
Theranostic Biomarkers for Schizophrenia

 ,
, 
Abstract
:
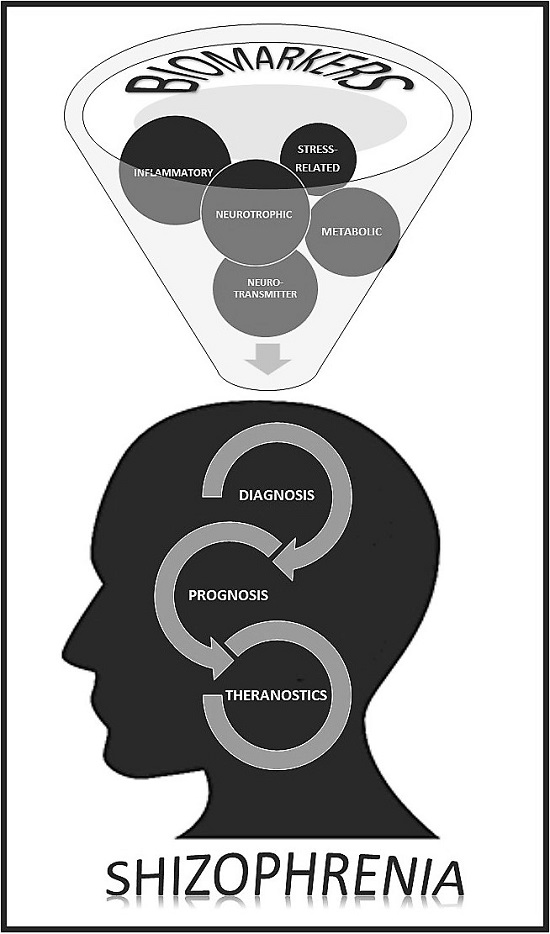{kind=link}
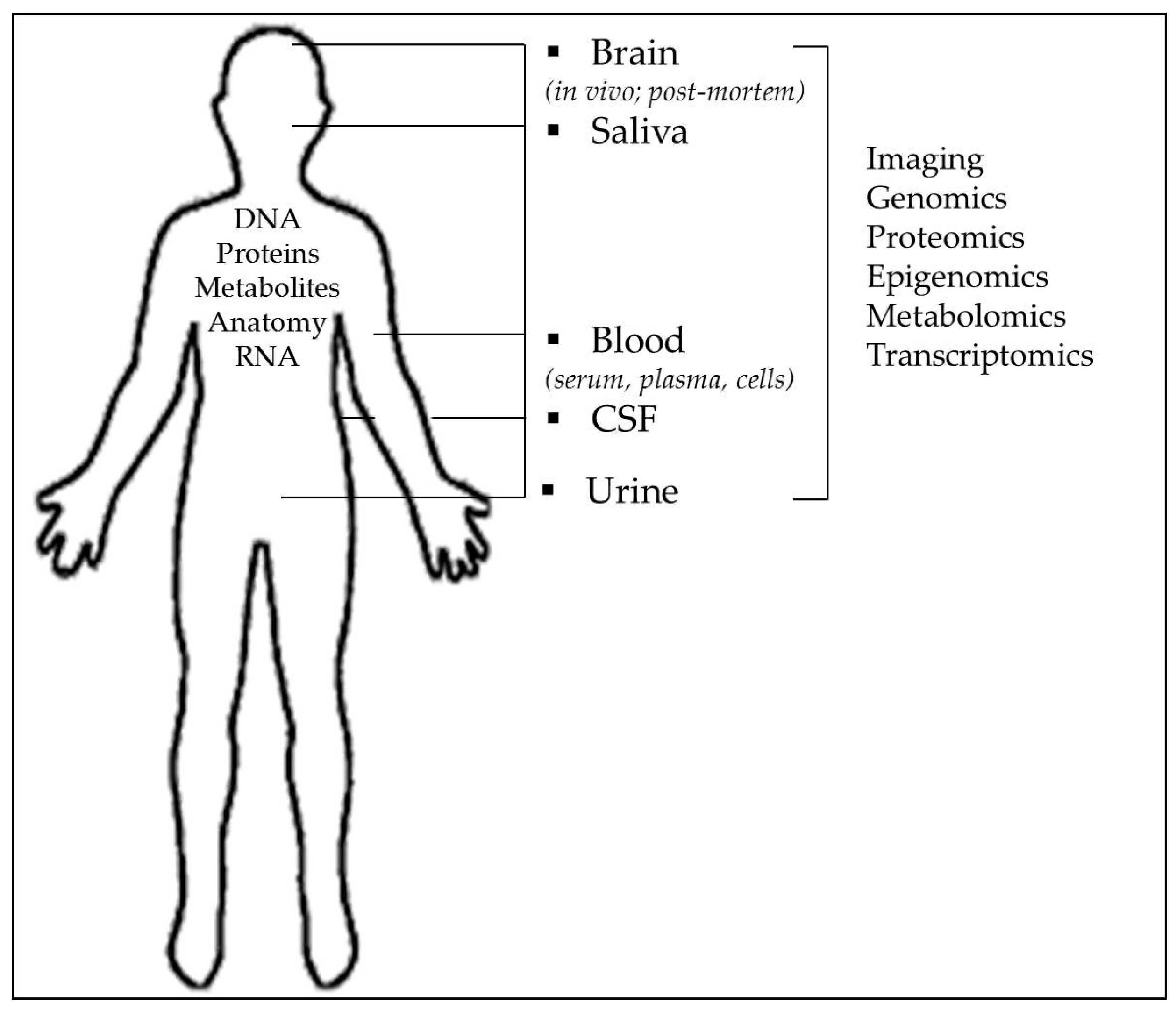{kind=link}
{kind=link}
1. Vulnerability and Resilience in Mental Disorders
2. Schizophrenia
3. Biomarkers
3.1. Inflammation Biomarkers in Schizophrenia
3.2. Neuroendocrine-Related Biomarkers in Schizophrenia
3.2.1. The HPA Axis-Related Biomarkers in Schizophrenia
3.2.2. Metabolic Biomarkers
3.3. Neurotrophins as Candidate Biomarkers in Schizophrenia
3.3.1. Brain-Derived Neurotrophic Factor
3.3.2. Other Neurotrophins
3.4. Neurotransmitter Biomarkers
3.4.1. Dopaminergic System
3.4.2. Serotonergic System
3.4.3. Norepinephrine System
3.4.4. Cholinergic System
3.4.5. Glutamatergic System
3.4.6. GABAergic System
4. Conclusions
Author Contributions
Conflicts of Interest
Abbreviations
| HPA | Hypothalamic–pituitary–adrenal |
| CRH | Corticotrophin releasing factor |
| ACTH | Adrenocorticotrophic hormone |
| CNS | Central nervous system |
| CSF | Cerebrospinal fluid |
| GWAS | Genome-wide association studies |
| MHC | Major histocompatibility complex |
| CRP | C-reactive protein |
| IL | Interleukins |
| PANSS | Positive and Negative Syndrome Scale |
| TNF | Tumor necrosis factor |
| TGF | Transforming growth factor |
| IFN | Interferon |
| sIL-2R | Soluble IL-2-receptor |
| CDL5 | Cluster of differentiation 5-like protein |
| CDL40 | cluster of differentiation 40 |
| NMDA | N-methyl-d-aspartate |
| NSAID | Nonsteroidal anti-inflammatory drugs |
| DST | Dexamethasone suppression test |
| CAR | Cortisol awakening response |
| SNPs | Single nucleotide polymorphisms |
| FKBP5 | FK506 binding protein-5 |
| CRHR1 | Corticotrophin-releasing hormone receptor 1 |
| GR | Glucocorticoid receptor |
| CRHBP | CRH binding protein |
| NC3R1 | BclI polymorphism in glucocorticoid receptor gene 11 |
| DHEA | dehydroepiandrosterone |
| DHEAS | DHEA sulphate |
| BMI | Body mass index |
| NGF | Nerve growth factor |
| BDNF | Brain-derived neurotrophic factor |
| NT-3 | Neurotrophin-3 |
| NT-4/5 | Neurotrophin-4/5 |
| p75NTR | Pan neurotrophin receptor |
| mRNA | messenger RNA |
| 5-HTR2 | Serotonergic receptor type 2 |
| DRD2 | Dopaminergic receptor type 2 |
| NGFR | NGF receptor |
| DA | Dopamine |
| DR | Dopamine receptor i.e., DRD2-dopamine receptor type D2 |
| PFC | Prefrontal cortex |
| TH | Tyrosine hydroxylase |
| DAT | Dopamine transporter |
| HVA | Homovanillic acid |
| COMT | Catechol-O-methyl transferase |
| LSD | Lysergic acid diethylamide |
| 5-HT | Serotonin |
| 5-HTT/SERT | Serotonin transporter |
| 5-HTR | Serotonin receptor i.e., 5-HTR1A-serotonin receptor type 1A |
| MHPG | 3-methoxy-4-hydroxyphenylglycol |
| NMDAR | N-methyl-d-aspartate receptor |
| mGluR | Metabotropic glutamate receptor i.e., mGluR3- Metabotropic glutamate receptor type 3 |
| GABA | Gamma-amino-butyric acid |
| GAD | Glutamic acid decarboxylase |
| GABR | GABA-A receptor gene i.e., GABRB2-GABA-A receptor beta2 subunit gene |
References
- Halldorsdottir, T.; Binder, E.B. Gene × Environment interactions: From molecular mechanisms to behavior. Annu. Rev. Psychol. 2017, 68, 215–241. [Google Scholar] [CrossRef] [PubMed]
- Kahn, R.S.; Sommer, I.E.; Murray, R.M.; Meyer-Lindenberg, A.; Weinberger, D.R.; Cannon, T.D.; O’Donovan, M.; Correll, C.U.; Kane, J.M.; van Os, J.; et al. Schizophrenia. Nat. Rev. Dis. Prim. 2015, 1, 15067. [Google Scholar] [CrossRef] [PubMed]
- Gispen-de Wied, C.C. Stress in schizophrenia: An integrative view. Eur. J. Pharmacol. 2000, 405, 375–384. [Google Scholar] [CrossRef]
- Mizuno, Y.; Wartelsteiner, F.; Frajo-Apor, B. Resilience research in schizophrenia: A review of recent developments. Curr. Opin. Psychiatry 2016, 29, 218–223. [Google Scholar] [CrossRef] [PubMed]
- McEwen, B.S. Allostasis and allostatic load: Implications for neuropsychopharmacology. Neuropsychopharmacology 2000, 22, 108–124. [Google Scholar] [CrossRef]
- Russo, S.J.; Murroughl, J.W.; Han, M.H.; Charney, D.S.; Nestler, E.J. Neurobiology of resilience. Nat. Neurosci. 2012, 15, 1475–1484. [Google Scholar] [CrossRef] [PubMed]
- Chan, M.K.; Krebs, M.-O.; Cox, D.; Guest, P.C.; Yolken, R.H.; Rahmoune, H.; Rothermundt, M.; Steiner, J.; Leweke, F.M.; van Beveren, N.J.M.; et al. Development of a blood-based molecular biomarker test for identification of schizophrenia before disease onset. Transl. Psychiatry 2015, 5, e601. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, C.; Mujica-Parodi, L.; Yale, S.; Leitman, D.; Malaspina, D. Could stress cause psychosis in individuals vulnerable to schizophrenia? CNS Spectr. 2002, 7, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Walker, E.F.; Diforio, D. Schizophrenia: A neural diathesis-stress model. Psychol. Rev. 1997, 104, 667–685. [Google Scholar] [CrossRef] [PubMed]
- Walker, E.; Mittal, V.; Tessner, K. Stress and the hypothalamic pituitary adrenal axis in the developmental course of schizophrenia. Annu. Rev. Clin. Psychol. 2008, 4, 189–216. [Google Scholar] [CrossRef] [PubMed]
- Pruessner, M.; Cullen, A.E.; Aas, M.; Walker, E.F. The neural diathesis-stress model of schizophrenia revisited: An update on recent findings considering illness stage and neurobiological and methodological complexities. Neurosci. Biobehav. Rev. 2016, 73, 191–218. [Google Scholar] [CrossRef] [PubMed]
- Hutson, P.H.; Clark, J.A.; Cross, A.J. CNS target identification and validation: Avoiding the valley of death or naive optimism? Annu. Rev. Pharmacol. Toxicol. 2017, 57, 171–187. [Google Scholar] [CrossRef] [PubMed]
- Weickert, C.S.; Weickert, T.W.; Pillai, A.; Buckley, P.F. Biomarkers in Schizophrenia: A Brief Conceptual Consideration. Dis. Markers 2013, 35, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Steiner, J.; Guest, P.C.; Rahmoune, H.; De-Souza, D.M. The application of multiplex biomarker techniques for improved stratification and treatment of schizophrenia patients. Methods Mol. Biol. 2017, 1546, 19–35. [Google Scholar] [PubMed]
- Chan, M.K.; Gottschalk, M.G.; Haenisch, F.; Tomasik, J.; Ruland, T.; Rahmoune, H.; Guest, P.C.; Bahn, S. Applications of blood-based protein biomarker strategies in the study of psychiatric disorders. Prog. Neurobiol. 2014, 122, 45–72. [Google Scholar] [CrossRef] [PubMed]
- Scarr, E.; Millan, M.J.; Bahn, S.; Bertolino, A.; Turck, C.W.; Kapur, S.; Möller, H.J.; Dean, B. Biomarkers for psychiatry: The journey from fantasy to fact, a report of the 2013 CINP Think Tank. Int. J. Neuropsychopharmacol. 2015, 18, pyv042. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.-Y.; Scarr, E.; Udawela, M.; Everall, I.; Chen, W.J.; Dean, B. Biomarkers in schizophrenia: A focus on blood based diagnostics and theranostics. World J. Psychiatry 2016, 6, 102–117. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Lee, S.; Chen, X. Nanoparticle-based theranostic agents. Adv. Drug Deliv. Rev. 2010, 62, 1064–1079. [Google Scholar] [CrossRef] [PubMed]
- Harris, L.W.; Pietsch, S.; Cheng, T.M.K.; Schwarz, E.; Guest, P.C.; Bahn, S. Comparison of peripheral and central schizophrenia biomarker profiles. PLoS ONE 2012, 7, e46368. [Google Scholar] [CrossRef] [PubMed]
- Yolken, R.H.; Torrey, E.F. Are some cases of psychosis caused by microbial agents? A review of the evidence. Mol. Psychiatry 2008, 13, 470–479. [Google Scholar] [CrossRef] [PubMed]
- Suvisaari, J.; Loo, B.M.; Saarni, S.E.; Haukka, J.; Perala, J.; Saarni, S.I.; Viertio, S.; Partti, K.; Lonnqvist, J.; Jula, A. Inflammation in psychotic disorders: A population-based study. Psychiatry Res. 2011, 189, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Chaudhuri, T.K. Role of c-reactive protein in schizophrenia: An overview. Psychiatry Res. 2014, 216, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Fineberg, A.M.; Ellman, L.M. Inflammatory cytokines and neurological and neurocognitive alterations in the course of schizophrenia. Biol. Psychiatry 2013, 73, 951–966. [Google Scholar] [CrossRef] [PubMed]
- Benros, M.E.; Nielsen, P.R.; Nordentoft, M.; Eaton, W.W.; Dalton, S.O.; Mortensen, P.B. Autoimmune diseases and severe infections as risk factors for schizophrenia: A 30-year population-based register study. Am. J. Psychiatry 2011, 168, 1303–1310. [Google Scholar] [CrossRef] [PubMed]
- Corvin, A.; Morris, D.W. Genome-wide association studies: Findings at the major histocompatibility complex locus in psychosis. Biol. Psychiatry 2014, 75, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Dickerson, F.; Stallings, C.; Origoni, A.; Vaughan, C.; Khushalani, S.; Yang, S.J.; Yolken, R. C-reactive protein is elevated in schizophrenia. Schizophr. Res. 2013, 143, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.J.; Culpepper, N.; Rapaport, M.H. C-reactive protein levels in schizophrenia: A review and meta-analysis. Clin. Schizophr. Relat. Psychoses 2014, 7, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.D.; Pristach, C.; Liu, E.Y.; Freudenreich, O.; Henderson, D.C.; Goff, D.C. Elevated serum levels of C-reactive protein are associated with more severe psychopathology in a subgroup of patients with schizophrenia. Psychiatry Res. 2007, 149, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Fawzi, M.H.; Fawzi, M.M.; Fawzi, M.M.; Said, N.S. C-reactive protein serum level in drug-free male egyptian patients with schizophrenia. Psychiatry Res. 2011, 190, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Vuksan-Cusa, B.; Sagud, M.; Jakovljevic, M. C-reactive protein and metabolic syndrome in patients with bipolar disorder compared to patients with schizophrenia. Psychiatr. Danub. 2010, 22, 275–277. [Google Scholar] [PubMed]
- Hope, S.; Melle, I.; Aukrust, P.; Steen, N.E.; Birkenaes, A.B.; Lorentzen, S.; Agartz, I.; Ueland, T.; Andreassen, O.A. Similar immune profile in bipolar disorder and schizophrenia: Selective increase in soluble tumor necrosis factor receptor I and von willebrand factor. Bipolar Disord. 2009, 11, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Baptista, T.; Davila, A.; El Fakih, Y.; Uzcategui, E.; Rangel, N.N.; Olivares, Y.; Galeazzi, T.; Vargas, D.; Pena, R.; Marquina, D.; et al. Similar frequency of abnormal correlation between serum leptin levels and bmi before and after olanzapine treatment in schizophrenia. Int. Clin. Psychopharmacol. 2007, 22, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.J.; Buckley, P.; Seabolt, W.; Mellor, A.; Kirkpatrick, B. Meta-analysis of cytokine alterations in schizophrenia: Clinical status and antipsychotic effects. Biol. Psychiatry 2011, 70, 663–671. [Google Scholar] [CrossRef] [PubMed]
- De Witte, L.; Tomasik, J.; Schwarz, E.; Guest, P.C.; Rahmoune, H.; Kahn, R.S.; Bahn, S. Cytokine alterations in first-episode schizophrenia patients before and after antipsychotic treatment. Schizophr. Res. 2014, 154, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, E.; Guest, P.C.; Rahmoune, H.; Martins-de-Souza, D.; Niebuhr, D.W.; Weber, N.S.; Cowan, D.N.; Yolken, R.H.; Spain, M.; Barnes, A.; et al. Identification of a blood-based biological signature in subjects with psychiatric disorders prior to clinical manifestation. World J. Biol. Psychiatry 2012, 13, 627–632. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, E.; Guest, P.C.; Steiner, J.; Bogerts, B.; Bahn, S. Identification of blood-based molecular signatures for prediction of response and relapse in schizophrenia patients. Transl. Psychiatry 2012, 2, e82. [Google Scholar] [CrossRef] [PubMed]
- Ellman, L.M.; Yolken, R.H.; Buka, S.L.; Torrey, E.F.; Cannon, T.D. Cognitive functioning prior to the onset of psychosis: The role of fetal exposure to serologically determined influenza infection. Biol. Psychiatry 2009, 65, 1040–1047. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.S.; Cohen, P.; Harkavy-Friedman, J.; Babulas, V.; Malaspina, D.; Gorman, J.M.; Susser, E.S. Prenatal rubella, premorbid abnormalities, and adult schizophrenia. Biol. Psychiatry 2001, 49, 473–486. [Google Scholar] [CrossRef]
- Suvisaari, J.; Haukka, J.; Tanskanen, A.; Hovi, T.; Lonnqvist, J. Association between prenatal exposure to poliovirus infection and adult schizophrenia. Am. J. Psychiatry 1999, 156, 1100–1102. [Google Scholar] [PubMed]
- Torrey, E.F.; Rawlings, R.; Waldman, I.N. Schizophrenic births and viral diseases in 2 states. Schizophr. Res. 1988, 1, 73–77. [Google Scholar] [CrossRef]
- Buka, S.L.; Tsuang, M.T.; Torrey, E.F.; Klebanoff, M.A.; Bernstein, D.; Yolken, R.H. Maternal infections and subsequent psychosis among offspring. Arch. Gen. Psychiatry 2001, 58, 1032–1037. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, P.B.; Norgaard-Pedersen, B.; Waltoft, B.L.; Sorensen, T.L.; Hougaard, D.; Yolken, R.H. Early infections of toxoplasma gondii and the later development of schizophrenia. Schizophr. Bull. 2007, 33, 741–744. [Google Scholar] [CrossRef] [PubMed]
- Patterson, P.H. Immune involvement in schizophrenia and autism: Etiology, pathology and animal models. Behav. Brain. Res. 2009, 204, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Buka, S.L.; Tsuang, M.T.; Torrey, E.F.; Klebanoff, M.A.; Wagner, R.L.; Yolken, R.H. Maternal cytokine levels during pregnancy and adult psychosis. Brain Behav. Immun. 2001, 15, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.S.; Hooton, J.; Schaefer, C.A.; Zhang, H.; Petkova, E.; Babulas, V.; Perrin, M.; Gorman, J.M.; Susser, E.S. Elevated maternal interleukin-8 levels and risk of schizophrenia in adult offspring. Am. J. Psychiatry 2004, 161, 889–895. [Google Scholar] [CrossRef] [PubMed]
- Ellman, L.M.; Deicken, R.F.; Vinogradov, S.; Kremen, W.S.; Poole, J.H.; Kern, D.M.; Tsai, W.Y.; Schaefer, C.A.; Brown, A.S. Structural brain alterations in schizophrenia following fetal exposure to the inflammatory cytokine interleukin-8. Schizophr. Res. 2010, 121, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Wright, I.C.; Rabe-Hesketh, S.; Woodruff, P.W.R.; David, A.S.; Murray, R.M.; Bullmore, E.T. Meta-analysis of regional brain volumes in schizophrenia. Am. J. Psychiatry 2000, 157, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Ezeoke, A.; Mellor, A.; Buckley, P.; Miller, B. A systematic, quantitative review of blood autoantibodies in schizophrenia. Schizophr. Res. 2013, 150, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Brey, R.L.; Holliday, S.I.; Saklad, A.R.; Navarrete, M.G.; Hermosillo-Romo, D.; Stallworth, C.L.; Valdez, C.R.; Escalante, A.; del Rincon, I.; Gronseth, G.; et al. Neuropsychiatric syndromes in lupus—Prevalence using standarized definitions. Neurology 2002, 58, 1214–1220. [Google Scholar] [CrossRef] [PubMed]
- Tsuang, M. Schizophrenia: Genes and environment. Biol. Psychiatry 2000, 47, 210–220. [Google Scholar] [CrossRef]
- Bergen, S.E.; O’Dushlaine, C.T.; Ripke, S.; Lee, P.H.; Ruderfer, D.M.; Akterin, S.; Moran, J.L.; Chambert, K.D.; Handsaker, R.E.; Backlund, L.; et al. Genome-wide association study in a swedish population yields support for greater CNV and MHC involvement in schizophrenia compared with bipolar disorder. Mol. Psychiatry 2012, 17, 880–886. [Google Scholar] [CrossRef] [PubMed]
- Beck, S.; Geraghty, D.; Inoko, H.; Rowen, L.; Aguado, B.; Bahram, S.; Campbell, R.D.; Forbes, S.A.; Guillaudeux, T.; Hood, L.; et al. Complete sequence and gene map of a human major histocompatibility complex. Nature 1999, 401, 921–923. [Google Scholar]
- Smoller, J.W.; Craddock, N.; Kendler, K.; Lee, P.H.; Neale, B.M.; Nurnberger, J.I.; Ripke, S.; Santangelo, S.; Sullivan, P.F.; Fanous, A.; et al. Identification of risk loci with shared effects on five major psychiatric disorders: A genome-wide analysis. Lancet 2013, 381, 1371–1379. [Google Scholar]
- Sinkus, M.L.; Adams, C.E.; Logel, J.; Freedman, R.; Leonard, S. Expression of immune genes on chromosome 6p21.3-22.1 in schizophrenia. Brain Behav. Immun. 2013, 32, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Sommer, I.E.; de Witte, L.; Begemann, M.; Kahn, R.S. Nonsteroidal anti-inflammatory drugs in schizophrenia: Ready for practice or a good start? A meta-analysis. J. Clin. Psychiatry 2012, 73, 414–419. [Google Scholar] [CrossRef] [PubMed]
- Laan, W.; Grobbee, D.E.; Selten, J.P.; Heijnen, C.J.; Kahn, R.S.; Burger, H. Adjuvant aspirin therapy reduces symptoms of schizophrenia spectrum disorders: Results from a randomized, double-blind, placebo-controlled trial. J. Clin. Psychiatry 2010, 71, 520–527. [Google Scholar] [CrossRef] [PubMed]
- El-Sisi, A.E.; Sokkar, S.S.; El-Sayad, M.E.; Ramadan, E.S.; Osman, E.Y. Celecoxib and ω-3 fatty acids alone and in combination with risperidone affect the behavior and brain biochemistry in amphetamine-induced model of schizophrenia. Biomed. Pharmacother. 2016, 82, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Mondelli, V.; Ciufolini, S.; Murri, M.B.; Bonaccorso, S.; Di Forti, M.; Giordano, A.; Marques, T.R.; Zunszain, P.A.; Morgan, C.; Murray, R.M.; et al. Cortisol and inflammatory biomarkers predict poor treatment response in first episode psychosis. Schizophr. Bull. 2015, 41, 1162–1170. [Google Scholar] [CrossRef] [PubMed]
- Fond, G.; Boyer, L.; Gaman, A.; Laouamri, H.; Attiba, D.; Richard, J.R.; Delavest, M.; Houenou, J.; Le Corvoisier, P.; Charron, D.; et al. Treatment with anti-toxoplasmic activity (TATA) for toxoplasma positive patients with bipolar disorders or schizophrenia: A cross-sectional study. J. Psychiatr. Res. 2015, 63, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Tourjman, V.; Kouassi, E.; Koue, M.E.; Rocchetti, M.; Fortin-Fournier, S.; Fusar-Poli, P.; Potvin, S. Antipsychotics’ effects on blood levels of cytokines in schizophrenia: A meta-analysis. Schizophr. Res. 2013, 151, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Song, X.Q.; Fan, X.D.; Li, X.; Zhang, W.; Gao, J.S.; Zhao, J.P.; Harrington, A.; Ziedonis, D.; Lv, L.X. Changes in pro-inflammatory cytokines and body weight during 6-month risperidone treatment in drug naive, first-episode schizophrenia. Psychopharmacology 2014, 231, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Hori, H.; Teraishi, T.; Sasayama, D.; Fujii, T.; Hattori, K.; Ishikawa, M.; Kunugi, H. Elevated cortisol level and cortisol/DHEAS ratio in schizophrenia as revealed by low-dose dexamethasone suppression test. Open Neuropsychopharmacol. J. 2012, 5, 18–24. [Google Scholar] [CrossRef]
- Jakovljevic, M.; Muck-Seler, D.; Pivac, N.; Crncevic, Z. Platelet 5-HT and plasma cortisol concentrations after dexamethasone suppression test in patients with different time course of schizophrenia. Neuropsychobiology 1998, 37, 142–145. [Google Scholar] [CrossRef] [PubMed]
- Mondelli, V.; Dazzan, P.; Hepgul, N.; Di Forti, M.; Aas, M.; D’Albenzio, A.; Di Nicola, M.; Fisher, H.; Handley, R.; Marques, T.R.; et al. Abnormal cortisol levels during the day and cortisol awakening response in first-episode psychosis: The role of stress and of antipsychotic treatment. Schizophr. Res. 2010, 116, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Muck-Seler, D.; Pivac, N.; Jakovljevic, M.; Brzovic, Z. Platelet serotonin, plasma cortisol, and dexamethasone suppression test in schizophrenic patients. Biol. Psychiatry 1999, 45, 1433–1439. [Google Scholar] [CrossRef]
- Girshkin, L.; Matheson, S.L.; Shepherd, A.M.; Green, M.J. Morning cortisol levels in schizophrenia and bipolar disorder: A meta-analysis. Psychoneuroendocrinology 2014, 49, 187–206. [Google Scholar] [CrossRef] [PubMed]
- Guest, P.C.; Schwarz, E.; Krishnamurthy, D.; Harris, L.W.; Leweke, F.M.; Rothermundt, M.; van Beveren, N.J.; Spain, M.; Barnes, A.; Steiner, J.; et al. Altered levels of circulating insulin and other neuroendocrine hormones associated with the onset of schizophrenia. Psychoneuroendocrinology 2011, 36, 1092–2096. [Google Scholar] [CrossRef] [PubMed]
- Murri, M.B.; Pariante, C.M.; Dazzan, P.; Hepgul, N.; Papadopoulos, A.S.; Zunszain, P.; Di Forti, M.; Murray, R.M.; Mondelli, V. Hypothalamic-pituitary-adrenal axis and clinical symptoms in first-episode psychosis. Psychoneuroendocrinology 2012, 37, 629–644. [Google Scholar] [CrossRef] [PubMed]
- Girshkin, L.; O’Reilly, N.; Quide, Y.; Teroganova, N.; Rowland, J.E.; Schofield, P.R.; Green, M.J. Diurnal cortisol variation and cortisol response to an MRI stressor in schizophrenia and bipolar disorder. Psychoneuroendocrinology 2016, 67, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Bradley, A.J.; Dinan, T.G. A systematic review of hypothalamic-pituitary-adrenal axis function in schizophrenia: Implications for mortality. J. Psychopharmacol. 2010, 24, 91–118. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Pickar, D.; Doran, A.; Wolkowitz, O.; Gallucci, W.; Chrousos, G.; Gould, P. The corticotropin-releasing hormone stimulation test in chronic schizophrenia. Am. J. Psychiatry 1986, 143, 1393–1397. [Google Scholar] [PubMed]
- Lammers, C.H.; Garcia-Borreguero, D.; Schmider, J.; Gotthardt, U.; Dettling, M.; Holsboer, F.; Heuser, I.J. Combined dexamethasone/corticotropin-releasing hormone test in patients with schizophrenia and in normal controls: II. Biol. Psychiatry 1995, 38, 803–807. [Google Scholar] [CrossRef]
- Gallagher, P.; Watson, S.; Smith, M.S.; Young, A.H.; Ferrier, I.N. Plasma cortisol-dehydroepiandrosterone (DHEA) ratios in schizophrenia and bipolar disorder. Schizophr. Res. 2007, 90, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Brenner, K.; Liu, A.; Laplante, D.P.; Lupien, S.; Pruessner, J.C.; Ciampi, A.; Joober, R.; King, S. Cortisol response to a psychosocial stressor in schizophrenia: Blunted, delayed, or normal? Psychoneuroendocrinology 2009, 34, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Jakovljevic, M.; Pivac, N.; Mihaljevic-Peles, A.; Mustapic, M.; Relja, M.; Ljubicic, D.; Marcinko, D.; Muck-Seler, D. The effects of olanzapine and fluphenazine on plasma cortisol, prolactin and muscle rigidity in schizophrenic patients: A double blind study. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2007, 1, 399–402. [Google Scholar] [CrossRef] [PubMed]
- Zannas, A.S.; Wiechmann, T.; Gassen, N.C.; Binder, E.B. Gene-stress-epigenetic regulation of FKBP5: Clinical and translational implications. Neuropsychopharmacology 2016, 41, 261–274. [Google Scholar] [CrossRef] [PubMed]
- Bevilacqua, L.; Carli, V.; Sarchiapone, M.; George, D.K.; Goldman, D.; Roy, A.; Enoch, M.A. Interaction between FKBP5 and childhood trauma and risk of aggressive behavior. Arch. Gen. Psychiatry 2012, 69, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Collip, D.; Myin-Germeys, I.; Wichers, M.; Jacobs, N.; Derom, C.; Thiery, E.; Lataster, T.; Simons, C.; Delespaul, P.; Marcelis, M.; et al. FKBP5 as a possible moderator of the psychosis-inducing effects of childhood trauma. Br. J. Psychiatry 2013, 202, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Pereira, M.J.; Palming, J.; Svensson, M.K.; Rizell, M.; Dalenback, J.; Hammar, M.; Fall, T.; Sidibeh, C.O.; Svensson, P.A.; Eriksson, J.W. FKBP5 expression in human adipose tissue increases following dexamethasone exposure and is associated with insulin resistance. Metabolism 2014, 63, 1198–1208. [Google Scholar] [CrossRef] [PubMed]
- De Luca, V.; Tharmalingam, S.; Zai, C.; Potapova, N.; Strauss, J.; Vincent, J.; Kennedy, J.L. J. Psychopharmacol. 2010, 24, 677–682. [PubMed]
- Ryan, M.C.; Thakore, J.H. Physical consequences of schizophrenia and its treatment: The metabolic syndrome. Life Sci. 2002, 71, 239–257. [Google Scholar] [CrossRef]
- De Hert, M.; Schreurs, W.; Vancampfort, D.; Van Winkel, R. Metabolic syndrome in people with schizophrenia: A review. World Psychiatry 2009, 8, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Akiibinu, M.O.; Ogundahunsi, O.A.; Ogunyemi, E.O. Inter-relationship of plasma markers of oxidative stress and thyroid hormones in schizophrenics. BMC Res. Notes 2012, 5, 169. [Google Scholar] [CrossRef] [PubMed]
- Vuksan Cusa, B.; Sagud, M.; Rados, I. The role of dehydroepiandrosterone (DHEA) in schizophrenia. Psychiatr. Danub. 2016, 28, 30–33. [Google Scholar] [PubMed]
- Matsumoto, T.; Rauskolb, S.; Polack, M.; Klose, J.; Kolbeck, R.; Korte, M.; Barde, Y.A. Biosynthesis and processing of endogenous BDNF: CNS neurons store and secrete BDNF, not pro-BDNF. Nat. Neurosci. 2008, 11, 131–133. [Google Scholar] [CrossRef] [PubMed]
- Huang, E.J.; Reichardt, L.F. Trk receptors: Roles in signal transduction. Annu. Rev. Biochem. 2003, 72, 609–642. [Google Scholar] [CrossRef] [PubMed]
- Marenco, S.; Weinberger, D.R. The neurodevelopmental hypothesis of schizophrenia: Following a trail of evidence from cradle to grave. Dev. Psychopathol. 2000, 12, 501–527. [Google Scholar] [CrossRef] [PubMed]
- Thompson Ray, M.; Weickert, C.S.; Wyatt, E.; Webster, M.J. Decreased BDNF, TrkB-TK+ and GAD67 mRNA expression in the hippocampus of individuals with schizophrenia and mood disorders. J. Psychiatry Neurosci. 2011, 36, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Durany, N.; Michel, T.; Zöchling, R.; Boissl, K.W.; Cruz-Sánchez, F.F.; Riederer, P.; Riederer, P.; Thome, J. Brain-derived neurotrophic factor and neurotrophin 3 in schizophrenic psychoses. Schizophr. Res. 2001, 52, 79–86. [Google Scholar] [CrossRef]
- Issa, G.; Wilson, C.; Terry, A.V., Jr.; Pillai, A. An inverse relationship between cortisol and BDNF level in schizophrenia: Data from human postmortem and animal studies. Neurobiol. Dis. 2010, 39, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Gurnay, M.E. Human platelets contain brain-derived neurotrophic factor. J. Neurosci. 1990, 10, 3469–3478. [Google Scholar] [PubMed]
- Colombo, E.; Bedogni, F.; Lorenzetti, I.; Landsberger, N.; Previtali, S.C.; Farina, C. Autocrine and immune cell-derived BDNF in human skeletal muscle: Implications for myogenesis and tissue regeneration. J. Pathol. 2013, 231, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.; Banks, W.A.; Fasold, M.B.; Bluth, J.; Kastin, A.J. Transport of brain-derived neurotrophic factor across the blood-brain barrier. Neuropharmacology 1998, 37, 1553–1561. [Google Scholar] [CrossRef]
- Nurjono, M.; Lee, J.; Chong, S.A. A review of brain-derived neurotrophic factor as a candidate biomarker in Schizophrenia. Clin. Psychopharmacol. Neurosci. 2012, 10, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Karege, F.; Schwald, M.; Cisse, M. Postnatal developmental profile of brain-derived neurotrophic factor in rat brain and platelets. Neurosci. Lett. 2002, 328, 261–264. [Google Scholar] [CrossRef]
- Klein, A.B.; Williamson, R.; Santini, M.A.; Clemmensen, C.; Ettrup, A.; Rios, M.; Knudsen, G.M.; Aznar, S. Blood BDNF concentrations reflect brain-tissue BDNF levels across species. Int. J. Neuropsychopharmacol. 2011, 14, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Wang, J.; Wang, B.; Yang, S.C.; Zhang, C.X.; Zheng, Y.L.; Li, Y.L.; Wang, N.; Yang, K.B.; Xiu, M.H.; et al. Decreased levels of serum brain-derived neurotrophic factor in drug-naïve first-episode schizophrenia: Relationship to clinical phenotypes. Psychopharmacol. Berl. 2009, 207, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Jindal, R.D.; Pillai, A.K.; Mahadik, S.P.; Eklund, K.; Montrose, D.M.; Keshavan, M.S. Decreased BDNF in patients with antipsychotic naïve first episode schizophrenia. Schizophr. Res. 2010, 119, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Pirildar, Ş.; Gönül, A.S.; Taneli, F.; Akdeniz, F. Low serum levels of brain-derived neurotrophic factor in patients with schizophrenia do not elevate after antipsychotic treatment. Prog. Neuropsychopharmacol. Biol. Psychiatry 2004, 28, 709–713. [Google Scholar] [CrossRef] [PubMed]
- Rizos, E.N.; Rontos, I.; Laskos, E.; Arsenis, G.; Michalopoulou, P.G.; Vasilopoulos, D.; Gournellis, R.; Lykouras, L. Investigation of serum BDNF levels in drug-naive patients with schizophrenia. Prog. Neuropsychopharmacol. Biol. Psychiatry 2008, 32, 1308–1311. [Google Scholar] [CrossRef] [PubMed]
- Grillo, R.W.; Ottoni, G.L.; Leke, R.; Souza, D.O.; Portela, L.V.; Lara, D.R. Reduced serum BDNF levels in schizophrenic patients on clozapine or typical antipsychotics. J. Psychiatr. Res. 2007, 41, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.L.; Zhou, D.F.; Cao, L.Y.; Zou, Y.Z.; Zhang, X.Y. Decreased BDNF in serum of patients with chronic schizophrenia on long-term treatment with antipsychotics. Neurosci. Lett. 2005, 382, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Toyooka, K.; Asama, K.; Watanabe, Y.; Muratake, T.; Takahashi, M.; Someya, T.; Nawa, H. Decreased levels of brain-derived neurotrophic factor in serum of chronic schizophrenic patients. Psychiatry Res. 2002, 110, 249–257. [Google Scholar] [CrossRef]
- Zhang, X.Y.; Tan, Y.L.; Zhou, D.F.; Cao, L.Y.; Wu, G.Y.; Xu, Q.; Shen, Y.; Haile, C.N.; Kosten, T.A.; Kosten, T.R. Serum BDNF levels and weight gain in schizophrenic patients on long-term treatment with antipsychotics. J. Psychiatr. Res. 2007, 41, 997–1004. [Google Scholar] [CrossRef] [PubMed]
- Xiu, M.H.; Hui, L.; Dang, Y.F.; Hou, T.D.; Zhang, C.X.; Zheng, Y.L.; Chen, D.C.; Kosten, T.R.; Zhang, X.Y. Decreased serum BDNF levels in chronic institutionalized schizophrenia on long-term treatment with typical and atypical antipsychotics. Prog. Neuropsychopharmacol. Biol. Psychiatry 2009, 33, 1508–1512. [Google Scholar] [CrossRef] [PubMed]
- Green, M.J.; Matheson, S.L.; Shepherd, A.; Weickert, C.S.; Carr, V.J. Brain-derived neurotrophic factor levels in schizophrenia: A systematic review with meta-analysis. Mol. Psychiatry 2011, 16, 960–972. [Google Scholar] [CrossRef] [PubMed]
- Pedrini, M.; Chendo, I.; Grande, I.; Lobato, M.I.; Belmontede-Abreu, P.S.; Lersch, C.; Walz, J.; Kauer-Sant’anna, M.; Kapczinski, F.; Gama, C.S. Serum brain-derived neurotrophic factor and clozapine daily dose in patients with schizophrenia: A positive correlation. Neurosci. Lett. 2011, 491, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Carlino, D.; Leone, E.; Di Cola, F.; Baj, G.; Marin, R.; Dinelli, G.; Tongiorgi, E.; De Vanna, M. Low serum truncated-BDNF isoform correlates with higher cognitive impairment in schizophrenia. J. Psychiatr. Res. 2011, 45, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Angelucci, F.; Mathe, A.A.; Aloe, L. Brain-derived neurotrophic factor and tyrosine kinase receptor TrkB in rat brain are significantly altered after haloperidol and risperidone administration. J. Neurosci. Res. 2000, 60, 783–794. [Google Scholar] [CrossRef]
- Dawson, N.M.; Hamid, E.H.; Egan, M.F.; Meredith, G.E. Changes in the pattern of brain-derived neurotrophic factor immunoreactivity in the rat brain after acute and subchronic haloperidol treatment. Synapse 2001, 39, 70–81. [Google Scholar] [CrossRef]
- Lipska, B.K.; Khaing, Z.Z.; Weickert, C.S.; Weinberger, D.R. BDNF mRNA expression in rat hippocampus and prefrontal cortex: Effects of neonatal ventral hippocampal damage and antipsychotic drugs. Eur. J. Neurosci. 2001, 14, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Parikh, V.; Khan, M.M.; Mahadik, S.P. Olanzapine counteracts reduction of brain-derived neurotrophic factor and TrkB receptors in rat hippocampus produced by haloperidol. Neurosci. Lett. 2004, 356, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Chlan-Fourney, J.; Ashe, P.; Nylen, K.; Juorio, A.V.; Li, X.M. Differential regulation of hippocampal BDNF mRNA by typical and atypical antipsychotic administration. Brain Res. 2002, 954, 11–20. [Google Scholar] [CrossRef]
- Egan, M.F.; Kojima, M.; Callicott, J.H.; Goldberg, T.E.; Kolachana, B.S.; Bertolino, A.; Zaitsev, E.; Gold, B.; Goldman, D.; Dean, M.; et al. The BDNF Val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell 2003, 112, 257–269. [Google Scholar] [CrossRef]
- Chen, Z.-Y.; Ieraci, A.; Teng, H.; Dall, H.; Meng, C.-X.; Herrera, D.G.; Nykjaer, A.; Hemp-stead, B.L.; Lee, F.S. Sortilin controls intracellular sorting of brain-derived neurotrophic factor to the regulated secretory pathway. J. Neurosci. 2005, 25, 6156–6166. [Google Scholar] [CrossRef] [PubMed]
- Dayem Ullah, A.Z.; Lemoine, N.R.; Chelala, C. SNPnexus: A web server for functional annotation of novel and publicly known genetic variants (2012 update). Nucleic Acids Res. 2012, 40, W65–W70. [Google Scholar] [CrossRef] [PubMed]
- Zakharyan, R.; Boyajyan, A.; Arakelyan, A.; Gevorgyan, A.; Mrazek, F.; Petrek, M. Functional variants of the genes involved in neurodevelopment and susceptibility to schizophrenia in an Armenian population. Hum. Immunol. 2011, 72, 746–748. [Google Scholar] [CrossRef] [PubMed]
- Gratacòs, M.; González, J.R.; Mercader, J.M.; de Cid, R.; Urretavizcaya, M.; Estivill, X. Brain-derived neurotrophic factor Val66Met and psychiatric disorders: Meta-analysis of case–control studies confirm association to substance-related disorders, eating disorders, and schizophrenia. Biol. Psychiatry 2007, 61, 911–922. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.N.; Thornton, A.E.; Lang, D.J.; MacEwan, G.W.; Ehmann, T.S.; Kopala, L.C.; Tee, K.; Shiau, G.; Voineskos, A.N.; Kennedy, J.L.; et al. Hippocampal volume and the brain-derived neurotrophic factor Val66Met polymorphism in first episode psychosis. Schizophr. Res. 2012, 134, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Neves-Pereira, M.; Cheung, J.K.; Pasdar, A.; Zhang, F.; Breen, G.; Yates, P.; Sinclair, M.; Crombie, C.; Walker, N.; St. Clair, D.M. BDNF gene is a risk factor for schizophrenia in a Scottish population. Mol. Psychiatry 2005, 10, 208–212. [Google Scholar] [CrossRef] [PubMed]
- Rosa, A.; Cuesta, M.J.; Fatjó-Vilas, M.; Peralta, V.; Zarzuela, A.; Fañanás, L. The Val66Met polymorphism of the brain-derived neurotrophic factor gene is associated with risk for psychosis: Evidence from a family-based association study. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2006, 141B, 135–138. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.-A.; Lu, R.-B.; Shy, M.-J.; Chang, C.-C.; Lee, M.-S.; Huang, S.-Y. Brain-derived neurotrophic factor Val66Met polymorphism: Association with psychopathological symptoms of schizophrenia? J. Neuropsychiatry Clin. Neurosci. 2009, 21, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Jönsson, E.G.; Edman-Ahlbom, B.; Sillen, A.; Gunnar, A.; Kulle, B.; Frigessi, A.; Vares, M.; Ekholm, B.; Wode-Helgodt, B.; Schumacher, J.; et al. Brain-derived neurotrophic factor gene (BDNF) variants and schizophrenia: An association study. Prog. Neuropsychopharmacol. Biol. Psychiatry 2006, 30, 924–933. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, K.; Ikeda, M.; Kishi, T.; Kitajima, T.; Yamanouchi, Y.; Kinoshita, Y.; Okochi, T.; Aleksic, B.; Tomita, M.; Okada, T.; et al. BDNF is not associated with schizophrenia: Data from a Japanese population study and meta-analysis. Schizophr. Res. 2009, 112, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.H.; Yan, Q.Z.; Yan, X.M.; Li, C.B.; Fang, H.; Zheng, Y.L.; Zhang, C.X.; Yao, H.J.; Chen da, C.; Xiu, M.H.; et al. The study of BDNF Val66Met polymorphism in Chinese schizophrenic patients. Prog. Neuropsychopharmacol. Biol. Psychiatry 2010, 34, 930–933. [Google Scholar] [CrossRef] [PubMed]
- Zintzaras, E. Brain-derived neurotrophic factor gene polymorphisms and schizophrenia: A meta-analysis. Psychiatr. Genet. 2007, 17, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Nunokawa, A.; Someya, T. Association of the BDNF C270T polymorphism with schizophrenia: Updated meta-analysis. Psychiatry Clin. Neurosci. 2013, 67, 123–125. [Google Scholar] [CrossRef] [PubMed]
- Nanko, S.; Kunugi, H.; Hirasawa, H.; Kato, N.; Nabika, T.; Kobayashi, S. Brain-derived neurotrophic factor gene and schizophrenia: Polymorphism screening and association analysis. Schizophr. Res. 2003, 62, 281–283. [Google Scholar] [PubMed]
- Szekeres, G.; Juhász, A.; Rimanóczy, A.; Kéri, S.; Janka, Z. The C270T polymorphism of the brain-derived neurotrophic factor gene is associated with schizophrenia. Schizophr. Res. 2003, 65, 15–18. [Google Scholar] [CrossRef]
- Xu, M.Q.; St Clair, D.; Ott, J.; Feng, G.Y.; He, L. Brain-derived neurotrophic factor gene C-270T and Val66Met functional polymorphisms and risk of schizophrenia: A moderate-scale population-based study and meta-analysis. Schizophr. Res. 2007, 91, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.J.; Yu, Y.W.; Lin, C.H.; Tsai, S.J. An association study of a brain-derived neurotrophic factor Val66Met polymorphism and clozapine response of schizophrenic patients. Neurosci. Lett. 2003, 349, 206–208. [Google Scholar] [CrossRef]
- Zai, G.C.; Chowdhury, N.I.; Tiwari, A.K.; Souza, R.P.; Lieberman, J.A.; Meltzer, H.Y.; Potkin, S.G.; Müller, D.J.; Kennedy, J.L. The role of brain-derived neurotrophic factor (BDNF) gene variants in antipsychotic response and antipsychotic-induced weight gain. Prog. Neuropsychopharmacol. Biol. Psychiatry 2012, 39, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Nikolac Perkovic, M.; Nedic Erjavec, G.; Zivkovic, M.; Sagud, M.; Uzun, S.; Mihaljevic-Peles, A.; Kozumplik, O.; Muck-Seler, D.; Pivac, N. Association between the brain-derived neurotrophic factor Val66Met polymorphism and therapeutic response to olanzapine in schizophrenia patients. Psychopharmacology 2014, 231, 3757–3764. [Google Scholar] [CrossRef] [PubMed]
- Mitjans, M.; Catalán, R.; Vázquez, M.; González-Rodríguez, A.; Penadés, R.; Pons, A.; Massana, G.; Munro, J.; Arranz, M.J.; Arias, B. Hypothalamic-pituitary-adrenal system, neurotrophic factors and clozapine response: Association with FKBP5 and NTRK2 genes. Pharmacogenet. Genom. 2015, 25, 274–277. [Google Scholar] [CrossRef] [PubMed]
- Terzic, T.; Kastelic, M.; Dolzan, V.; Plesnicar, B.K. Genetic variability testing of neurodevelopmental genes in schizophrenic patients. J. Mol. Neurosci. 2015, 56, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.Q.; St Clair, D.; Feng, G.Y.; Lin, Z.G.; He, G.; Li, X.; He, L. BDNF gene is a genetic risk factor for schizophrenia and is related to the chlorpromazine-induced extrapyramidal syndrome in the Chinese population. Pharmacogenet. Genom. 2008, 18, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.Y.; Zhou, D.F.; Wu, G.Y.; Cao, L.Y.; Tan, Y.L.; Haile, C.N.; Li, J.; Lu, L.; Kosten, T.A.; Kosten, T.R. BDNF levels and genotype are associated with antipsychotic-induced weight gain in patients with chronic schizophrenia. Neuropsychopharmacology 2008, 33, 2200–2205. [Google Scholar] [CrossRef] [PubMed]
- Tsai, A.; Liou, Y.J.; Hong, C.J.; Wu, C.L.; Tsai, S.J.; Bai, Y.M. Association study of brain-derived neurotrophic factor gene polymorphisms and body weight change in schizophrenic patients under long-term atypical antipsychotic treatment. Neuromol. Med. 2011, 13, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Cargnin, S.; Massarotti, A.; Terrazzino, S. BDNF Val66Met and clinical response to antipsychotic drugs: A systematic review and meta-analysis. Eur. Psychiatry 2016, 33, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Martinowich, K.; Hattori, D.; Wu, H.; Fouse, S.; He, F.; Hu, Y.; Fan, G.; Sun, Y.E. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science 2003, 302, 890–893. [Google Scholar] [CrossRef] [PubMed]
- Varendi, K.; Kumar, A.; Härma, M.A.; Andressoo, J.O. miR-1, miR-10b, miR-155, and miR-191 are novel regulators of BDNF. Cell. Mol. Life Sci. 2014, 71, 4443–4456. [Google Scholar] [CrossRef] [PubMed]
- Boulle, F.; van den Hove, D.L.; Jakob, S.B.; Rutten, B.P.; Hamon, M.; van Os, J.; Lesch, K.P.; Lanfumey, L.; Steinbusch, H.W.; Kenis, G. Epigenetic regulation of the BDNF gene: Implications for psychiatric disorders. Mol. Psychiatry 2012, 17, 584–596. [Google Scholar] [CrossRef] [PubMed]
- Ikegame, T.; Bundo, M.; Murata, Y.; Kasai, K.; Kato, T.; Iwamoto, K. DNA methylation of the BDNF gene and its relevance to psychiatric disorders. J. Hum. Genet. 2013, 58, 434–438. [Google Scholar] [CrossRef] [PubMed]
- Mitchelmore, C.; Gede, L. Brain derived neurotrophic factor: Epigenetic regulation in psychiatric disorders. Brain Res. 2014, 1586, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Mill, J.; Tang, T.; Kaminsky, Z.; Khare, T.; Yazdanpanah, S.; Bouchard, L.; Jia, P.; Assadzadeh, A.; Flanagan, J.; Schumacher, A.; et al. Epigenomic profiling reveals DNA-methylation changes associated with major psychosis. Am. J. Hum. Genet. 2008, 82, 696–711. [Google Scholar] [CrossRef] [PubMed]
- Ikegame, T.; Bundo, M.; Sunaga, F.; Asai, T.; Nishimura, F.; Yoshikawa, A.; Kawamura, Y.; Hibino, H.; Tochigi, M.; Kakiuchi, C.; et al. DNA methylation analysis of BDNF gene promoters in peripheral blood cells of schizophrenia patients. Neurosci. Res. 2013, 77, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Kordi-Tamandani, D.M.; Sahranavard, R.; Torkamanzehi, A. DNA methylation and expression profiles of the brain-derived neurotrophic factor (BDNF) and dopamine transporter (DAT1) genes in patients with schizophrenia. Mol. Biol. Rep. 2012, 39, 10889–10893. [Google Scholar] [CrossRef] [PubMed]
- Melas, P.A.; Rogdaki, M.; Ösby, U.; Schalling, M.; Lavebratt, C.; Ekström, T.J. Epigenetic aberrations in leukocytes of patients with schizophrenia: Association of global DNA methylation with antipsychotic drug treatment and disease onset. FASEB J. 2012, 26, 2712–2718. [Google Scholar] [CrossRef] [PubMed]
- Guidotti, A.; Dong, E.; Kundakovic, M.; Satta, R.; Grayson, D.R.; Costa, E. Characterization of the action of antipsychotic subtypes on valproateinduced chromatin remodeling. Trends Pharmacol. Sci. 2009, 30, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Nanko, S.; Hattori, M.; Kuwata, S.; Sasaki, T.; Fukuda, R.; Dai, X.Y.; Yamaguchi, K.; Shibata, Y.; Kazamatsuri, H. Neurotrophin-3 gene polymorphism associated with schizophrenia. Acta Psychiatr. Scand. 1994, 89, 390–392. [Google Scholar] [CrossRef] [PubMed]
- Dawson, E.; Powell, J.F.; Sham, P.C.; Nöthen, M.; Crocq, M.A.; Propping, P.; Körner, J.; Rietschel, M.; van Os, J.; Wright, P.; et al. An association study of a neurotrophin-3 (NT-3) gene polymorphism with schizophrenia. Acta Psychiatr. Scand. 1995, 92, 425–428. [Google Scholar] [CrossRef] [PubMed]
- Nimganokar, V.l.; Zhang, X.R.; Brar, J.S.; DeLeo, M.; Ganguli, R. Lack of association of schizophrenia with the neurotrophin-3 gene locus. Acta Psychiatr. Scand. 1995, 92, 464–466. [Google Scholar] [CrossRef]
- Jŏnsson, E.; Brené, S.; Zhang, X.R.; Nimgaonkar, V.L.; Tylec, A.; Schalling, M.; Sedvall, G. Schizophrenia and neurotrophin-3 alleles. Acta Psychiatr. Scand. 1997, 95, 414–419. [Google Scholar] [CrossRef] [PubMed]
- Hattori, M.; Nanko, S. Association of neurotrophin-3 gene variant with severe forms of schizophrenia. Biochem. Biophys. Res. Commun. 1995, 209, 513–518. [Google Scholar] [CrossRef] [PubMed]
- Xiong, P.; Zeng, Y.; Zhu, Z.; Tan, D.; Xu, F.; Lu, J.; Wan, J.; Ma, M. Reduced NGF serum levels and abnormal P300 event-related potential in first episode schizophrenia. Schizophr. Res. 2010, 119, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Xiong, P.; Zeng, Y.; Wan, J.; Xiaohan, D.H.; Tan, D.; Lu, J.; Xu, F.; Li, H.Y.; Zhu, Z.; Ma, M. The role of NGF and IL-2 serum level in assisting the diagnosis in first episode schizophrenia. Psychiatry Res. 2011, 189, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Zakharyan, R.; Atshemyan, S.; Gevorgyan, A.; Boyajyan, A. Nerve growth factor and its receptor in schizophrenia. BBA Clin. 2014, 1, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, A.; Waters, N.; Carlsson, M.L. Neurotransmitter interactions in schizophrenia-therapeutic implications. Eur. Arch. Psychiatry Clin. Neurosci. 1999, 249, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Howes, O.D.; Kapur, S. The dopamine hypothesis of schizophrenia: Version III—The final common pathway. Schizophr. Bull. 2009, 35, 549–562. [Google Scholar] [CrossRef] [PubMed]
- Kapur, S. Psychosis as a state of aberrant salience: A framework linking biology, phenomenology, and pharmacology in schizophrenia. Am. J. Psychiatry 2003, 160, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Moore, H.; West, A.R.; Grace, A.A. The regulation of forebrain dopamine transmission: Relevance to the pathophysiology and psychopathology of schizophrenia. Biol. Psychiatry 1999, 46, 40–55. [Google Scholar] [CrossRef]
- Seeman, P. Dopamine receptors and the dopamine hypothesis of schizophrenia. Synapse 1987, 1, 133–152. [Google Scholar] [CrossRef] [PubMed]
- Hietala, J.; Syvälahti, E.; Vuorio, K.; Räkköläinen, V.; Bergman, J.; Haaparanta, M.; Solin, O.; Kuoppamäki, M.; Kirvelä, O.; Ruotsalainen, U.; et al. Presynaptic dopamine function in striatum of neuroleptic-naive schizophrenic patients. Lancet 1995, 346, 1130–1131. [Google Scholar] [CrossRef]
- Laruelle, M. Imaging dopamine transmission in schizophrenia. A review and meta-analysis. Q. J. Nucl. Med. 1998, 42, 211–221. [Google Scholar] [PubMed]
- Laruelle, M.; Abi-Dargham, A. Dopamine as the wind of the psychotic fire: New evidence from brain imaging studies. J. Psychopharmacol. 1999, 13, 358–371. [Google Scholar] [CrossRef] [PubMed]
- Laruelle, M.; Abi-Dargham, A.; Gil, R.; Kegeles, L.; Innis, R. Increased dopamine transmission in schizophrenia: Relationship to illness phases. Biol. Psychiatry 1999, 46, 56–72. [Google Scholar] [CrossRef]
- Glenthoj, B.Y.; Mackeprang, T.; Svarer, C.; Rasmussen, H.; Pinborg, L.H.; Friberg, L.; Baaré, W.; Hemmingsen, R.; Videbaek, C. Frontal dopamine D(2/3) receptor binding in drug-naive first-episode schizophrenic patients correlates with positive psychotic symptoms and gender. Biol. Psychiatry 2006, 60, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Kestler, L.P.; Walker, E.; Vega, E.M. Dopamine receptors in the brains of schizophrenia patients: A meta-analysis of the findings. Behav. Pharmacol. 2001, 12, 355–371. [Google Scholar] [CrossRef] [PubMed]
- Kellendonk, C.; Simpson, E.H.; Polan, H.J.; Malleret, G.; Vronskaya, S.; Winiger, V.; Moore, H.; Kandel, E.R. Transient and selective overexpression of dopamine D2 receptors in the striatum causes persistent abnormalities in prefrontal cortex functioning. Neuron 2006, 49, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Seeman, P.; Niznik, H.B. Dopamine receptors and transporters in Parkinson’s disease and schizophrenia. FASEB J. 1990, 4, 2737–2744. [Google Scholar] [PubMed]
- Creese, I.; Burt, D.R.; Snyder, S.H. Dopamine receptor binding predicts clinical and pharmacological potencies of antischizophrenic drugs. Science 1976, 192, 481–483. [Google Scholar] [CrossRef] [PubMed]
- Howes, O.D.; Egerton, A.; Allan, V.; McGuire, P.; Stokes, P.; Kapur, S. Mechanisms underlying psychosis and antipsychotic treatment response in schizophrenia: Insights from PET and SPECT imaging. Curr. Pharm. Des. 2009, 15, 2550–2559. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, S.; Duncan, G.E.; Marx, C.E.; Lieberman, J.A. Treatments for schizophrenia: A critical review of pharmacology and mechanisms of action of antipsychotic drugs. Mol. Psychiatry 2005, 10, 79–104. [Google Scholar] [CrossRef] [PubMed]
- Abi-Dargham, A.; Kegeles, L.S.; Zea-Ponce, Y.; Mawlawi, O.; Martinez, D.; Mitropoulou, V.; O’Flynn, K.; Koenigsberg, H.W.; Van Heertum, R.; Cooper, T.; et al. Striatal amphetamine-induced dopamine release in patients with schizotypal personality disorder studied with single photon emission computed tomography and [123I] iodobenzamide. Biol. Psychiatry 2004, 55, 1001–1006. [Google Scholar] [CrossRef] [PubMed]
- Laruelle, M.; Abi-Dargham, A.; van Dyck, C.H.; Gil, R.; D’Souza, C.D.; Erdos, J.; McCance, E.; Rosenblatt, W.; Fingado, C.; Zoghbi, S.S.; et al. Single photon emission computerized tomography imaging of amphetamine-induced dopamine release in drug-free schizophrenic subjects. Proc. Natl. Acad. Sci. USA 1996, 93, 9235–9240. [Google Scholar] [CrossRef] [PubMed]
- Breier, A.; Su, T.P.; Saunders, R.; Carson, R.E.; Kolachana, B.S.; de Bartolomeis, A.; Weinberger, D.R.; Weisenfeld, N.; Malhotra, A.K.; Eckelman, W.C.; et al. Schizophrenia is associated with elevated amphetamine-induced synaptic dopamine concentrations: Evidence from a novel positron emission tomography method. Proc. Natl. Acad. Sci. USA 1997, 94, 2569–2574. [Google Scholar] [CrossRef] [PubMed]
- Abi-Dargham, A. Do we still believe in the dopamine hypothesis? New data bring new evidence. Int. J. Neuropsychopharmacol. 2004, 7 (Suppl. 1), S1–S5. [Google Scholar] [CrossRef] [PubMed]
- Abi-Dargham, A.; Moore, H. Prefrontal DA transmission at D1 receptors and the pathology of schizophrenia. Neuroscientist 2003, 9, 404–416. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, P.; Smith, L.; Farde, L.; Härnryd, C.; Sedvall, G.; Wiesel, F.A. Lack of apparent antipsychotic effect of the D1-dopamine receptor antagonist SCH39166 in acutely ill schizophrenic patients. Psychopharmacol. Berl. 1995, 121, 309–316. [Google Scholar] [CrossRef]
- Okubo, Y.; Suhara, T.; Sudo, Y.; Toru, M. Possible role of dopamine D1 receptors in schizophrenia. Mol. Psychiatry 1997, 2, 291–292. [Google Scholar] [CrossRef] [PubMed]
- Davis, K.L.; Kahn, R.S.; Ko, G.; Davidson, M. Dopamine in schizophrenia: A review and reconceptualization. Am. J. Psychiatry 1991, 148, 1474–1486. [Google Scholar] [PubMed]
- Meyer-Lindenberg, A.; Miletich, R.S.; Kohn, P.D.; Esposito, G.; Carson, R.E.; Quarantelli, M.; Weinberger, D.R.; Berman, K.F. Reduced prefrontal activity predicts exaggerated striatal dopaminergic function in schizophrenia. Nat. Neurosci. 2002, 5, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Sesack, S.R.; Carr, D.B. Selective prefrontal cortex inputs to dopamine cells: Implications for schizophrenia. Physiol. Behav. 2002, 77, 513–517. [Google Scholar] [CrossRef]
- Stahl, S.M. Describing an atypical antipsychotic: Receptor binding and its role in pathophysiology. Prim. Care Companion J. Clin. Psychiatry 2003, 5, 9–13. [Google Scholar]
- Harrison, P.J.; Owen, M.J. Genes for schizophrenia? Recent findings and their pathophysiological implications. Lancet 2003, 361, 417–419. [Google Scholar] [CrossRef]
- Berry, N.; Jobanputra, V.; Pal, H. Molecular genetics of schizophrenia: A critical review. J. Psychiatry Neurosci. 2003, 28, 415–429. [Google Scholar] [PubMed]
- Howes, O.D.; McCutcheon, R.; Owen, M.J.; Murray, R.M. The Role of Genes, Stress, and Dopamine in the Development of Schizophrenia. Biol. Psychiatry 2017, 81, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Ohara, K.; Nakamura, Y.; Xie, D.W.; Ishigaki, T.; Deng, Z.L.; Tani, K.; Zhang, H.Y.; Kondo, N.; Liu, J.C.; Miyasato, K.; et al. Polymorphisms of dopamine D2-like (D2, D3, and D4) receptors in schizophrenia. Biol. Psychiatry 1996, 40, 1209–1217. [Google Scholar] [CrossRef]
- Talkowski, M.E.; Bamne, M.; Mansour, H.; Nimgaonkar, V.L. Dopamine genes and schizophrenia: Case closed or evidence pending? Schizophr. Bull. 2007, 33, 1071–1081. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, M.; Yamanouchi, Y.; Kinoshita, Y.; Kitajima, T.; Yoshimura, R.; Hashimoto, S.; O’Donovan, M.C.; Nakamura, J.; Ozaki, N.; Iwata, N. Variants of dopamine and serotonin candidate genes as predictors of response to risperidone treatment in first-episode schizophrenia. Pharmacogenomics 2008, 9, 1437–1443. [Google Scholar] [CrossRef] [PubMed]
- Lencz, T.; Robinson, D.G.; Xu, K.; Ekholm, J.; Sevy, S.; Gunduz-Bruce, H.; Woerner, M.G.; Kane, J.M.; Goldman, D.; Malhotra, A.K. DRD2 promoter region variation as a predictor of sustained response to antipsychotic medication in first-episode schizophrenia patients. Am. J. Psychiatry 2006, 163, 529–531. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, G.P.; Yao, Z.; Zhang, X.; Sun, J.; Zhang, Z. Pharmacogenetics of treatment in first-episode schizophrenia: D3 and 5-HT2C receptor polymorphisms separately associate with positive and negative symptom response. Eur. Neuropsychopharmacol. 2005, 15, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Zalsman, G.; Frisch, A.; Lev-Ran, S.; Martin, A.; Michaelovsky, E.; Bensason, D.; Gothelf, D.; Nahshoni, E.; Tyano, S.; Weizman, A. DRD4 exon III polymorphism and response to risperidone in Israeli adolescents with schizophrenia: A pilot pharmacogenetic study. Eur. Neuropsychopharmacol. 2003, 13, 183–185. [Google Scholar] [CrossRef]
- Buttarelli, F.R.; Fanciulli, A.; Pellicano, C.; Pontieri, F.E. The dopaminergic system in peripheral blood lymphocytes: From physiology to pharmacology and potential applications to neuropsychiatric disorders. Curr. Neuropharmacol. 2011, 9, 278–288. [Google Scholar] [PubMed]
- Dean, B.; Kulkarni, J.; Copolov, D.L.; Shrikanthan, P.; Malone, V.; Hill, C. Dopamine uptake by platelets from subjects with schizophrenia: A correlation with the delusional state of the patient. Psychiatry Res. 1992, 41, 17–24. [Google Scholar] [CrossRef]
- Liu, L.; Jia, F.; Yuan, G.; Chen, Z.; Yao, J.; Li, H.; Fang, C. Tyrosine hydroxylase, interleukin-1β and tumor necrosis factor-α are overexpressed in peripheral blood mononuclear cells from schizophrenia patients as determined by semi-quantitative analysis. Psychiatry Res. 2010, 176, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Yuan, G.; Cheng, Z.; Zhang, G.; Liu, X.; Zhang, H. Identification of the mRNA expression status of the dopamine D2 receptor and dopamine transporter in peripheral blood lymphocytes of schizophrenia patients. PLoS ONE 2013, 8, e75259. [Google Scholar] [CrossRef] [PubMed]
- Marazziti, D.; Catena Dell’osso, M.; Baroni, S.; Masala, I.; Dell’Osso, B.; Consoli, G.; Giannaccini, G.; Betti, L.; Lucacchini, A. Alterations of the dopamine transporter in resting lymphocytes of patients with different psychotic disorders. Psychiatry Res. 2010, 175, 54–57. [Google Scholar] [CrossRef] [PubMed]
- Arrúe, A.; Dávila, R.; Zumárraga, M.; Basterreche, N.; González-Torres, M.A.; Goienetxea, B.; Zamalloa, M.I.; Anguiano, J.B.; Guimón, J. GABA and homovanillic acid in the plasma of Schizophrenic and bipolar I patients. Neurochem. Res. 2010, 35, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Luykx, J.J.; Bakker, S.C.; Lentjes, E.; Neeleman, M.; Strengman, E.; Mentink, L.; DeYoung, J.; de Jong, S.; Sul, J.H.; Eskin, E.; et al. Genome-wide association study of monoamine metabolite levels in human cerebrospinal fluid. Mol. Psychiatry 2014, 19, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Siever, L.J.; Amin, F.; Coccaro, E.F.; Bernstein, D.; Kavoussi, R.J.; Kalus, O.; Horvath, T.B.; Warne, P.; Davidson, M.; Davis, K.L. Plasma homovanillic acid in schizotypal personality disorder. Am. J. Psychiatry 1991, 148, 1246–1248. [Google Scholar] [PubMed]
- Sumiyoshi, T.; Kurachi, M.; Kurokawa, K.; Yotsutsuji, T.; Uehara, T.; Itoh, H.; Saitoh, O. Plasma homovanillic acid in the prodromal phase of schizophrenia. Biol. Psychiatry 2000, 47, 428–433. [Google Scholar] [CrossRef]
- Baeza, I.; Castro-Fornieles, J.; Deulofeu, R.; de la Serna, E.; Goti, J.; Salvà, J.; Bernardo, M. Plasma homovanillic acid differences in clinical subgroups of first episode schizophrenic patients. Psychiatry Res. 2009, 168, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Koreen, A.R.; Lieberman, J.; Alvir, J.; Mayerhoff, D.; Loebel, A.; Chakos, M.; Amin, F.; Cooper, T. Plasma homovanillic acid levels in first-episode schizophrenia. Psychopathology and treatment response. Arch. Gen. Psychiatry 1994, 51, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Nagaoka, S.; Iwamoto, N.; Arai, H. First-episode neuroleptic free schizophrenics: Concentrations of monoamines and their metabolites in plasma and their correlations with clinical responses to haloperidol treatment. Biol. Psychiatry 1997, 41, 857–864. [Google Scholar] [CrossRef]
- Bondy, B.; Ackenheil, M.; Elbers, R.; Fröhler, M. Binding of 3H-spiperone to human lymphocytes: A biological marker in schizophrenia? Psychiatry Res. 1985, 15, 41–48. [Google Scholar] [CrossRef]
- Zvara, A.; Szekeres, G.; Janka, Z.; Kelemen, J.Z.; Cimmer, C.; Sántha, M.; Puskás, L.G. Over-expression of dopamine D2 receptor and inwardly rectifying potassium channel genes in drug-naive schizophrenic peripheral blood lymphocytes as potential diagnostic markers. Dis. Markers 2005, 21, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Boneberg, E.M.; von Seydlitz, E.; Pröpster, K.; Watzl, H.; Rockstroh, B.; Illges, H. D3 dopamine receptor mRNA is elevated in T cells of schizophrenic patients whereas D4 dopamine receptor mRNA is reduced in CD4+ -T cells. J. Neuroimmunol. 2006, 173, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.N.; Barlas, C.; Saeedi, H.; Mishra, R.K. Effect of loxapine on peripheral dopamine-like and serotonin receptors in patients with schizophrenia. J. Psychiatry Neurosci. 2003, 28, 39–47. [Google Scholar] [PubMed]
- Ilani, T.; Ben-Shachar, D.; Strous, R.D.; Mazor, M.; Sheinkman, A.; Kotler, M.; Fuchs, S. A peripheral marker for schizophrenia: Increased levels of D3 dopamine receptor mRNA in blood lymphocytes. Proc. Natl. Acad. Sci. USA 2001, 98, 625–628. [Google Scholar] [CrossRef] [PubMed]
- Kwak, Y.T.; Koo, M.S.; Choi, C.H.; Sunwoo, I. Change of dopamine receptor mRNA expression in lymphocyte of schizophrenic patients. BMC Med. Genet. 2001, 2, 3. [Google Scholar] [CrossRef]
- Vogel, M.; Pfeifer, S.; Schaub, R.T.; Grabe, H.J.; Barnow, S.; Freyberger, H.J.; Cascorbi, I. Decreased levels of dopamine D3 receptor mRNA in schizophrenic and bipolar patients. Neuropsychobiology 2004, 50, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Bondy, B.; Ackenheil, M.; Birzle, W.; Elbers, R.; Frohler, M. Catecholamines and their receptors in blood: Evidence for alterations in schizophrenia. Biol. Psychiatry 1984, 19, 1377–1393. [Google Scholar] [PubMed]
- Grodzicki, J.; Pardo, M.; Schved, G.; Schlosberg, A.; Fuchs, S.; Kanety, H. Differences in [3H]-spiperone binding to peripheral blood lymphocytes from neuroleptic responsive and nonresponsive schizophrenic patients. Biol. Psychiatry 1990, 27, 1327–1330. [Google Scholar] [CrossRef]
- Egan, M.F.; Goldberg, T.E.; Kolachana, B.S.; Callicott, J.H.; Mazzanti, C.M.; Straub, R.E.; Goldman, D.; Weinberger, D.R. Effect of COMT Val108/158 Met genotype on frontal lobe function and risk for schizophrenia. Proc. Natl. Acad. Sci. USA 2001, 98, 6917–6922. [Google Scholar] [CrossRef] [PubMed]
- Williams, H.J.; Owen, M.J.; O’Donovan, M.C. Is COMT a susceptibility gene for schizophrenia? Schizophr. Bull. 2007, 33, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Owen, M.J.; Williams, N.M.; O’Donovan, M.C. The molecular genetics of schizophrenia: New findings promise new insights. Mol. Psychiatry 2004, 9, 14–27. [Google Scholar] [CrossRef] [PubMed]
- Williams, N.M.; Owen, M.J. Genetic abnormalities of chromosome 22 and the development of psychosis. Curr. Psychiatry Rep. 2004, 6, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Lachman, H.M.; Papolos, D.F.; Saito, T.; Yu, Y.M.; Szumlanski, C.L.; Weinshilboum, R.M. Human catechol-O-methyltransferase pharmacogenetics: Description of a functional polymorphism and its potential application to neuropsychiatric disorders. Pharmacogenetics 1996, 6, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Tunbridge, E.M.; Harrison, P.J.; Weinberger, D.R. Catechol-omethyltransferase, cognition, and psychosis: Val158Met and beyond. Biol. Psychiatry 2006, 60, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Da Silva Alves, F.; Figee, M.; van Amelsvoort, T.; Veltman, D.; de Haan, L. The revised dopamine hypothesis of schizophrenia: Evidence from pharmacological MRI studies with atypical antipsychotic medication. Psychopharmacol. Bull. 2008, 41, 121–132. [Google Scholar] [PubMed]
- Ohmori, O.; Shinkai, T.; Kojima, H.; Terao, T.; Suzuki, T.; Mita, T.; Abe, K. Association study of a functional catechol-O-methyltransferase gene polymorphism in Japanese schizophrenics. Neurosci. Lett. 1998, 243, 109–112. [Google Scholar] [CrossRef]
- Glatt, S.J.; Faraone, S.V.; Tsuang, M.T. Association between a functional catechol O-methyltransferase gene polymorphism and schizophrenia: Meta-analysis of case-control and family-based studies. Am. J. Psychiatry 2003, 160, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Sham, P.C.; Vallada, H.; Xie, T.; Tang, X.; Murray, R.M.; Liu, X.; Collier, D.A. Preferential transmission of the high activity allele of COMT in schizophrenia. Psychiatr. Genet. 1996, 6, 131–133. [Google Scholar] [CrossRef] [PubMed]
- Shifman, S.; Bronstein, M.; Sternfeld, M.; Pisanté-Shalom, A.; Lev-Lehman, E.; Weizman, A.; Reznik, I.; Spivak, B.; Grisaru, N.; Karp, L.; et al. A highly significant association between a COMT haplotype and schizophrenia. Am. J. Hum. Genet. 2002, 71, 1296–1302. [Google Scholar] [CrossRef] [PubMed]
- Strous, R.D.; Bark, N.; Parsia, S.S.; Volavka, J.; Lachman, H.M. Analysis of a functional catechol-O-methyltransferase gene polymorphism in schizophrenia: Evidence for association with aggressive and antisocial behavior. Psychiatry Res. 1997, 69, 71–77. [Google Scholar] [CrossRef]
- Nunokawa, A.; Watanabe, Y.; Muratake, T.; Kaneko, N.; Koizumi, M.; Someya, T. No associations exist between five functional polymorphisms in the catechol-O-methyltransferase gene and schizophrenia in a Japanese population. Neurosci. Res. 2007, 58, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Semwal, P.; Prasad, S.; Varma, P.G.; Bhagwat, A.M.; Deshpande, S.N.; Thelma, B.K. Candidate gene polymorphisms among North Indians and their association with schizophrenia in a case-control study. J. Genet. 2002, 81, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.B.; Zhang, C.S.; Gu, N.F.; Li, X.W.; Sun, W.W.; Wang, H.Y.; Feng, G.Y.; St Clair, D.; He, L. Catechol-O-methyltransferase gene Val/Met functional polymorphism and risk of schizophrenia: A large-scale association study plus meta-analysis. Biol. Psychiatry 2005, 57, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.G.; Joo, Y.; Kim, B.; Chung, S.; Kim, H.L.; Lee, I.; Choi, B.; Kim, C.; Song, K. Association of Ala72Ser polymorphism with COMT enzyme activity and the risk of schizophrenia in Koreans. Hum. Genet. 2005, 116, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Allen, N.C.; Bagade, S.; McQueen, M.B.; Ioannidis, J.P.; Kavvoura, F.K.; Khoury, M.J.; Tanzi, R.E.; Bertram, L. Systematic meta analyses and field synopsis of genetic association studies in schizophrenia: The SzGene database. Nat. Genet. 2008, 40, 827–834. [Google Scholar] [CrossRef] [PubMed]
- Chien, Y.L.; Liu, C.M.; Fann, C.S.; Liu, Y.L.; Hwu, H.G. Association of the 3′ region of COMT with schizophrenia in Taiwan. J. Formos. Med. Assoc. 2009, 108, 301–309. [Google Scholar] [CrossRef]
- Gupta, M.; Bhatnagar, P.; Grover, S.; Kaur, H.; Baghel, R.; Bhasin, Y.; Chauhan, C.; Verma, B.; Manduva, V.; Mukherjee, O.; et al. Association studies of catechol-O-methyltransferase (COMT) gene with schizophrenia and response to antipsychotic treatment. Pharmacogenomics 2009, 10, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Pal, P.; Mihanovic, M.; Molnar, S.; Xi, H.; Sun, G.; Guha, S.; Jeran, N.; Tomljenović, A.; Malnar, A.; Missoni, S.; et al. Association of tagging single nucleotide polymorphisms on 8 candidate genes in dopaminergic pathway with schizophrenia in Croatian population. Croat. Med. J. 2009, 50, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Prata, D.P.; Mechelli, A.; Fu, C.H.; Picchioni, M.; Toulopoulou, T.; Bramon, E.; Walshe, M.; Murray, R.M.; Collier, D.A.; McGuire, P. Epistasis between the DAT 3′ UTR VNTR and the COMT Val158Met SNP on cortical function in healthy subjects and patients with schizophrenia. Proc. Natl. Acad. Sci. USA 2009, 106, 13600–13605. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.Y.; Chen, Q.; Sust, S.; Buckholtz, J.W.; Meyers, J.D.; Egan, M.F.; Mattay, V.S.; Meyer-Lindenberg, A.; Weinberger, D.R.; Callicott, J.H. Epistasis between catechol-O-methyltransferase and type II metabotropic glutamate receptor 3 genes on working memory brain function. Proc. Natl. Acad. Sci. USA 2007, 104, 12536–12541. [Google Scholar] [CrossRef] [PubMed]
- Abdolmaleky, H.M.; Cheng, K.H.; Faraone, S.V.; Wilcox, M.; Glatt, S.J.; Gao, F.; Smith, C.L.; Shafa, R.; Aeali, B.; Carnevale, J.; et al. Hypomethylation of MB-COMT promoter is a major risk factor for schizophrenia and bipolar disorder. Hum. Mol. Genet. 2006, 15, 3132–3145. [Google Scholar] [CrossRef] [PubMed]
- Nishioka, M.; Bundo, M.; Koike, S.; Takizawa, R.; Kakiuchi, C.; Araki, T.; Kasai, K.; Iwamoto, K. Comprehensive DNA methylation analysis of peripheral blood cells derived from patients with first-episode schizophrenia. J. Hum. Genet. 2013, 58, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Nohesara, S.; Ghadirivasfi, M.; Mostafavi, S.; Eskandari, M.R.; Ahmadkhaniha, H.; Thiagalingam, S.; Abdolmaleky, H.M. DNA hypomethylation of MB-COMT promoter in the DNA derived from saliva in schizophrenia and bipolar disorder. J. Psychiatr. Res. 2011, 45, 1432–1438. [Google Scholar] [CrossRef] [PubMed]
- Dingemanse, J.; Jorga, K.M.; Schmitt, M.; Gieschke, R.; Fotteler, B.; Zürcher, G.; Da Prada, M.; van Brummelen, P. Integrated pharmacokinetics and pharmacodynamics of the novel catechol-O-methyltransferase inhibitor tolcapone during first administration to humans. Clin. Pharmacol. Ther. 1995, 57, 508–517. [Google Scholar] [CrossRef]
- Ferreira, J.J.; Almeida, L.; Cunha, L.; Ticmeanu, M.; Rosa, M.M.; Januário, C.; Mitu, C.E.; Coelho, M.; Correia-Guedes, L.; Morgadinho, A.; et al. Effects of nebicapone on levodopa pharmacokinetics, catechol-O-methyltransferase activity, and motor fluctuations in patients with Parkinson disease. Clin. Neuropharmacol. 2008, 31, 2–18. [Google Scholar] [CrossRef] [PubMed]
- Forsberg, M.; Lehtonen, M.; Heikkinen, M.; Savolainen, J.; Jarvinen, T.; Mannisto, P.T. Pharmacokinetics and pharmacodynamics of entacapone and tolcapone after acute and repeated administration: A comparative study in the rat. J. Pharmacol. Exp. Ther. 2003, 304, 498–506. [Google Scholar] [CrossRef] [PubMed]
- Kiss, L.E.; Ferreira, H.S.; Torrão, L.; Bonifácio, M.J.; Palma, P.N.; Soares-da-Silva, P.; Learmonth, D.A. Discovery of a long acting, peripherally selective inhibitor of catechol-O-methyltransferase. J. Med. Chem. 2010, 53, 3396–3411. [Google Scholar] [CrossRef] [PubMed]
- Mannisto, P.T.; Kaakkola, S. Catechol-Omethyltransferase [COMT]: Biochemistry, molecular biology, pharmacology, and clinical efficacy of the new selective COMT inhibitors. Pharmacol. Rev. 1999, 51, 593–628. [Google Scholar] [PubMed]
- Onofrj, M.; Thomas, A.; Iacono, D.; Di Iorio, A.; Bonanni, L. Switch over from tolcapone to entacapone in severe Parkinson’s disease patients. Eur. Neurol. 2001, 46, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Zürcher, G.; Colzi, A.; Da Prada, M. Ro 40-7592: Inhibition of COMT in rat brain and extracerebral tissues. J. Neural Transm. Suppl. 1990, 32, 375–380. [Google Scholar] [PubMed]
- Tunbridge, E.M.; Bannerman, D.M.; Sharp, T.; Harrison, P.J. Catechol-O-methyltransferase inhibition improves set-shifting performance and elevates stimulated dopamine release in the rat prefrontal cortex. J. Neurosci. 2004, 24, 5331–5335. [Google Scholar] [CrossRef] [PubMed]
- Illi, A.; Kampman, O.; Hänninen, K.; Anttila, S.; Mattila, K.M.; Katila, H.; Rontu, R.; Hurme, M.; Lehtimäki, T.; Leinonen, E. Catechol-O-methyltransferase val108/158met genotype and response to antipsychotic medication in schizophrenia. Hum. Psychopharmacol. 2007, 22, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Bertolino, A.; Caforio, G.; Blasi, G.; Rampino, A.; Nardini, M.; Weinberger, D.R.; Dallapiccola, B.; Sinibaldi, L.; Douzgou, S. COMT Val158Met polymorphism predicts negative symptoms response to treatment with olanzapine in schizophrenia. Schizophr. Res. 2007, 95, 253–255. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.Y.; Xu, X.F.; Shi, Z.Y.; Yang, J.Z.; Liu, H.; Xu, H.H. Interaction of catechol-O-methyltransferase (COMT) Val108/158 Met genotype and risperidone treatment in Chinese Han patients with schizophrenia. Psychiatry Res. 2010, 176, 94–95. [Google Scholar] [CrossRef] [PubMed]
- Molero, P.; Ortuno, F.; Zalacain, M.; Patino-Garcia, A. Clinical involvement of catechol-O-methyltransferase polymorphisms in schizophrenia spectrum disorders: Influence on the severity of psychotic symptoms and on the response to neuroleptic treatment. Pharmacogenom. J. 2007, 7, 418–426. [Google Scholar] [CrossRef] [PubMed]
- Weickert, T.W.; Goldberg, T.E.; Mishara, A.; Apud, J.A.; Kolachana, B.S.; Egan, M.F.; Weinberger, D.R. Catechol-O-methyltransferase val108/158met genotype predicts working memory response to antipsychotic medications. Biol. Psychiatry 2004, 56, 677–682. [Google Scholar] [CrossRef] [PubMed]
- Woodward, N.D.; Jayathilake, K.; Meltzer, H.Y. COMT val108/158met genotype, cognitive function, and cognitive improvement with clozapine in schizophrenia. Schizophr. Res. 2007, 90, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Nolan, K.A.; Czobor, P.; Citrome, L.L.; Krakowski, M.; Lachman, H.M.; Kennedy, J.L.; Ni, X.; Lieberman, J.; Chakos, M.; Volavka, J. Catechol-O-methyltransferase and monoamine oxidase-A polymorphisms and treatment response to typical and atypical neuroleptics. J. Clin. Psychopharmacol. 2006, 26, 338–340. [Google Scholar] [CrossRef] [PubMed]
- Pelayo-Terán, J.M.; Pérez-Iglesias, R.; Vázquez-Bourgon, J.; Mata, I.; Carrasco-Marín, E.; Vázquez-Barquero, J.L.; Crespo-Facorro, B. Catechol-O-methyltransferase Val158Met polymorphism and negative symptoms after acute antipsychotic treatment in first-episode non-affective psychosis. Psychiatry Res. 2011, 185, 286–289. [Google Scholar] [CrossRef] [PubMed]
- Yamanouchi, Y.; Iwata, N.; Suzuki, T.; Kitajima, T.; Ikeda, M.; Ozaki, N. Effect of DRD2, 5-HT2A, and COMT genes on antipsychotic response to risperidone. Pharmacogenom. J. 2003, 3, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Hagen, K.; Stovner, L.J.; Skorpen, F.; Pettersen, E.; Zwart, J.A. COMT genotypes and use of antipsychotic medication: Linking population-based prescription database to the HUNT study. Pharmacoepidemiol. Drug Saf. 2008, 17, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Inada, T.; Nakamura, A.; Iijima, Y. Relationship between catechol-O-methyltransferase polymorphism and treatment resistant schizophrenia. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2003, 120, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Fijal, B.A.; Kinon, B.J.; Kapur, S.; Stauffer, V.L.; Conley, R.R.; Jamal, H.H.; Kane, J.M.; Witte, M.M.; Houston, J.P. Candidate-gene association analysis of response to risperidone in African-American and white patients with schizophrenia. Pharmacogenom. J. 2009, 9, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Abi-Dargham, A. Alterations of serotonin transmission in schizophrenia. Int. Rev. Neurobiol. 2007, 78, 133–164. [Google Scholar] [PubMed]
- Meltzer, H.Y.; Li, Z.; Kaneda, Y.; Ichikawa, J. Serotonin receptors: Their key role in drugs to treat schizophrenia. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2003, 27, 1159–1172. [Google Scholar] [CrossRef] [PubMed]
- Kapur, S.; Remington, G. Serotonin-dopamine interaction and its relevance to schizophrenia. Am. J. Psychiatry 1996, 153, 466–476. [Google Scholar] [PubMed]
- Stephan, K.E.; Friston, K.J.; Frith, C.D. Dysconnection in schizophrenia: From abnormal synaptic plasticity to failures of self-monitoring. Schizophr. Bull. 2009, 35, 509–527. [Google Scholar] [CrossRef] [PubMed]
- Dean, B. A predicted cortical serotonergic/cholinergic/GABAergic interface as a site of pathology in schizophrenia. Clin. Exp. Pharmacol. Physiol. 2001, 28, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Boyer, P.; Phillips, J.L.; Rousseau, F.L.; Ilivitsky, S. Hippocampal abnormalities and memory deficits: New evidence of a strong pathophysiological link in schizophrenia. Brain Res. Rev. 2007, 54, 92–112. [Google Scholar] [CrossRef] [PubMed]
- Puig, M.; Gener, T. Serotonin modulation of prefronto-hippocampal rhythms in health and disease. ACS Chem. Neurosci. 2015, 6, 1017–1025. [Google Scholar] [CrossRef] [PubMed]
- Svob Strac, D.; Pivac, N.; Mück-Seler, D. The serotonergic system and cognitive function. Transl. Neurosci. 2016, 7, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Joyce, J.N.; Shane, A.; Lexow, N.; Winokur, A.; Casanova, M.F.; Kleinman, J.E. Serotonin uptake sites and serotonin receptors are altered in the limbic system of schizophrenics. Neuropsychopharmacology 1993, 8, 315–336. [Google Scholar] [CrossRef] [PubMed]
- Laruelle, M.; Abi-Dargham, A.; Casanova, M.F.; Toti, R.; Weinberger, D.R.; Kleinman, J.E. Selective abnormalities of prefrontal serotonergic receptors in schizophrenia: A postmortem study. Arch. Gen. Psychiatry 1993, 50, 810–818. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, S.; Arnone, D.; Cappai, A.; Howes, O. Alterations in the serotonin system in schizophrenia: A systematic review and metaanalysis of postmortem and molecular imaging studies. Neurosci. Biobehav. Rev. 2014, 45, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, J.; Ishii, H.; Bonaccorso, S.; Fowler, W.L.; O’Laughlin, I.A.; Meltzer, H.Y. 5-HT2A and D2 receptor blockade increases cortical DA release via 5-HT1A receptor activation: A possible mechanism of atypical antipsychotic-induced cortical dopamine release. J. Neurochem. 2001, 76, 1521–1531. [Google Scholar] [CrossRef] [PubMed]
- López-Figueroa, A.L.; Norton, C.S.; López-Figueroa, M.O.; Armellini-Dodel, D.; Burke, S.; Akil, H.; López, J.F.; Watson, S.J. Serotonin 5-HT1A, 5-HT1B, and 5-HT2A receptor mRNA expression in subjects with major depression, bipolar disorder, and schizophrenia. Biol. Psychiatry 2004, 55, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Okubo, Y.; Suhara, T.; Suzuki, K.; Kobayashi, K.; Inoue, O.; Terasaki, O.; Someya, Y.; Sassa, T.; Sudo, Y.; Matsushima, E.; et al. Serotonin 5-HT2 receptors in schizophrenic patients studied by positron emission tomography. Life Sci. 2000, 66, 2455–2464. [Google Scholar] [CrossRef]
- Rasmussen, H.; Erritzoe, D.; Andersen, R.; Ebdrup, B.H.; Aggernaes, B.; Oranje, B.; Kalbitzer, J.; Madsen, J.; Pinborg, L.H.; Baaré, W.; et al. Decreased frontal serotonin 2A receptor binding in antipsychotic-naive patients with first-episode schizophrenia. Arch. Gen. Psychiatry 2010, 67, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Abi-Dargham, A.; Laruelle, M.; Lipska, B.; Jaskiw, G.E.; Wong, D.T.; Robertson, D.W.; Weinberger, D.R.; Kleinman, J.E. Serotonin 5-HT3 receptors in schizophrenia: A postmortem study of the amygdala. Brain Res. 1993, 616, 53–57. [Google Scholar] [CrossRef]
- Dean, B.; Tomaskovic-Crook, E.; Opeskin, K.; Keks, N.; Copolov, D. No change in the density of the serotonin1A receptor, the serotonin 4 receptor or the serotonin transporter in the dorsolateral prefrontal cortex from subjects with schizophrenia. Neurochem. Int. 1999, 34, 109–115. [Google Scholar] [CrossRef]
- Lennertz, L.; Wagner, M.; Frommann, I.; Schulze-Rauschenbach, S.; Schuhmacher, A.; Kühn, K.U.; Pukrop, R.; Klosterkötter, J.; Wölwer, W.; Gaebel, W.; et al. A coding variant of the novel serotonin receptor subunit 5-HT3E influences sustained attention in schizophrenia patients. Eur. Neuropsychopharmacol. 2010, 20, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Iwata, N.; Kitamura, Y.; Kitajima, T.; Yamanouchi, Y.; Ikeda, M.; Nishiyama, T.; Kamatani, N.; Ozaki, N. Association of a haplotype in the serotonin 5-HT4 receptor gene (HTR4) with Japanese schizophrenia. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2003, 121B, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Dean, B.; Pavey, G.; Thomas, D.; Scarr, E. Cortical serotonin7, 1D and 1F receptors: Effects of schizophrenia, suicide and antipsychotic drug treatment. Schizophr. Res. 2006, 88, 265–274. [Google Scholar] [CrossRef] [PubMed]
- East, S.Z.; Burnet, P.W.; Kerwin, R.W.; Harrison, P.J. An RT-PCR study of 5-HT6 and 5-HT7 receptor mRNAs in the hippocampal formation and prefrontal cortex in schizophrenia. Schizophr. Res. 2002, 57, 15–26. [Google Scholar] [CrossRef]
- Ikeda, M.; Iwata, N.; Kitajima, T.; Suzuki, T.; Yamanouchi, Y.; Kinoshita, Y.; Ozaki, N. Positive association of the serotonin 5-HT7 receptor gene with schizophrenia in a Japanese population. Neuropsychopharmacology 2006, 31, 866–871. [Google Scholar] [CrossRef] [PubMed]
- Roth, B.L.; Craigo, S.C.; Choudhary, M.S.; Uluer, A.; Monsma, F.J., Jr.; Shen, Y.; Meltzer, H.Y.; Sibley, D.R. Binding of typical and atypical antipsychotic agents to 5-hydroxytryptamine-6 and 5-hydroxytryptamine-7 receptors. J. Pharmacol. Exp. Ther. 1994, 268, 1403–1410. [Google Scholar]
- Ertugrul, A.; Ucar, G.; Basar, K.; Demir, B.; Yabanoglu, S.; Ulug, B. Influence of clozapine on platelet serotonin, Monoamino oxidase and plasma serotonin levels. Psychiatry Res. 2007, 149, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Muck-Seler, D.; Pivac, N.; Jakovljevic, M. Sex differences, season of birth and platelet 5-HT levels in schizophrenic patients. J. Neural Transm. 1999, 106, 337–347. [Google Scholar] [PubMed]
- Muck-Seler, D.; Pivac, N.; Mustapic, M.; Crncevic, Z.; Jakovljevic, M.; Sagud, M. Platelet serotonin and plasma prolactin and cortisol in healthy, depressed and schizophrenic women. Psychiatry Res. 2004, 127, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Van der Heijden, F.M.; Tuinier, S.; Fekkes, D.; Sijben, A.E.; Kahn, R.S.; Verhoeven, W.M. Atypical antipsychotics and the relevance of glutamate and serotonin. Eur. Neuropsychopharmacol. 2004, 14, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Kaneda, Y.; Fujii, A.; Nagamine, I. Platelet serotonin concentrations in medicated schizophrenic patients. Prog. Neuropsychopharmacol. Biol. Psychiatry 2001, 25, 983–992. [Google Scholar] [CrossRef]
- Arranz, M.J.; de Leon, J. Pharmacogenetics and pharmacogenomics of schizophrenia: A review of last decade of research. Mol. Psychiatry 2007, 12, 707–747. [Google Scholar] [CrossRef] [PubMed]
- Baou, M.; Boumba, V.A.; Petrikis, P.; Rallis, G.; Vougiouklakis, T.; Mavreas, V. A review of genetic alterations in the serotonin pathway and their correlation with psychotic diseases and response to atypical antipsychotics. Schizophr. Res. 2016, 170, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.-P.; Malhotra, A.K. Pharmacogenetics and Antipsychotics: Therapeutic Efficacy and Side Effects Prediction. Expert Opin. Drug Metab. Toxicol. 2011, 7, 9–37. [Google Scholar] [CrossRef] [PubMed]
- Abdolmaleky, H.M.; Faraone, S.V.; Glatt, S.J.; Tsuang, M.T. Meta-analysis of association between the T102C polymorphism of the 5HT2a receptor gene and schizophrenia. Schizophr. Res. 2004, 67, 53–62. [Google Scholar] [CrossRef]
- Williams, J.; Spurlock, G.; McGuffin, P.; Mallet, J.; Nöthen, M.M.; Gill, M.; Aschauer, H.; Nylander, P.O.; Macciardi, F.; Owen, M.J. Association between schizophrenia and T102C polymorphism of the 5-hydroxytryptamine type 2a-receptor gene. European Multicentre Association Study of Schizophrenia (EMASS) Group. Lancet 1996, 347, 1294–1296. [Google Scholar] [CrossRef]
- Vázquez-Bourgon, J.; Arranz, M.J.; Mata, I.; Pelayo-Terán, J.M.; Pérez-Iglesias, R.; Medina-González, L.; Carrasco-Marín, E.; Vázquez-Barquero, J.L.; Crespo-Facorro, B. Serotonin transporter polymorphisms and early response to antipsychotic treatment in first episode of psychosis. Psychiatry Res. 2010, 175, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, G.P. Weight gain, antipsychotic drug treatment and pharmacogenomics. Pharmacogenomics 2002, 3, 567–570. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, G.P.; Zhang, Z.; Zhang, X. Polymorphism of the promoter region of the serotonin 5-HT(2C) receptor gene and clozapine-induced weight gain. Am. J. Psychiatry 2003, 160, 677–679. [Google Scholar] [CrossRef] [PubMed]
- Templeman, L.A.; Reynolds, G.P.; Arranz, B.; San, L. Polymorphisms of the 5-HT2C receptor and leptin genes are associated with antipsychotic drug-induced weight gain in Caucasian subjects with a first-episode psychosis. Pharmacogenet. Genom. 2005, 15, 195–200. [Google Scholar] [CrossRef]
- Zhang, J.P.; Lencz, T.; Zhang, R.X.; Nitta, M.; Maayan, L.; John, M.; Robinson, D.G.; Fleischhacker, W.W.; Kahn, R.S.; Ophoff, R.A.; et al. Pharmacogenetic Associations of Antipsychotic Drug-Related Weight Gain: A Systematic Review and Meta-analysis. Schizophr. Bull. 2016, 42, 1418–1437. [Google Scholar] [CrossRef] [PubMed]
- Güzey, C.; Scordo, M.G.; Spina, E.; Landsem, V.M.; Spigset, O. Antipsychotic-induced extrapyramidal symptoms in patients with schizophrenia: Associations with dopamine and serotonin receptor and transporter polymorphisms. Eur. J. Clin. Pharmacol. 2007, 63, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Al Hadithy, A.F.; Ivanova, S.A.; Pechlivanoglou, P.; Semke, A.; Fedorenko, O.; Kornetova, E.; Ryadovaya, L.; Brouwers, J.R.; Wilffert, B.; Bruggeman, R.; et al. Tardive dyskinesia and DRD3, HTR2A and HTR2C gene polymorphisms in Russian psychiatric inpatients from Siberia. Prog. Neuropsychopharmacol. Biol. Psychiatry 2009, 33, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Knol, W.; van Marum, R.J.; Jansen, P.A.; Strengman, E.; Al Hadithy, A.F.; Wilffert, B.; Schobben, A.F.; Ophoff, R.A.; Egberts, T.C. Genetic variation and the risk of haloperidol-related parkinsonism in elderly patients: A candidate gene approach. J. Clin. Psychopharmacol. 2013, 33, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Ghadirivasfi, M.; Nohesara, S.; Ahmadkhaniha, H.R.; Eskandari, M.B.; Mostafavi, S.; Thiagalingam, S. Hypomethylation of the serotonin receptor type 2A gene (HTR2A) at T102C plymorphic site in DNA derived from the sliva of patients with schizophrenia and bipolar disorder. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2011, 156, 536–545. [Google Scholar] [CrossRef] [PubMed]
- Dunlop, J.; Marquis, K.L.; Lim, H.K.; Leung, L.; Kao, J.; Cheesman, C.; Rosenzweig-Lipson, S. Pharmacological profile of the 5-HT(2C) receptor agonist WAY-163909; therapeutic potential in multiple indications. CNS Drug Rev. 2006, 12, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Siuciak, J.A.; Chapin, D.S.; McCarthy, S.A.; Guanowsky, V.; Brown, J.; Chiang, P.; Marala, R.; Patterson, T.; Seymour, P.A.; Swick, A.; et al. CP-809,101, a selective 5-HT2C agonist, shows activity in animal models of antipsychotic activity. Neuropharmacology 2007, 52, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.J.; Kang, W.H.; Li, Q.; Wang, X.Y.; Yao, S.M.; Ma, A.Q. Beneficial effects of ondansetron as an adjunct to haloperidol for chronic, treatment resistant schizophrenia: A double-blind, randomized, placebo-controlled study. Schizophr. Res. 2006, 88, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, E.S.; Neumaier, J.F. 5-HT6 receptors: A novel target for cognitive enhancement. Pharmacol. Ther. 2005, 108, 320–333. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, P.J. Is elevated norepinephrine an etiological factor in some cases of schizophrenia? Psychiatry Res. 2014, 30, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Kemali, D.; Maj, M.; Galderisi, S.; Grazia Ariano, M.; Starace, F. Factors associated with increased noradrenaline levels in schizophrenic patients. Prog. Neuropsychopharmacol. Biol. Psychiatry 1990, 14, 49–59. [Google Scholar] [CrossRef]
- Lechin, F.; van der Dijs, B. Noradrenergic hypothesis of schizophrenia. Prog. Neuropsychopharmacol. Biol. Psychiatry 2005, 29, 777–778. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Ozawa, N.; Shinba, T.; Hoshino, T.; Yoshii, M. Possible noradrenergic dysfunction in schizophrenia. Brain Res. Bull. 1994, 35, 529–543. [Google Scholar] [CrossRef]
- Yamamoto, K.; Hornykiewicz, O. Proposal for a noradrenaline hypothesis of schizophrenia. Prog. Neuropsychopharmacol. Biol. Psychiatry 2004, 28, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Gattaz, W.F.; Waldmeier, P.; Beckmann, H. CSF monoamine metabolites in schizophrenic patients. Acta Psychiatr. Scand. 1982, 66, 350–360. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.H.; Chen, T.Y.; Lin, S.K.; Lung, F.W.; Lin, W.L.; Hu, W.H.; Yeh, E.K. Plasma catecholamine metabolites in schizophrenics: Evidence for the two-subtype concept. Biol. Psychiatry 1990, 27, 510–518. [Google Scholar] [CrossRef]
- Kaneko, M.; Honda, K.; Kanno, T.; Horikoshi, R.; Manome, T.; Watanabe, A.; Kumashiro, H. Plasma free 3-methoxy-4-hydroxyphenylglycol in acute schizophrenics before and after treatment. Neuropsychobiology 1992, 25, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Elman, I.; Goldstein, D.S.; Green, A.I.; Eisenhofer, G.; Folio, C.J.; Holmes, C.S.; Pickar, D.; Breier, A. Effects of risperidone on the peripheral noradrenegic system in patients with schizophrenia: A comparison with clozapine and placebo. Neuropsychopharmacology 2002, 27, 293–300. [Google Scholar] [CrossRef]
- Friedman, J.I.; Adler, D.N.; Davis, K.L. The role of norepinephrine in the pathophysiology of cognitive disorders: Potential applications to the treatment of cognitive dysfunction in schizophrenia and Alzheimer’s disease. Biol. Psychiatry 1999, 46, 1243–1252. [Google Scholar] [CrossRef]
- Wallace, T.L.; Bertrand, D. Neuronal α7 Nicotinic Receptors as a Target for the Treatment of Schizophrenia. Int. Rev. Neurobiol. 2015, 124, 79–111. [Google Scholar] [PubMed]
- Tani, M.; Akashi, N.; Hori, K.; Konishi, K.; Kitajima, Y.; Tomioka, H.; Inamoto, A.; Hirata, A.; Tomita, A.; Koganemaru, T.; et al. Anticholinergic Activity and Schizophrenia. Neurodegener. Dis. 2015, 15, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Mexal, S.; Berger, R.; Logel, J.; Ross, R.G.; Freedman, R.; Leonard, S. Differential regulation of α7 nicotinic receptor gene (CHRNA7) expression in schizophrenic smokers. J. Mol. Neurosci. 2010, 40, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Perl, O.; Ilani, T.; Strous, R.D.; Lapidus, R.; Fuchs, S. The α7 nicotinic acetylcholine receptor in schizophrenia: Decreased mRNA levels in peripheral blood lymphocytes. FASEB J. 2003, 17, 1948–1950. [Google Scholar] [PubMed]
- Freedman, R.; Hall, M.; Adler, L.E.; Leonard, S. Evidence in postmortem brain tissue for decreased numbers of hippocampal nicotinic receptors in schizophrenia. Biol. Psychiatry 1995, 38, 22–33. [Google Scholar] [CrossRef]
- Court, J.; Spurden, D.; Lloyd, S.; McKeith, I.; Ballard, C.; Cairns, N.; Kerwin, R.; Perry, R.; Perry, E. Neuronal nicotinic receptors in dementia with Lewy bodies and schizophrenia: Alpha-bungarotoxin and nicotine binding in the thalamus. J. Neurochem. 1999, 73, 1590–1597. [Google Scholar] [CrossRef] [PubMed]
- Guan, Z.Z.; Zhang, X.; Blennow, K.; Nordberg, A. Decreased protein level of nicotinic receptor α7 subunit in the frontal cortex from schizophrenic brain. Neuroreport 1999, 10, 1779–1782. [Google Scholar] [CrossRef] [PubMed]
- Freedman, R.; Olincy, A.; Ross, R.G.; Waldo, M.C.; Stevens, K.E.; Adler, L.E.; Leonard, S. The genetics of sensory gating deficits in schizophrenia. Curr. Psychiatry Rep. 2003, 5, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, M.S.; Christensen, D.Z.; Hansen, H.H.; Redrobe, J.P.; Mikkelsen, J.D. α(7) Nicotinic acetylcholine receptor activation prevents behavioral and molecular changes induced by repeated phencyclidine treatment. Neuropharmacology 2009, 56, 1001–1009. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, M.S.; Hansen, H.H.; Timmerman, D.B.; Mikkelsen, J.D. Cognitive improvement by activation of α7 nicotinic acetylcholine receptors: From animal models to human pathophysiology. Curr. Pharm. Des. 2010, 16, 323–343. [Google Scholar] [CrossRef] [PubMed]
- Carruthers, S.P.; Gurvich, C.T.; Rossell, S.L. The muscarinic system, cognition and schizophrenia. Neurosci. Biobehav. Rev. 2015, 55, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Crook, J.M.; Tomaskovic-Crook, E.; Copolov, D.L.; Dean, B. Decreased muscarinic receptor binding in subjects with schizophrenia: A study of the human hippocampal formation. Biol. Psychiatry 2000, 48, 381–388. [Google Scholar] [CrossRef]
- Mancama, D.; Arranz, M.J.; Landau, S.; Kerwin, R. Reduced expression of the muscarinic 1 receptor cortical subtype in schizophrenia. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2003, 119, 2–6. [Google Scholar] [CrossRef] [PubMed]
- Melancon, B.J.; Tarr, J.C.; Panarese, J.D.; Wood, M.R.; Lindsley, C.W. Allosteric modulation of the M1 muscarinic acetylcholine receptor: Improving cognition and a potential treatment for schizophrenia and Alzheimer’s disease. Drug Discov. Today 2013, 18, 1185–1199. [Google Scholar] [CrossRef] [PubMed]
- Cherlyn, S.Y.; Woon, P.S.; Liu, J.J.; Ong, W.Y.; Tsai, G.C.; Sim, K. Genetic association studies of glutamate, GABA and related genes in schizophrenia and bipolar disorder: A decade of advance. Neurosci. Biobehav. Rev. 2010, 34, 958–977. [Google Scholar] [CrossRef] [PubMed]
- Harrison, P.J.; West, V.A. Six degrees of separation: On the prior probability that schizophrenia susceptibility genes converge on synapses, glutamate and NMDA receptors. Mol. Psychiatry 2006, 11, 981–983. [Google Scholar] [CrossRef] [PubMed]
- Neeman, G.; Blanaru, M.; Bloch, B.; Kremer, I.; Ermilov, M.; Javitt, D.C.; Heresco-Levy, U. Relation of plasma glycine, serine, and homocysteine levels to schizophrenia symptoms and medication type. Am. J. Psychiatry 2005, 162, 1738–1740. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, L.K.; Linderholm, K.R.; Engberg, G.; Paulson, L.; Blennow, K.; Lindström, L.H.; Nordin, C.; Karanti, A.; Persson, P.; Erhardt, S. Elevated levels of kynurenic acid in the cerebrospinal fluid of male patients with schizophrenia. Schizophr. Res. 2005, 80, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Steullet, P.; Neijt, H.C.; Cuenod, M.; Do, K.Q. Synaptic plasticity impairment and hypofunction of NMDA receptors induced by glutathione deficit: Relevance to schizophrenia. Neuroscience 2006, 137, 807–819. [Google Scholar] [CrossRef] [PubMed]
- Egerton, A.; Reid, L.; McGregor, S.; Cochran, S.M.; Morris, B.J.; Pratt, J.A. Subchronic and chronic PCP treatment produces temporally distinct deficits in attentional set shifting and prepulse inhibition in rats. Psychopharmacol. Berl. 2008, 198, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Javitt, D.C.; Zukin, S.R. Recent advances in the phencyclidine model of schizophrenia. Am. J. Psychiatry 1991, 148, 1301–1308. [Google Scholar] [PubMed]
- Malhotra, A.K.; Pinals, D.A.; Weingartner, H.; Sirocco, K.; Missar, C.D.; Pickar, D.; Breier, A. NMDA receptor function and human cognition: The effects of ketamine in healthy volunteers. Neuropsychopharmacology 1996, 14, 301–307. [Google Scholar] [CrossRef]
- Kegeles, L.S.; Abi-Dargham, A.; Zea-Ponce, Y.; Rodenhiser-Hill, J.; Mann, J.J.; Van Heertum, R.L.; Cooper, T.B.; Carlsson, A.; Laruelle, M. Modulation of amphetamine-induced striatal dopamine release by ketamine in humans: Implications for schizophrenia. Biol. Psychiatry 2000, 48, 627–640. [Google Scholar] [CrossRef]
- Javitt, D.C.; Balla, A.; Burch, S.; Suckow, R.; Xie, S.; Sershen, H. Reversal of phencyclidine-induced dopaminergic dysregulation by N-methyl-Daspartate receptor/glycine-site agonists. Neuropsychopharmacology 2004, 29, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Akbarian, S.; Sucher, N.J.; Bradley, D.; Tafazzoli, A.; Trinh, D.; Hetrick, W.P.; Potkin, S.G.; Sandman, C.A.; Bunney, W.E., Jr.; Jones, E.G. Selective alterations in gene expression for NMDA receptor subunits in prefrontal cortex of schizophrenics. J. Neurosci. 1996, 16, 19–30. [Google Scholar] [PubMed]
- Coyle, J.T.; Tsai, G. NMDA receptor function, neuroplasticity, and the pathophysiology of schizophrenia. Int. Rev. Neurobiol. 2004, 59, 491–515. [Google Scholar] [PubMed]
- Kornhuber, J.; Mack-Burkhardt, F.; Riederer, P.; Hebenstreit, G.F.; Reynolds, G.P.; Andrews, H.B.; Beckmann, H. [3H]MK-801 binding sites in postmortem brain regions of schizophrenic patients. J. Neural Transm. 1989, 77, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Berk, M.; Plein, H.; Csizmadia, T. Supersensitive platelet glutamate receptors as a possible peripheral marker in schizophrenia. Int. Clin. Psychopharmacol. 1999, 14, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Javitt, D.C.; Spencer, K.M.; Thaker, G.K.; Winterer, G.; Hajós, M. Neurophysiological biomarkers for drug development in schizophrenia. Nat. Rev. Drug Discov. 2008, 7, 68–83. [Google Scholar] [CrossRef] [PubMed]
- Tsai, G.; Passani, L.A.; Slusher, B.S.; Carter, R.; Baer, L.; Kleinman, J.E.; Coyle, J.T. Abnormal excitatory neurotransmitter metabolism in schizophrenic brains. Arch. Gen. Psychiatry 1995, 52, 829–836. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Kornhuber, H.H.; Schmid-Burgk, W.; Holzmüller, B. Low cerebrospinal fluid glutamate in schizophrenic patients and a new hypothesis on schizophrenia. Neurosci. Lett. 1980, 20, 379–382. [Google Scholar] [CrossRef]
- Sumiyoshi, T.; Jin, D.; Jayathilake, K.; Lee, M.; Meltzer, H.Y. Prediction of the ability of clozapine to treat negative symptoms from plasma glycine and serine levels in schizophrenia. Int. J. Neuropsychopharmacol. 2005, 8, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K.; Engberg, G.; Shimizu, E.; Nordin, C.; Lindström, L.H.; Iyo, M. Reduced d-serine to total serine ratio in the cerebrospinal fluid of drug naive schizophrenic patients. Prog. Neuropsychopharmacol. Biol. Psychiatry 2005, 29, 767–769. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Viggiano, A.; Monda, M.; De Luca, V. Peripheral glutamate levels in schizophrenia: Evidence from a meta-analysis. Neuropsychobiology 2014, 70, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Uzbay, T.; Goktalay, G.; Kayir, H.; Eker, S.S.; Sarandol, A.; Oral, S.; Buyukuysal, L.; Ulusoy, G.; Kirli, S. Increased plasma agmatine levels in patients with schizophrenia. J. Psychiatr. Res. 2013, 47, 1054–1060. [Google Scholar] [CrossRef] [PubMed]
- Bartha, R.; Williamson, P.C.; Drost, D.J.; Malla, A.; Carr, T.J.; Cortese, L.; Canaran, G.; Rylett, R.J.; Neufeld, R.W. Measurement of glutamate and glutamine in the medial prefrontal cortex of never-treated schizophrenic patients and healthy controls by proton magnetic resonance spectroscopy. Arch. Gen. Psychiatry 1997, 54, 959–965. [Google Scholar] [CrossRef] [PubMed]
- Ohnuma, T.; Sakai, Y.; Maeshima, H.; Hatano, T.; Hanzawa, R.; Abe, S.; Kida, S.; Shibata, N.; Suzuki, T.; Arai, H. Changes in plasma glycine, l-serine, and d-serine levels in patients with schizophrenia as their clinical symptoms improve: Results from the Juntendo University Schizophrenia Projects (JUSP). Prog. Neuropsychopharmacol. Biol. Psychiatry 2008, 32, 1905–1912. [Google Scholar] [CrossRef] [PubMed]
- Yamamori, H.; Hashimoto, R.; Fujita, Y.; Numata, S.; Yasuda, Y.; Fujimoto, M.; Ohi, K.; Umeda-Yano, S.; Ito, A.; Ohmori, T.; et al. Changes in plasma d-serine, l-serine, and glycine levels in treatment-resistant schizophrenia before and after clozapine treatment. Neurosci. Lett. 2014, 582, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Ohnuma, T.; Sakai, Y.; Maeshima, H.; Higa, M.; Hanzawa, R.; Kitazawa, M.; Hotta, Y.; Katsuta, N.; Takebayashi, Y.; Shibata, N.; et al. No correlation between plasma NMDA-related glutamatergic amino acid levels and cognitive function in medicated patients with schizophrenia. Int. J. Psychiatry Med. 2012, 44, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Heresco-Levy, U.; Javitt, D.C.; Ebstein, R.; Vass, A.; Lichtenberg, P.; Bar, G.; Catinari, S.; Ermilov, M. d-serine efficacy as add-on pharmacotherapy to risperidone and olanzapine for treatment-refractory schizophrenia. Biol. Psychiatry 2005, 57, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Lane, H.Y.; Chang, Y.C.; Liu, Y.C.; Chiu, C.C.; Tsai, G.E. Sarcosine or d-serine add-on treatment for acute exacerbation of schizophrenia: A randomized, double-blind, placebo-controlled study. Arch. Gen. Psychiatry 2005, 62, 1196–1204. [Google Scholar] [CrossRef] [PubMed]
- Tuominen, H.J.; Tiihonen, J.; Wahlbeck, K. Glutamatergic drugs for schizophrenia. Cochrane Database Syst. Rev. 2006, 2, CD003730. [Google Scholar]
- Chaki, S.; Hikichi, H. Targeting of metabotropic glutamate receptors for the treatment of schizophrenia. Curr. Pharm. Des. 2011, 17, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Conn, P.J.; Lindsley, C.W.; Jones, C.K. Activation of metabotropic glutamate receptors as a novel approach for the treatment of schizophrenia. Trends Pharmacol. Sci. 2009, 30, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Patil, S.T.; Zhang, L.; Martenyi, F.; Lowe, S.L.; Jackson, K.A.; Andreev, B.V.; Avedisova, A.S.; Bardenstein, L.M.; Gurovich, I.Y.; Morozova, M.A.; et al. Activation of mGlu2/3 receptors as a new approach to treat schizophrenia: A randomized Phase 2 clinical trial. Nat. Med. 2007, 13, 1102–1107. [Google Scholar] [CrossRef] [PubMed]
- Akbarian, S.; Kim, J.J.; Potkin, S.G.; Hagman, J.O.; Tafazzoli, A.; Bunney, W.E.; Jones, E.G. Gene expression for Glutamic acid decarboxylase is reduced without loss of neurons in prefrontal cortex of schizophrenics. Arch. Gen. Psychiatry 1995, 52, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Benes, F.M.; Berretta, S. GABAergic interneurons: Implications for understanding schizophrenia and bipolar disorder. Neuropsychopharmacology 2001, 25, 1–27. [Google Scholar] [CrossRef]
- Benes, F.M.; Sorensen, I.; Vincent, S.L.; Bird, E.D.; Sathi, M. Increased density of glutamate-immunoreactive vertical processes in superficial laminae in cingulate cortex of schizophrenic brain. Cereb. Cortex 1992, 2, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Burgos, G.; Hashimoto, T.; Lewis, D.A. Alterations of cortical GABA neurons and network oscillations in schizophrenia. Curr. Psychiatry Rep. 2010, 12, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Guidotti, A.; Auta, J.; Davis, J.M.; Dong, E.; Grayson, D.R.; Veldic, M.; Zhang, X.; Costa, E. GABAergic dysfunction in schizophrenia: New treatment strategies on the horizon. Psychopharmacol. Berl. 2005, 180, 191–205. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.A.; Hashimoto, T.; Volk, D.W. Cortical inhibitory neurons and schizophrenia. Nat. Rev. Neurosci. 2005, 6, 312–324. [Google Scholar] [CrossRef] [PubMed]
- Sequeira, P.A.; Martin, M.V.; Vawter, M.P. The first decade and beyond of transcriptional profiling in schizophrenia. Neurobiol. Dis. 2012, 45, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Costa, E.; Chen, Y.; Dong, E.; Grayson, D.R.; Kundakovic, M.; Maloku, E.; Ruzicka, W.; Satta, R.; Veldic, M.; Zhubi, A.; et al. GABAergic promoter hypermethylation as a model to study the neurochemistry of schizophrenia vulnerability. Expert Rev. Neurother. 2009, 9, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Grayson, D.R.; Guidotti, A. The dynamics of DNA methylation in schizophrenia and related psychiatric disorders. Neuropsychopharmacology 2013, 38, 138–166. [Google Scholar] [CrossRef] [PubMed]
- Guidotti, A.; Auta, J.; Chen, Y.; Davis, J.M.; Dong, E.; Gavin, D.P.; Grayson, D.R.; Matrisciano, F.; Pinna, G.; Satta, R.; et al. Epigenetic GABAergic targets in schizophrenia and bipolar disorder. Neuropharmacology 2011, 60, 1007–1016. [Google Scholar] [CrossRef] [PubMed]
- Veldic, M.; Guidotti, A.; Maloku, E.; Davis, J.M.; Costa, E. In psychosis, cortical interneurons overexpress DNA-methyltransferase I. Proc. Natl. Acad. Sci. USA 2005, 102, 2152–2157. [Google Scholar] [CrossRef] [PubMed]
- Veldic, M.; Kadriu, B.; Maloku, E.; Agis-Balboa, R.C.; Guidotti, A.; Davis, J.M.; Costa, E. Epigenetic mechanisms expressed in basal ganglia GABAergic neurons differentiate schizophrenia from bipolar disorder. Schizophr. Res. 2007, 91, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Ruzicka, W.B.; Zhubi, A.; Veldic, M.; Grayson, D.R.; Costa, E.; Guidotti, A. Selective epigenetic alteration of layer I GABAergic neurons isolated from prefrontal cortex of schizophrenia patients using laser-assisted microdissection. Mol. Psychiatry 2007, 4, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Arion, D.; Unger, T.; Maldonado-Avilés, J.G.; Morris, H.M.; Volk, D.W.; Mirnics, K.; Lewis, D.A. Alterations in GABA-related transcriptome in the dorsolateral prefrontal cortex of subjects with schizophrenia. Mol. Psychiatry 2008, 13, 147–161. [Google Scholar] [CrossRef] [PubMed]
- Gavin, D.P.; Akbarian, S. Epigenetic and post-transcriptional dysregulation of gene expression in schizophrenia and related disease. Neurobiol. Dis. 2012, 46, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Zhubi, A.; Veldic, M.; Puri, N.V.; Kadriu, B.; Caruncho, H.; Loza, I.; Sershen, H.; Lajtha, A.; Smith, R.C.; Guidotti, A.; et al. An upregulation of DNA-methyltransferase 1 and 3A expressed in telencephalic GABAergic neurons of schizophrenia patients is also detected in peripheral blood lymphocytes. Schizophr. Res. 2009, 111, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Akbarian, S.; Huang, H.S. Molecular and cellular mechanisms of altered GAD1/GAD67 expression in schizophrenia and related disorders. Brain Res. Rev. 2006, 52, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Dracheva, S.; Elhakem, S.L.; McGurk, S.R.; Davis, K.L.; Haroutunian, V. GAD67 and GAD65 mRNA and protein expression in cerebrocortical regions of elderly patients with schizophrenia. J. Neurosci. Res. 2004, 76, 581–592. [Google Scholar] [CrossRef] [PubMed]
- Guidotti, A.; Auta, J.; Davis, J.M.; Di Giorgi-Gerevini, V.; Dwivedi, Y.; Grayson, D.R.; Impagnatiello, F.; Pandey, G.; Pesold, C.; Sharma, R.; et al. Decrease in reelin and glutamic acid decarboxylase67 (GAD67) expression in schizophrenia and bipolar disorder: A postmortem brain study. Arch. Gen. Psychiatry 2000, 57, 1061–1069. [Google Scholar] [CrossRef] [PubMed]
- Mirnics, K.; Middleton, F.A.; Marquez, A.; Lewis, D.A.; Levitt, P. Molecular characterization of schizophrenia viewed by microarray analysis of gene expression in prefrontal cortex. Neuron 2000, 28, 53–67. [Google Scholar] [CrossRef]
- Straub, R.E.; Lipska, B.K.; Egan, M.F.; Goldberg, T.E.; Callicott, J.H.; Mayhew, M.B.; Vakkalanka, R.K.; Kolachana, B.S.; Kleinman, J.E.; Weinberger, D.R. Allelic variation in GAD1 (GAD67) is associated with schizophrenia and influences cortical function and gene expression. Mol. Psychiatry 2007, 12, 854–869. [Google Scholar] [CrossRef] [PubMed]
- Duncan, C.E.; Webster, M.J.; Rothmond, D.A.; Bahn, S.; Elashoff, M.; Shannon Weickert, C. Prefrontal GABA(A) receptor alpha-subunit expression in normal postnatal human development and schizophrenia. J. Psychiatr. Res. 2010, 44, 673–681. [Google Scholar] [CrossRef] [PubMed]
- Lo, W.S.; Harano, M.; Gawlik, M.; Yu, Z.; Chen, J.; Pun, F.W.; Tong, K.L.; Zhao, C.; Ng, S.K.; Tsang, S.Y.; et al. GABRB2 association with schizophrenia: Commonalities and differences between ethnic groups and clinical subtypes. Biol. Psychiatry 2007, 61, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Ritsner, M.; Modai, I.; Gibel, A.; Leschiner, S.; Silver, H.; Tsinovoy, G.; Weizman, A.; Gavish, M. Decreased platelet peripheral-type benzodiazepine receptors in persistently violent schizophrenia patients. J. Psychiatr. Res. 2003, 37, 549–556. [Google Scholar] [CrossRef]
- Ramsey, J.M.; Cooper, J.D.; Penninx, B.W.J.H.; Bahn, S. Variation in serum biomarkers with sex and female hormonal status: Implications for clinical tests. Sci. Rep. 2016, 6, 26947. [Google Scholar] [CrossRef] [PubMed]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 4th ed.; Text Revision (DSM-IV TR); American Psychiatric Association: Arlington County, WV, USA, 2000; Available online: http://www.psych.org/MainMenu/Research/DSMIV.aspx (accessed on 23 March 2017).
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Publishing Inc.: Washington, DC, USA, 2013; Available online: https://psicovalero.files.wordpress.com/2014/06/dsm-v-manual-diagnc3b3stico-y-estadc3adstico-de-los-trastornos-mentales.pdf (accessed on 23 March 2017).
- Rajiv Tandon, R.; Gaebel, W.; Barch, D.M.; Bustillo, J.; Gur, R.-E.; Heckers, S.; Malaspina, D.; Owen, M.J.; Schultz, S.; Tsuang, M.; et al. Definition and description of schizophrenia in the DSM-5. Schizophr. Res. 2013, 150, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, E.; Guest, P.C.; Rahmoune, H.; Harris, L.W.; Wang, L.; Leweke, F.M.; Rothermundt, M.; Bogerts, B.; Koethe, D.; Kranaster, L.; et al. Identification of a biological signature for schizophrenia in serum. Mol. Psychiatry 2012, 17, 494–502. [Google Scholar] [CrossRef] [PubMed]
- Pillai, A.; Buckley, P.F. Reliable biomarkers and predictors of schizophrenia and its treatment. Psychiatr. Clin. N. Am. 2012, 35, 645–659. [Google Scholar] [CrossRef] [PubMed]
- Fond, G.; d’Albis, M.A.; Jamain, S.; Tamouza, R.; Arango, C.; Fleischhacker, W.W.; Glenthøj, B.; Leweke, M.; Lewis, S.; McGuire, P.; et al. The promise of biological markers for treatment response in first-episode psychosis: A systematic review. Schizophr. Bull. 2015, 41, 559–573. [Google Scholar] [CrossRef] [PubMed]
- Stober, G.; Ben-Shachar, D.; Cardon, M.; Falkai, P.; Fonteh, A.N.; Gawlik, M.; Glenthoj, B.Y.; Grunblatt, E.; Jablensky, A.; Kim, Y.K.; et al. Schizophrenia: From the brain to peripheral markers. A consensus paper of the WFSBP task force on biological markers. World J. Biol. Psychiatry 2009, 10, 127–155. [Google Scholar] [CrossRef] [PubMed]
- Landek-Salgado, M.A.; Faust, T.E.; Sawa, A. Molecular substrates of schizophrenia: Homeostatic signaling to connectivity. Mol. Psychiatry 2016, 21, 10–28. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perkovic, M.N.; Erjavec, G.N.; Strac, D.S.; Uzun, S.; Kozumplik, O.; Pivac, N. Theranostic Biomarkers for Schizophrenia. Int. J. Mol. Sci. 2017, 18, 733. https://doi.org/10.3390/ijms18040733
Perkovic MN, Erjavec GN, Strac DS, Uzun S, Kozumplik O, Pivac N. Theranostic Biomarkers for Schizophrenia. International Journal of Molecular Sciences. 2017; 18(4):733. https://doi.org/10.3390/ijms18040733
Chicago/Turabian StylePerkovic, Matea Nikolac, Gordana Nedic Erjavec, Dubravka Svob Strac, Suzana Uzun, Oliver Kozumplik, and Nela Pivac. 2017. "Theranostic Biomarkers for Schizophrenia" International Journal of Molecular Sciences 18, no. 4: 733. https://doi.org/10.3390/ijms18040733
APA StylePerkovic, M. N., Erjavec, G. N., Strac, D. S., Uzun, S., Kozumplik, O., & Pivac, N. (2017). Theranostic Biomarkers for Schizophrenia. International Journal of Molecular Sciences, 18(4), 733. https://doi.org/10.3390/ijms18040733