From basic research to cancer treatment clinics. J. Clin. Oncol. 2002, 20, 2595–2601. [Google Scholar] [CrossRef] [PubMed]
- Vijayalaxmi; Reiter, R.J.; Herman, T.S.; Thomas, C.R., Jr. Melatonin as a radioprotective agent: A review. Int. J. Radiat. Oncol. Biol. Phys. 2004, 59, 639–653. [Google Scholar] [CrossRef] [PubMed]
- Zetner, D.; Andersen, L.P.; Rosenberg, J. Melatonin as protection against radiation injury: A systematic review. Drug Res. 2016, 66, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Menendez-Pelaez, A.; Reiter, R.J. Distribution of melatonin in mammalian tissues: Relative importance of nuclear versus cytosolic localization. J. Pineal Res. 1993, 15, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Venegas, C.; Garcia, J.A.; Escames, G.; Ortiz, F.; Lopez, A.; Doerrier, C.; Garcia-Corzo, L.; Lopez, L.C.; Reiter, R.J.; Acuna-Castroviejo, D. Extrapineal melatonin: Analysis of its subcellular distribution and daily fluctuations. J. Pineal Res. 2012, 52, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Acuna-Castroviejo, D.; Escames, G.; Venegas, C.; Diaz-Casado, M.E.; Rosales-Corral, S.A.; Tan, D.X.; Reiter, R.J. Extrapineal melatonin: Sources, regulation and potential functions. Cell. Mol. Life Sci. 2014, 71, 2997–3025. [Google Scholar] [CrossRef] [PubMed]
- Shao, B.; Mao, L.; Qu, N.; Wang, Y.F.; Gao, H.Y.; Li, F.; Qin, L.; Shao, J.; Huang, C.H.; Xu, D.; et al. Mechanisms of synergistic DNA damage induced by hydroquinone metabolite of brominated phenolic environmental pollutants and Cu (II): Formation of DNA-Cu complex and site-specific production of hydroxyl radicals. Free Radic. Biol. Med. 2017, 104, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Roginskaya, M.; Moore, T.J.; Ampadu-Boateng, D.; Razskazovskiy, Y. Efficacy and site specificity of hydrogen abstraction from DNA 2-deoxyribose by carbonate radicals. Free Radic. Res. 2015, 49, 1431–1437. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.X.; Manchester, L.C.; Reiter, R.J.; Plummer, B.F. Cyclic 3-hydroxymelatonin: A melatonin metabolite generated as a result of hydroxyl radical scavenging. Neurosignals 1999, 8, 70–74. [Google Scholar] [CrossRef]
- Hardeland, R.; Tan, D.X.; Reiter, R.J. Kynuramines, metabolites of melatonin and other indoles: The resurrection of an almost forgotten class of biogenic amines. J. Pineal Res. 2009, 47, 109–126. [Google Scholar] [CrossRef] [PubMed]
- Galano, A.; Tan, D.X.; Reiter, R.J. On the free radical scavenging activities of melatonin’s metabolites, AFMK and AMK. J. Pineal Res. 2013, 54, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Tan, D.X.; Galano, A. Melatonin reduces lipid peroxidation and membrane viscosity. Front. Physiol. 2014, 5, 377. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Tan, D.X.; Galano, A. Melatonin: Exceeding expectations. Physiology 2014, 29, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Manchester, L.C.; Coto-Montes, A.; Boga, J.A.; Andersen, L.P.H.; Zhou, Z.; Galano, A.; Vriend, J.; Tan, D.X.; Reiter, R.J. Melatonin: An ancient molecule that makes oxygen metabolically tolerable. J. Pineal Res. 2015, 59, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Barlow-Walden, L.R.; Reiter, R.J.; Abe, M.; Pablos, M.I.; Menendez-Pelaez, A. Melatonin stimulates brain glutathione peroxidase activity. Neurochem. Int. 1995, 26, 497–502. [Google Scholar] [CrossRef]
- Pablos, M.I.; Agapito, M.T.; Gutierrez, R.; Recio, J.M.; Reiter, R.J.; Barlow-Walden, L.R.; Acuna-Castroviejo, D.; Menendez-Pelaez, A. Melatonin stimulates the activity of the detoxifying enzyme glutathione peroxidase in several tissues of chicks. J. Pineal Res. 1995, 19, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Pablos, M.I.; Reiter, R.J.; Ortiz, G.G.; Guerrero, J.M.; Agapito, M.T.; Chuang, J.I.; Sewerynek, E. Rhythms of glutathione peroxidase and glutathione reductase in the brain of chick and their inhibition by light. Neurochem. Int. 1998, 32, 69–75. [Google Scholar] [CrossRef]
- Reiter, R.J.; Tan, D.X.; Osuna, C.; Gitto, E. Actions of melatonin in the reduction of oxidative stress: A review. J. Biomed. Sci. 2000, 7, 444–458. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, C.; Mayo, J.C.; Sainz, R.M.; Antolin, I.; Herrora, F.; Martin, V.; Reiter, R.J. Regulation of antioxidant enzymes: A significant role for melatonin. J. Pineal Res. 2004, 36, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Sliwinski, T.; Rozej, W.; Morawiec-Bajda, A.; Morawiec, Z.; Reiter, R.J.; Blasiak, J. Protective action of melatonin oxidative DNA damage—Chemical inactivation versus base-excision repair. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2007, 634, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Mayo, J.C.; Tan, D.X.; Sainz, R.M.; Alatorre-Jimenez, M.; Oin, L. Melatonin as an antioxidant: Under promises but over delivers. J. Pineal Res. 2016, 61, 253–278. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Tan, D.X.; Korkmaz, A. The disaster in Japan: Utility of melatonin in providing protection against ionizing radiation. J. Pineal Res. 2011, 50, 357–358. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Korkmaz, A.; Ma, S.; Rosales-Corral, S.A.; Tan, D.X. Melatonin protection from low-level ionizing radiation. Mutat. Res. 2012, 751, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.; O’Malley, M.; Nocera, A. Ensuring the safety of surgical teams when managing casualties of a radiological dirty bomb. Injury 2010, 41, 938–942. [Google Scholar] [CrossRef] [PubMed]
- Dropkin, G. Low dose radiation risks for women surviving the a-bombs in Japan: Generalized additive model. Environ. Health 2016, 15, 112. [Google Scholar] [CrossRef] [PubMed]
- Suman, S.; Rodriguez, O.C.; Winters, T.A.; Fornace, A.J., Jr.; Albanese, C.; Datta, K. Therapeutic and space radiation exposure of mouse brain causes impaired DNA repair response and premature senescence by chronic oxidant production. Aging 2013, 5, 607–622. [Google Scholar] [CrossRef] [PubMed]
- Cheema, A.K.; Suman, S.; Kaur, P.; Singh, R.; Fornace, A.J., Jr.; Datta, K. Long-term differential changes in mouse intestinal metabolomics after y and heavy ion radiation exposure. PLoS ONE 2014, 9, e87079. [Google Scholar] [CrossRef] [PubMed]
- Freitas, I.; Vairetti, M.; Ferrigno, A.; Bertone, V.; Guarnaschelli, C.; Rizzo, V.; Boncompagni, E.; Reiter, R.J. Rationale for the use of melatonin as a protective agent against cosmic radiation and ischemia-reperfusion damage in long term spaceflight. J. Br. Interplanet. Soc. 2006, 59, 124–129. [Google Scholar]
- Cardinali, D.P.; Srinivasan, V.; Brzezinski, A.; Brown, G.M. Melatonin and its analogs in insomnia and depression. J. Pineal Res. 2012, 52, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Hardeland, R.; Madrid, J.A.; Tan, D.X.; Reiter, R.J. Melatonin, the circadian multioscillator system and health: The need for detailed analyses of peripheral melatonin signaling. J. Pineal Res. 2012, 52, 139–166. [Google Scholar] [CrossRef] [PubMed]
- Vriend, J.; Reiter, R.J. Melatonin feedback on clock genes: A theory involving the proteasome. J. Pineal Res. 2015, 58, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Maria, S.; Witt-Enderby, P.A. Melatonin effects on bone: Potential use for the prevention and treatment of osteopenia, osteoporosis, and periodontal disease and for use in bone-grafting procedures. J. Pineal Res. 2014, 56, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Amstrup, A.K.; Sikjaer, T.; Heickendorff, L.; Mosekilde, L.; Rejnmark, L. Melatonin improves bone mineral density at the femoral neck in postmenopausal women with osteopenia: A randomized, controlled trial. J. Pineal Res. 2015, 59, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Kim, J.H.; Jin, Y.; Lee, S.; Park, K.; Lee, Y.; Chang, K.T.; Hong, Y. Melatonin treatment combined with treadmill exercise accelerates muscular adaptation through early inhibition of CHOP-mediated autophagy in the gastrocnemius of rats with intra-articular collagenase-induced knee laxity. J. Pineal Res. 2014, 56, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Da Silva Borges, L.; Dermargos, A.; Pinto da Silva Junior, E.; Weimann, E.; Lambertucci, R.H.; Hatanaka, E. Melatonin decrease muscular oxidative stress and inflammation induced by strenuous exercise and stimulates growth factor synthesis. J. Pineal Res. 2015, 58, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Coto-Montes, A.; Boga, J.A.; Tan, D.X.; Reiter, R.J. Melatonin as a potential agent in the treatment of sarcopenia. Int. J. Mol. Sci. 2016, 17, 1771. [Google Scholar] [CrossRef] [PubMed]
- Dauchy, R.T.; Wren, M.S.; Dauchy, E.M.; Hanifin, J.P.; Jablonski, M.R.; Warfield, B.; Brainard, G.C.; Hill, S.M.; Mao, L.; Dupepe, L.M.; et al. Effects of spectral transmittance through standard laboratory cages on circadian metabolism and physiology of nude rats. J. Am. Assoc. Lab. Anim. Sci. 2013, 52, 146–156. [Google Scholar] [PubMed]
- Brainard, G.C.; Hanifin, J.P.; Warfield, B.; Stone, M.K.; James, M.E.; Ayers, M.; Kubey, A.; Byrne, B.; Rollag, M.D. Short-wavelength enrichment of polychromatic light enhances human melatonin suppression potency. J. Pineal Res. 2015, 58, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Durante, M.; Cucinotta, F.A. Heavy ion carcinogenesis and human space exploration. Nat. Rev. Cancer 2008, 8, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Galadari, S.; Rahman, A.; Pallichankandy, S.; Thayyullathil, F. Reactive oxygen species and cancer paradox: To promote or suppress? Free Radic. Biol. Med. 2017, 104, 144–164. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Feng, W.; Wang, S.; Zhang, X.; Zhang, W.; He, M.; Zhang, X.; Wu, T.; Guo, H. Essential metals zinc, selenium and strontium protect against chromosome damage caused by polycyclic aromatic hydrocarbon exposures. Environ. Sci. Technol. 2016, 50, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Hou, X.Y.; Wei, Y.; Thai, P.; Choi, F. Biomarkers of the health outcomes associated with ambient particular matter exposure. Sci. Total Environ. 2017, 579, 1446–1459. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Manchester, L.C.; Tan, D.X. Neurotoxins: Free radical mechanisms and melatonin protection. Curr. Neuropharmacol. 2010, 8, 194–210. [Google Scholar] [CrossRef] [PubMed]
- Assi, M.A.; Hezmee, M.N.; Haron, A.W.; Sabri, M.Y.; Rajion, M.A. The detrimental effects of lead on human and animal health. Vet. World 2016, 9, 660–671. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Tan, D.X.; Sainz, R.M.; Mayo, J.C.; Lopez-Burillo, S. Melatonin: Reducing the toxicity and increasing the efficacy of drugs. J. Pharm. Pharmacol. 2002, 54, 1299–1321. [Google Scholar] [CrossRef] [PubMed]
- Korkina, L. Metabolic and redox barriers in the skin exposed to drugs and xenobiotics. Expert Opin. Drug Metab. Toxicol. 2016, 12, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Asghari, M.H.; Moloudizargari, M.; Bahadar, H.; Abdollahi, M. A review of the protective effect of melatonin is pesticide-induce toxicity. Expert Opin. Drug Metab. Toxicol. 2016, 29, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Martinez, M.A.; Dai, M.; Chen, D.; Ares, I.; Romero, A.; Castellano, V.; Martinez, M.; Rodriguez, J.L.; Martinez-Larranaga, M.R.; Anadon, A.; Yuan, Z. Permethrin-induced oxidative stress and toxicity and metabolism: A review. Environ. Res. 2016, 149, 86–104. [Google Scholar] [CrossRef] [PubMed]
- Saybasili, M.; Yuksel, G.; Haklar, G.; Yalcin, A.S. Effect of mitochondrial electron transport chain inhibitors on superoxide radical generation in rat hippocampal and striatal slices. Antioxid. Redox Signal. 2001, 3, 1099–1104. [Google Scholar] [CrossRef] [PubMed]
- Staniek, K.; Gille, L.; Kozlov, A.V.; Nohl, H. Mitochondrial superoxide radical formation is controlled by electron bifurcation to the high and low potential pathways. Free Radic. Res. 2002, 36, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Muller, F.L.; Liu, Y.; van Remmen, H. Complex III releases superoxide to both sides of the inner mitochondrial membrane. J. Biol. Chem. 2004, 279, 49064–49073. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Fang, P.; Mai, J.; Choi, E.T.; Wang, H.; Yang, X.F. Targeting mitochondrial oxygen species as a novel therapy for inflammatory diseases and cancers. J. Hematol. Oncol. 2013, 6, 19. [Google Scholar] [CrossRef] [PubMed]
- Newsholme, P.; Cruzal, V.F.; Keane, K.M.; Carlessi, R.; de Bittencourt, P.I., Jr. Molecular mechanisms of ROS production and oxidative stress in diabetes. Biochem. J. 2016, 473, 4527–4550. [Google Scholar] [CrossRef] [PubMed]
- Mutlu, E.; Gao, L.; Collins, L.B.; Walker, N.J.; Hartwell, H.J.; Olson, J.R.; Sun, W.; Gold, A.; Ball, L.M.; Swenberg, J.A. Polychlorinated biphenyls induce oxidative DNA adducts in female Sprague-Dawley rats. Chem. Res. Toxicol. 2016, 29, 1335–1344. [Google Scholar] [CrossRef] [PubMed]
- Jou, M.J.; Peng, T.I.; Hsu, L.F.; Jou, S.B.; Reiter, R.J.; Yang, L.M.; Chiao, C.C.; Li, Y.F.; Chen, C.C. Visualization of melatonin’s multiple mitochondrial levels of protection against mitochondrial Ca2+-mediated permeability transition and beyond in rat brain astrocytes. J. Pineal Res. 2010, 48, 20–38. [Google Scholar] [CrossRef] [PubMed]
- Lowes, D.A.; Webster, M.R.; Murphy, M.P.; Galley, H.F. Antioxidants that protect mitochondria reduce interleukin-6 and oxidative stress, improve mitochondrial function, and reduce biochemical markers of organ dysfunction in a rat model of acute sepsis. Br. J. Anaesth. 2013, 110, 472–480. [Google Scholar] [CrossRef] [PubMed]
- Ramis, M.R.; Esteban, S.; Miralles, A.; Tan, D.X.; Reiter, R.J. Protective effects of melatonin and mitochondria-targeted antioxidants against oxidative stress: A review. Curr. Med. Chem. 2015, 22, 2690–2711. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.X.; Manchester, L.C.; Qin, L.; Reiter, R.J. Melatonin: A mitochondrial targeting molecule involving mitochondrial protection and dynamics. Int. J. Mol. Sci. 2016, 17, 2124. [Google Scholar] [CrossRef] [PubMed]
- Hevia, D.; Gonzalez-Menendez, P.; Quiros-Gonzalez, I.; Miar, A.; Rodriguez-Garcia, A.; Tan, D.X.; Reiter, R.J.; Mayo, J.C.; Sainz, R.M. Melatonin uptake through glucose transporters: A new target for melatonin inhibition of cancer. J. Pineal Res. 2015, 58, 234–250. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Yang, Y.; Fan, C.; Han, J.; Wang, D.; Di, S.; Hu, W.; Liu, D.; Li, X.; Reiter, R.J.; Yan, X. Melatonin as a potential anticarcinogen for non-small-cell cancer. Oncotarget 2016, 7, 46768–46784. [Google Scholar] [CrossRef] [PubMed]
- Hardeland, R. Antioxidative protection by melatonin: Multiplicity of mechanisms from radical detoxification to radical avoidance. Endocrine 2005, 27, 119–130. [Google Scholar] [CrossRef]
- Galano, A.; Medina, M.E.; Tan, D.X.; Reiter, R.J. Melatonin and its metabolites as copper chelating agents and their role in inhibiting oxidative stress: A physicochemical analysis. J. Pineal Res. 2015, 58, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.X.; Hardeland, R.; Manchester, L.C.; Galano, A.; Reiter, R.J. Cyclic-3-hydroxymelatonin (C3OHM), a potent antioxidant, scavenges free radicals and suppresses oxidative reactions. Curr. Med. Chem. 2014, 21, 1557–1565. [Google Scholar] [CrossRef] [PubMed]
- Brouha, B.; Badge, R.; Schustak, J.; Lutz-Prigge, S.; Farley, A.H.; Moran, J.V.; Kayazian, H.H., Jr. Active L1 transposons in the human genome. Am. J. Hum. Genet. 2002, 71, 410. [Google Scholar]
- Brouha, B.; Schustak, J.; Badge, R.M.; Lutz-Prigge, S.; Farley, A.H.; Moran, J.V.; Kazazian, H.H., Jr. Hot L1s account of the bulk of retrotransposition in the human population. Proc. Natl. Acad. Sci. USA 2003, 100, 5280–5285. [Google Scholar] [CrossRef] [PubMed]
- Evrony, G.D.; Cai, X.; Lee, E.; Hills, L.B.; Elhosary, P.C.; Lehmann, H.S.; Parker, L.L.; Atabay, K.D.; Gilmore, E.C.; Poduri, A.; et al. Single-neuron sequencing analysis of L1 retrotransposition and somatic mutation in the human brain. Cell 2012, 15, 483–496. [Google Scholar] [CrossRef] [PubMed]
- Belancio, V.P.; Blask, D.E.; Deininger, P.; Hill, S.M.; Jazwinski, S.M. The aging clock and circadian control of metabolism and genomic stability. Front. Genet. 2015, 5, 445–463. [Google Scholar] [CrossRef] [PubMed]
- De Haro, D.; Kines, K.J.; Sokolowski, M.; Dauchy, R.T.; Streva, V.A.; Hill, S.M.; Hanifin, J.P.; Brainard, G.C.; Blask, D.E.; Belancio, V.P. Regulation of L1 expression and retro-transposition by melatonin and its receptor: Implications for cancer risk associated with light exposure at night. Nucleic Acids Res. 2014, 42, 7694–7707. [Google Scholar] [CrossRef] [PubMed]
- Belancio, V.P.; Roy-Engel, A.M.; Deininger, P.L. All y’all need to know ‘bout retroelements in cancer. Semin. Cancer Biol. 2010, 20, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Belancio, V.P.; Roy-Engel, A.M.; Pochampally, R.R.; Deininger, J.P. Somatic expression of LINE-1 elements in human tissues. Nucleic Acids Res. 2010, 38, 3909–3922. [Google Scholar] [CrossRef] [PubMed]
- Howard, S.M.; Yanez, D.A.; Stark, J.M. DNA damage response factors from diverse pathways including DNA crosslink repair, mediate alternative end joining. PLoS Genet. 2015, 11, e1004943. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Peng, G. Non-coding RNAs: An emerging player in DNA damage response. Mutat. Res. 2015, 763, 202–211. [Google Scholar] [CrossRef] [PubMed]
- Vriend, L.E.; Krawczyk, P.M. Nick-initiated homologous recombination: Protecting the genome, one strand at a time. DNA Repair 2017, 50, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Gherardini, L.; Sharma, A.; Capobianco, E.; Cinti, C. Targeting cancer with epi-drugs: A precision medicine perspective. Curr. Pharm. Biotechnol. 2016, 17, 856–865. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, M.; Kastan, M.B. The DNA damage response: Implication for tumor responses to radiation and chemotherapy. Annu. Rev. Med. 2015, 66, 129–143. [Google Scholar] [CrossRef] [PubMed]
- Majidinia, M.; Yousefi, B. DNA damage response regulation by microRNAs as a therapeutic target in cancer. DNA Repair 2017, 47, 1–11. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, M.J. Targeting the DNA damage response in cancer. Mol. Cell 2015, 60, 547–560. [Google Scholar] [CrossRef] [PubMed]
- Pearl, L.H.; Schiery, A.C.; Ward, S.E.; Al-Lazibani, B.; Pearl, F.M. Therapeutic opportunities with the DNA damage response. Nat. Rev. Cancer 2015, 15, 166–180. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Gao, Z.; Li, H.; Zhang, B.; Wang, G.; Zhang, Q.; Pei, D.; Zheng, J. DNA damage response—A double-edged sword in cancer prevention and cancer therapy. Cancer Lett. 2015, 358, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Harper, J.W.; Elledge, S.J. The DNA damage response: Ten years later. Mol. Cell 2007, 28, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.F.; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. The DNA damage response and cancer therapy. Nature 2012, 481, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.Y.; Lin, X.; Mao, L.Z.; Ge, W.H.; Zhang, L.M.; Huang, Y.L.; Gu, J. Neuroprotection by melatonin against ischemic neuronal injury associated with modulation of DNA damage and repair in the rat following transient cerebral ischemia. J. Pineal Res. 2002, 33, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Blasiak, J.; Synowiec, E.; Tarnawska, J.; Czarny, P.; Poplawski, T.; Reiter, R.J. Dental methacrylates may exert genotoxic effects via the oxidative induction of DNA double strand breaks and the inhibition of their repair. Mol. Biol. Rep. 2012, 39, 7487–7496. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Alfaro, M.; Hernandez-Cortes, D.; Wrobél, K.; Crug-Jimenez, G.; Rivera-Leyvax, J.C.; Pina-Zentella, R.M.; Carabez-Trejo, A. Effect of melatonin administration on DNA damage and repair responses in lymphocytes of rats subchronically exposed to lead. Mutat. Res. 2012, 742, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Grant, S. New insights into checkpoint kinase 1 in the DNA damage response signaling network. Clin. Cancer Res. 2010, 16, 376–383. [Google Scholar] [CrossRef] [PubMed]
- Mayo, J.C.; Sainz, R.M.; Gonzalez-Menendez, P.; Cepas, V.; Tan, D.X.; Reiter, R.J. Melatonin and sirtuins: A “not-so unexpected” relationship. J. Pineal Res. 2017, 62. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Fan, M.; Chen, Y.; Zhao, Q.; Song, C.; Yan, Y.; Jin, Y.; Huang, Z.; Lin, C.; Mu, J. Melatonin induces cell apoptosis in AGS cells through the activation of JNK and p58 MAPK and the suppression of nuclear factor-κB: A novel therapeutic implication for gastric cancer. Cell. Physiol. Biochem. 2015, 37, 2323–2338. [Google Scholar] [CrossRef] [PubMed]
- Xin, Z.; Jiang, S.; Jiang, P.; Yan, X.; Fan, C.; Di, S.; Wu, G.; Yang, Y.; Reiter, R.J.; Ji, G. Melatonin as a treatment for gastrointestinal cancer: A review. J. Pineal Res. 2015, 58, 375–387. [Google Scholar] [CrossRef] [PubMed]
- Tuntapakul, S.; Kitkhuandee, A.; Kanpittaya, J.; Johns, J.; Johns, N.P. Pineal calcification is associated with pediatric primary brain tumor. Asia Pac. J. Clin. Oncol. 2016, 12, e405–e410. [Google Scholar] [CrossRef] [PubMed]
- Buswell, R.S. The pineal gland and neoplasia. Lancet 1975, 1, 34–35. [Google Scholar] [CrossRef]
- Karamali, R.A.; Horrobin, D.F.; Ghagur, T. Role of the pineal gland in the aetiology and treatment of breast cancer. Lancet 1978, 2, 1002. [Google Scholar]
- Bartsch, H.; Bartsch, C. Effect of melatonin on experimental tumors under different photoperiods and time of administration. J. Neural Transm. 1981, 52, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Lapin, V.; Frowein, A. Effects of growing tumor on melatonin levels in rats. J. Neural Transm. 1981, 52, 343. [Google Scholar] [CrossRef]
- Blask, D.E. The pineal gland: An oncostatic gland? In The Pineal Gland; Reiter, R.E., Ed.; Naven Press: New York, NY, USA, 1984; pp. 253–284. [Google Scholar]
- Stevens, R.J. Working against our endogenous circadian clock: Breast cancer and electric lighting in the modern society. Mutat. Res. 2009, 680, 106–108. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Wan, J.; Zeng, K.; Tony, M.; Lee, A.L.; Ding, J.; Chen, Q. The reduction in circulating melatonin level may contribute to the pathogenesis of ovarian cancer: A retrospective study. J. Cancer 2016, 7, 831–836. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Barcello, E.; Mediavilla, M.D.; Tan, D.X.; Reiter, R.J. Clinical uses of melatonin: Evolution of human trials. Curr. Med. Chem. 2010, 17, 2070–2095. [Google Scholar] [CrossRef]
- Sookprasert, A.; Johns, N.P.; Phunmanee, A.; Pongthai, P.; Cheawchanwattana, A.; Johns, J.; Konsil, J.; Plaimee, P.; Porasuphatana, S.; Jitpimolmard, S. Melatonin in patients with cancer receiving chemotherapy: A randomized, double-blind placebo-controlled trial. Anticancer Res. 2014, 34, 7327–7337. [Google Scholar] [PubMed]
- Tatani, M.; Sansone, L.; Limana, F.; Arcangeli, T.; de Santis, E.; Polese, M.; Fini, M.; Russo, M.A. The interplay of reactive oxygen species, hypoxia, inflammation and sirtuins in cancer initiation and progression. Oxid. Med. Cell. Longev. 2016, 2016, 3907147. [Google Scholar]
- Prasad, S.; Gupta, S.C.; Tyagi, A.K. Reactive oxygen species (ROS) and cancer: Role of antioxidative nutraceuticals. Cancer Lett. 2016, 387, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Fresno Vara, J.A.; Casado, E.; de Castro, J.; Cejas, P.; Belda-Investa, C.; Gonzales-Baron, M. PI3K/AKT signaling pathway and cancer. Cancer Treat. Rev. 2004, 30, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signaling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.; Lee, S.R.; Yang, K.S.; Ahn, Y.; Kim, Y.J.; Stadlman, E.R.; Rhee, S.G. Reversible oxidation and inactivation of the tumor suppressor PTEN in cells stimulated with peptide growth factors. Proc. Natl. Acad. Sci. USA 2004, 101, 16419–16424. [Google Scholar] [CrossRef] [PubMed]
- Peshavariya, H.; Dusting, G.J.; Jiang, F.; Halmos, L.R.; Sobey, C.G.; Drummond, G.R.; Selemidis, S. NADPH oxidase isoform selective regulation of endothelial cell proliferation and survival. Naunyn Schmiedeberg’s Arch. Pharmacol. 2009, 380, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Potente, M.; Gerhardt, H.; Carmeliet, P. Basic and therapeutic aspects of angiogenesis. Cell 2011, 146, 873–887. [Google Scholar] [CrossRef] [PubMed]
- Madesh, M.; Hajnoczky, G. VDAC-dependent permeabilization of the outer mitochondrial membrane by superoxide induces rapid and massive cytochrome c release. J. Cell Biol. 2001, 155, 1003–1015. [Google Scholar] [CrossRef] [PubMed]
- Ordonez, R.; Fernandez, A.; Prieto-Dominguez, N.; Martinez, L.; Garcia-Ruiz, C.; Fernandez-Checa, J.C.; Mauriz, J.L.; Gonzalez-Gallego, J. Ceramide metabolism regulates autophagy and apoptotic cell death induced by melatonin in liver cancer cells. J. Pineal Res. 2015, 59, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Ishdorj, G.; Gibson, S.B. Reactive oxygen species regulation of autophagy in cancer: Implications for cancer treatment. Free Radic. Biol. Med. 2012, 53, 1399–1410. [Google Scholar] [CrossRef] [PubMed]
- Thayyullathil, F.; Rahman, A.; Pallichankandy, S.; Patel, S.; Galadari, S. ROS-dependent prostate apoptosis response-4 (Par-4) up-regulation and ceramide generation are the prime signaling events associated with curcumin-induced autophagic cell death in malignant glioma. FEBS Open Biol. 2014, 4, 763–776. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Kroemer, G. Necroptosis: A specialized pathway of programmed necrosis. Cell 2008, 135, 1161–1163. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantely, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of non-apoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Terada, Y.; Ogura, J.; Tsujimoto, T.; Kuwayama, K.; Koizumi, T.; Sasaki, H.; Maruyama, H.; Kobayashi, M.; Yamaguchi, H.; Iseki, K. Intestinal P-glycoprotein expression is multimodally regulated by intestinal ischemia-reperfusion. J. Pharm. Pharmacol. Sci. 2014, 17, 266–276. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell. Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J. Melatonin: The chemical expression of darkness. Mol. Cell. Endocrinol. 1991, 79, C153–C158. [Google Scholar] [CrossRef]
- Claustrat, B.; Brun, H.; Chagot, G. The basic physiology and pathophysiology of melatonin. Sleep Med. Rev. 2005, 9, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Jasser, S.A.; Blask, D.E.; Brainard, G.C. Light during darkness and cancer: Relationships in circadian photoreception and tumor biology. Cancer Causes Control 2006, 17, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Stevens, R.G.; Blask, D.E.; Brainard, G.C.; Hansen, J.; Lockley, S.W.; Provencio, I.; Rea, M.S.; Reinlib, I. Meeting report: The role of environmental lighting and circadian disruption in cancer and other diseases. Environ. Health Perspect. 2007, 115, 1357–1362. [Google Scholar] [CrossRef] [PubMed]
- Hill, S.M.; Blask, D.E.; Xiang, S.; Yuan, L.; Mao, L.; Dauchy, R.T.; Dauchy, E.M.; Frash, T.; Duplesis, T. Melatonin and associated signaling pathways that control normal breast epithelium and breast cancer. J. Mammary Gland Biol. Neoplasia 2011, 16, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.M.; Min, K.J.; Kwon, T.K. Melatonin-mediated Bim upregulation and cyclooxygenase-2 (COX-2) down-regulation enhances tunicamycin-induced apoptosis in MDA-MB-231 cells. J. Pineal Res. 2015, 58, 310–320. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Tan, D.X.; Korkmaz, A.; Erren, T.C.; Piekarski, C.; Tamura, H.; Manchester, L.C. Light-at-night, chronodisruption, melatonin suppression and cancer risk: A review. Crit. Rev. Oncog. 2007, 13, 303–328. [Google Scholar] [CrossRef] [PubMed]
- Erren, T.C.; Pape, H.G.; Reiter, R.J.; Piekarski, D. Chronodisruption and cancer. Naturwissenschaften 2008, 95, 367–382. [Google Scholar] [CrossRef] [PubMed]
- Erren, T.C.; Reiter, R.J. Revisiting chronodisruption: When the physiological nexus between internal and external times splits in humans. Naturwissenschaften 2013, 100, 291–293. [Google Scholar] [CrossRef] [PubMed]
- Greene, M.W. Circadian rhythms and tumor growth. Cancer Lett. 2012, 318, 115–123. [Google Scholar] [CrossRef] [PubMed]
- You, P.; Wood, P.A.; Xiong, Y.; Kobayashi, M.; Du-Quiton, J.; Hrushesky, W.J. Daily coordination of cancer growth and circadian clock gene expression. Breast Cancer Res. Treat. 2005, 91, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Baydon, L.; Roka, F.; Petit, L.; de Coppet, P.; Tirsot, M.; Barrett, P.; Morgan, P.J.; Nanoff, C.; Strosberg, A.D.; Jackers, R. Dual signaling of human Mel1a melatonin receptors via Gi2, Gi3, and Gq/11 proteins. Mol. Endocrinol. 1999, 13, 2025–2038. [Google Scholar]
- Kiefer, T.; Yuan, L.; Ram, P.; Hill, S.M. Melatonin inhibits estrogen receptor transactivation and cAMP levels in breast cancer cells. Breast Cancer Res. Treat. 2002, 71, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Dinit, V.; Korf, H.W. Impact of melatonin receptors on pCREB and clock-gene protein levels in the murine retina. Cell Tissue Res. 2007, 330, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Lai, L.; Yuan, L.; Cheng, Q.; Dong, C.; Mao, L.; Hill, S.M. Alteration of the MT1 melatonin receptor gene and its expression in primary human breast tumors and breast cancer cell lines. Breast Cancer Res. Treat. 2009, 118, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Pozo, D.; Reiter, R.J.; Calvo, J.R.; Guerrero, J.M. Inhibition of cerebellar nitric oxide synthase and cyclic AMP production by melatonin via complex formation with calmodulin. J. Cell. Biochem. 1997, 65, 430–442. [Google Scholar] [CrossRef]
- Dai, J.; Inscho, E.W.; Yuan, L.; Hill, S.M. Modulation of intracellular calcium and calmodulin by melatonin in MCF-7 human breast cancer cells. J. Pineal Res. 2002, 32, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Del Rio, B.; Garcia Pedrero, J.M.; Martinez-Campa, C.; Zuazua, P.; Lazo, P.S.; Ramos, S. Melatonin, an endogenous-specific inhibitor of estrogen receptor α via calmodulin. J. Clin. Oncol. 2004, 279, 38294–38302. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Barcelo, E.J.; Mediavilla, M.D.; Alonso-Gonzalez, C.; Rueda, N. Breast cancer therapy based on melatonin. Recent Pat. Endocr. Metab. Immune Drug Discov. 2012, 6, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Blask, D.E.; Wilson, S.T.; Zalaton, F. Physiological melatonin inhibition of human breast cancer cell growth in vitro: Evidence for a glutathione mediated pathway. Cancer Res. 1997, 57, 1909–1914. [Google Scholar] [PubMed]
- Calogero, R.A.; Cordero, F.; Forni, G.; Cavallo, F. Inflammation and breast cancer. Inflammatory component of mammary carcinogenesis in ErbB2 transgenic mice. Breast Cancer Res. 2007, 9, 211. [Google Scholar] [CrossRef] [PubMed]
- Korkmaz, A.; Sanchez-Barcelo, E.J.; Tan, D.X.; Reiter, R.J. Role of melatonin in the epigenetic regulation of breast cancer. Breast Cancer Res. Treat. 2009, 115, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Tamarkin, L.; Cohen, M.; Roselle, D.; Reichert, C.; Lippman, M.; Chabner, B. Melatonin inhibition and pinealectomy enhancement of 7, 12-dimethylbenz(a)anthracene-induced mammary tumors in the rat. Cancer Res. 1981, 41, 4432–4443. [Google Scholar] [PubMed]
- Blask, D.E.; Pelletier, D.B.; Hill, S.M.; Lemus-Wilson, A.; Grosso, D.S.; Wilson, S.T.; Wise, M.E. Pineal melatonin inhibition of tumor progression in the N-nitroso-N-methylurea model of mammary carcinogenesis: Potential involvement of antiestrogenic mechanisms in vivo. J. Cancer Res. Clin. Oncol. 1991, 117, 526–533. [Google Scholar] [CrossRef] [PubMed]
- Teplitzky, S.R.; Kiefer, T.L.; Cheng, Q.; Dwivedi, P.D.; Moroz, K.; Myers, L.; Anderson, M.B.; Collins, A.; Dai, J.; Yuan, I. Chemoprevention of NMU-induced rat mammary carcinoma with the combination of melatonin and 9-cis-retinoic acid. Cancer Lett. 2001, 168, 155–163. [Google Scholar] [CrossRef]
- Mao, L.; Yuan, L.; Xiang, S.; Zeringue, S.B.; Dauchy, R.T.; Blask, D.E.; Hauch, A.; Hill, S.M. Molecular deficiency(ies) in MT1 melatonin signaling pathway underlies the melatonin-unresponsive phenotype in MDA-MB-231 human breast cancer cells. J. Pineal Res. 2014, 56, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J. The pineal gland and melatonin in relation to aging: A summary of the theories and of the data. Exp. Gerontol. 1995, 30, 199–212. [Google Scholar] [CrossRef]
- Nooshinfar, E.; Safaroghli-Azar, A.; Bashash, D.; Akbari, M.E. Melatonin: An inhibitory agent in breast cancer. Breast Cancer 2017, 24, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Vriend, J.; Reiter, R.J. Melatonin and the von Hippel-Lindau/HIF-1 oxygen sensing mechanism: A review. Biochem. Biophys. Acta 2016, 1865, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.; Yuan, L.; Slakey, L.M.; Jones, F.E.; Burow, M.E.; Hill, S.M. Inhibition of breast cancer cell invasion by melatonin mediated through regulation of the p38 mitogen-activated protein kinase signaling pathways. Breast Cancer Res. 2010, 12, R107. [Google Scholar] [CrossRef] [PubMed]
- Santoro, R.; Mori, F.; Marani, M.; Grosso, G.; Cambria, M.A.; Blandino, G.; Muti, P.; Strano, S. Blockage of melatonin receptors impairs p53-mediated prevention of DNA damage accumulation. Carcinogenesis 2013, 34, 1051–1061. [Google Scholar] [CrossRef] [PubMed]
- Cos, S.; Blask, D.E.; Lemus-Wilson, S.; Hill, A.B. Effects of melatonin on the cell cycle kinetics and “estrogen rescue” of MCF-7 human breast cancer cells in culture. J. Pineal Res. 1991, 10, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Campa, C.; Gonzalez, A.; Mediavilla, M.D.; Alonso-Gonzalez, C.; Alvarez-Garcia, V.; Sanchez-Barcelo, E.J.; Cos, S. Melatonin inhibits aromatase promoter expression by regulating cyclooxygenases expression and activity in breast cancer cells. Br. J. Cancer 2009, 101, 1613–1619. [Google Scholar] [CrossRef] [PubMed]
- Blask, D.E.; Dauchy, R.T.; Sauer, L.A. Putting cancer to sleep at night: The neuroendocrine/circadian melatonin signal. Endocrine 2005, 27, 179–188. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Dauchy, R.T.; Sauer, L.A. Preparation of “tissue-isolated” rat tumors for perfusion: A new surgical technique that preserves blood flow. Lab. Anim. Sci. 1986, 36, 678–681. [Google Scholar] [PubMed]
- Blask, D.E.; Dauchy, R.T.; Dauchy, E.M.; Mao, L.; Hill, S.M.; Greene, M.W.; Belancio, V.P.; Sauer, L.A.; Davidson, L. Light exposure at night disrupts host/cancer circadian regulatory dynamics: Impact on the Warburg effect, lipid signaling and tumor growth prevention. PLoS ONE 2014, 9, e102776. [Google Scholar] [CrossRef] [PubMed]
- Vriend, J.; Reiter, R.J. Breast cancer cells: Modulation by melatonin and the ubiquitin-proteasome system—A review. Mol. Cell. Endocrinol. 2015, 417, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Menashe, J. Managing and avoiding bortezomib toxicity. Commun. Oncol. 2007, 4, 480–484. [Google Scholar] [CrossRef]
- Oz, E.; Ilhan, M.N. Effects of melatonin in reducing the toxic effects of melatonin on doxorubicin. Mol. Cell. Biochem. 2006, 286, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Konturek, P.C.; Konturek, S.J.; Celinski, K.; Slomka, M.; Cichoz-lock, H.; Bielanski, W.; Reiter, R.J. Role of melatonin in mucosal gastroprotection against aspirin-induced gastric lesions in humans. J. Pineal Res. 2010, 48, 418–423. [Google Scholar] [CrossRef]
- Wang, D.; Wei, Y.; Wang, T.; Wan, X.; Yang, C.S.; Reiter, R.J.; Zhang, J. Melatonin attenuates (−)-epigallocatehin-3-gallate-triggered hepatotoxicity without compromizing its downregulation of hepatic gluconeogenic and ipogenic gene in mice. J. Pineal Res. 2015, 59, 497–507. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Barcelo, E.; Mediavilla, M.D.; Vriend, J.; Reiter, R.J. Constitutive photomorphogenesis protein 1 (COP1) and COP9 signalosome, evolutionarily conserved photomorphogenic proteins as possible targets of melatonin. J. Pineal Res. 2016, 61, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.M.; Woo, S.H.; Oh, S.T.; Hong, S.E.; Chae, T.B.; Ye, S.K.; Kim, E.K.; Seong, M.K.; Kim, H.A.; Noh, W.C.; et al. Melatonin enhances arsenic trioxide-induced cell death via sustained upregulation of Redd1 expression in breast cancer cells. Mol. Cell. Endocrinol. 2016, 422, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.H.; Sohn, E.J.; Shin, E.A.; Lee, D.; Kim, B.; Jung, D.B.; Kim, J.H.; Yun, M.; Lee, H.J.; Park, Y.K.; et al. Melatonin suppresses the expression of 45S preribasomal RNA and upstream binding factor and enhances the antitumor activity of puromycin in MDA-MB-231 breast cancer cells. Evid. Based Complement. Altern. Med. 2013, 2013, 879746. [Google Scholar] [CrossRef] [PubMed]
- Margheri, M.; Pacini, N.; Tani, A.; Nasi, D.; Squcco, R.; Dama, A.; Marsha, E.; Francini, F.; Zecchi-Orlandini, S.; Fornighli, L. Combined effects of melatonin ad all-trans retinoir acid and somatostatin on breast cancer cell proliferation and death: Molecular basis for the anticancer effects of these molecules. Eur. J. Pharmacol. 2012, 681, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Kosar, P.A.; Razirogler, M.; Ovey, I.S.; Ciq, B. Synergic effects of doxorubicin and melatonin on apoptosis and mitochondrial oxidative stress in MCF-7 breast cancer cells: Involvement of TRPV1 channels. J. Membr. Biol. 2016, 249, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Gonzalez, C.; Gonzalez, A.; Martinez-Campa, C.; Gomez-Arozemena, J.; Cos, S. Melatonin sensitizes human breast cancer cells to ionizing radiation by downregulating proteins involved in double-strand DNA break repair. J. Pineal Res. 2015, 58, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Gonzalez, C.; Gonzalez, A.; Martinez-Campa, C.; Menendez-Menendez, J.; Gomez-Aroyamena, J.; Garcia-Vidal, A.; Cos, S. Melatonin enhancement of the radiosensitivity of human breast cancer cells is associated with the modulation of proteins involved in estrogen biosynthesis. Cancer Lett. 2016, 37, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Bizzarri, M.; Proietti, S.; Cucina, A.; Reiter, R.J. Molecular mechanisms of the pro-apoptotic actions of melatonin in cancer: A review. Expert Opin. Ther. Targets 2013, 17, 1483–1496. [Google Scholar] [CrossRef] [PubMed]
- Koshiyama, M.; Matsumura, N.; Konishi, I. Recent concepts of ovarian carcinogenesis: Type I and type II. Biomed. Res. Int. 2014, 2014, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.; Noishdham, D.; Jemal, A. Cancer statistics, 2012. CA Cancer J. Clin. 2012, 62, 10–29. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Jeong, S.J.; Kim, B.; Yun, S.M.; Choi, D.Y.; Kim, S.H. Melatonin synergistically enhances cisplatin-induced apoptosis via the dephosphorylation of ERK/p90 ribosomal S6 kinase/heat shock protein-27 in SK-OV-3 cells. J. Pineal Res. 2012, 52, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Chuffa, L.G.; Fioruci-Fontanelli, B.A.; Mendes, L.O.; Favaro, W.J.; Pinheiro, P.F.; Martinez, M.; Martinez, F.E. Characterization of chemically-induced ovarian carcinoma in an ethanol-preferring rat model: Influence of long-term melatonin treatment. PLoS ONE 2013, 8, e81676. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, G.M.; Martinez, M.; Camargo, I.C.; Domeniconi, R.F.; Martinez, F.E.; Chuffa, L.G. Melatonin attenuates Her-2, p38 MAPK, p-AKT, and mTOR levels in ovarian carcinoma of ethanol-preferring rats. J. Cancer 2014, 5, 728–735. [Google Scholar] [CrossRef] [PubMed]
- Chuffa, L.G.; Fioruci-Fontanelli, B.A.; Mendes, L.O.; Ferriera Silva, F.R.; Martinez, M.; Favaro, W.J.; Domeniconi, R.F.; Pinheiro, P.F.; Delayari Dos Santos, L.; Martinez, F.E. Melatonin attenuates the TLR4-mediated inflammatory response thorough MyD88- and TRIF-dependent signaling pathways in an in vivo model of ovarian cancer. BMC Cancer 2015, 15, 34. [Google Scholar] [CrossRef] [PubMed]
- Chuffa, L.G.; Alves, M.S.; Martinez, M.; Camargo, I.C.; Pinheiro, P.F.; Domeniconi, R.F.; Junior, L.A.; Martinez, F.E. Apoptosis is triggered by melatonin in an in vivo model of ovarian carcinoma. Endocr. Relat. Cancer 2016, 23, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Chuffa, L.G.; Junior, L.A.; Seiva, F.R.; Martinez, M.; Domeniconi, R.F.; Pinheiro, P.F.; dos Santos, L.D.; Martinez, F.E. Quantitative proteomic profiling reveals that diverse metabolic pathways are influenced by melatonin in an in vivo model of ovarian carcinoma. J. Prot. Res. 2016, 15, 3872–3882. [Google Scholar] [CrossRef] [PubMed]
- Dauchy, R.T.; Blask, D.E.; Dauchy, E.M.; Davidson, L.K.; Tirrell, P.C.; Greene, M.W.; Tirrell, R.P.; Hill, C.R.; Sauer, L.A. Antineoplastic effects of melatonin in a rare malignancy of mesenchymal origins: Melatonin receptor-mediated inhibition of signal transduction, lisoleic acid metabolism and growth in tissue-isolated leiomyosarcoma xenografts. J. Pineal Res. 2009, 47, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.; Dauchy, R.T.; Blask, D.E.; Dauchy, E.M.; Slakey, L.M.; Brimer, S.; Yuan, L.; Xiang, S.; Hauch, A.; Smith, K.; et al. Melatonin suppression of aerobic glycolysis (Warburg effect), survival signaling and metastasis in human leiomyosarcoma. J. Pineal Res. 2016, 60, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Berlin, J.; Benson, A.B. Chemotherapy: Gemcitabine remains the standard of care for pancreatic cancer. Nat. Rev. Clin. Oncol. 2010, 7, 135–157. [Google Scholar] [CrossRef] [PubMed]
- Saif, M.W.; Black, G.; Roy, S.; Bell, D.; Russo, S.; Eloubeidi, M.A.; Steg, A.; Johnson, M.R.; Zelterman, D.; Diasio, R.B. Phase II study of capecitabine with concomitant radiotherapy for patients with locally advanced pancreatic cancer: Up-regulation of thymidine phosphorylase. Cancer J. 2007, 13, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Uguz, A.C.; Cig, B.; Espino, J.; Bejerano, I.; Naziraglu, M.; Rodriguez, A.B.; Pariente, J.A. Melatonin potentiates chemotherapy induced cytotoxicity and apoptosis in rat pancreatic tumor cells. J. Pineal Res. 2012, 53, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Rabelo, J.F.; Vasquez, R.; Perea, M.D.; Cruz, A.; Gonzalez, R.; Romero, A.; Munoz-Villanueva, M.C.; Tunez, I.; Montilla, P.; Muntane, J.; et al. Beneficial properties of melatonin in an experimental model of pancreatic cancer. J. Pineal Res. 2007, 43, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Rabelo, J.F.; Vasquez, R.; Arjona, A.; Perea, D.; Montilla, P.; Tunez, I.; Muntane, J.; Padillo, J. Improvement of capecitabine antitumor activity by melatonin in pancreatic cancer. Pancreas 2011, 40, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Leja-Szpak, A.; Jaworek, J.; Pierzchalski, P.; Reiter, R.J. Melatonin induces pro-apoptotic signaling pathway in human pancreatic carcinoma cells (PANC-1). J. Pineal Res. 2010, 49, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Perez, V.I.; Cortez, L.A.; Lew, C.M.; Rodriguez, M.; Webb, C.R.; Van Remmen, H.; Chaudhuri, A.; Qi, W.; Lee, S.; Bokov, A.; et al. Thioredoxin 1 overexpression extends mainly the earlier part of life span in mice. J. Gerontol. A Biol. Sci. Med. Sci. 2011, 66, 1286–1299. [Google Scholar] [CrossRef] [PubMed]
- Padillo, F.J.; Ruiz-Rabelo, J.F.; Cruz, A.; Perea, M.D.; Tasset, I.; Montilla, P.; Tunez, I.; Muntane, J. Melatonin and celecoxib improve the outcomes in hamsters with experimental pancreatic cancer. J. Pineal Res. 2010, 49, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Lissoni, P.; Brivio, F.; Fumagalli, L.; Messina, G.; Vigora, L.; Parolini, D.; Calciago, M.; Rovelli, F. Neuroimmunomodulation in medical oncology: Application of psychoneuroimmunology with subcutaneous low dose IL-2 and the pineal hormone melatonin in patients with untreatable metastatic solid tumors. Anticancer Res. 2008, 28, 1377–1381. [Google Scholar] [PubMed]
- Augello, C.; Caruso, L.; Maggioni, M. Inhibitors of apoptosis proteins (IAPs) expression and their prognostic significance in hepatocellular carcinoma. BMC Cancer 2009, 9, 125. [Google Scholar] [CrossRef] [PubMed]
- Gyrd-Hansen, M.; Meier, P. IAPs: From caspase inhibitors to modulators of KF-κB, inflammation and cancer. Nat. Rev. Cancer 2010, 10, 561–574. [Google Scholar] [CrossRef] [PubMed]
- Mannhold, R.; Fulda, S.; Carosati, E. IAP antagonists: Promising candidates for cancer therapy. Drug Discov. Today 2010, 15, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Straub, C.S. Targeting IAPs as an approach to anti-cancer therapy. Curr. Top. Med. Chem. 2011, 11, 291–316. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Sun, G.; Ma, T.; Zhong, F.; Wei, W. Melatonin overcomes apoptosis in human hepatocellular carcinoma by targeting survivin and XIAP. J. Pineal Res. 2013, 55, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Hoffman, K.; Gao, C.; Petrulionis, M.; Herr, I.; Schemmer, P. Melatonin promotes sorafenib induced apoptosis through synergistic activation of JNK/c-Jun pathway in human hepatocellular carcinoma. J. Pineal Res. 2017, 62. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.M.; Zhang, Y. Melatonin: A well-documented antioxidant with conditional pro-oxidant actions. J. Pineal Res. 2014, 57, 131–146. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.S.; Lai, F.P.; Lo, R.K.; Voyno-Yasenetskaya, T.A.; Stanbridge, E.J.; Wong, Y.H. Melatonin MT1 and MT2 receptors stimulate c-Jun N-terminal kinase via pertursis toxin-sensitive and insensitive G-proteins. Cell. Signal. 2002, 14, 249–257. [Google Scholar] [CrossRef]
- Wei, J.Y.; Li, W.M.; Zhou, L.L.; Lee, Q.N.; He, W. Melatonin induces apoptosis of colorectal cancer cells through HDAC4 nuclear impact mediated by Ca MKII inactivation. J. Pineal Res. 2015, 58, 429–431. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Wen, J.; Lee, Y.; Lee, S.; Park, K.; Chang, K.T.; Hong, Y. Melatonin treatment induces interplay of apoptosis, autophagy, and senescence in human colorectal cancer cells. J. Pineal Res. 2014, 56, 264–274. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Tan, D.X.; Burkhardt, S.; Manchester, L.C. Melatonin in plants. Nutr. Rev. 2001, 59, 286–290. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Tan, D.X.; Zhou, Z.; Cruz, M.H.; Fuentes-Broto, L.; Galano, A. Phytomelatonin: Assisting plants to survive and thrive. Molecules 2015, 20, 7396–7437. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.X.; Zanghi, B.M.; Manchester, L.C.; Reiter, R.J. Melatonin identified in meats and other food stuffs: Potential nutritional impact. J. Pineal Res. 2014, 57, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Leon, J.; Casado, J.; Jimenez Ruiz, S.M.; Zurita, M.S.; Gonzalez-Puga, C.; Rejon, J.D.; Gila, A.; Munoz de Rueda, P.; Pavon, E.J.; Reiter, R.J.; et al. Melatonin reduces endothelin-1 expression and secretion in colon cancer cells through the inactivation of FoxO-1 and NF-κβ. J. Pineal Res. 2014, 56, 415–426. [Google Scholar] [CrossRef] [PubMed]
- Bender, E. Epidemiology: The dominant malignancy. Nature 2014, 513, S2–S3. [Google Scholar] [CrossRef] [PubMed]
- Plaimee, P.; Weerapreeyakyul, N.; Barusrux, S.; Johns, N.P. Melatonin potentiates cisplatin-induced apoptosis and cell cycle arrest in human lung adenocarcinoma cells. Cell Prolif. 2015, 48, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Anisimov, V.N.; Popovich, I.G.; Zabezhinski, M.A.; Anisimov, S.V.; Vesnushkin, G.M.; Vinogradova, I.A. Melatonin as antioxidant, geroprotector and anticarcinogen. Biochem. Biophys. Acta 2006, 1757, 573–589. [Google Scholar] [CrossRef] [PubMed]
- Vesnushkin, G.M.; Plotnikova, N.A.; Semenchenko, A.V.; Anisimov, V.N. Melatonin inhibits urethane-induced carcinogenesis tumors in murine lung. Vopr. Onkol. 2006, 52, 164–168. [Google Scholar] [PubMed]
- Stoimenov, I.; Helleday, T. PCNA on the crossroad of cancer. Biochem. Soc. Trans. 2009, 37, 605–613. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.; Pan, Y.; Yang, Y.; Di, S.; Jiang, S.; Ma, Z.; Li, T.; Zhang, Z.; Li, W.; Li, X.; et al. HDAC1 inhibition by melatonin leads to suppression of lung adenocarcinoma cells via induction of oxidative stress and activation of apoptotic pathways. J. Pineal Res. 2015, 59, 321–333. [Google Scholar] [CrossRef] [PubMed]
- Mehta, V. Radiation pneumonitis and pulmonary fibrosis in non-small-cell lung cancer: Pulmonary function, prediction and prevention. Int. J. Radiat. Oncol. Biol. Phys. 2005, 63, 5–25. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Cai, L.; Jiang, P.; Wang, J.; Gao, L.; Feng, H.; Wang, C.; Pan, H.; Yang, Y. SIRT1 inhibition by melatonin exerts antitumor activity in human osteosarcoma cells. Eur. J. Pharmacol. 2013, 715, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Xu, Y.; Reiter, R.J. Melatonin inhibits the proliferation of human osteosarcoma cell line MG-63. Bone 2013, 55, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Xu, Y.; Reiter, R.J.; Pan, Y.; Chen, D.; Liu, Y.; Pu, X.; Jiang, L.; Li, Z. Inhibition of ERK1/2 signaling pathway is involved in melatonin’s antiproliferative effect of human MG-63 osteosarcoma cells. Cell. Physiol. Biochem. 2016, 39, 2297–2307. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Hao, A.; Li, X.; Du, Z.; Li, H.; Wang, H.; Yang, H.; Fang, Z. Melatonin inhibits tumorigenicity of glioblastoma stem-like cells via AKT-EZH2-STAT3 signalling axis. J. Pineal Res. 2016, 61, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Martin, V.; Sanchez-Sanchez, A.M.; Puente-Moncada, N.; Gomez-Lobo, M.; Alvarez-Vega, M.A.; Antolin, I.; Rodriguez, C. Involvement of autophagy in melatonin-induced cytotoxicity in glioma-initating cells. J. Pineal Res. 2014, 57, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, Q.; Wang, F.; Ling, E.A.; Liu, S.; Wang, L.; Yang, Y.; Yao, L.; Chen, X.; Wang, F.; et al. Melatonin antagonizes hypoxia-mediated glioblastoma cell migration and invasion via inhibition of HIF-1α. J. Pineal Res. 2013, 55, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Quintana, C.; Cabrera, J.; Perdome, J.; Estevez, F.; Loro, J.F.; Reiter, R.J.; Quintana, J. Melatonin enhances hyperthermia-induced apoptotic cell death in human leukemia cells. J. Pineal Res. 2016, 61, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Toraya-Brown, S.; Fiering, S. Local tumor hyperthermia as immunotherapy for metastatic cancer. Int. J. Hyperth. 2014, 30, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Datta, N.R.; Grobholz, R.; Puric, E. Enchanced tumor regression in patient with liposarcoma treatment with radiotherapy and hyperthermia: Hint for dynamic immunomodulation of hyperthermia. Int. J. Hyperth. 2015, 20, 1–4. [Google Scholar]
- Srinivasan, V.; Spence, D.W.; Pandi-Perumal, S.R.; Trakht, I.; Cardinali, D.P. Therapeutic actions of melatonin in cancer: Possible mechanisms. Integr. Cancer Ther. 2008, 7, 189–203. [Google Scholar] [CrossRef] [PubMed]
- Tai, S.Y.; Huang, S.P.; Bao, B.Y.; Wu, M.T. Urinary melatonin-sulfate/cortisol ratio and the presence of prostate cancer: A case-control study. Sci. Rep. 2016, 6, 29606. [Google Scholar] [CrossRef] [PubMed]
- Shiu, S.Y.; Law, I.C.; Lau, K.W.; Tam, P.C.; Yip, A.W.; Ng, W.T. Melatonin slowed the early biochemical progression of hormone-refractory prostate cancer in a patient whose prostate tumor tissue expressed MT1 receptor subtype. J. Pineal Res. 2003, 35, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Shiu, S.Y. Towards rational and evidence-based use of melatonin in prostate cancer prevention and treatment. J. Pineal Res. 2007, 43, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Marelli, M.M.; Limonta, P.; Maggi, R.; Motta, M.; Moretti, R.M. Growth-inhibitory activity of melatonin on human androgen-independent DU 145 prostate cancer cells. Prostate 2000, 45, 238–344. [Google Scholar] [CrossRef]
- Xi, S.C.; Tam, P.C.; Brown, G.M.; Pang, S.F.; Shiu, S.Y. Potential involvement of mt1 receptor and attenuated sex steroid-induced calcium influx in the direct anti-proliferative action of melatonin on androgen-responsive LNCaP human prostate cancer cells. J. Pineal Res. 2000, 29, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Xi, S.C.; Siu, S.W.; Fong, S.W.; Shiu, S.Y. Inhibition of androgen-sensitive LNCaP prostate cancer growth in vivo by melatonin: Association of antiproliferative action of the pineal hormone with mt1 receptor protein expression. Prostate 2001, 46, 52–61. [Google Scholar] [CrossRef]
- Siu, S.W.; Lau, K.W.; Tam, P.C.; Shiu, S.Y. Melatonin and prostate cancer cell proliferation: Interplay with castration, epidermal growth factor, and androgen sensitivity. Prostate 2002, 52, 106–122. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.H.; Yoo, Y.M. Melatonin induces apoptotic cell death via p53 in LNCaP cells. Korean J. Physiol. Pharmacol. 2010, 14, 365–369. [Google Scholar] [CrossRef] [PubMed]
- Moretti, R.M.; Marelli, M.M.; Maggi, R.; Dondi, D.; Motta, M.; Limonta, P. Antiproliferative action of melatonin on human prostate cancer LNCaP cells. Oncol. Rep. 2000, 7, 347–351. [Google Scholar] [CrossRef] [PubMed]
- Bonomini, F.R.P.; Borsani, E.; Castrezzati, S.; Fraschini, F.; Lonati, C.; Finati, E.; Samaja, M. Prostate cancer and novel ways to deliver melatonin. Ital. J. Anat. Embryol. 2013, 118, 3. [Google Scholar]
- Paroni, R.; Terraneo, L.; Bonomini, F.; Finati, E.; Virgili, E.; Bianciardi, P.; Favero, G.; Fraschini, F.; Reiter, R.J.; Rezzani, R.; et al. Antitumour activity of melatonin in a mouse model of human prostate cancer: Relationship with hypoxia signalling. J. Pineal Res. 2014, 57, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Sohn, E.J.; Won, G.; Lee, J.; Lee, S.; Kim, S.H. Upregulation of miRNA3195 and miRNA374b mediates the anti-angiogenic properties of melatonin in hypoxic PC-3 prostate cancer cells. J. Cancer 2015, 6, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Nishida, N.; Yano, H.; Nishida, T.; Kamura, T.; Kojiro, M. Angiogenesis in cancer. Vasc. Health Risk Manag. 2006, 2, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Garcia, A.; Mayo, J.C.; Hevia, D.; Quiros-Gonzalez, I.; Navarro, M.; Sainz, R.M. Phenotypic changes caused by melatonin increased sensitivity of prostate cancer cells to cytokine-induced apoptosis. J. Pineal Res. 2013, 54, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Sainz, R.M.; Reiter, R.J.; Tan, D.X.; Roldan, F.; Natarajan, M.; Quiros, I.; Hevia, D.; Rodriguez, C.; Mayo, J.C. Critical role of glutathione in melatonin enhancement of tumor necrosis factor and ionizing radiation-induced apoptosis in prostate cancer cells in vitro. J. Pineal Res. 2008, 45, 258–370. [Google Scholar] [CrossRef] [PubMed]
- Ge, D.; Dauchy, R.T.; Liu, S.; Zhang, Q.; Mao, L.; Dauchy, E.M.; Blask, D.E.; Hill, S.M.; Rowan, B.G.; Brainard, G.C.; et al. Insulin and IGF1 enhance IL-17-induced chemokine expression through a GSK3B-dependent mechanism: A new target for melatonin’s anti-inflammatory action. J. Pineal Res. 2013, 55, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Neri, B.; de Leonardis, V.; Gemelli, M.T.; di Loro, F.; Mottola, A.; Ponchietti, R.; Raugei, A.; Cini, G. Melatonin as biological response modifier in cancer patients. Anticancer Res. 1998, 18, 1329–1332. [Google Scholar] [PubMed]
- Sainz, R.M.; Mayo, J.C.; Tan, D.X.; Leon, J.; Manchester, L.; Reiter, R.J. Melatonin reduces prostate cancer cell growth leading to neuroendocrine differentiation via a receptor and PKA independent mechanism. Prostate 2005, 63, 29–43. [Google Scholar] [CrossRef] [PubMed]
- Mayo, J.C.; Hevia, D.; Quiros-Gonzalez, I.; Rodriguez-Garcia, A.; Gonzalez-Menendez, P.; Cepas, V.; Gonzalez-Pola, I.; Sainz, R.M. IGFBP3 and MAPK/ERK signaling mediates melatonin-induced antitumor activity in prostate cancer. J. Pineal Res. 2017, 62. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Niu, J.; Huang, J. Neuroendocrine differentiation in prostate cancer. Am. J. Transl. Res. 2009, 1, 148–162. [Google Scholar] [PubMed]
- Berman-Booty, L.D.; Knudsen, K.E. Models of neuroendocrine prostate cancer. Endocr. Relat. Cancer 2015, 22, R33–R49. [Google Scholar] [CrossRef] [PubMed]
- Tam, C.W.; Mo, C.W.; Yao, K.M.; Shiu, S.Y. Signaling mechanisms of melatonin in antiproliferation of hormone-refractory 22Rv1 human prostate cancer cells: Implications for prostate cancer chemoprevention. J. Pineal Res. 2007, 42, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Shiu, S.Y.; Pang, B.; Tam, C.W.; Yao, K.M. Signal transduction of receptor-mediated antiproliferative action of melatonin on human prostate epithelial cells involves dual activation of Galpha(s) and Galpha(q) proteins. J. Pineal Res. 2010, 49, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Tam, C.W.; Chan, K.W.; Liu, V.W.; Pang, B.; Yao, K.M.; Shiu, S.Y. Melatonin as a negative mitogenic hormonal regulator of human prostate epithelial cell growth: Potential mechanisms and clinical significance. J. Pineal Res. 2008, 45, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Lonergan, P.E.; Tindall, D.J. Androgen receptor signaling in prostate cancer development and progression. J. Carcinog. 2011, 10, 20. [Google Scholar] [PubMed]
- Lupowitz, Z.; Rimler, A.; Zisapel, N. Evaluation of signal transduction pathways mediating the nuclear exclusion of the androgen receptor by melatonin. Cell. Mol. Life Sci. 2001, 58, 2129–2135. [Google Scholar] [CrossRef] [PubMed]
- Rimler, A.; Culig, Z.; Levy-Rimler, G.; Lupowitz, Z.; Klocker, H.; Matzkin, H.; Bartsch, G.; Zisapel, N. Melatonin elicits nuclear exclusion of the human androgen receptor and attenuates its activity. Prostate 2001, 49, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Rimler, A.; Culig, Z.; Lupowitz, Z.; Zisapel, N. Nuclear exclusion of the androgen receptor by melatonin. J. Steroid Biochem. Mol. Biol. 2002, 81, 77–84. [Google Scholar] [CrossRef]
- Joo, S.S.; Yoo, Y.M. Melatonin induces apoptotic death in LNCaP cells via p38 and JNK pathways: Therapeutic implications for prostate cancer. J. Pineal Res. 2009, 47, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Park, J.W.; Hwang, M.S.; Suh, S.I.; Baek, W.K. Melatonin down-regulates HIF-1 α expression through inhibition of protein translation in prostate cancer cells. J. Pineal Res. 2009, 46, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Hudson, C.C.; Liu, M.; Chiang, G.G.; Otterness, D.M.; Loomis, D.C.; Kaper, F.; Giaccia, A.J.; Abraham, R.T. Regulation of hypoxia-inducible factor 1α expression and function by the mammalian target of rapamycin. Mol. Cell. Biol. 2002, 22, 7004–7014. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.T.; Blask, D.E.; Lemus-Wilson, A.M. Melatonin augments the sensitivity of MCF-7 human breast cancer cells to tamoxifen in vitro. J. Clin. Endocrinol. Metab. 1992, 75, 669–670. [Google Scholar] [CrossRef] [PubMed]
- Dauchy, R.T.; Xiang, S.; Mao, L.; Brimer, S.; Wren, M.A.; Yuan, L.; Anbalagan, M.; Hauch, A.; Frasch, T.; Rowan, B.G.; et al. Circadian and melatonin disruption by exposure to light at night drives intrinsic resistance to tamoxifen therapy in breast cancer. Cancer Res. 2014, 74, 4099–5110. [Google Scholar] [CrossRef] [PubMed]
- Dauchy, R.T.; Blask, D.E.; Sauer, L.A.; Brainard, G.C.; Krause, J.A. Dim light during darkness stimulates tumor progression by enhancing tumor fatty acid uptake and metabolism. Cancer Lett. 1999, 144, 131–136. [Google Scholar] [CrossRef]
- Blask, D.E.; Hill, S.M.; Dauchy, R.T.; Xiang, S.; Yuan, L.; Duplessis, T.; Mao, L.; Dauchy, E.; Sauer, L. Circadian regulation of molecular, dietary, and metabolic signaling mechanisms of human breast cancer growth by the nocturnal melatonin signal and the consequence of its disruption by light at night. J. Pineal Res. 2011, 51, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Becerra, R.; Santos, N.; Diaz, L.; Camacho, J. Mechanisms of resistance to endocrine therapy in breast cancer: Focus on signaling pathways, miRNAs and genetically based resistance. Int. J. Mol. Sci. 2012, 13, 108–145. [Google Scholar] [CrossRef] [PubMed]
- Browne, B.C.; Hochgrafe, F.; Wu, J.; Millar, E.K.; Baraaclough, J.; Stone, A. Global characterization of signaling networks associated with tamoxifen resistance in breast cancer. FEBS J. 2013, 280, 5237–5257. [Google Scholar] [CrossRef] [PubMed]
- Hiscox, S.; Morgan, L.; Green, T.P.; Barrow, D.; Gee, J.; Nicholson, R.I. Elevated Src activity promotes cellular invasion and motility in tamoxifen resistant breast cancer cells. Breast Cancer Res. Treat. 2006, 97, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Keshet-Sitton, A.; Or-Chen, K.I.; Yitzhak, S.; Tzabary, I.; Haim, A. Can avoiding light at night reduce the risk of breast cancer? Integr. Cancer Ther. 2016, 15, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Martin, V.; Garica-Santos, G.; Rodriguez-Blanco, J.; Casado-Zapico, S.; Sanchez-Sanchez, A.; Antolin, I.; Medina, M.; Rodriguez, C. Melatonin sensitizes human malignant glioma cells against TRAIL-induced cell death. Cancer Lett. 2010, 287, 216–223. [Google Scholar] [CrossRef] [PubMed]
- Casado-Zapico, S.; Rodriguez-Blanco, J.; Garcia-Santos, G.; Martin, V.; Sanchez-Sanchez, A.M.; Antolin, I.; Rodrigeuez, C. Synergistic antitumor effect of melatonin with sevral chemotherapeutic drugs on human Ewing sarcoma cancer cells: Potentiation of the extrinsic apoptotic pathway. J. Pineal Res. 2010, 48, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Xiang, S.; Dauchy, R.T.; Hauch, A.; Mao, L.; Yuan, L.; Wren, M.A.; Belancio, V.P.; Mondal, D.; Frasch, T.; Blask, D.E.; et al. Doxorubicin resistance in breast cancer is driven by light at night-induced disruption of the circadian melatonin signal. J. Pineal Res. 2015, 59, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Coghlin, C.; Murray, G.I. Current and emerging concepts in tumour metastasis. J. Pathol. 2010, 222, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Su, S.C.; Hsieh, M.J.; Yang, W.E.; Chung, W.H.; Reiter, R.J.; Yang, S.F. Cancer metastasis: Mechanisms of inhibition by melatonin. J. Pineal Res. 2017, 62, e12370. [Google Scholar] [CrossRef] [PubMed]
- Cavallaro, U.; Christofori, G. Cell adhesion in tumor invasion and metastasis: Loss of the glue is not enough. Biochim. Biophys. Acta 2001, 1552, 39–45. [Google Scholar] [CrossRef]
- Cos, S.; Fernandez, R.; Guezmes, A.; Sanchez-Barcelo, E.J. Influence of melatonin on invasive and metastatic properties of MCF-7 human breast cancer cells. Cancer Res. 1998, 58, 4383–4390. [Google Scholar] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Beavon, I.R. The E-cadherin-catenin complex in tumour metastasis: Structure, function and regulation. Eur. J. Cancer 2000, 36, 1607–1620. [Google Scholar] [CrossRef]
- Onder, T.T.; Gupta, P.B.; Mani, S.A.; Yang, J.; Lander, E.S.; Weinberg, R.A. Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res. 2008, 68, 3645–3654. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.M.; Lin, W.Y.; Shen, C.C.; Pan, H.C.; Keh-Bin, W.; Chen, Y.C.; Jan, Y.J.; Lai, D.W.; Tang, S.C.; Tien, H.R.; et al. Melatonin set out to ER stress signaling thwarts epithelial mesenchymal transition and peritoneal dissemination via calpain-mediated C/EBPβ and NF-κB cleavage. J. Pineal Res. 2016, 60, 142–154. [Google Scholar] [CrossRef] [PubMed]
- Martin, T.A.; Jiang, W.G. Loss of tight junction barrier function and its role in cancer metastasis. Biochim. Biophys. Acta 2009, 1788, 872–891. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Gui, S.; Zhou, Q.; Wang, Y. Melatonin inhibits the migration of human lung adenocarcinoma A549 cell lines involving JNK/mAPK pathway. PLoS ONE 2014, 9, e101132. [Google Scholar] [CrossRef] [PubMed]
- Du, D.; Xu, F.; Yu, L.; Zhang, C.; Lu, X.; Yuan, H.; Huang, Q.; Zhang, F.; Bao, H.; Jia, L.; et al. The tight junction protein, occludin, regulates the directional migration of epithelial cells. Dev. Cell 2010, 18, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O. Integrins: Bidirectional, allosteric signaling machines. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef]
- Xu, C.S.; Wang, Z.F.; Huang, X.D.; Dai, L.M.; Cao, C.J.; Li, Z.Q. Involvement of ROS-αvβ3 integrin-FAK/Pyk2 in the inhibitory effect of melatonin on U251 glioma cell migration and invasion under hypoxia. J. Transl. Med. 2015, 13, 95. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.G.; Li, Y.; Li, L.; Kan, X. Overexpression of osteopontin and integrin alphav in laryngeal and hypopharyngeal carcinomas associated with differentiation and metastasis. J. Cancer Res. Clin. Oncol. 2011, 137, 1613–1618. [Google Scholar] [CrossRef] [PubMed]
- Van den Hoogen, C.; van der Horst, G.; Cheung, H.; Buijs, J.T.; Pelger, R.C.; van der Pluijm, G. Integrin α expression is required for the acquisition of a metastatic stem/progenitor cell phenotype in human prostate cancer. Am. J. Pathol. 2011, 179, 2559–2568. [Google Scholar] [CrossRef] [PubMed]
- Psaila, B.; Lyden, D. The metastatic niche: Adapting the foreign soil. Nat. Rev. Cancer 2009, 9, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Swarnakar, S.; Paul, S.; Singh, L.P.; Reiter, R.J. Matrix metalloproteinases in health and disease: Regulation by melatonin. J. Pineal Res. 2011, 50, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Ho, H.Y.; Lin, C.W.; Chien, M.H.; Reiter, R.J.; Su, S.C.; Hsieh, Y.H.; Yang, S.F. Melatonin suppresses TPA-induced metastasis by downregulating matrix metalloproteinase-9 expression through JNK/SP-1 signaling in nasopharyngeal carcinoma. J. Pineal Res. 2016, 61, 479–492. [Google Scholar] [CrossRef] [PubMed]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell. Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; Ji, H.; Feng, N.; Zhang, Y.; Yang, X.; Andersson, P.; Sun, Y.; Tritsaris, K.; Hansen, A.J.; Dissing, S.; et al. Collaborative interplay between FGF-2 and VEGF-C promotes lymphangiogenesis and metastasis. Proc. Natl. Acad. Sci. USA 2012, 109, 15894–15899. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y. Opinion: Emerging mechanisms of tumour lymphangiogenesis and lymphatic metastasis. Nat. Rev. Cancer 2005, 5, 735–743. [Google Scholar] [CrossRef] [PubMed]
- Cross, M.J.; Claesson-Welsh, L. FGF and VEGF function in angiogenesis: Signalling pathways, biological responses and therapeutic inhibition. Trends Pharmacol. Sci. 2001, 22, 201–207. [Google Scholar] [CrossRef]
- Rudra, D.S.; Pal, U.; Maiti, N.C.; Reiter, R.J.; Swarnakar, S. Melatonin inhibits matrix metalloproteinase-9 activity by binding to its active site. J. Pineal Res. 2013, 54, 398–405. [Google Scholar] [CrossRef] [PubMed]
- Yeh, C.M.; Lin, C.W.; Yang, J.S.; Yang, W.E.; Su, S.C.; Yang, S.F. Melatonin inhibits TPA-induced oral cancer cell migration by suppressing matrix metalloproteinase-9 activation through the histone acetylation. Oncotarget 2016, 7, 21952–21967. [Google Scholar] [PubMed]
- Lin, Y.W.; Lee, L.M.; Lee, W.J.; Chu, C.Y.; Tan, P.; Yang, Y.C.; Chen, W.Y.; Yang, S.F.; Hsiao, M.; Chien, M.H. Melatonin inhibits MMP-9 transactivation and renal cell carcinoma metastasis by suppressing Akt-MAPKs pathway and NF-κB DNA-binding activity. J. Pineal Res. 2016, 60, 277–290. [Google Scholar] [CrossRef] [PubMed]
- Borin, T.F.; Arbab, A.S.; Gelaleti, G.B.; Ferreira, L.C.; Moschetta, M.G.; Jardim-Perassi, B.V.; Iskander, A.S.; Varma, N.R.; Shankar, A.; Coimbra, V.B.; et al. Melatonin decreases breast cancer metastasis by modulating Rho-associated kinase protein-1 expression. J. Pineal Res. 2016, 60, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Goncalves Ndo, N.; Rodrigues, R.V.; Jardim-Perassi, B.V.; Moschetta, M.G.; Lopes, J.R.; Colombo, J.; Zuccari, D.A. Molecular markers of angiogenesis and metastasis in lines of oral carcinoma after treatment with melatonin. Anticancer Agents Med. Chem. 2014, 14, 1302–1311. [Google Scholar] [CrossRef] [PubMed]
- Shook, D.; Keller, R. Mechanisms, mechanics and function of epithelial-mesenchymal transitions in early development. Mech. Dev. 2003, 120, 1351–1383. [Google Scholar] [CrossRef] [PubMed]
- Heerboth, S.; Housman, G.; Leary, M.; Longacre, M.; Byler, S.; Lapinska, K.; Willbanks, A.; Sarkar, S. EMT and tumor metastasis. Clin. Transl. Med. 2015, 4, 6. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.C.; Fattet, L.; Yang, J. The forces behind EMT and tumor metastasis. Cell Cycle 2015, 14, 2387–2388. [Google Scholar] [CrossRef] [PubMed]
- Acloque, H.; Adams, M.S.; Fishwick, K.; Bronner-Fraser, M.; Nieto, M.A. Epithelial-mesenchymal transitions: The importance of changing cell state in development and disease. J. Clin. Investig. 2009, 119, 1438–1449. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A. Epithelial plasticity: A common theme in embryonic and cancer cells. Science 2013, 342, 1234850. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Ten Berge, D.; Koole, W.; Fuerer, C.; Fish, M.; Eroglu, E.; Nusse, R. Wnt signaling mediates self-organization and axis formation in embryoid bodies. Cell Stem Cell 2008, 3, 508–518. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.; Dauchy, R.T.; Blask, D.E.; Slakey, L.M.; Xiang, S.; Yuan, L.; Dauchy, E.M.; Shan, B.; Brainard, G.C.; Hanifin, J.P.; et al. Circadian gating of epithelial-to-mesenchymal transition in breast cancer cells via melatonin-regulation of GSK3β. Mol. Endocrinol. 2012, 26, 1808–1820. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.; Summers, W.; Xiang, S.; Yuan, L.; Dauchy, R.T.; Reynolds, A.; Wren-Dail, M.A.; Pointer, D.; Frasch, T.; Blask, D.E.; et al. Melatonin represses metastasis in Her2-positive human breast cancer cells by suppressing RSK2 expression. Mol. Cancer Res. 2016, 14, 1159–1169. [Google Scholar] [CrossRef] [PubMed]
- Furuya, M.; Yonemitsu, Y.; Aoki, I., III. Angiogenesis: Complexity of tumor vasculature and microenvironment. Curr. Pharm. Des. 2009, 15, 1854–1867. [Google Scholar] [CrossRef] [PubMed]
- Otrock, Z.K.; Mahfouz, R.A.; Makarem, J.A.; Shamseddine, A.I. Understanding the biology of angiogenesis: Review of the most important molecular mechanisms. Blood Cells Mol. Dis. 2007, 39, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Saharinen, P.; Eklund, L.; Pulkki, K.; Bono, P.; Alitalo, K. VEGF and angiopoietin signaling in tumor angiogenesis and metastasis. Trends Mol. Med. 2011, 17, 347–362. [Google Scholar] [CrossRef] [PubMed]
- Sia, D.; Alsinet, C.; Newell, P.; Villanueva, A. VEGF signaling in cancer treatment. Curr. Pharm. Des. 2014, 20, 2834–2842. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.J.; Choi, J.S.; Kang, I.; Kim, K.W.; Jeong, C.H.; Jeong, J.W. Melatonin suppresses tumor progression by reducing angiogenesis stimulated by HIF-1 in a mouse tumor model. J. Pineal Res. 2013, 54, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Sohn, E.J.; Won, G.; Lee, J.; Lee, S.; Kim, S.H. Upregulation of miRNA3195 and miRNA374b Mediates the Anti-Angiogenic Properties of Melatonin in Hypoxic PC-3 Prostate Cancer Cells. J. Cancer 2015, 6, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.X.; Liu, H.; Xu, L.; Zhang, H.; Zhou, R.X. Involvement of nuclear receptor RZR/RORγ in melatonin-induced HIF-1α inactivation in SGC-7901 human gastric cancer cells. Oncol. Rep. 2015, 34, 2541–2546. [Google Scholar] [CrossRef] [PubMed]
- Bagnato, A.; Spinella, F. Emerging role of endothelin-1 in tumor angiogenesis. Trends Endocrinol. Metab. 2003, 14, 44–50. [Google Scholar] [CrossRef]
- Cavallo, F.; de Giovanni, C.; Nanni, F.; Forni, G.; Lollini, P.L. 2011: The immune hallmarks of cancer. Cancer Immunol. Immunother. 2011, 60, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwell, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Greten, F.R.; Echmann, L.; Greten, T.F.; Park, J.M.; Li, Z.W.; Egan, L.J.; Kagnoff, M.F.; Karin, M. IKKβ links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 2004, 118, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Pikarsky, E.; Porat, R.M.; Stein, I.; Abramovitch, R.; Amit, S.; Kasem, S.; Gutkovich-Pyest, E.; Urieli-Shoval, S.; Galun, E.; Ben-Neriah, Y. NF-κB functions as a tumor promoter in inflammation-associated cancer. Nature 2004, 431, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Mohan, N.; Sadeghi, K.; Reiter, R.J.; Melt, M.L. The neurohormone melatonin inhibits cytokine, mitogen and ionizing radiation induce NF-κB. Biochem. Mol. Biol. Int. 1995, 37, 1063–1070. [Google Scholar] [PubMed]
- Chuang, J.I.; Natarajan, M.; Meltz, M.L.; Reiter, R.J. Effect of melatonin on NF-κB DNA-binding activity in the spleen. Cell Biol. Int. 1996, 20, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Hosseinzadeh, A.; Kamrava, S.K.; Joghataei, M.T.; Darabi, R.; Shakeri-Zadeh, A.; Shahriari, M.; Reiter, R.J.; Ghaznavi, H.; Mehrzadi, S. Apoptosis signaling pathways in osteoarthritis and possible protective role of melatonin. J. Pineal Res. 2016, 61, 411–425. [Google Scholar] [CrossRef] [PubMed]
- Carrillo-Vico, A.; Guerro, J.M.; Lardone, P.J.; Reiter, R.J. A review of the multiple actions of melatonin on the immune system. Endocrine 2005, 27, 189–200. [Google Scholar] [CrossRef]
- Tuli, H.S.; Kashyap, D.; Sharma, A.K.; Sandhu, S.S. Molecular aspects of melatonin (MLT)-induced therapeutic effects. Life Sci. 2015, 135, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Chao, H.; Li, Z.; Xu, X.; Liu, Y.; Hou, L.; Liu, N.; Ji, J. Melatonin attenuates traumatic brain injury-induced inflammation: A possible role for mitophagy. J. Pineal Res. 2016, 61, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Mincheva-Nilsson, L.; Baranov, V. Cancer exosomes and NKG2D receptor-ligand interactions: Impairing NKG2D-mediated cytotoxity and anti-tumor immune surveillance. Semin. Cancer Biol. 2014, 28, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, C.; Wang, S.; Wang, Z.; Jiang, J.; Wong, W.; Li, X.; Chen, J.; Liu, K.; Li, C.; et al. Exosomes derived from hypoxic oral squamous cell carcinoma cells deliver miR-21 to normal cells to elicit a prometastatic phenotype. Cancer Res. 2016, 76, 1770–1780. [Google Scholar] [CrossRef] [PubMed]
- Iorgulescu, J.B.; Ivan, M.E.; Safaee, M.; Parsa, A.T. The limited capacity of malignant glioma-derived exosomes to suppress peripheral immune effectors. J. Neuroimmunol. 2016, 290, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Garfall, A.L.; Stadmauer, E.A. Cellular and vaccine immunotherapy for multiple myoloma. Am. Soc. Hematol. Educ. Program. 2016, 2016, 521–527. [Google Scholar]
- Abd-Elhafeez, H.H.; Mokhtar, D.M.; Hassan, A.H. Effect of melatonin on telocytes in the seminal vesicle of the Soay ram: An immunohistochemical ultrastructural and morphometrical study. Cells Tissues Organs 2017, 203, 29–54. [Google Scholar] [CrossRef] [PubMed]
- Maschio-Signorini, L.B.; Gelaleti, G.B.; Morschetta, M.G.; Barin, T.F.; Jardin-Perassi, B.V.; Lopes, J.R.; Lacerda, J.Z.; Roela, R.A.; Bordin, N.A.; Correa, L.A.; et al. Melatonin regulates angiogenic and inflammatory proteins in MDA-MB-231 cell line and in co-culture with cancer-associated fibroblasts. Anticancer Agents Med. Chem. 2016, 16, 1474–1484. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.L.; Tang, H.M.; Mak, K.H.; Hu, S.; Wang, S.S.; Wong, K.M.; Wong, C.S.T.; Wu, H.Y.; Law, H.T.; Liu, K.; et al. Cell survival, DNA damage, and oncogenic transformation after a transient and reversible apoptotic response. Mol. Biol. Cell 2012, 23, 2240–2252. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, F.; Acuna-Castroviejo, D.; Doerrier, C.; Dayoub, J.C.; Lopez, L.C.; Venegas, C.; Garcia, J.A.; Lopez, A.; Volt, H.; Luna-Sanchez, M.; et al. Melatonin blunts the mitochondrial/NLR3 connection and protects against radiation-induced oral mucositis. J. Pineal Res. 2015, 58, 34–49. [Google Scholar] [CrossRef] [PubMed]
- Kilic, U.; Kilic, E.; Tuzcu, Z.; Tuzcu, M.; Ozercan, I.H.; Yilmaz, O.; Sahin, F.; Sahin, K. Melatonin suppresses cisplatin-induced nephrotoxicity via activation of Nrf-2/HO-1 pathway. Nutr. Metab. 2013, 10, 7. [Google Scholar] [CrossRef] [PubMed]
- Rezzani, R.; Rodella, L.F.; Fraschini, F.; Gasco, M.R.; Demartini, G.; Musicanti, C.; Reiter, R.J. Melatonin delivery in solid lipid nanoparticles: Prevention of cyclosporine A induced cardiac damage. J. Pineal Res. 2009, 46, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Yeung, H.M.; Hung, M.W.; Lau, C.F.; Fung, M.L. Cardioprotective effects of melatonin against myocardial injuries induced by chronic intermittent hypoxia in rats. J. Pineal Res. 2015, 58, 12–25. [Google Scholar] [CrossRef] [PubMed]
- Nduhirabandi, F.; Lamont, K.; Albertyn, Z.; Opie, L.H.; Lacour, S. Role of toll-like receptor 4 in melatonin-induced cardiprotection. J. Pineal Res. 2016, 60, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Simko, F.; Baka, T.; Raulis, L.; Reiter, R.J. Elevated heart rate and nondipping heartrate as potential targets for melatonin: A review. J. Pineal Res. 2016, 61, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Pir, J.F.; Hau, J.N.; Wei, F.P.; Xue, Q.; Zhang, F.; Peng, C.F.; Yang, Y.; Tian, Y.; Feng, J.; Du, J.; et al. Melatonin attenuates postmyocardial infarction injury via increasing Tam 70 expression. J. Pineal Res. 2017, 62, e12371. [Google Scholar]
- Boutin, J.A. Quinone reductase 2 as a promising target of melatonin therapeutic actions. Expert Opin. Ther. Targets 2016, 20, 303–317. [Google Scholar] [CrossRef] [PubMed]
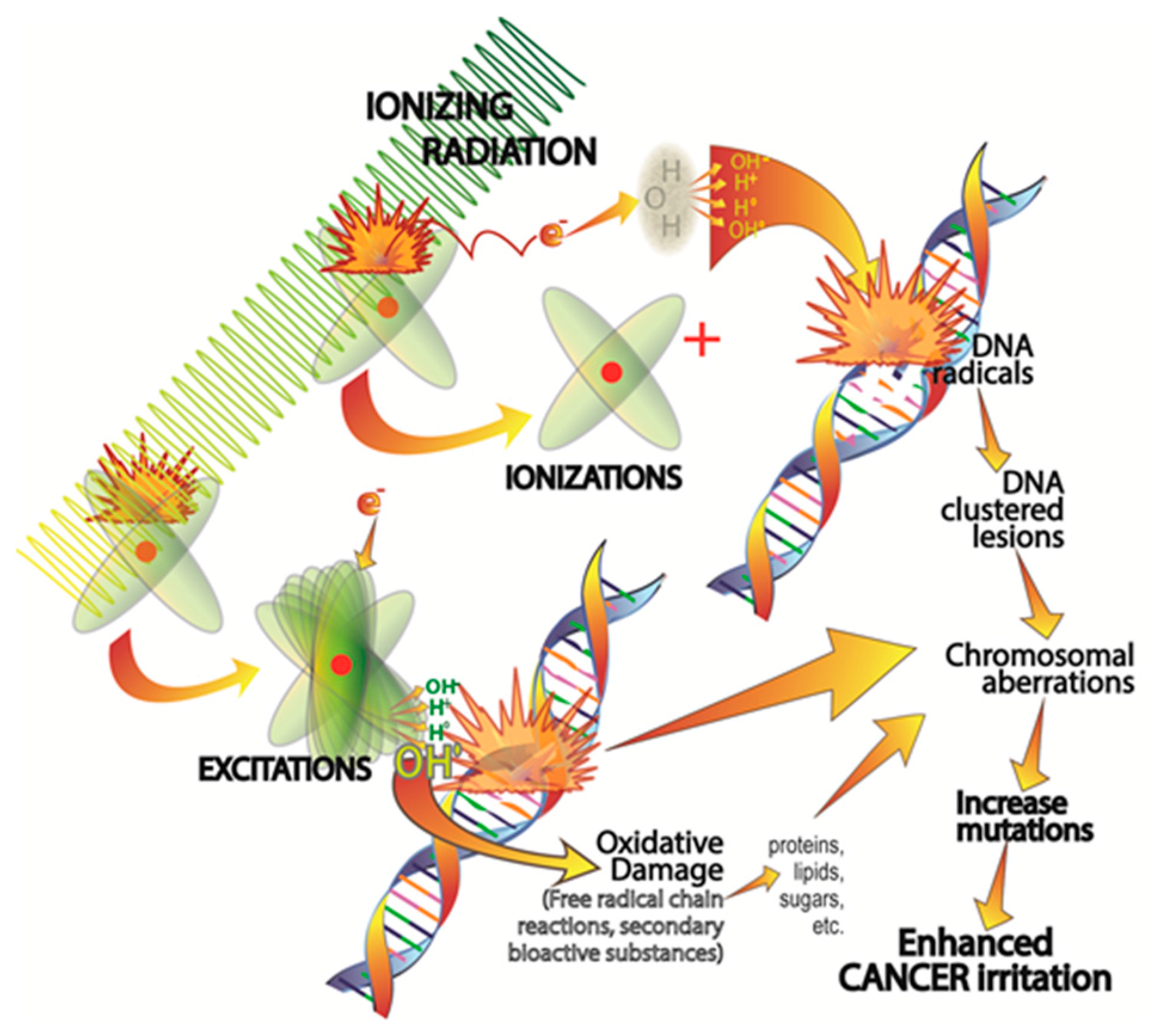
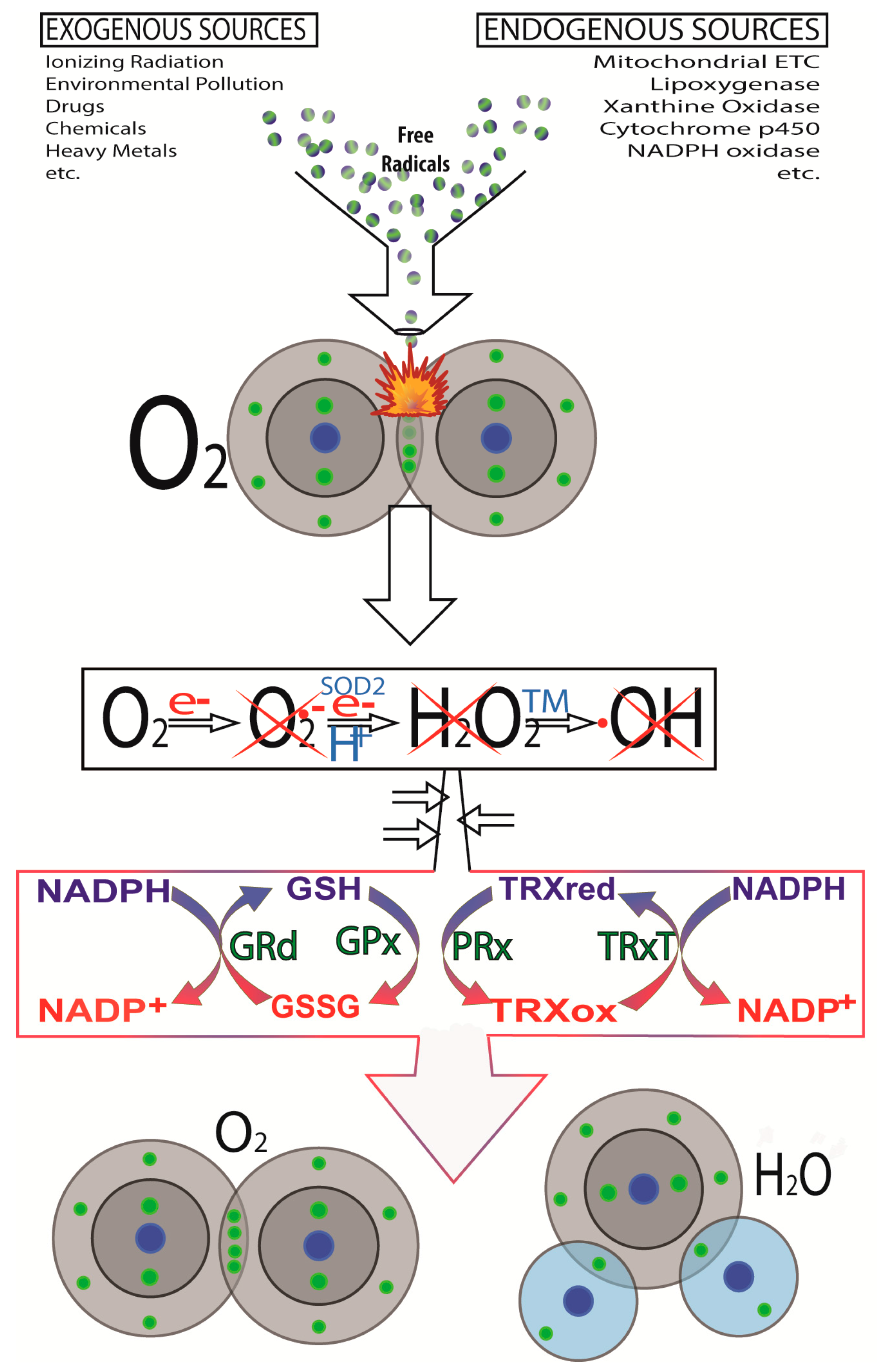
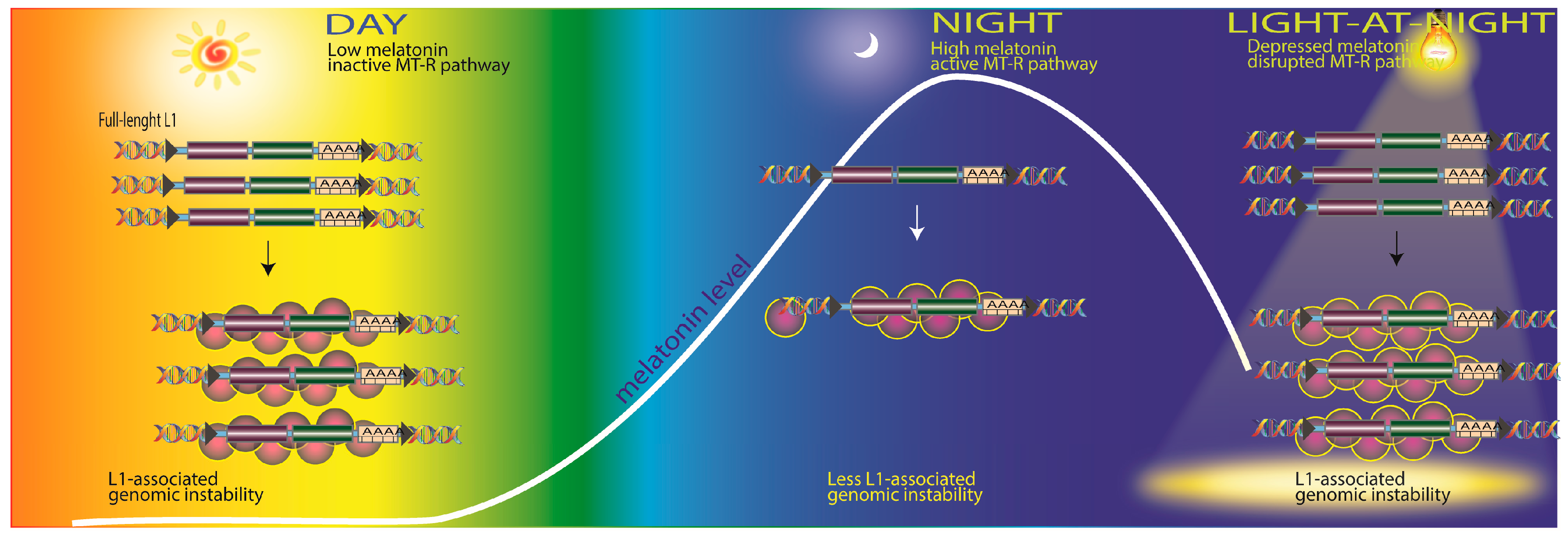
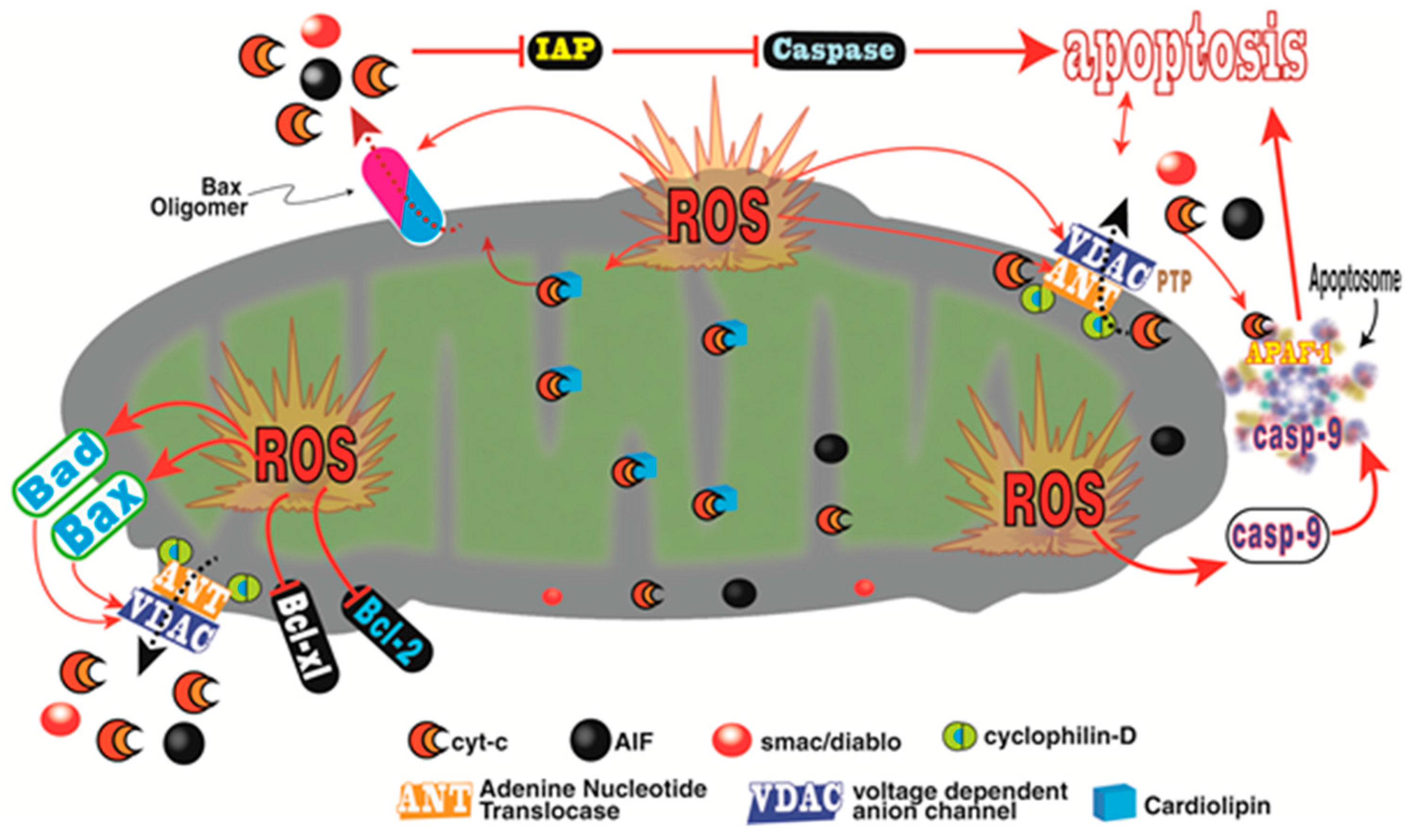
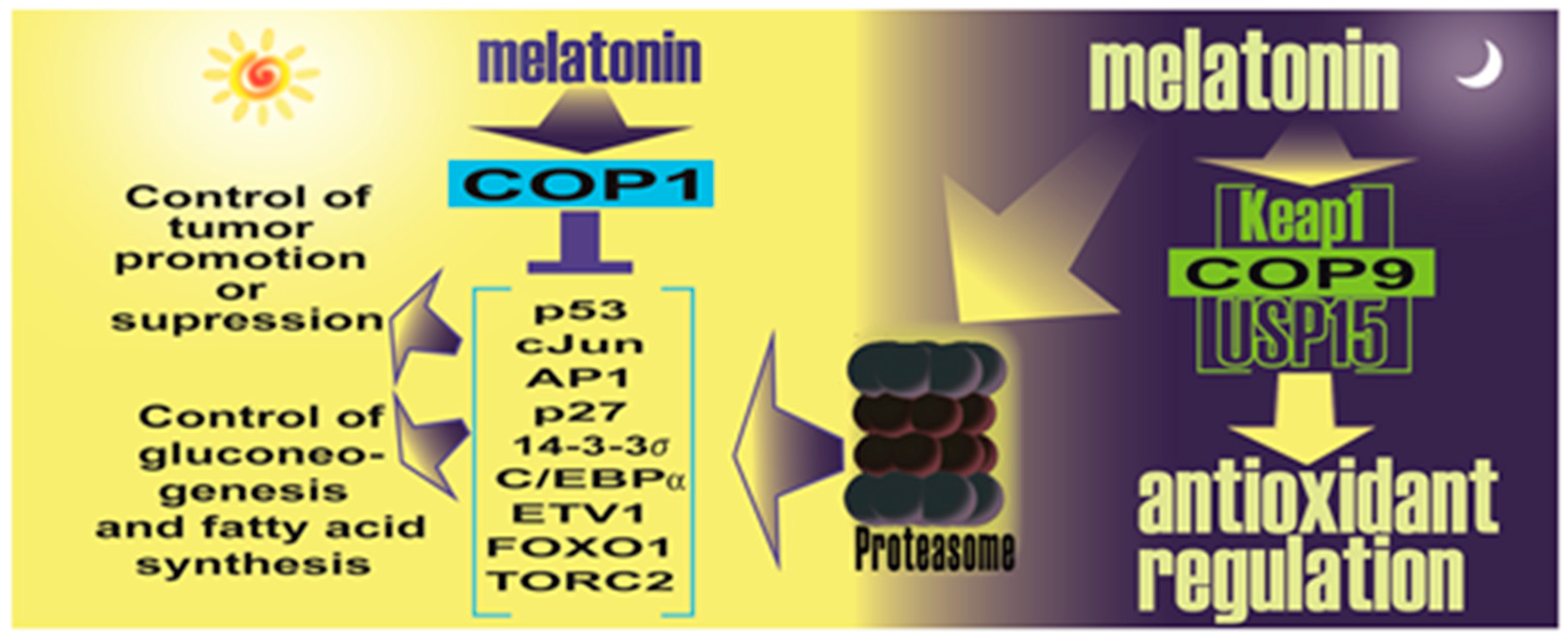
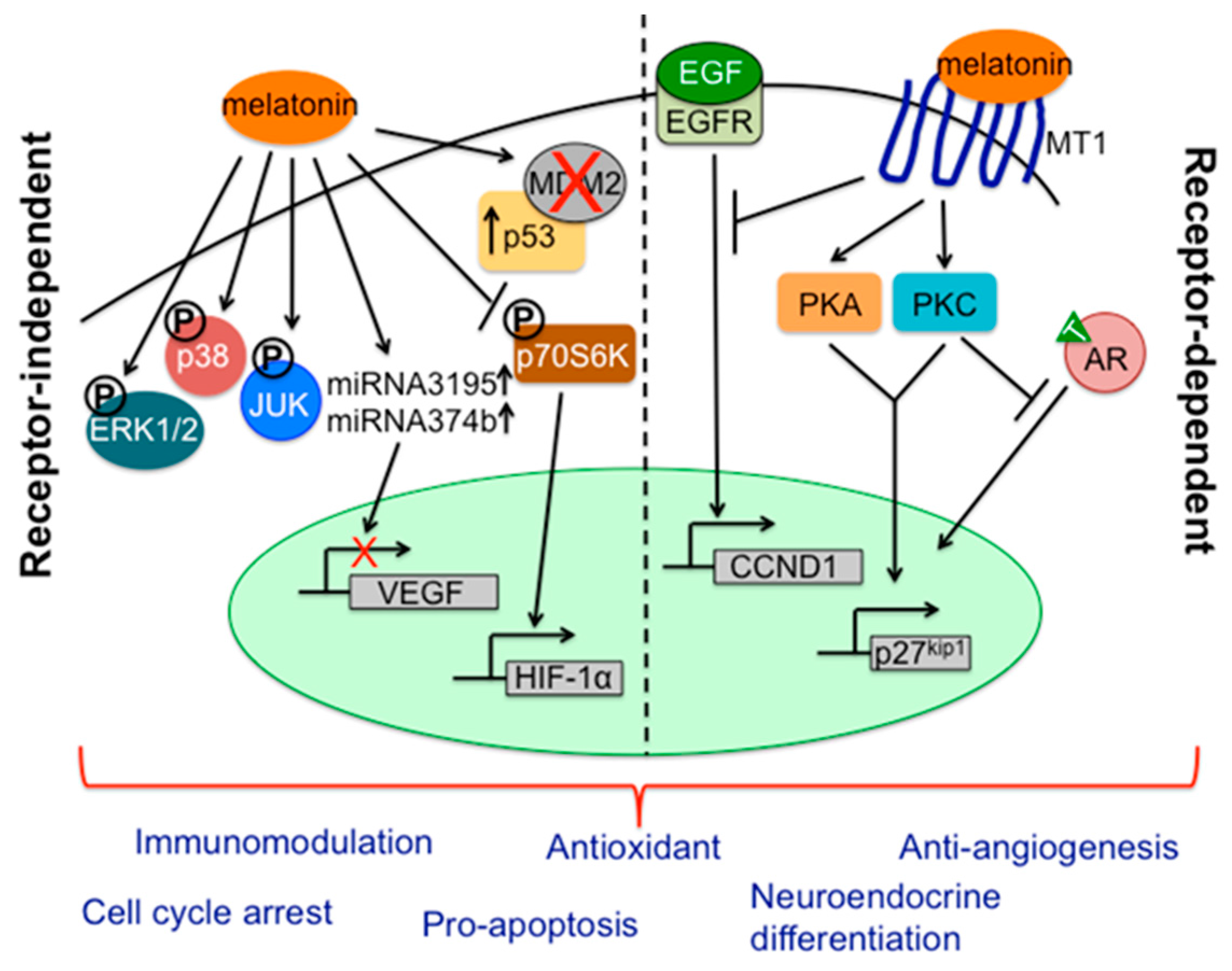
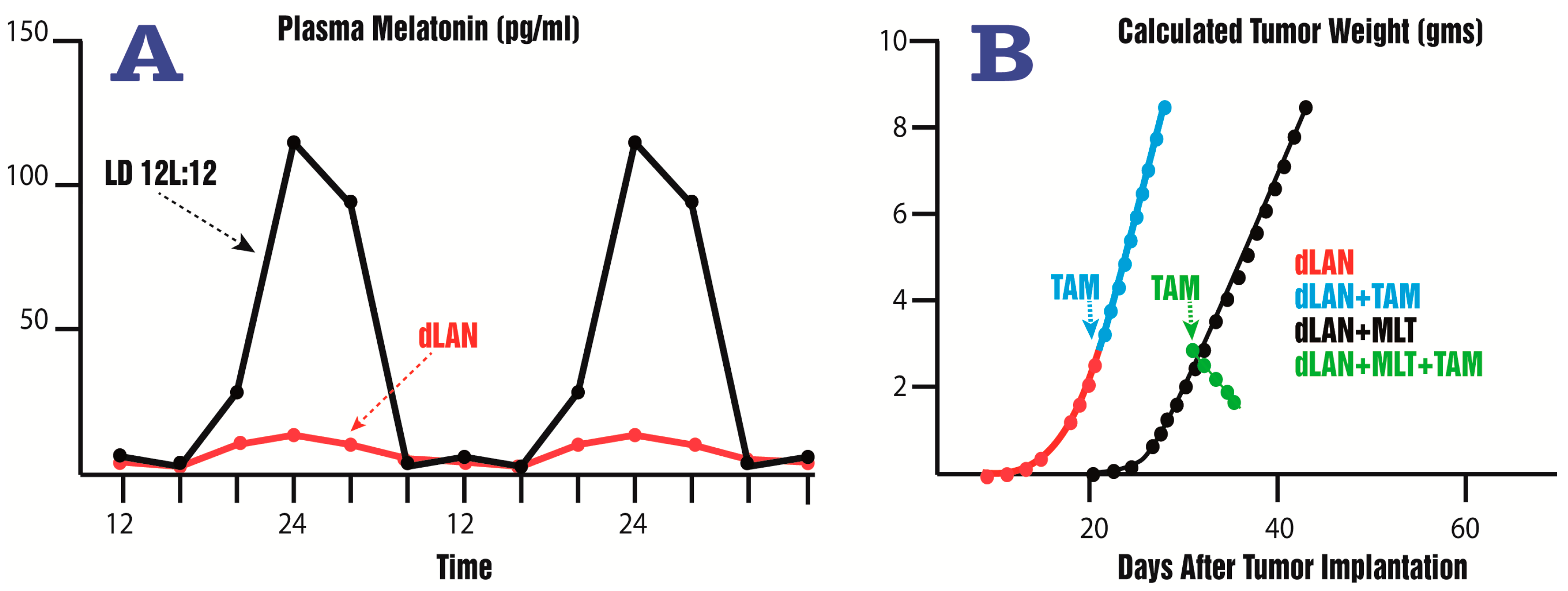
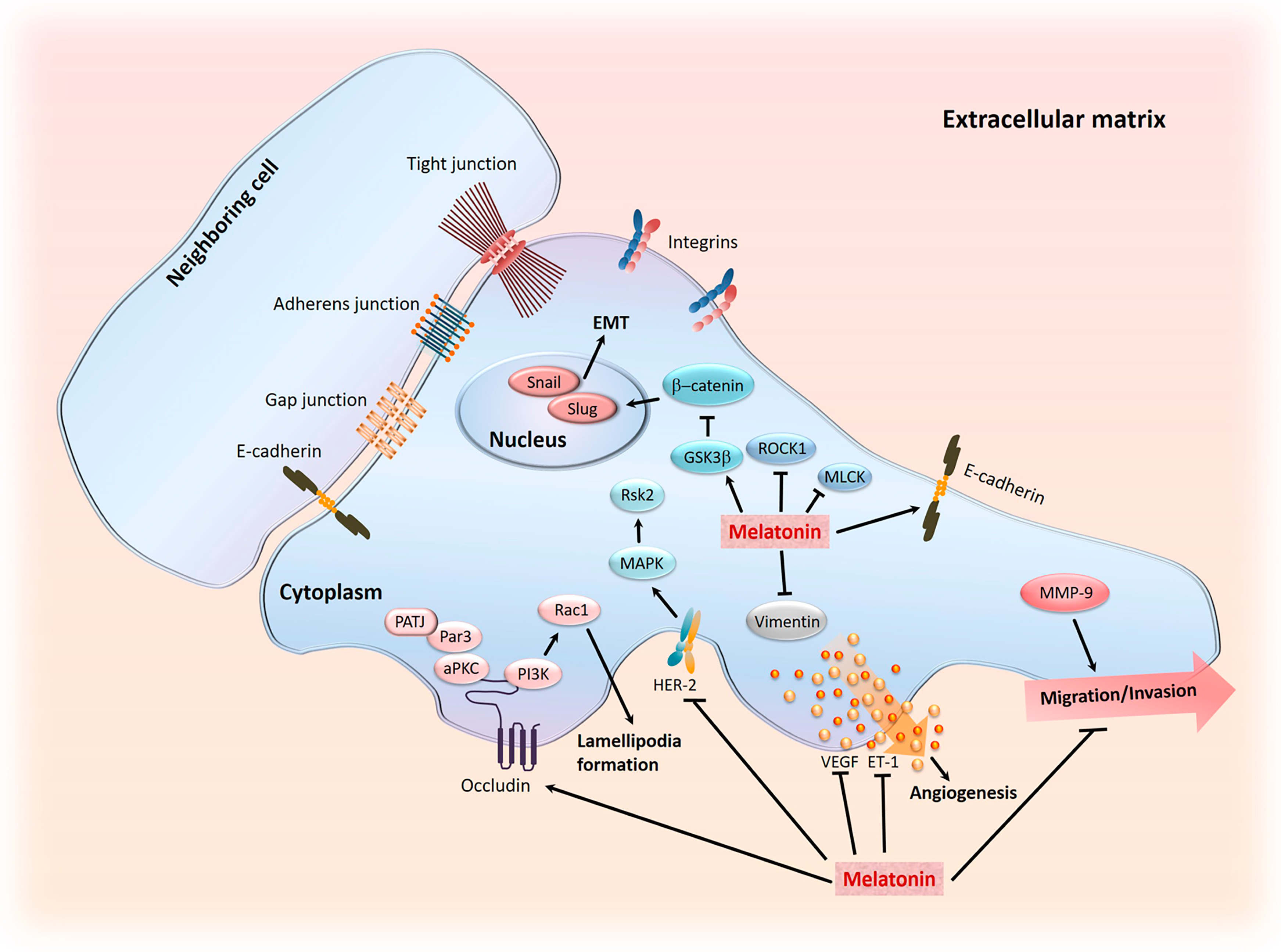
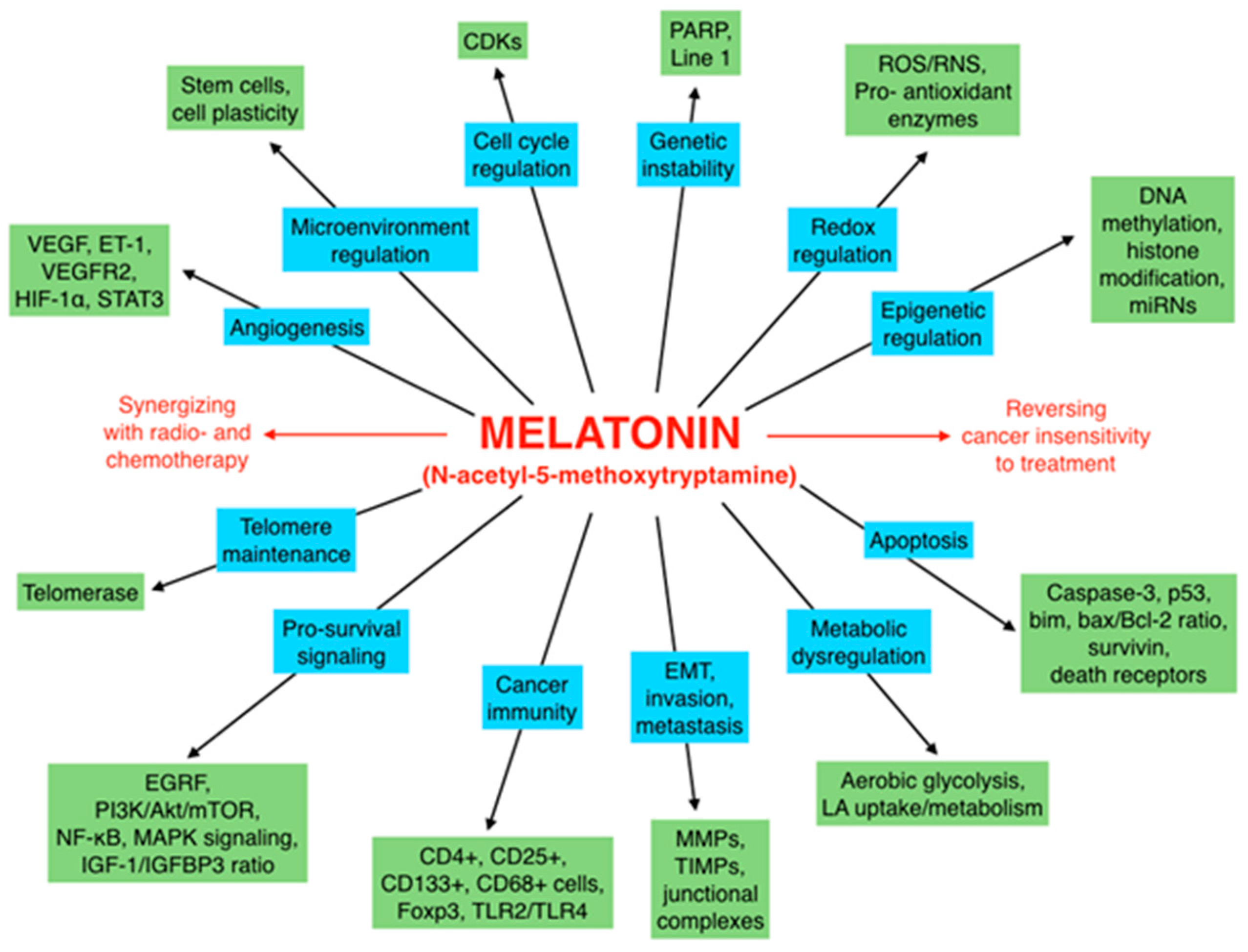
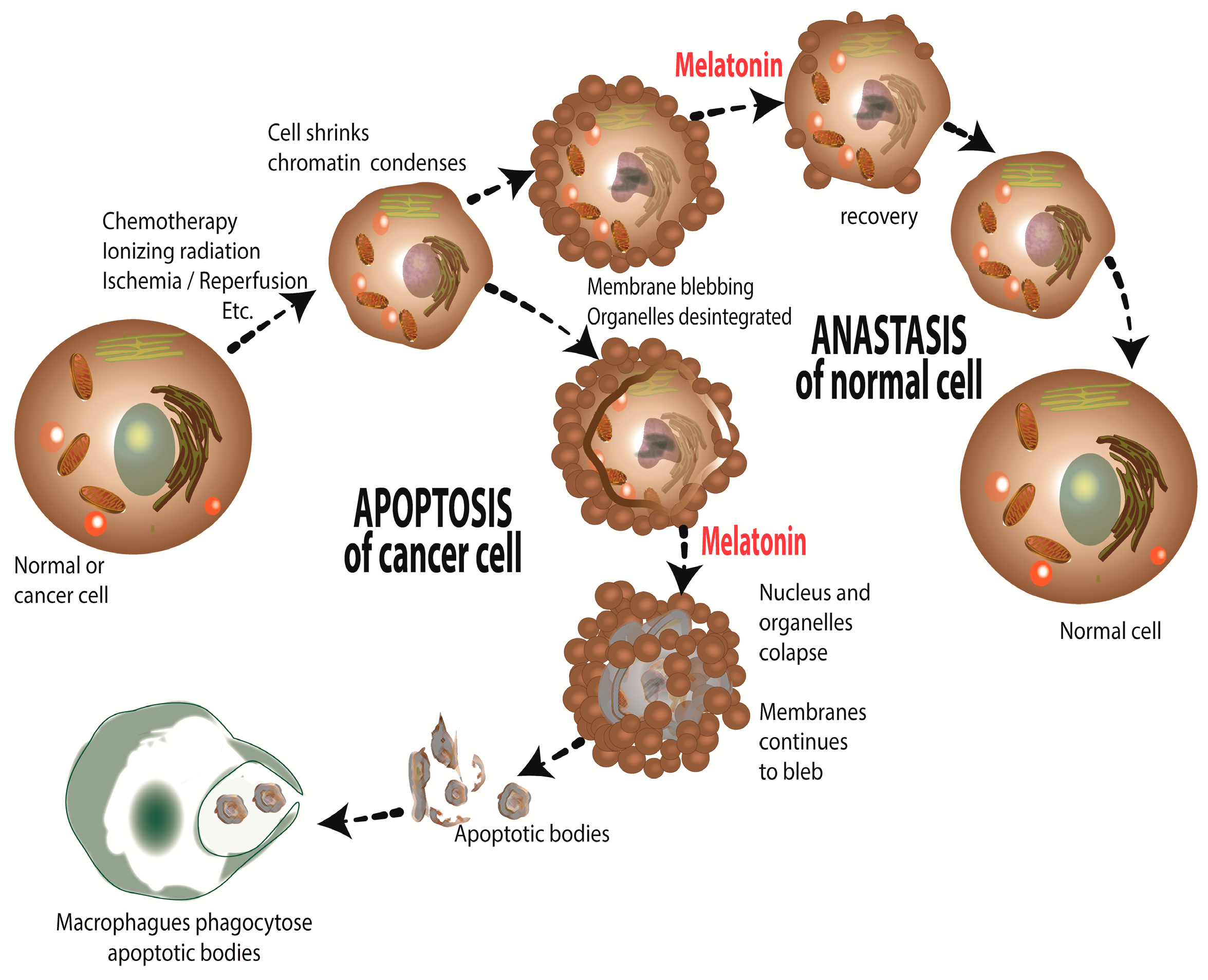

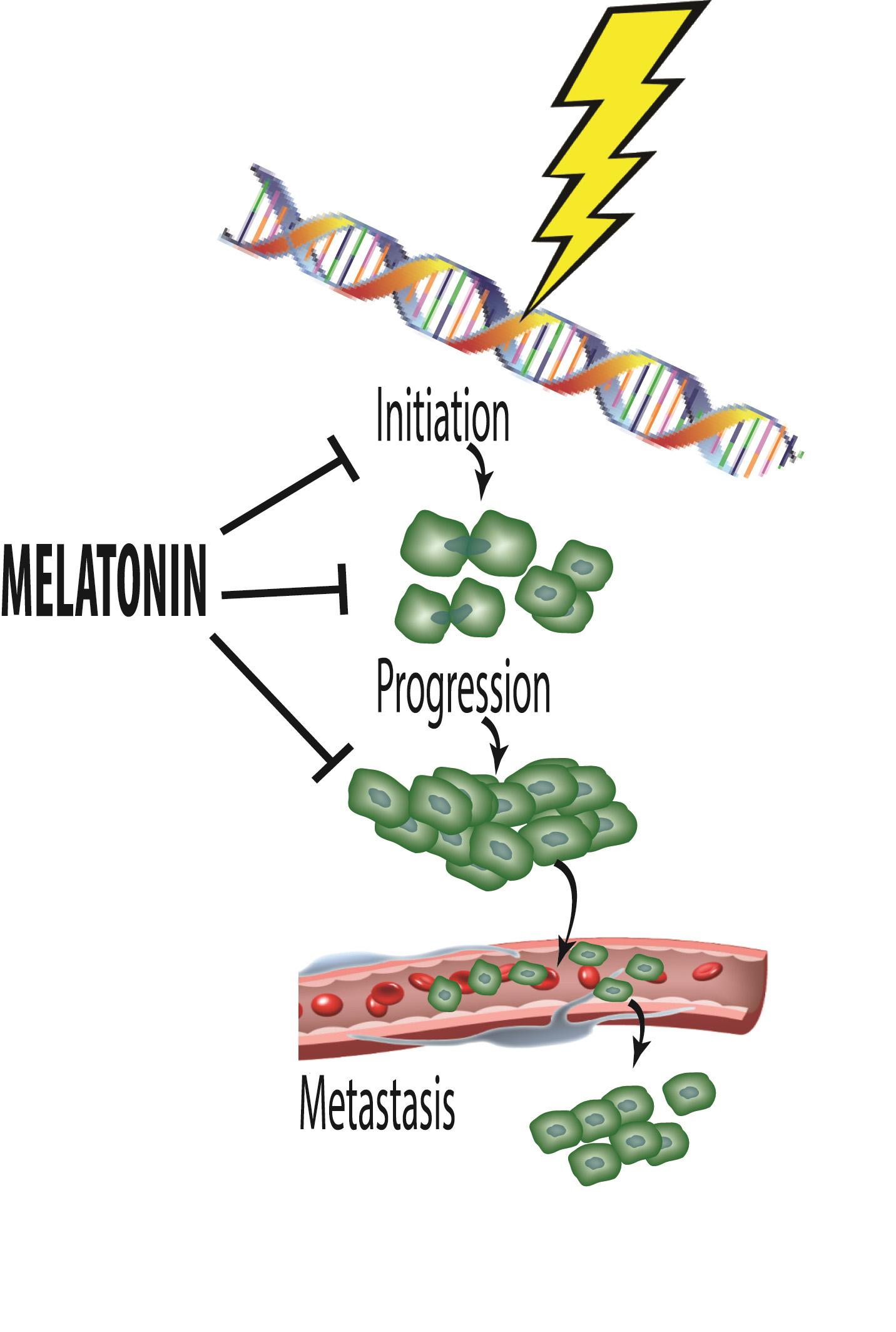{kind=link}
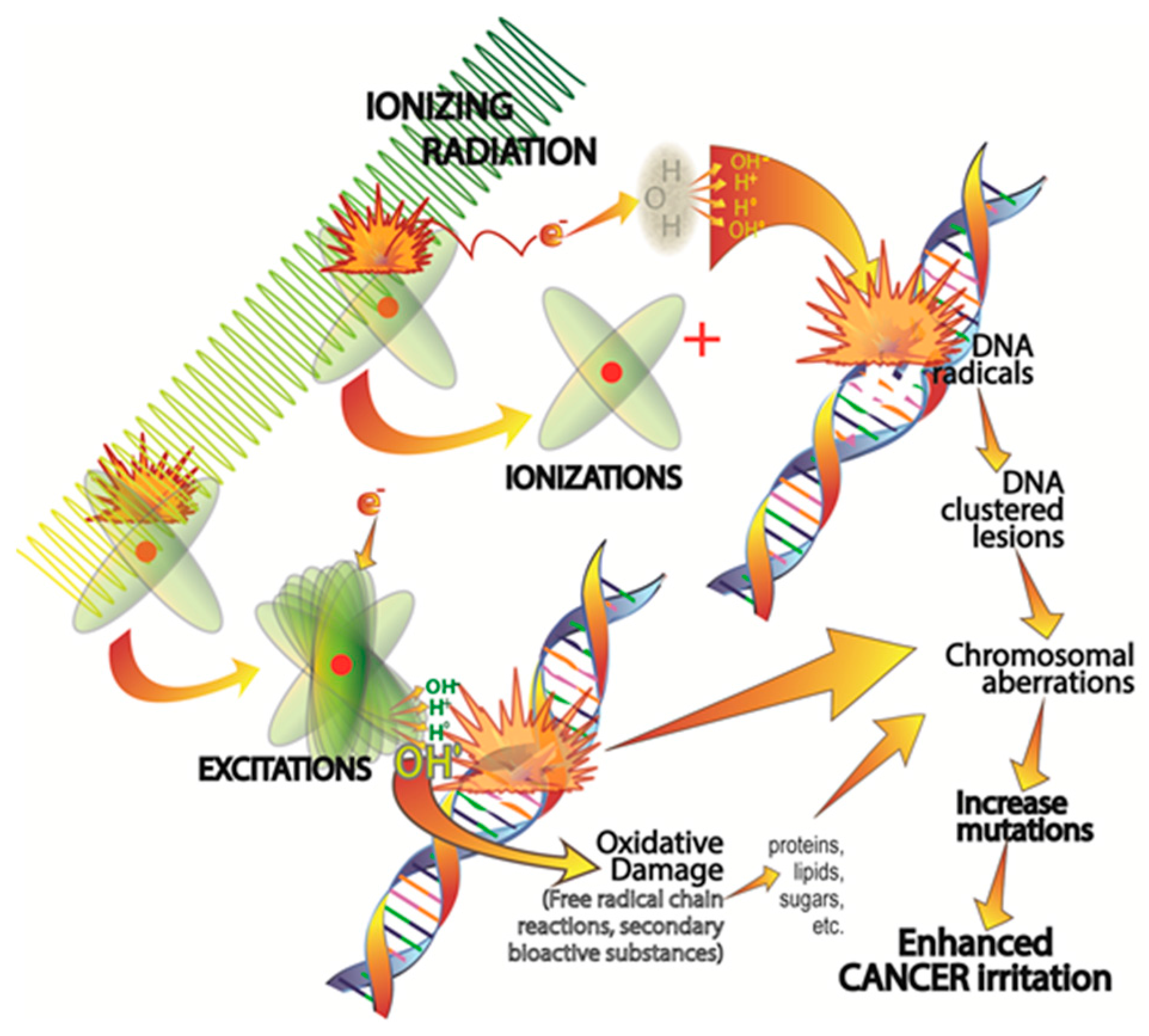{kind=link}
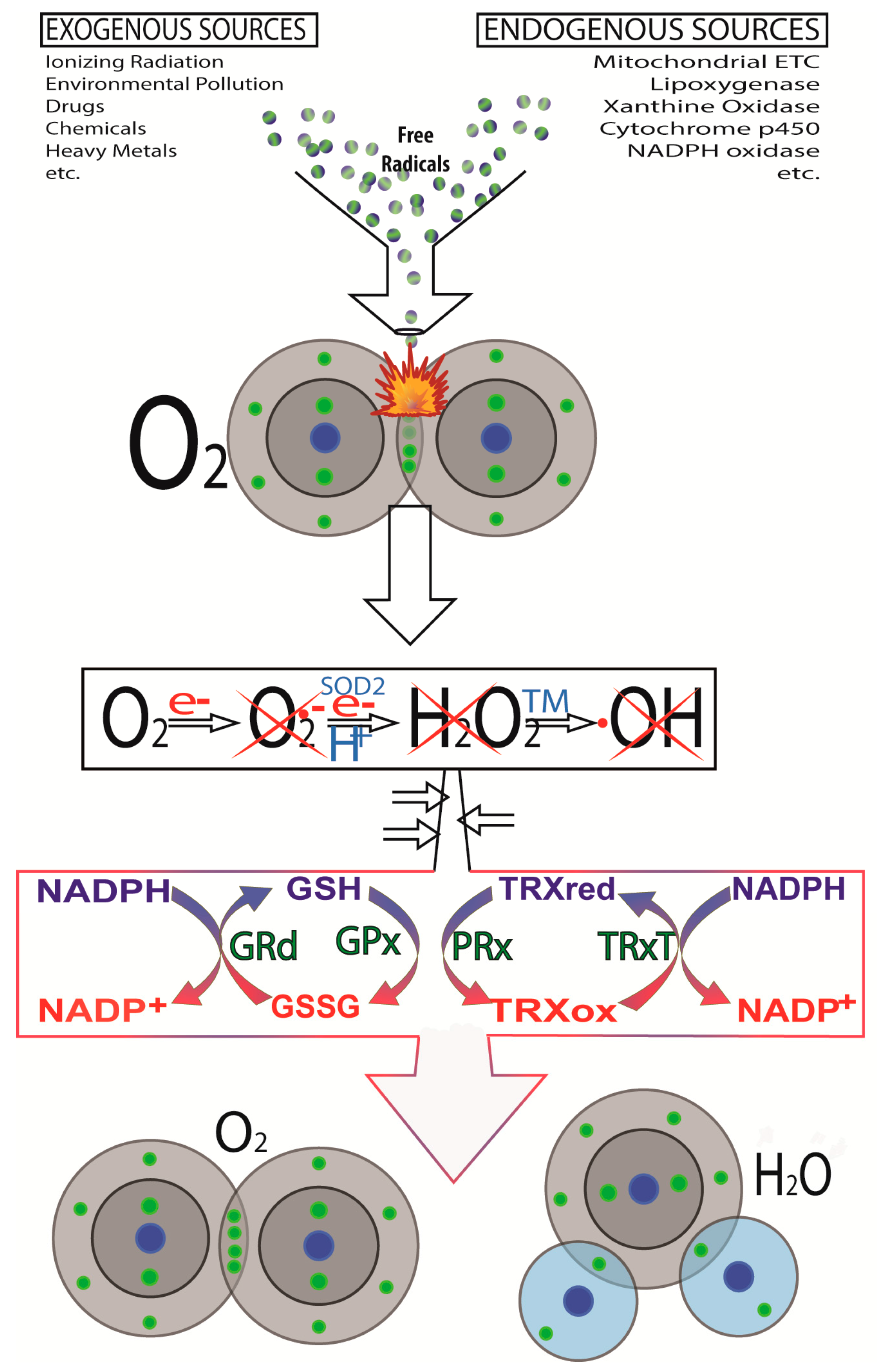{kind=link}
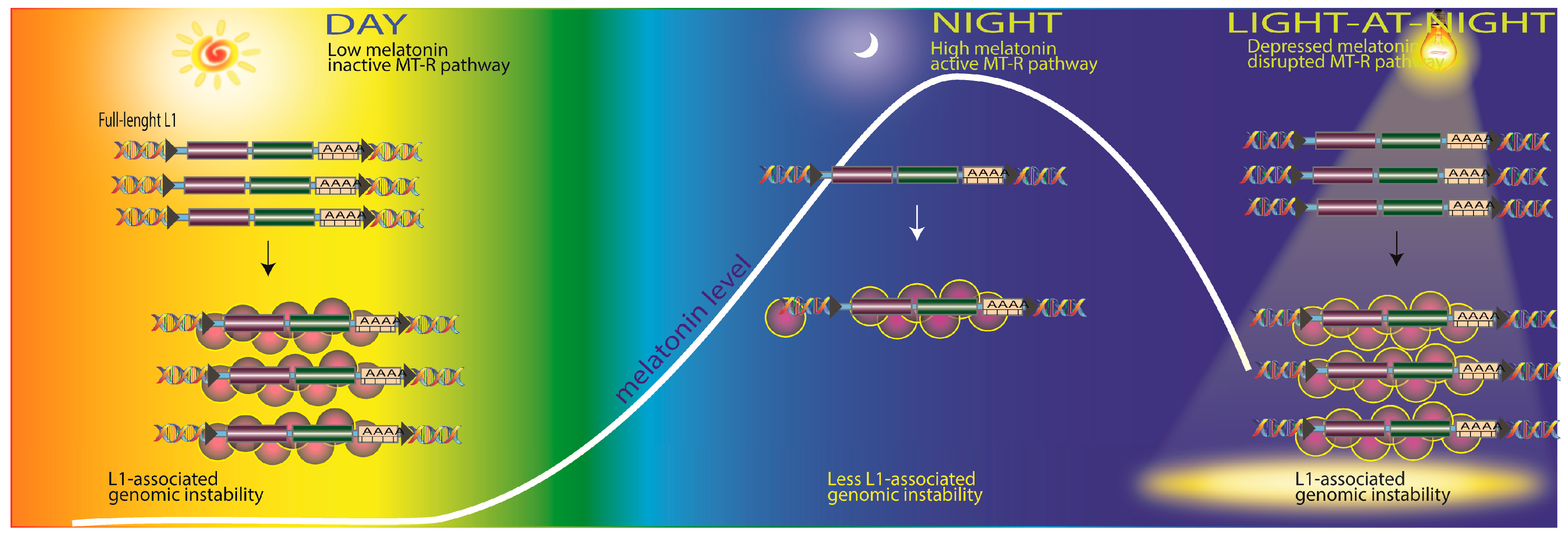{kind=link}
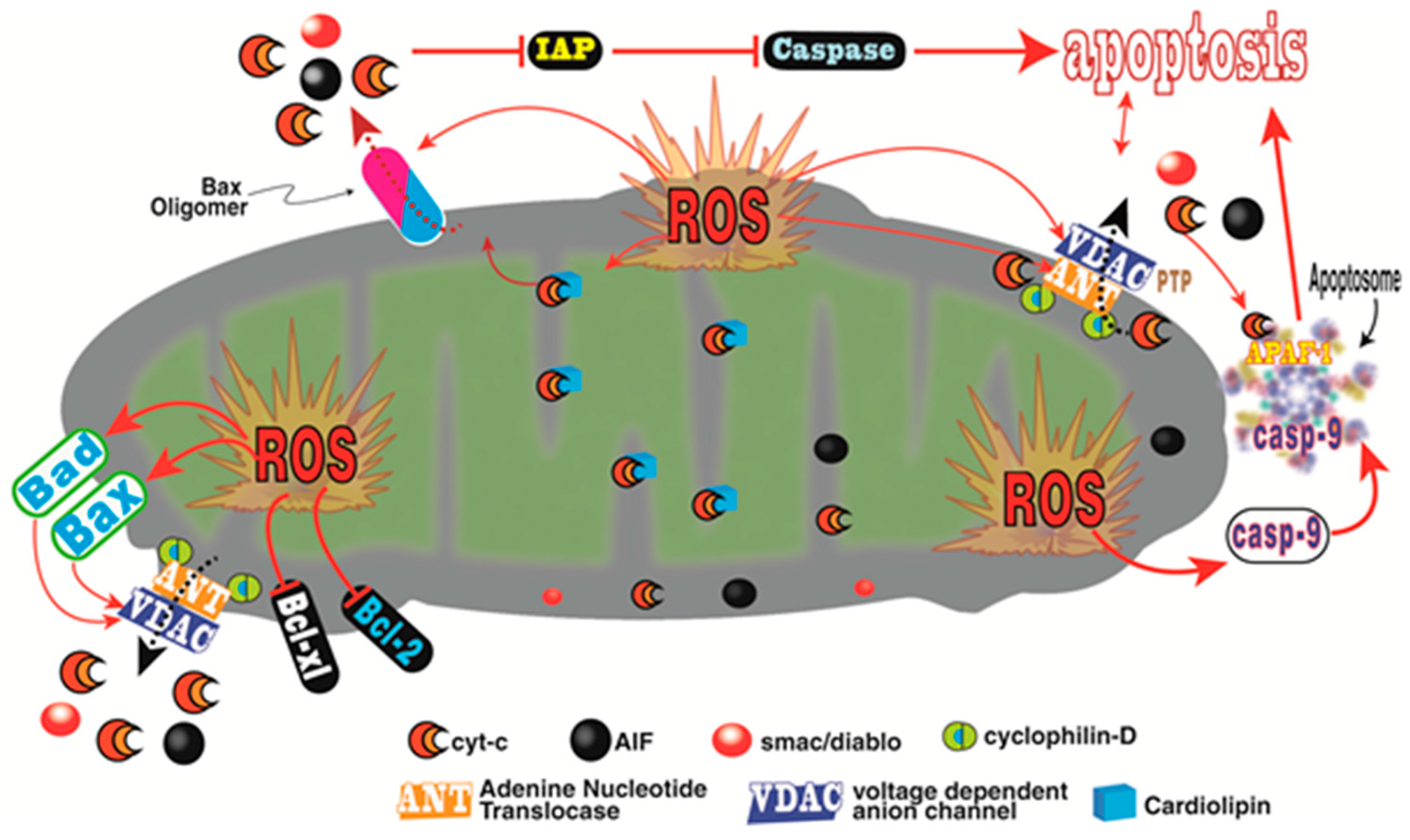{kind=link}
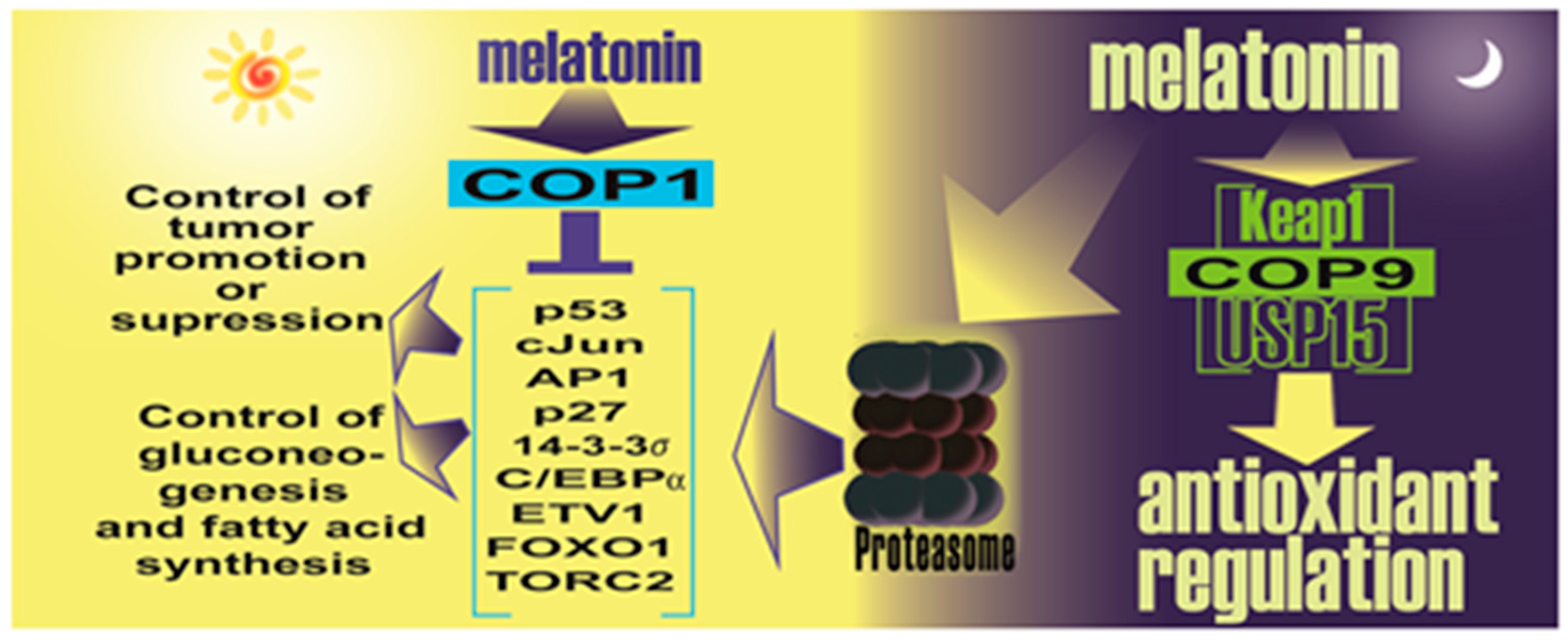{kind=link}
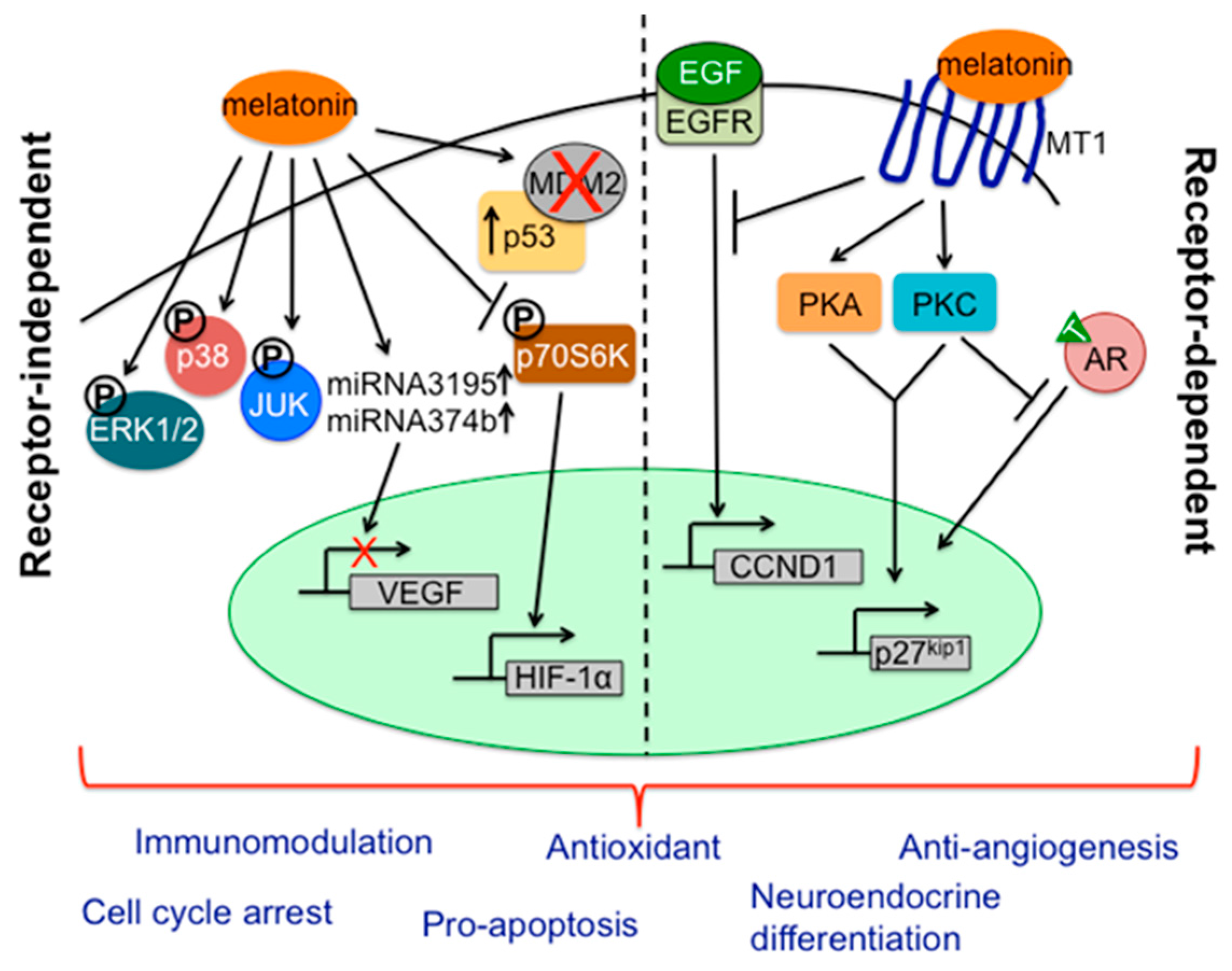{kind=link}
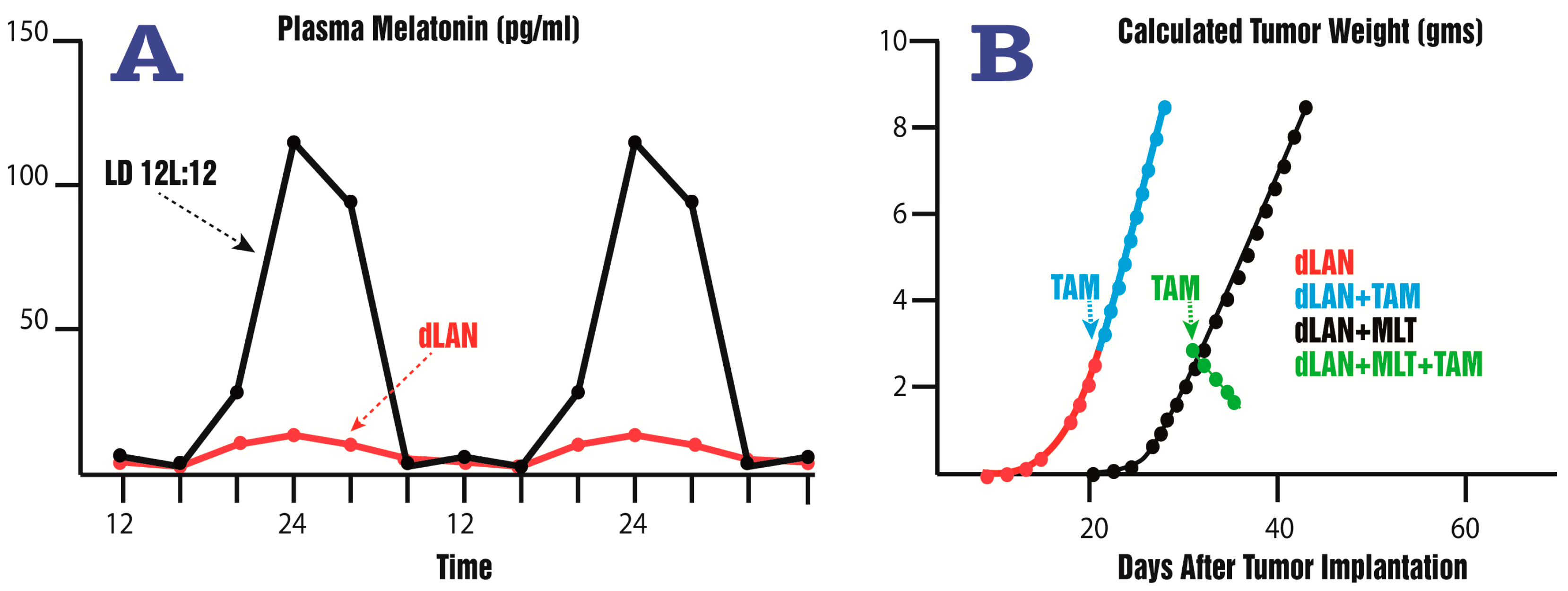{kind=link}
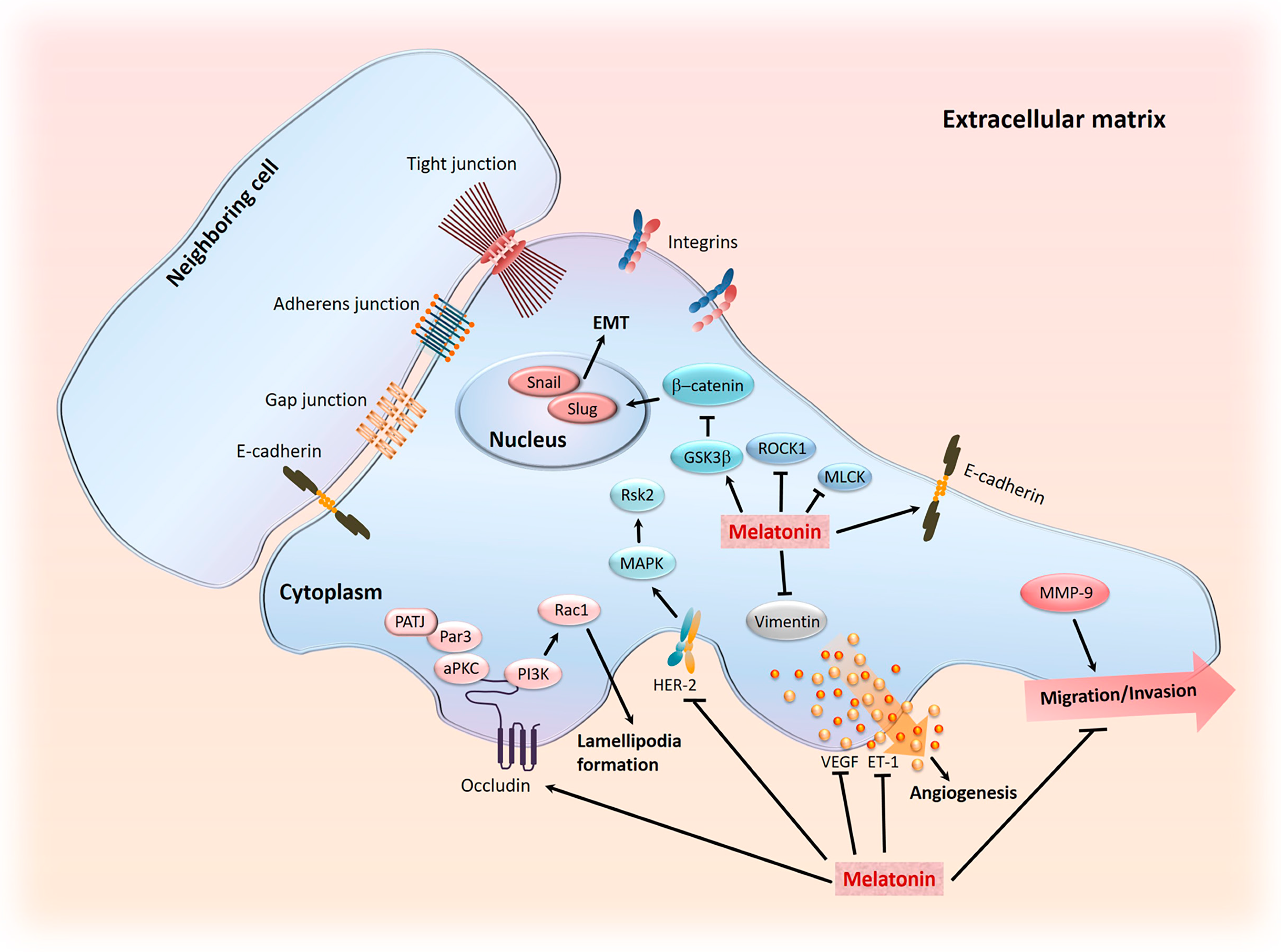{kind=link}
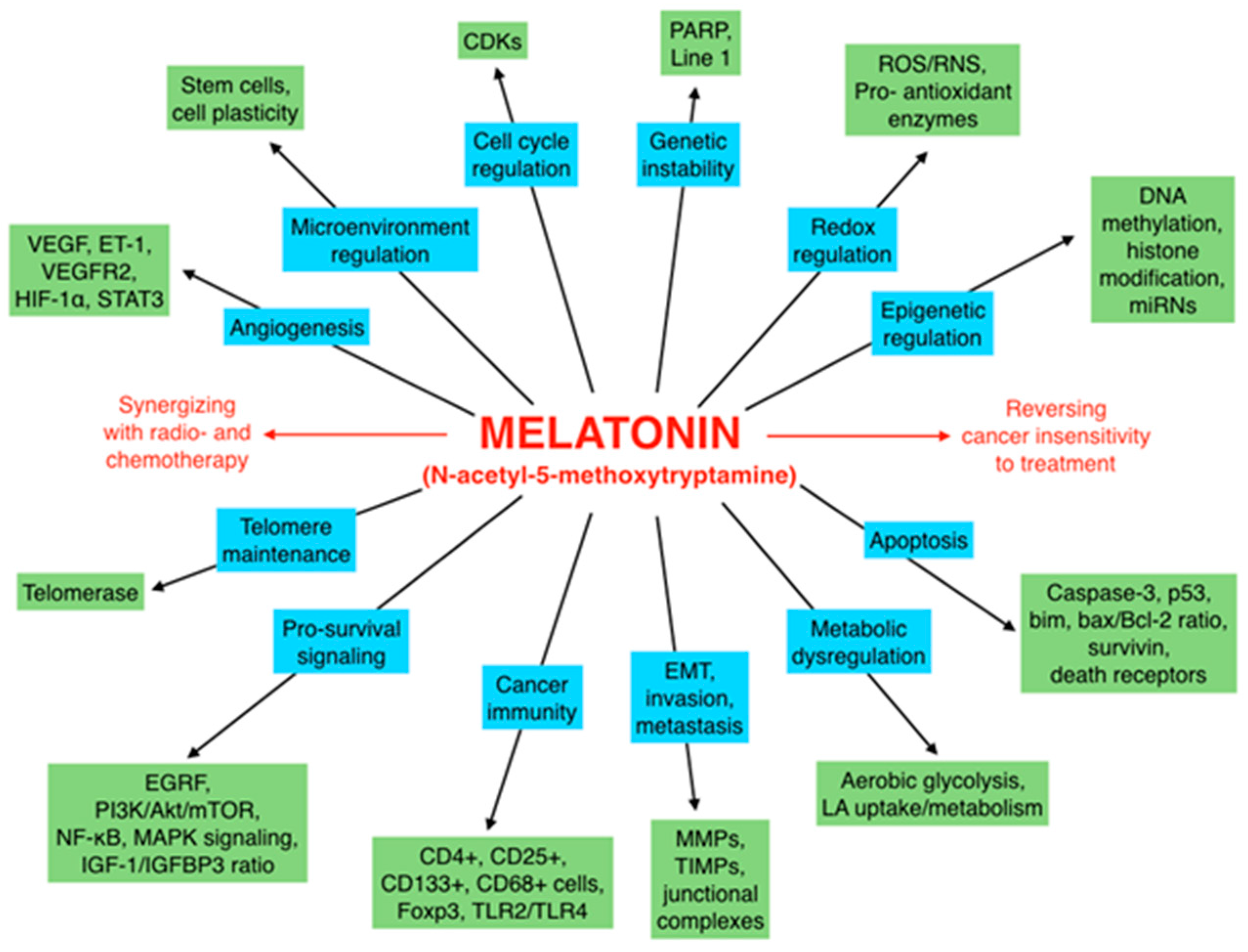{kind=link}
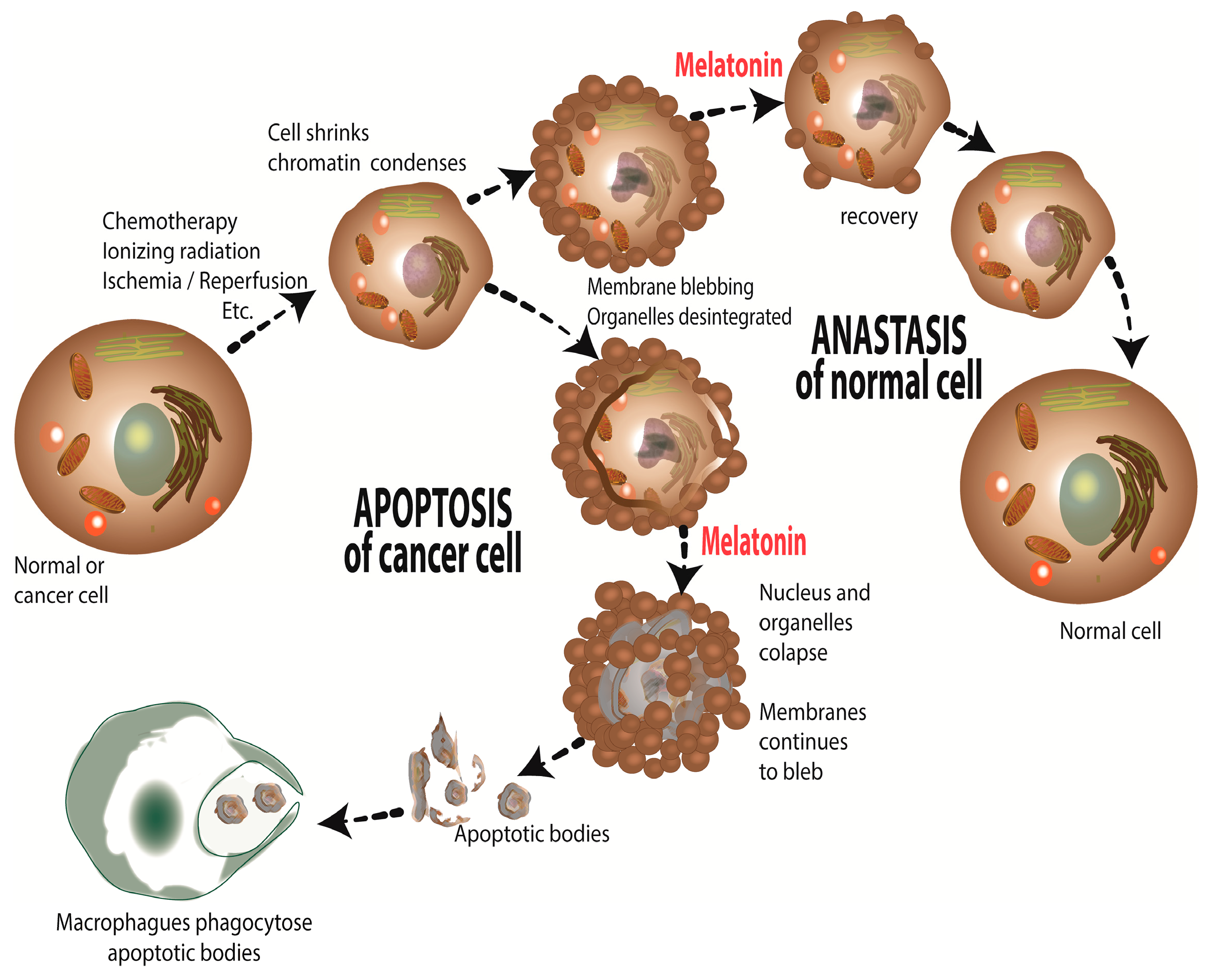{kind=link}
| Cancer Type | Cells Resistant to | Proposed Melatonin Action | Reference |
|---|---|---|---|
| Human glioma (A172 and U87MG cells) | Death receptor ligand (TRAIL) | Increased death receptor (DR5) sensitivity | [315] |
| Human Ewing sarcoma (SK-N-Mc cells) | Vincristine or ifosofamide | Potentiation of extrinsic apoptotic pathway | [316] |
| Hepatocellular carcinoma (tissue samples from cancer patients) | Various chemotherapies or radiotherapies | Inhibition of survivin and XIAP | [249] |
| Human lung adenocarcinoma (SK-LU-1 cells) | Cisplatin | Cell cycle arrest in S phase | [260] |
| Human breast cancer xenografts (MCF-7 cells) | Doxorubicin | Circadian regulated kinase inhibition | [317] |
| Human breast cancer xenografts (MCF-7 cells) | Tamoxifen | Circadian regulated kinase inhibition | [308] |
| Human breast cancer (MCF-7 cells) | Ionizing radiation | Downregulation of DNA repair | [222] |
| Human breast cancer (MCF-7 cells) | Ionizing radiation | Modulation of p53 | [223] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reiter, R.J.; Rosales-Corral, S.A.; Tan, D.-X.; Acuna-Castroviejo, D.; Qin, L.; Yang, S.-F.; Xu, K. Melatonin, a Full Service Anti-Cancer Agent: Inhibition of Initiation, Progression and Metastasis. Int. J. Mol. Sci. 2017, 18, 843. https://doi.org/10.3390/ijms18040843
Reiter RJ, Rosales-Corral SA, Tan D-X, Acuna-Castroviejo D, Qin L, Yang S-F, Xu K. Melatonin, a Full Service Anti-Cancer Agent: Inhibition of Initiation, Progression and Metastasis. International Journal of Molecular Sciences. 2017; 18(4):843. https://doi.org/10.3390/ijms18040843
Chicago/Turabian StyleReiter, Russel J., Sergio A. Rosales-Corral, Dun-Xian Tan, Dario Acuna-Castroviejo, Lilan Qin, Shun-Fa Yang, and Kexin Xu. 2017. "Melatonin, a Full Service Anti-Cancer Agent: Inhibition of Initiation, Progression and Metastasis" International Journal of Molecular Sciences 18, no. 4: 843. https://doi.org/10.3390/ijms18040843
APA StyleReiter, R. J., Rosales-Corral, S. A., Tan, D.-X., Acuna-Castroviejo, D., Qin, L., Yang, S.-F., & Xu, K. (2017). Melatonin, a Full Service Anti-Cancer Agent: Inhibition of Initiation, Progression and Metastasis. International Journal of Molecular Sciences, 18(4), 843. https://doi.org/10.3390/ijms18040843