
In Silico and In Vitro Analysis of Interaction between Ximelagatran and Human Leukocyte Antigen (HLA)-DRB1*07:01

Abstract
:1. Introduction
2. Results
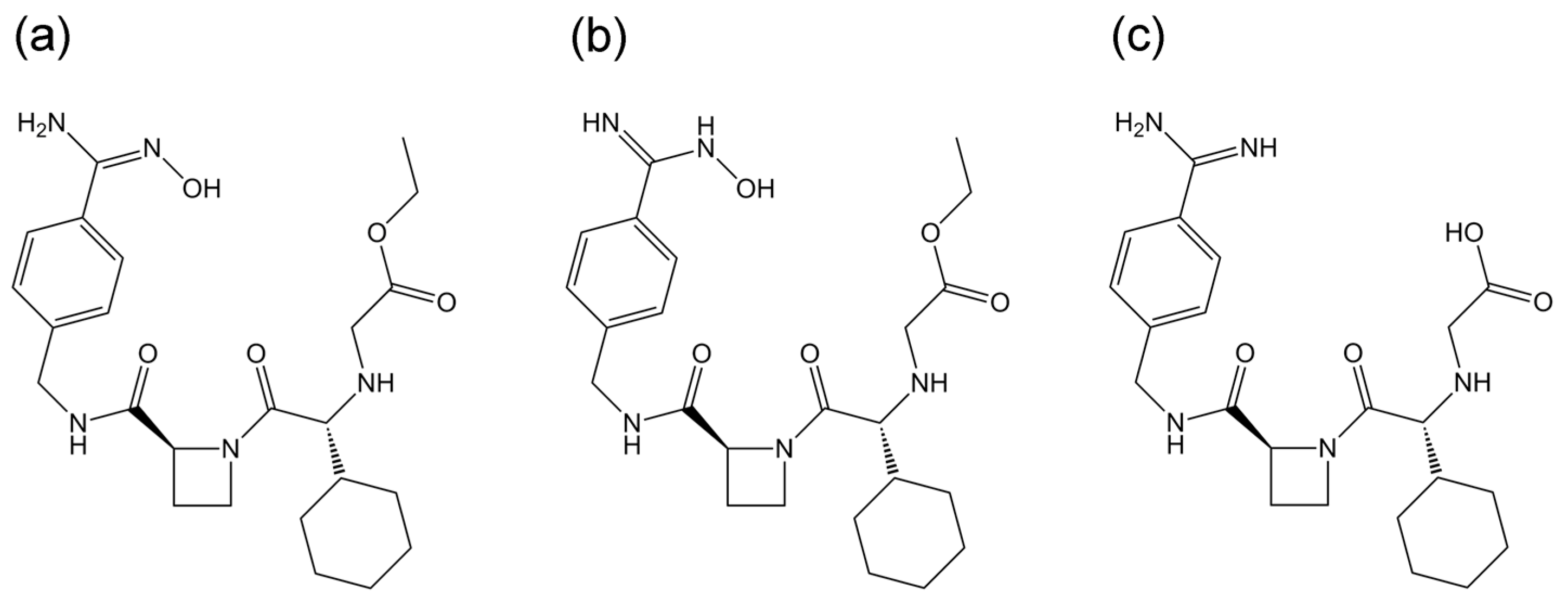
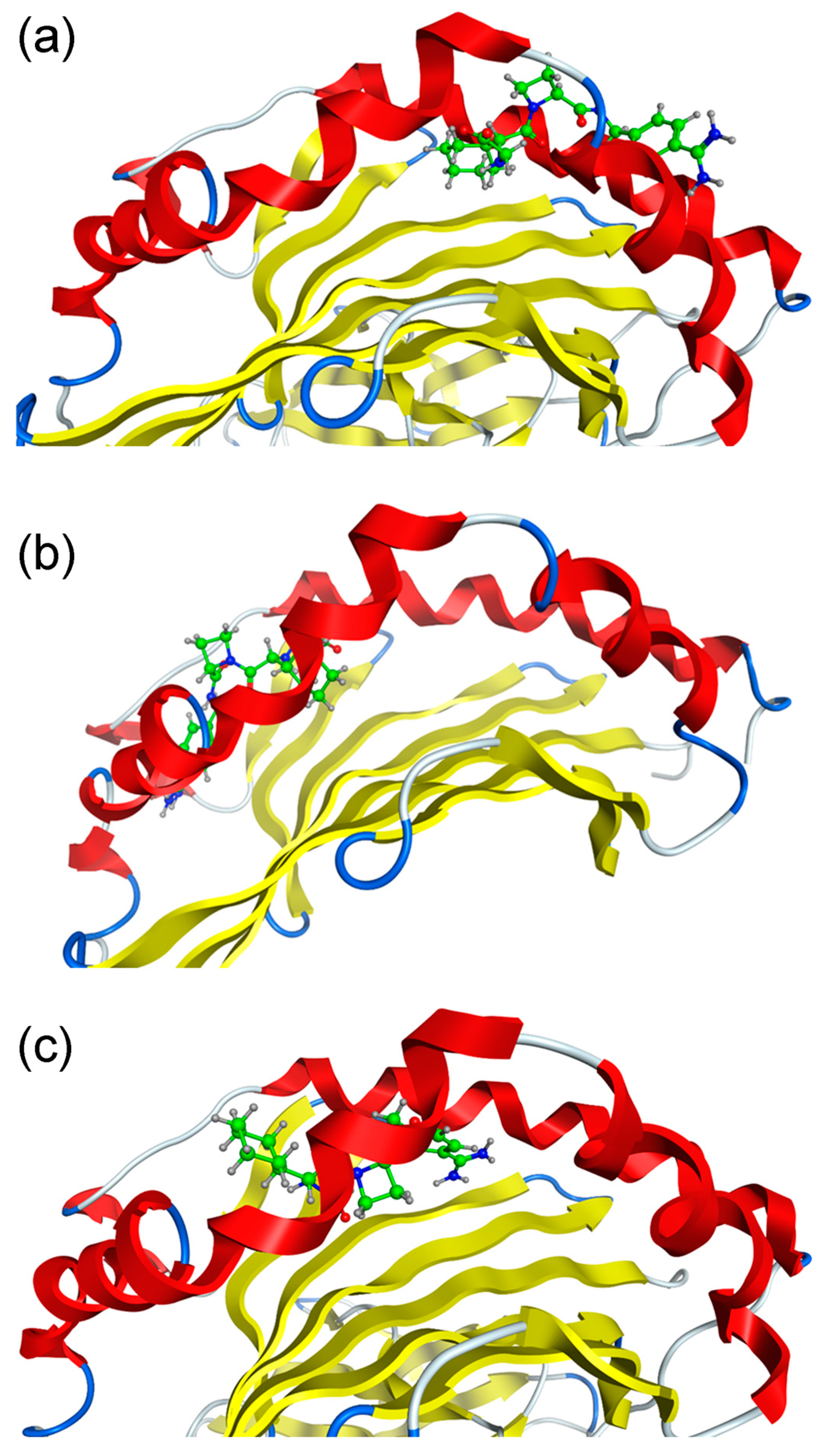
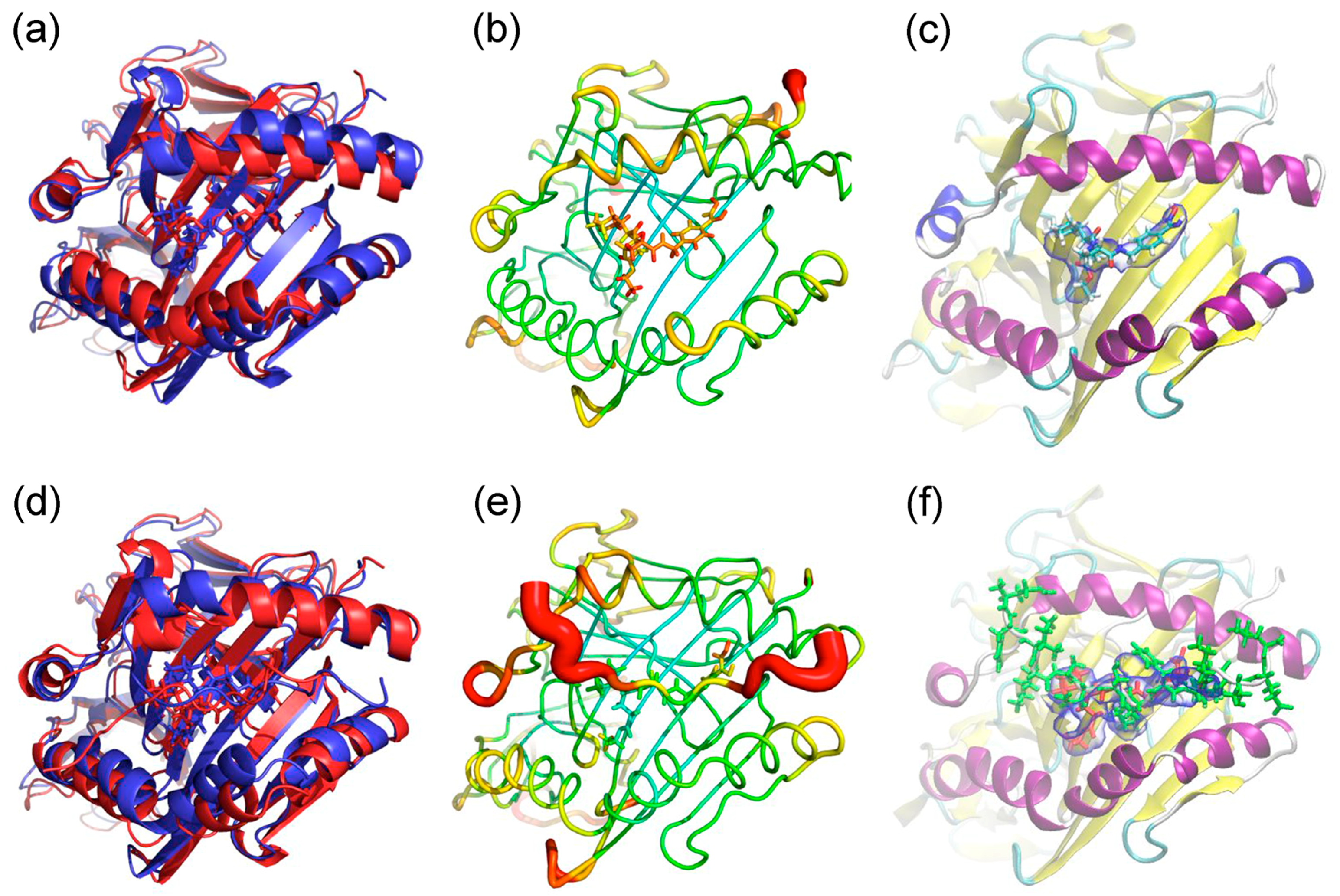
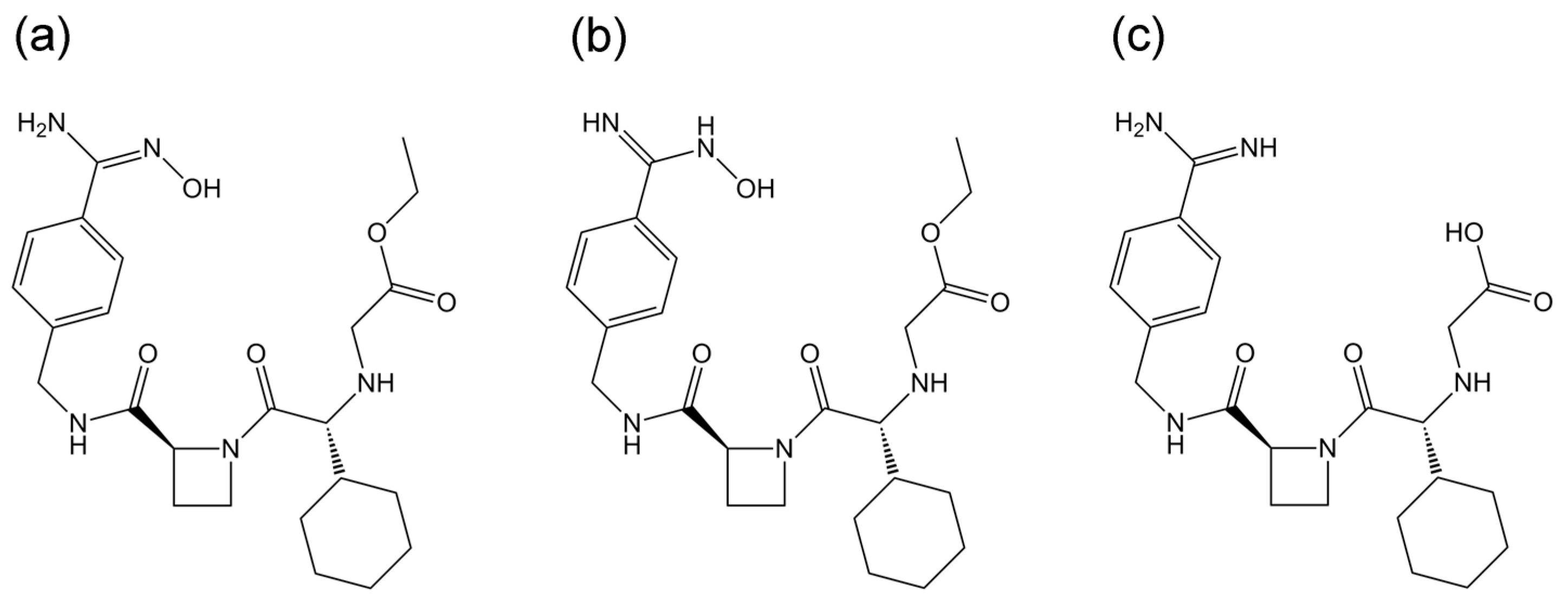
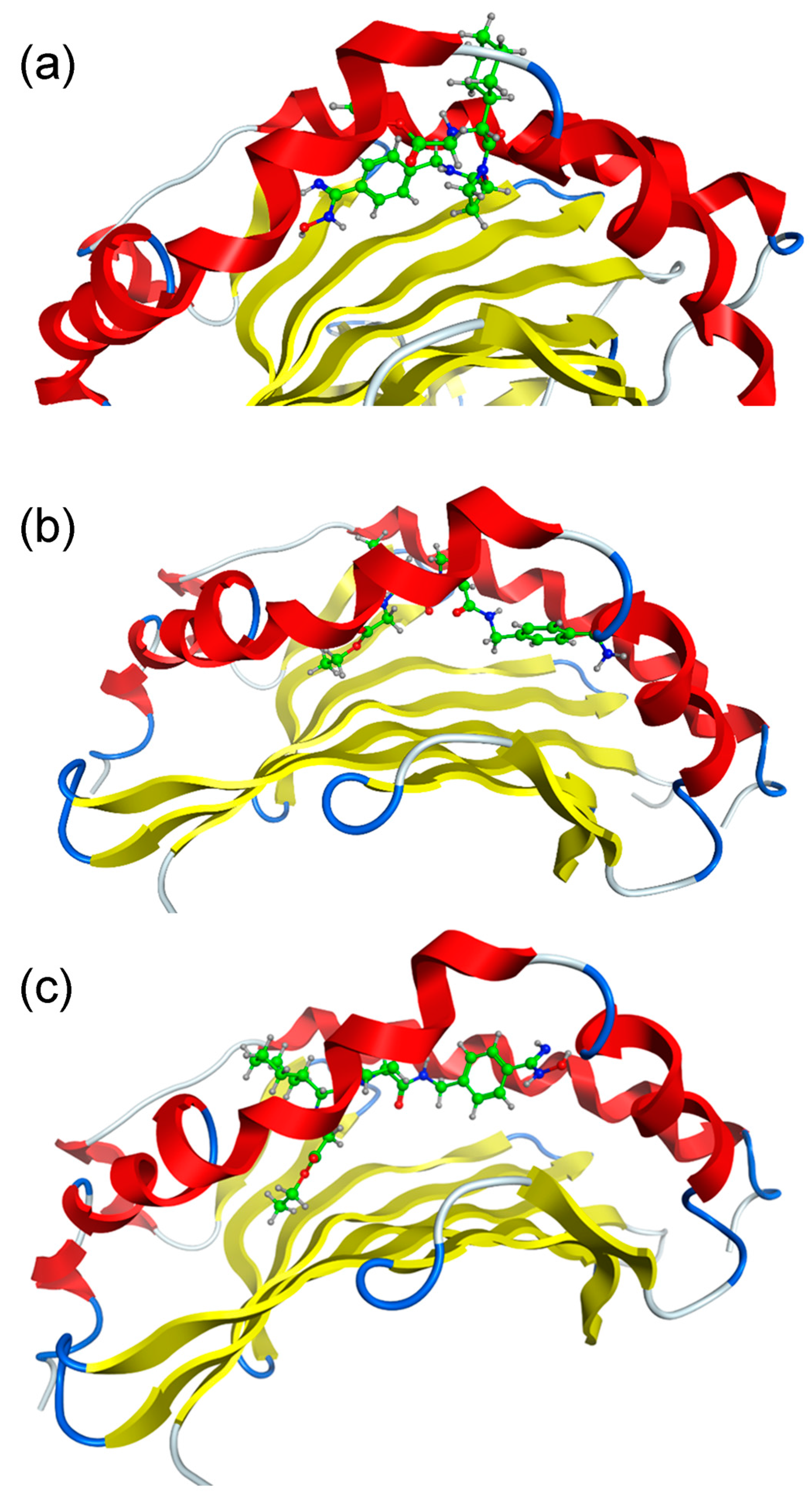
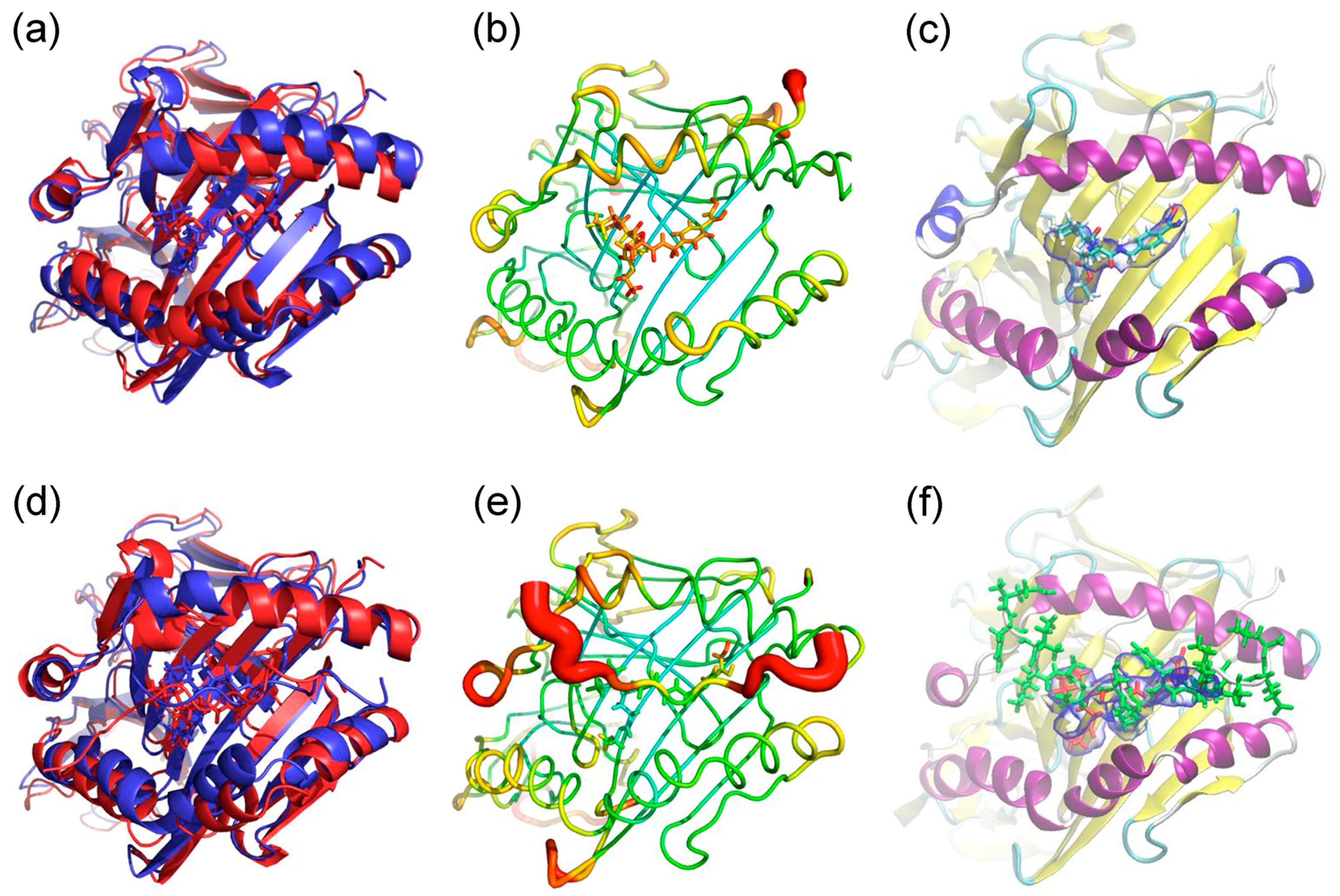
2.1. Docking Simulations
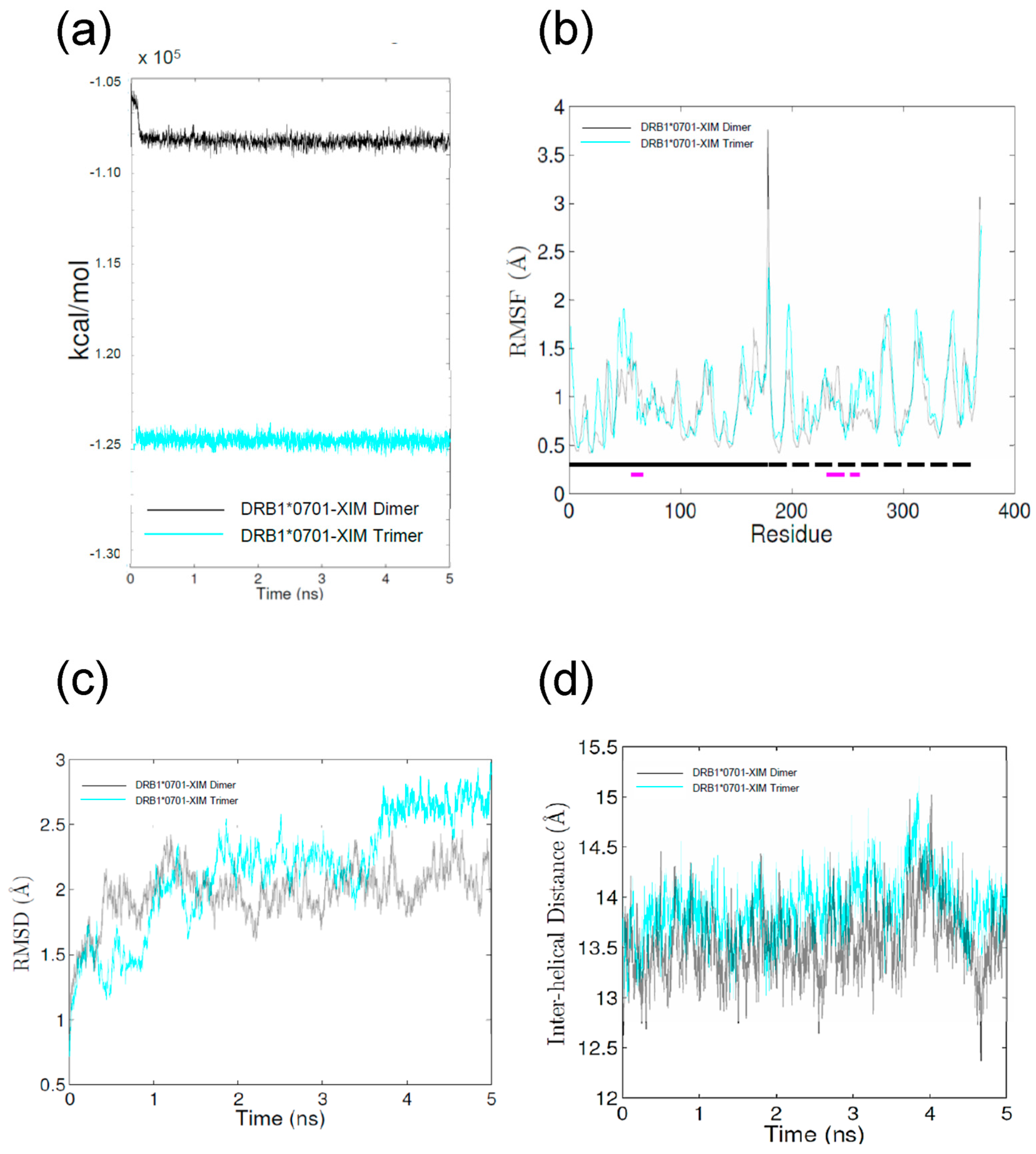
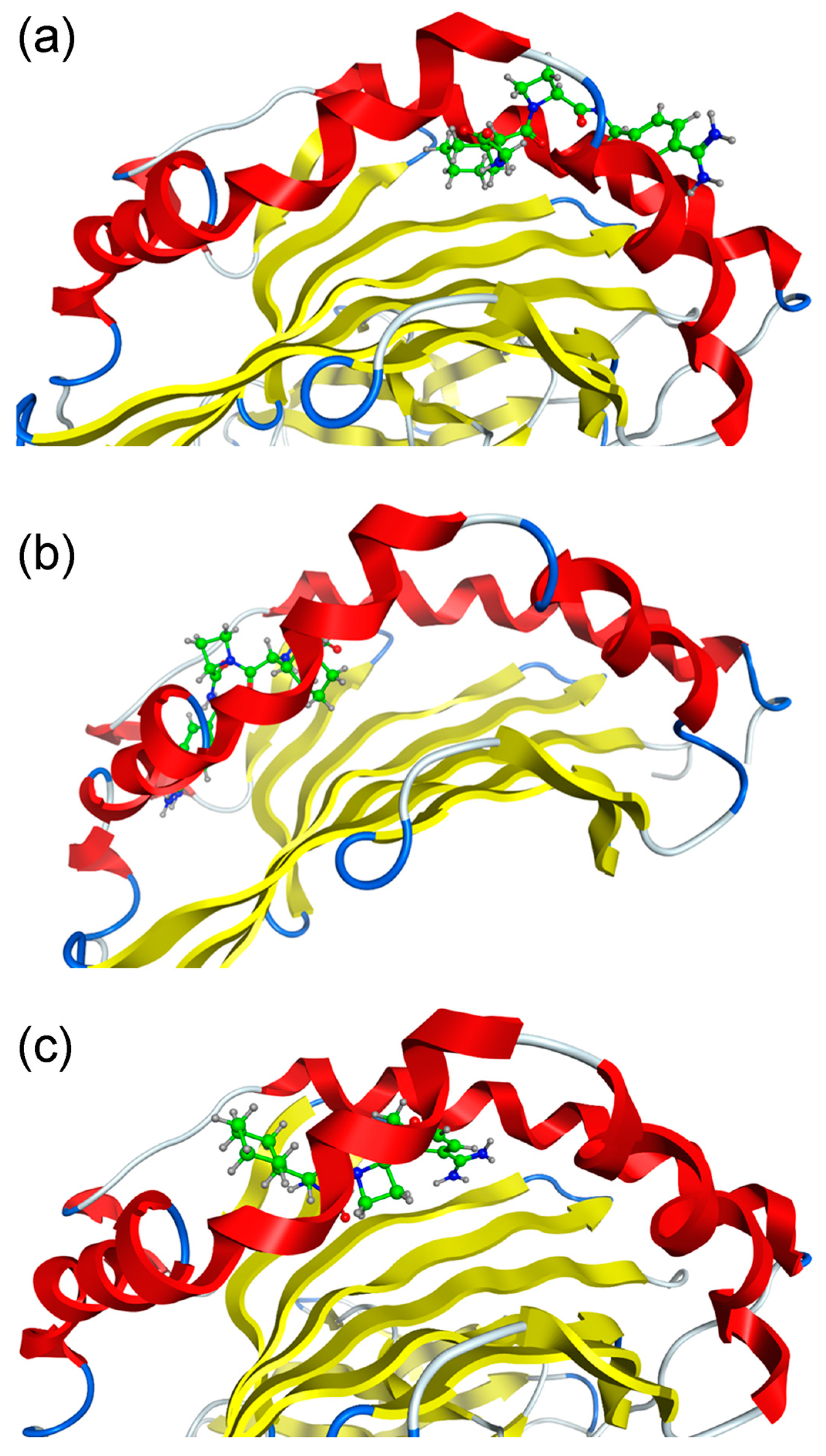
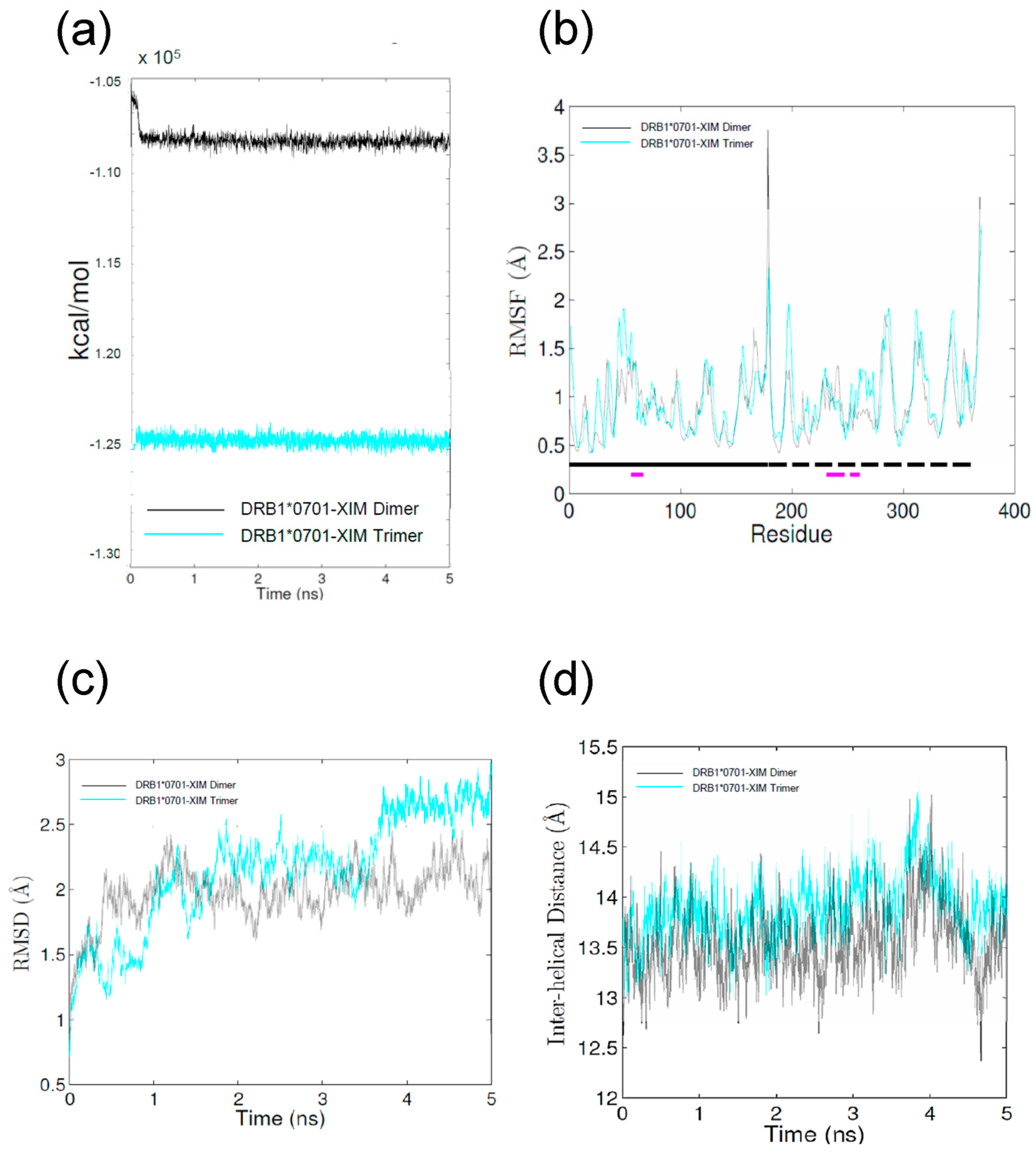
2.2. Molecular Dynamics (MD) Simulations
2.3. Human Leukocyte Antigen (HLA) Class II Binding Assay and Liquid Chromatography Tandem Mass Spectrometry (LC-MS/MS) Detection of Ximelagatran Bound to HLA-DR Molecules
3. Discussion
4. Materials and Methods
4.1. Docking Simulations
4.2. MD Simulations
4.3. HLA Class II Binding Assay and LC-MS/MS Detection of Ximelagatran Bound to HLA-DR Molecules
4.4. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| HLA | human leukocyte antigen |
| GBVI/WSA_dG | generalized-Born volume integral/weighted surface area |
| MD | molecular dynamics |
| LC-MS/MS | liquid chromatography tandem mass spectrometry |
| DMSO | dimethyl sulfoxide |
| IDT | idiosyncratic drug toxicity |
| ALT | alanine aminotransferase |
| RMSD | root mean-square-deviation |
| RMSF | root mean-square-fluctuation |
| TT | tetanus toxoid |
References
- Uetrecht, J.; Naisbitt, D.J. Idiosyncratic adverse drug reactions: Current concepts. Pharmacol. Rev. 2013, 65, 779–808. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.M.; Larrey, D.; Olsson, R.; Lewis, J.H.; Keisu, M.; Auclert, L.; Sheth, S. Hepatic findings in long-term clinical trials of ximelagatran. Drug Saf. 2005, 28, 351–370. [Google Scholar] [CrossRef] [PubMed]
- Kindmark, A.; Jawaid, A.; Harbron, C.G.; Barratt, B.J.; Bengtsson, O.F.; Andersson, T.B.; Carlsson, S.; Cederbrant, K.E.; Gibson, N.J.; Armstrong, M.; et al. Genome-wide pharmacogenetic investigation of a hepatic adverse event without clinical signs of immunopathology suggests an underlying immune pathogenesis. Pharmacogenom. J. 2008, 8, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Pichler, W.J.; Beeler, A.; Keller, M.; Lerch, M.; Posadas, S.; Schmid, D.; Spanou, Z.; Zawodniak, A.; Gerber, B. Pharmacological interaction of drugs with immune receptors: The p-i concept. Allergol. Int. 2006, 55, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.; Marcaida, M.J.; Eriksson, K.K.; Jamin, H.; Fontana, S.; Pichler, W.J.; Yerly, D. Oxypurinol directly and immediately activates the drug-specific T cells via the preferential use of HLA-B*58:01. J. Immunol. 2014, 192, 2984–2993. [Google Scholar] [CrossRef] [PubMed]
- Illing, P.T.; Vivian, J.P.; Dudek, N.L.; Kostenko, L.; Chen, Z.; Bharadwaj, M.; Miles, J.J.; Kjer-Nielsen, L.; Gras, S.; Williamson, N.A.; et al. Immune self-reactivity triggered by drug-modified HLA-peptide repertoire. Nature 2012, 486, 554–558. [Google Scholar] [CrossRef] [PubMed]
- Höpner, S.; Dickhaut, K.; Hofstätter, M.; Krämer, H.; Rückerl, D.; Söderhäll, J.A.; Gupta, S.; Marin-Esteban, V.; Kühne, R.; Freund, C.; et al. Small organic compounds enhance antigen loading of class II major histocompatibility complex proteins by targeting the polymorphic P1 pocket. J. Biol. Chem. 2006, 281, 38535–38542. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Höpner, S.; Rupp, B.; Günther, S.; Dickhaut, K.; Agarwal, N.; Cardoso, M.C.; Kühne, R.; Wiesmüller, K.H.; Jung, G.; et al. Anchor side chains of short peptide fragments trigger ligand-exchange of class II MHC molecules. PLoS ONE 2008, 3, e1814. [Google Scholar] [CrossRef] [PubMed]
- Zaheer-ul-Haq; Khan, W. Molecular and structural determinants of adamantyl susceptibility to HLA-DRs allelic variants: An in silico approach to understand the mechanism of MLEs. J. Comput. Aided Mol. Des. 2011, 25, 81–101. [Google Scholar]
- Hirasawa, M.; Hagihara, K.; Okudaira, N.; Izumi, T. The possible mechanism of idiosyncratic lapatinib-induced liver injury in patients carrying human leukocyte antigen-DRB1*07:01. PLoS ONE 2015, 10, e0130928. [Google Scholar] [CrossRef] [PubMed]
- Corbeil, C.R.; Williams, C.I.; Labute, P.; Spraggs, C.F. Variability in docking success rates due to dataset preparation. J. Comput. Aided Mol. Des. 2012, 26, 775–786. [Google Scholar] [CrossRef] [PubMed]
- Isogai, H.; Hirayama, N. In silico analysis of interactions between nevirapine-related compounds, HLA-B*14:02 and T-cell receptor. Chem-Bio Inform. J. 2016, 16, 9–12. [Google Scholar] [CrossRef]
- Osabe, M.; Tohkin, M.; Hirayama, N. In silico Analysis of Interactions between HLA-B*58:01 and Allopurinol-related Compounds. Chem-Bio Inform. J. 2016, 16, 1–4. [Google Scholar] [CrossRef]
- Eriksson, U.G.; Bredberg, U.; Hoffmann, K.J.; Thuresson, A.; Gabrielsson, M.; Ericsson, H.; Ahnoff, M.; Gislén, K.; Fager, G.; Gustafsson, D. Absorption, distribution, metabolism, and excretion of ximelagatran, an oral direct thrombin inhibitor, in rats, dogs, and humans. Drug Metab. Dispos. 2003, 31, 294–305. [Google Scholar] [CrossRef] [PubMed]
- Norcross, M.A.; Luo, S.; Lu, L.; Boyne, M.T.; Gomarteli, M.; Rennels, A.D.; Woodcock, J.; Margulies, D.H.; McMurtrey, C.; Vernon, S.; et al. Abacavir induces loading of novel self-peptides into HLA-B*57: 01: An autoimmune model for HLA-associated drug hypersensitivity. AIDS 2011, 26, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, F.C.; Koetzle, T.F.; Williams, G.J.; Meyer, E.F., Jr.; Brice, M.D.; Rodgers, J.R.; Kennard, O.; Shimanouchi, T.; Tasumi, M. The Protein Data Bank: A computer-based archival file for macromolecular structures. J. Mol. Biol. 1977, 112, 535–542. [Google Scholar] [CrossRef]
- Amari, S.; Kataoka, R.; Ikegami, T.; Hirayama, N. HLA-modeler: Automated homology modeling of human leukocyte antigens. Int. J. Med. Chem. 2013, 690513. [Google Scholar] [CrossRef] [PubMed]
- Goto, J.; Kataoka, R.; Muta, H.; Hirayama, N. ASEDock-docking based on alpha spheres and excluded volumes. J. Chem. Inf. Model. 2008, 48, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Chemical Computing Group Inc. Molecular Operating Environment (MOE); Chemical Computing Group Inc.: Montreal, QC, Canada, 2017. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Steere, A.C.; Klitz, W.; Drouin, E.E.; Falk, B.A.; Kwok, W.W.; Nepom, G.T.; Baxter-Lowe, L.A. Antibiotic-refractory Lyme arthritis is associated with HLA-DR molecules that bind a Borrelia burgdorferi peptide. J. Exp. Med. 2006, 203, 961–971. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HLA Allele | Compounds | ||
|---|---|---|---|
| Ximelagatran | Melagatran | ||
| Tautomer | GBVI/WSA_dG (kcal/mol) | GBVI/WSA_dG (kcal/mol) | |
| DRB1*01:01 | Hydroxylamine | −11.88 | −9.91 |
| DRB1*07:01 | Oxime | −11.24 | −10.41 |
| DRB1*15:01 | Hydroxylamine | −10.99 | −10.70 |
| HLA Allele | DRB1*01:01 | DRB1*07:01 | DRB1*15:01 |
|---|---|---|---|
| % of DMSO control | 105.7 ± 4.4 | 91.1 ± 13.4 | 112.3 ± 8.4 |
| HLA Alleles | DRB1*01:01 | DRB1*07:01 | DRB1*15:01 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Incubation No. | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 |
| HLA | − | + | + | − | + | + | − | + | + |
| Ligand peptide | + | − | + | + | − | + | + | − | + |
| Concentration (nM) | 0.11 ± 0.01 | 0.17 ± 0.03 | 0.13 ± 0.01 | 0.13 ± 0.01 | 0.17 ± 0.03 | 0.14 ± 0.01 | 0.14 ± 0.03 | 0.23 ± 0.09 | 0.14 ± 0.02 |
| p-Value | n/a | 0.006 (vs. 1) | 0.023 (vs. 2) | n/a | 0.087 (vs. 4) | 0.211 (vs. 5) | n/a | 0.084 (vs. 7) | 0.076 (vs. 8) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hirasawa, M.; Hagihara, K.; Abe, K.; Ando, O.; Hirayama, N. In Silico and In Vitro Analysis of Interaction between Ximelagatran and Human Leukocyte Antigen (HLA)-DRB1*07:01. Int. J. Mol. Sci. 2017, 18, 694. https://doi.org/10.3390/ijms18040694
Hirasawa M, Hagihara K, Abe K, Ando O, Hirayama N. In Silico and In Vitro Analysis of Interaction between Ximelagatran and Human Leukocyte Antigen (HLA)-DRB1*07:01. International Journal of Molecular Sciences. 2017; 18(4):694. https://doi.org/10.3390/ijms18040694
Chicago/Turabian StyleHirasawa, Makoto, Katsunobu Hagihara, Koji Abe, Osamu Ando, and Noriaki Hirayama. 2017. "In Silico and In Vitro Analysis of Interaction between Ximelagatran and Human Leukocyte Antigen (HLA)-DRB1*07:01" International Journal of Molecular Sciences 18, no. 4: 694. https://doi.org/10.3390/ijms18040694
APA StyleHirasawa, M., Hagihara, K., Abe, K., Ando, O., & Hirayama, N. (2017). In Silico and In Vitro Analysis of Interaction between Ximelagatran and Human Leukocyte Antigen (HLA)-DRB1*07:01. International Journal of Molecular Sciences, 18(4), 694. https://doi.org/10.3390/ijms18040694