
Biglycan- and Sphingosine Kinase-1 Signaling Crosstalk Regulates the Synthesis of Macrophage Chemoattractants



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
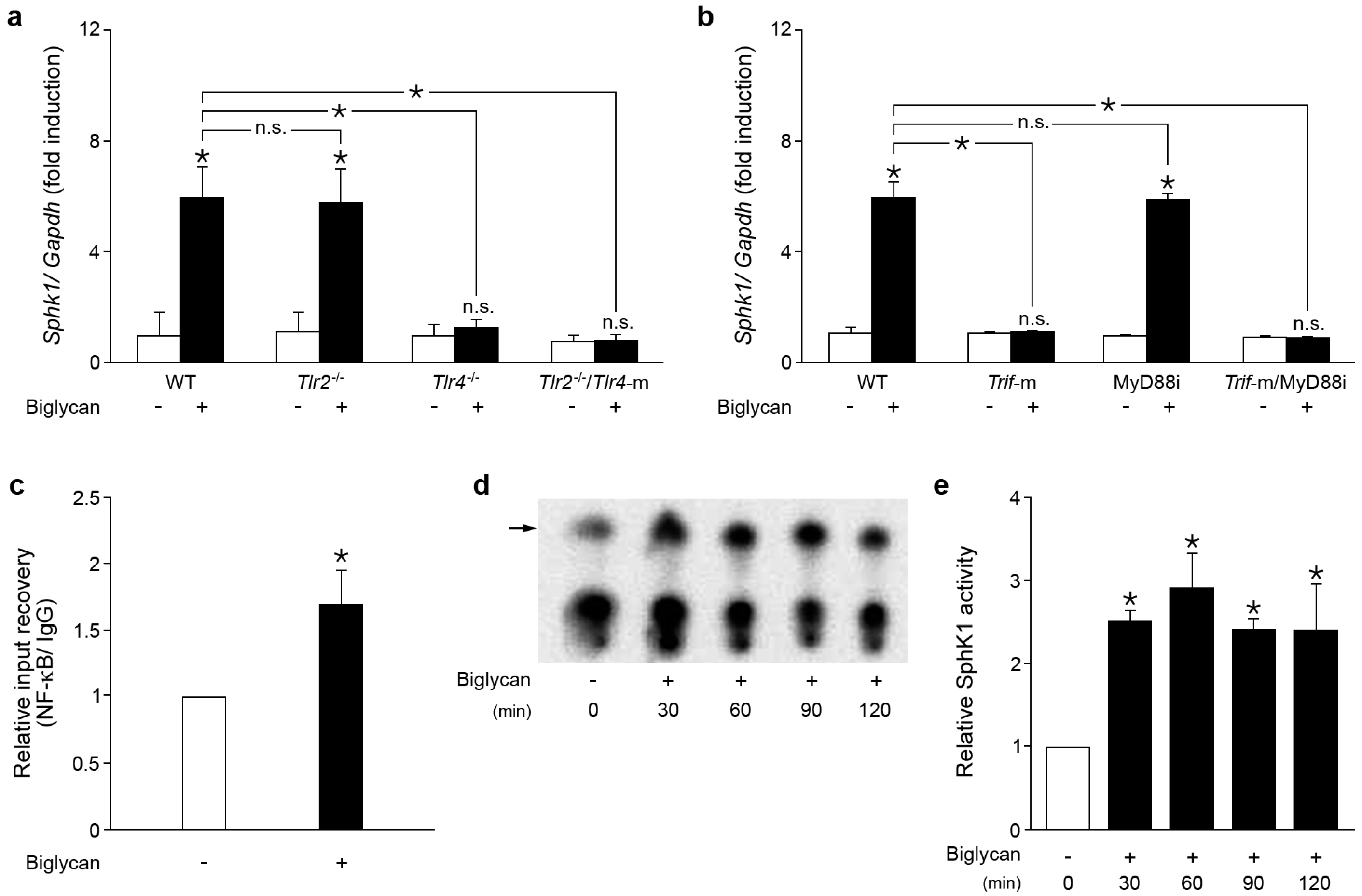
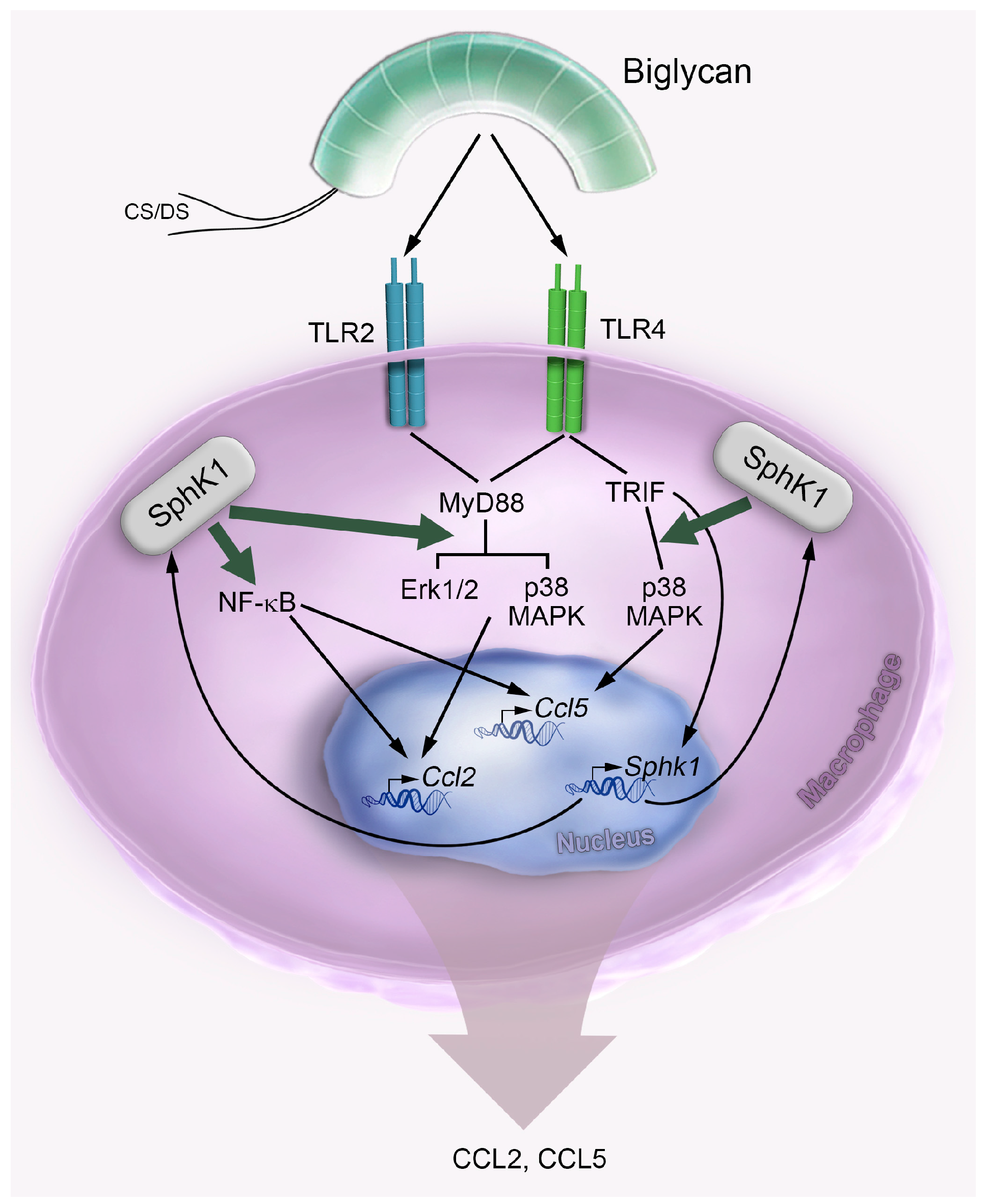
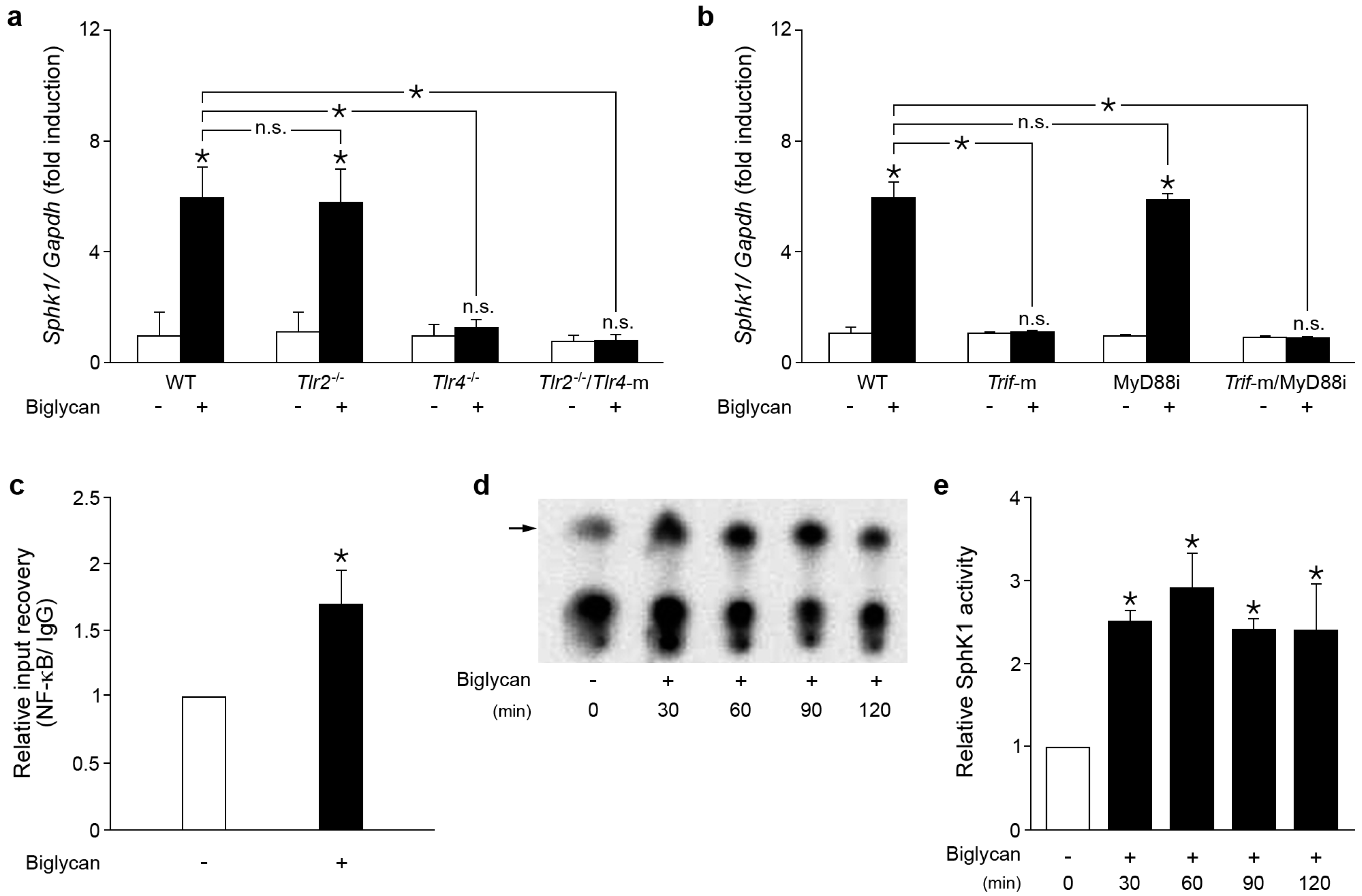
2.1. Biglycan Triggers the Expression and Activity of Sphk1 in Mouse Peritoneal Macrophages
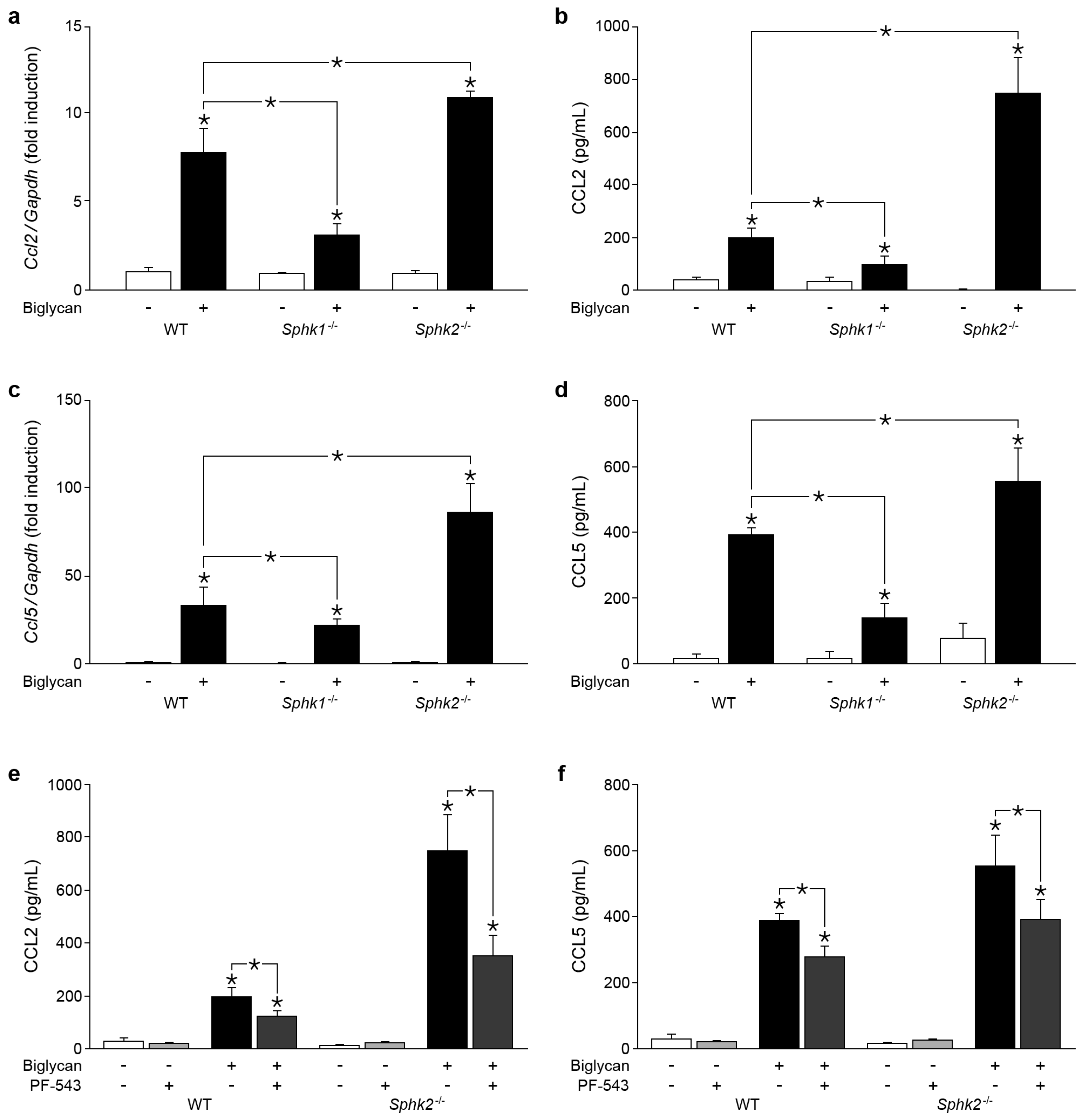
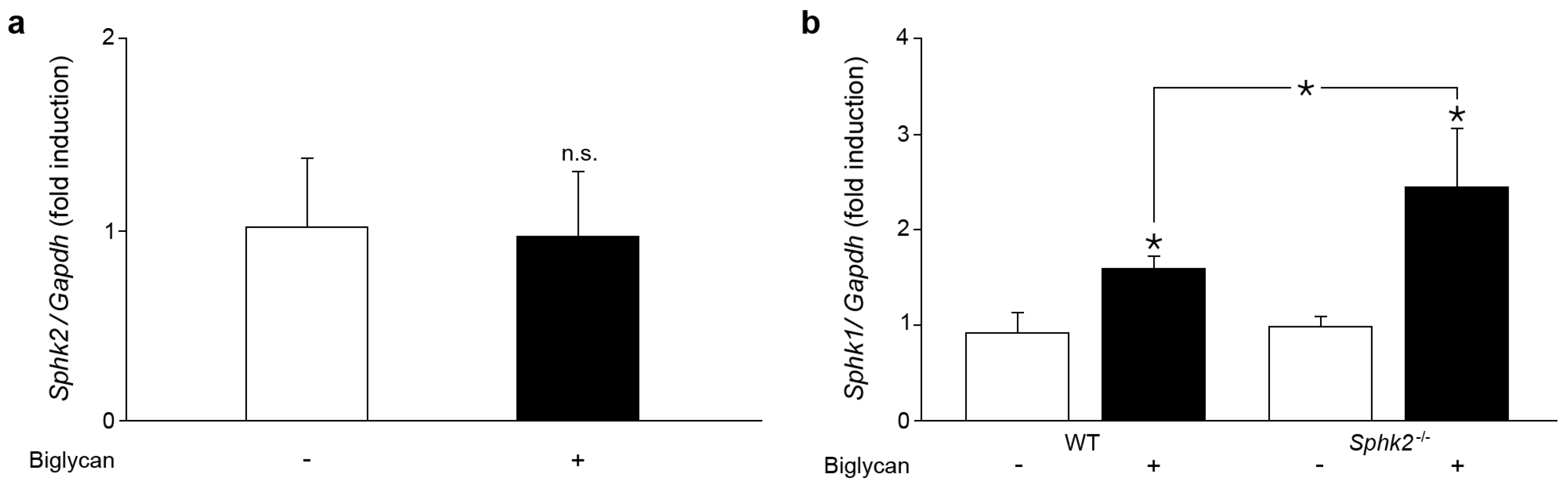
2.2. Sphk2 Deficiency Potentiates Biglycan Triggered Sphk1 mRNA Expression
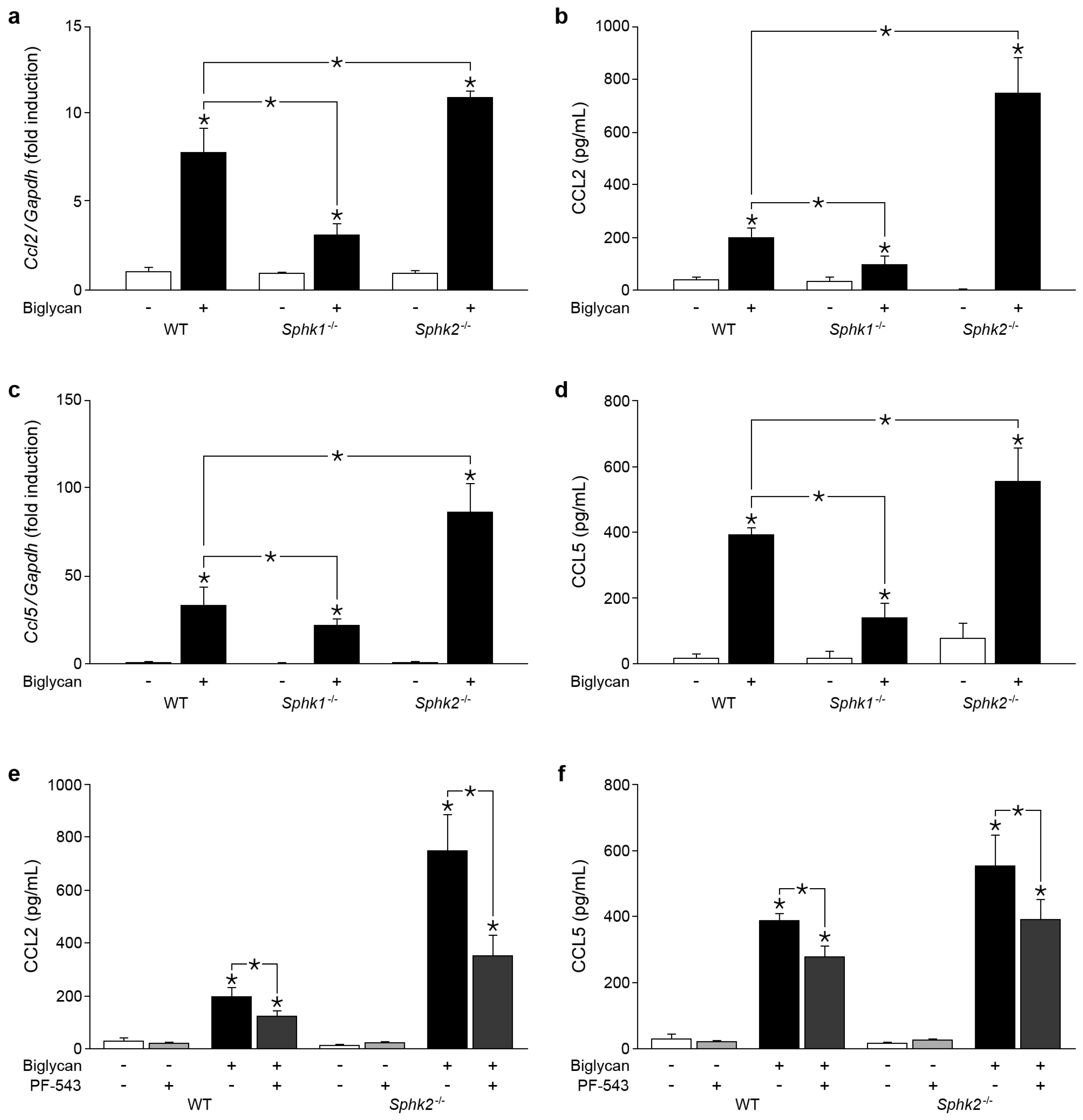
2.3. Biglycan Triggers CCL2 and CCL5 Production in a SphK1-Dependent Manner
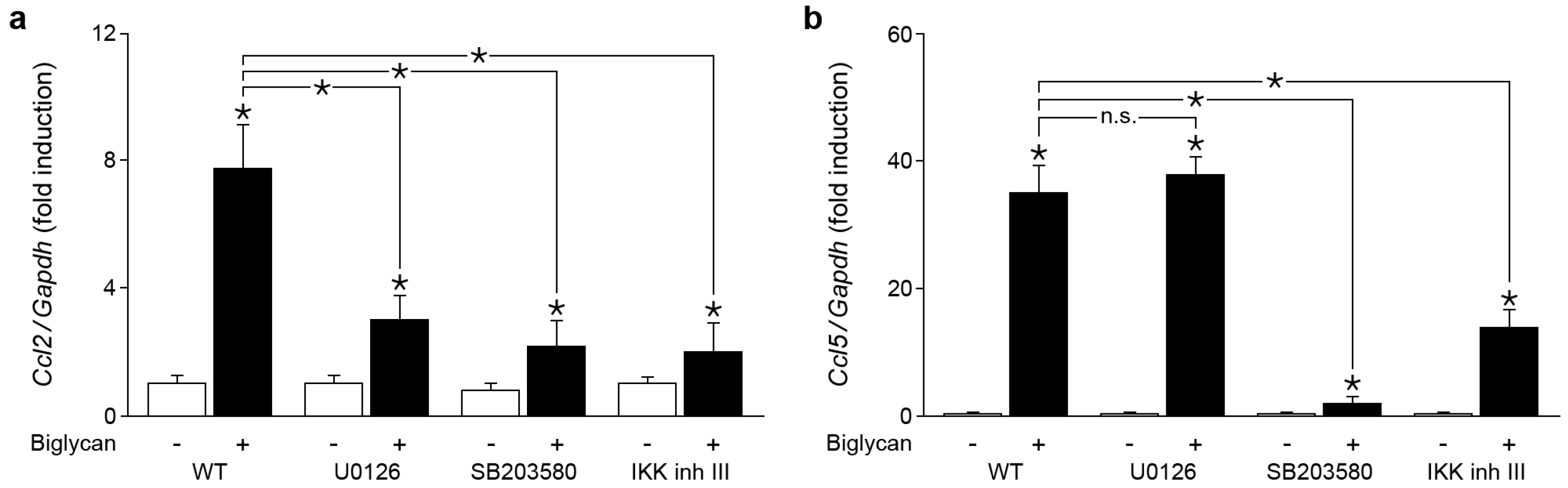
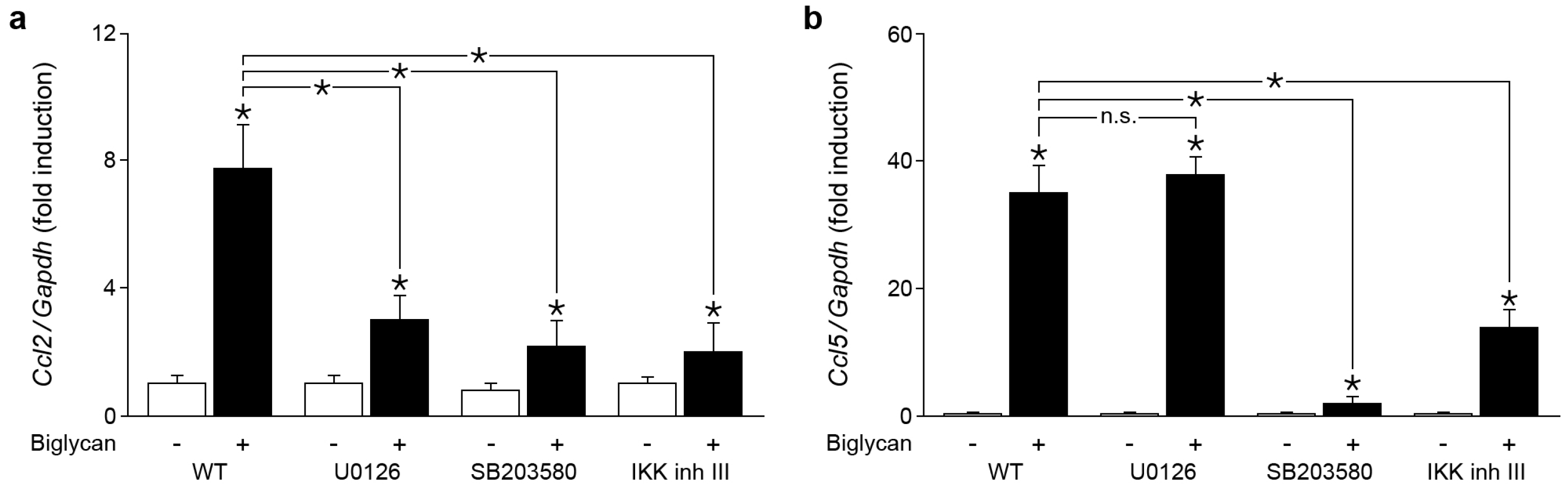
2.4. Biglycan Triggers Expression of Ccl2 via NF-κB, Erk1/2 and p38 MAPK, While Ccl5 Expression is Induced through NF-κB and p38 MAPK
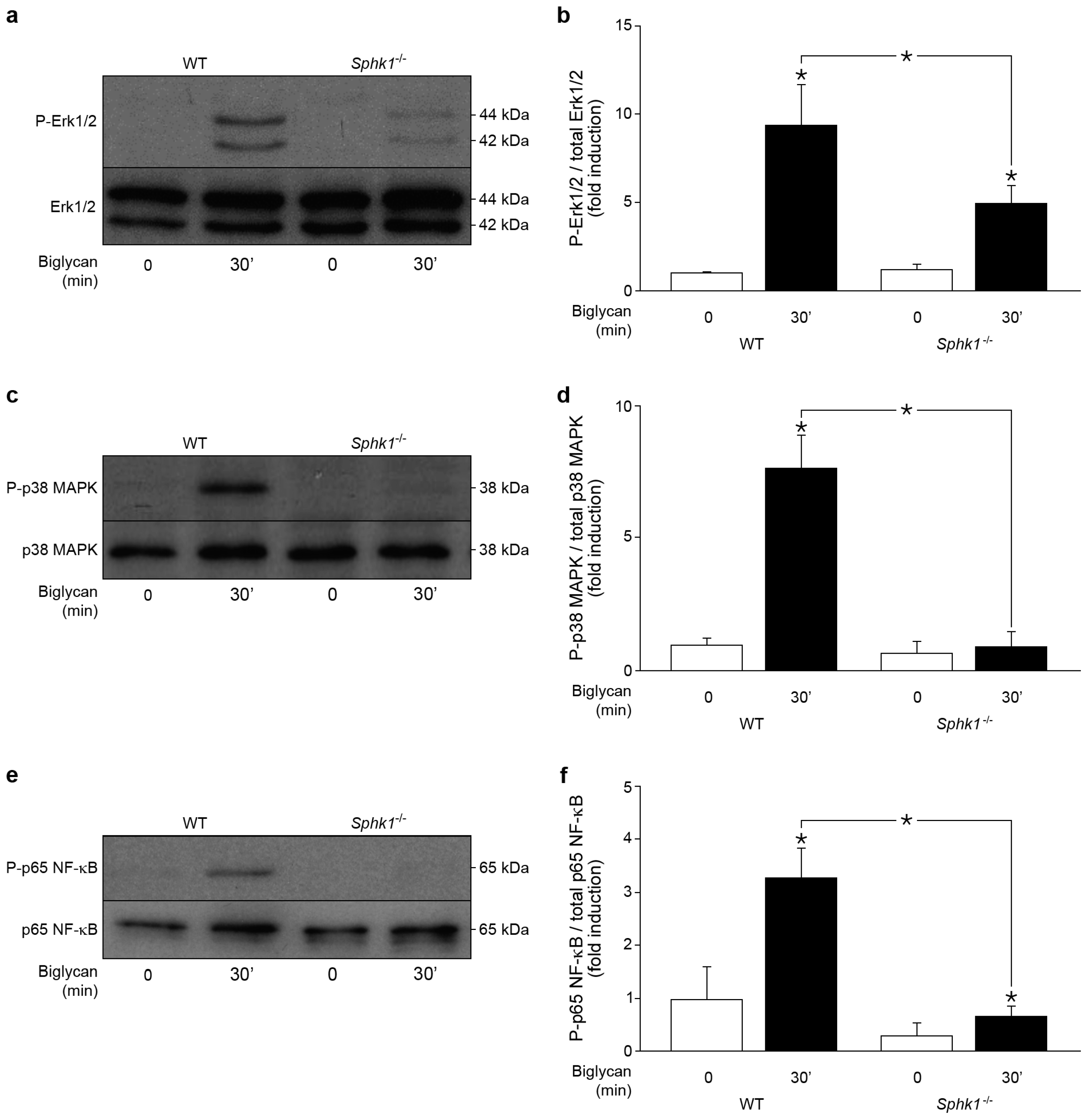
2.5. Control of Biglycan-Dependent Erk1/2, p38 MAPK and NF-κB Subunit p65 by SphK1 in Macrophages
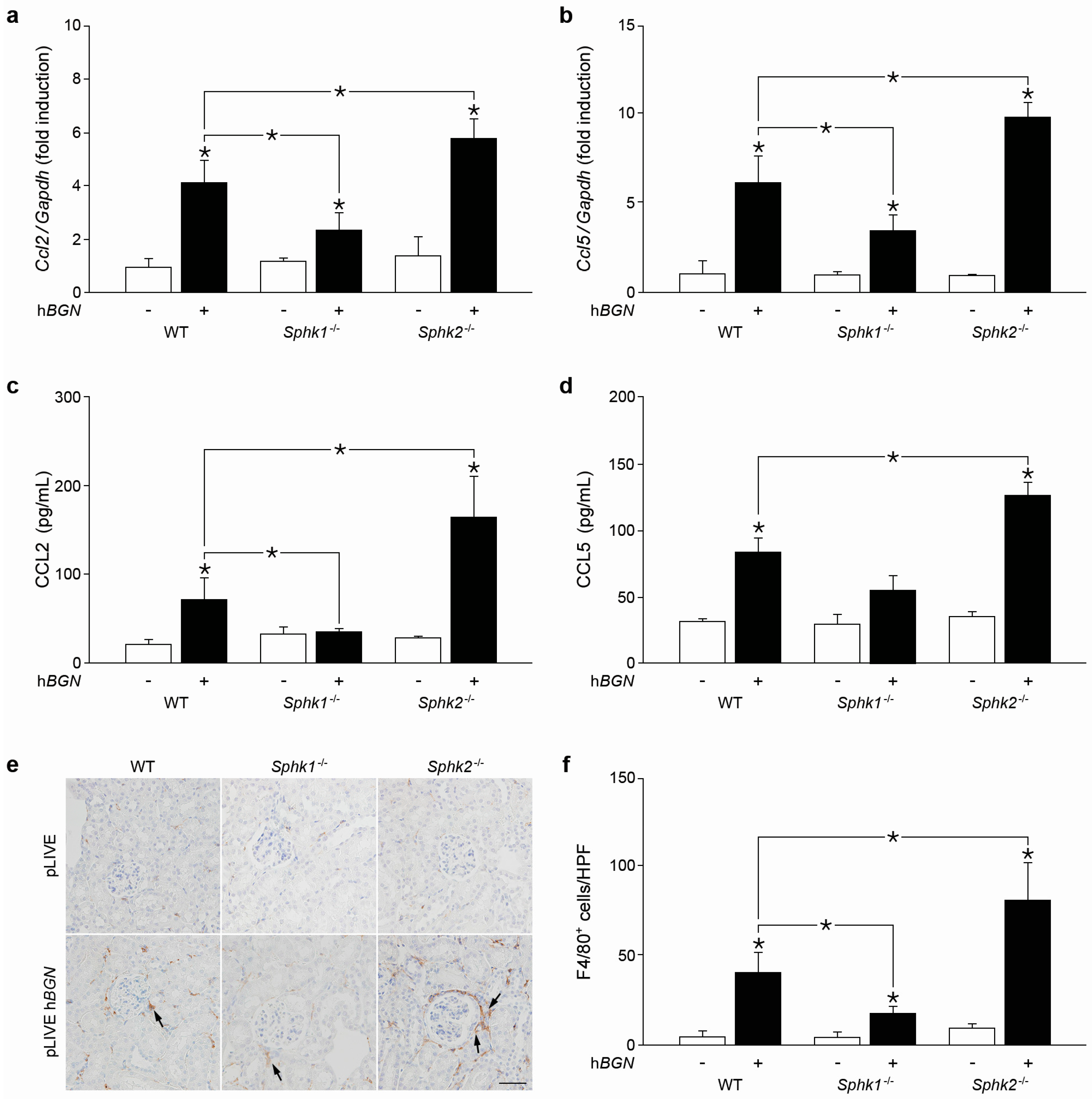
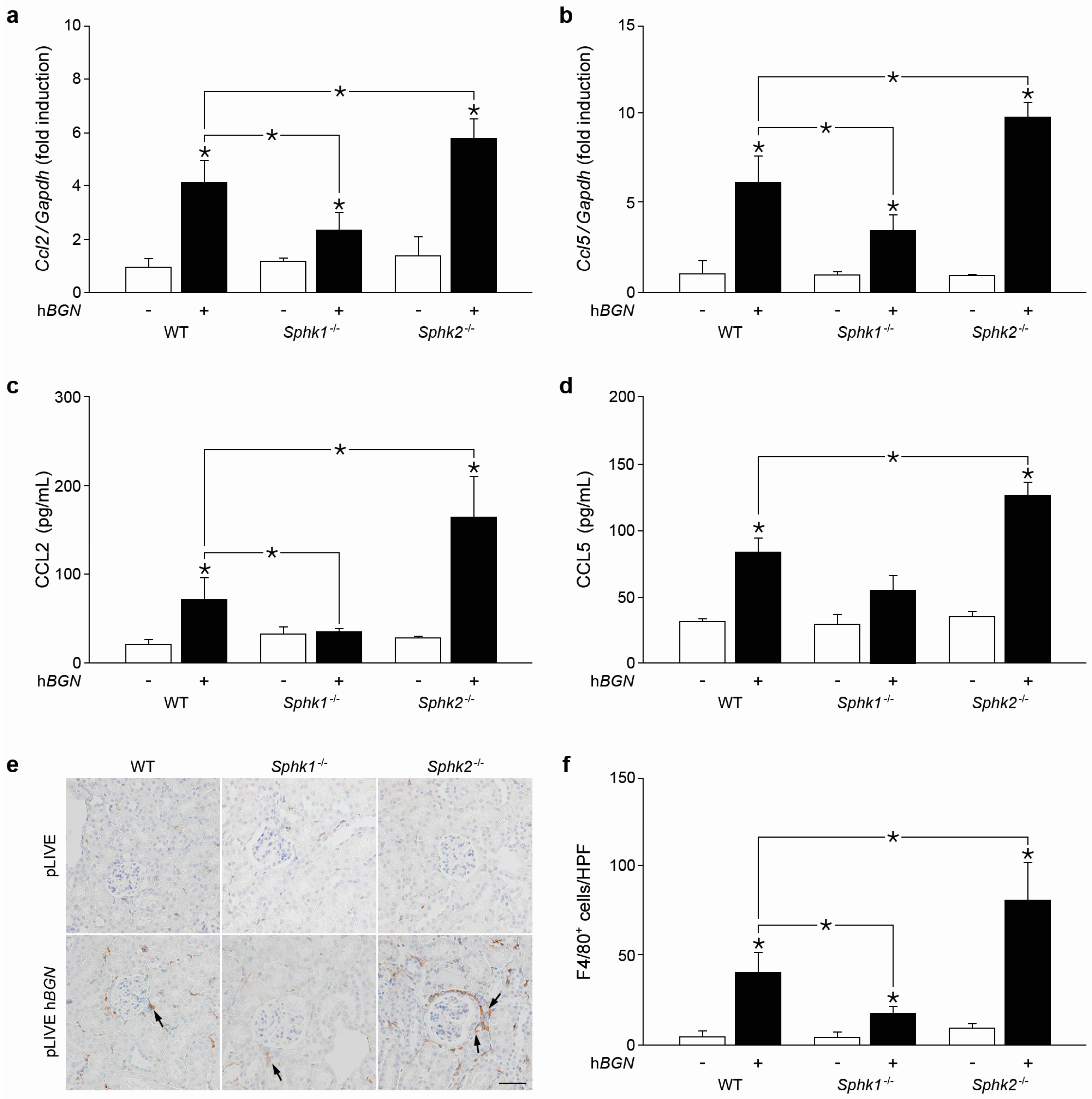
2.6. Soluble Biglycan Triggers Renal Expression of Ccl2 and Ccl5 and Macrophage Recruitment into the Kidney in Sphk1-Dependent Manner
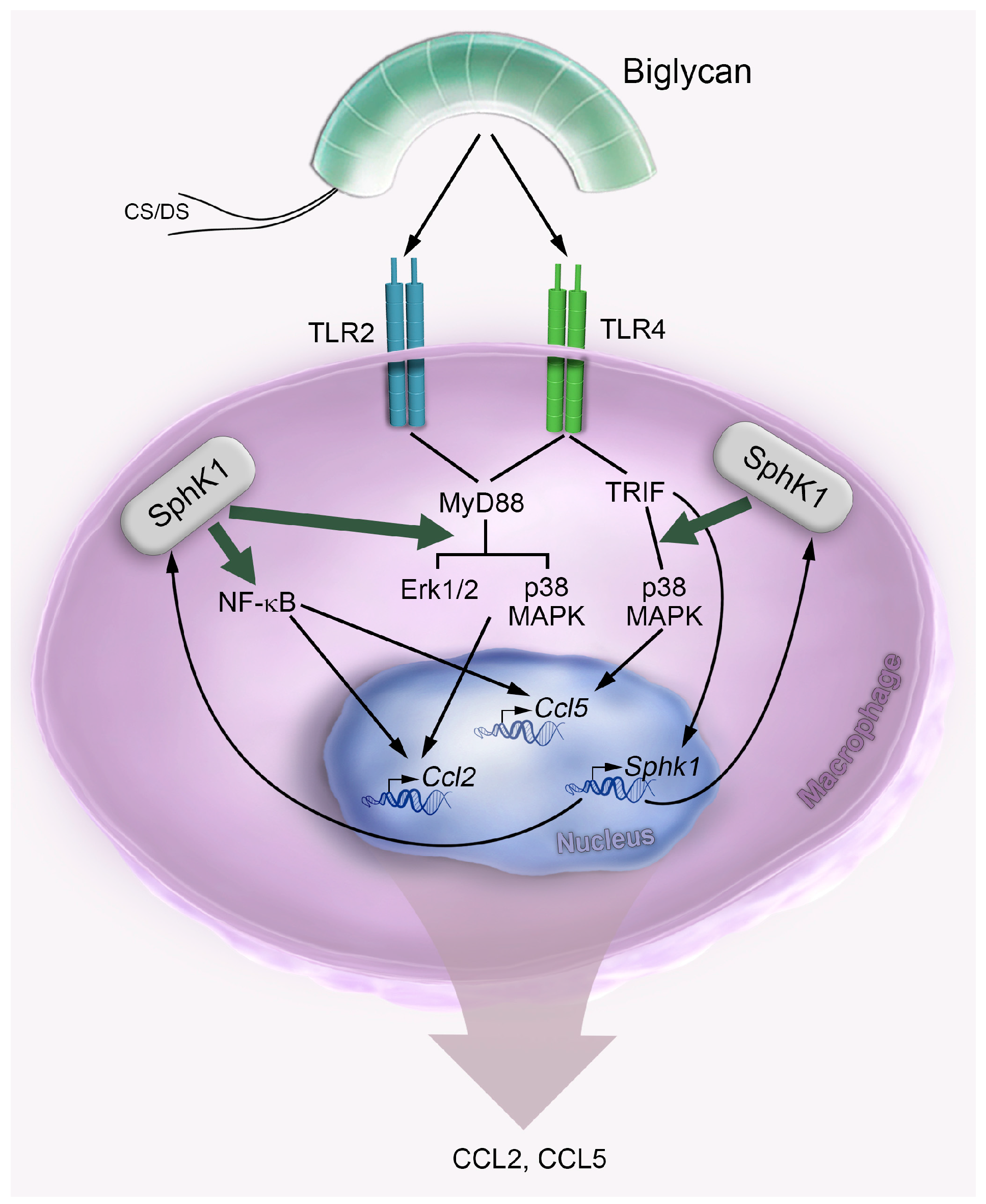
3. Discussion
4. Materials and Methods
4.1. Animal Experiments
4.2. In Vivo Transfection
4.3. Immunohistochemistry
4.4. Cell Culture and Stimulation
4.5. RNA Isolation and Quantitative Real-Time PCR
4.6. Chromatin Immunoprecipitation
4.7. Western Blot and ELISA
4.8. Determination of Sphingosine Kinase Activity
4.9. Statistics
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| CCL | chemokine (C-C motif) ligand |
| CXCL | chemokine (C-X-C motif) ligand |
| DAMP | damage-associated molecular patterns |
| ECM | extracellular matrix |
| IL-1 | interleukin-1 |
| LPS | lipopolysaccharide |
| MAPK | mitogen-activated protein kinase |
| MyD88 | myeloid differentiation primary response protein |
| NF-κB | nuclear factor κ-light-chain-enhancer of activated B-cells |
| S1P | sphingosine-1 phosphate |
| SphK | sphingosine kinase |
| TLR | Toll-like receptor |
| TNF | tumor necrosis factor |
| TRIF | Toll/IL-1R domain-containing adaptor inducing IFN-β |
| TSS | transcription start site |
| WT | wild-type |
References
- Schaefer, L. Complexity of danger: The diverse nature of damage-associated molecular patterns. J. Biol. Chem. 2014, 289, 35237–35245. [Google Scholar] [CrossRef] [PubMed]
- Iozzo, R.V.; Schaefer, L. Proteoglycan form and function: A comprehensive nomenclature of proteoglycans. Matrix Biol. 2015, 42, 11–55. [Google Scholar] [CrossRef] [PubMed]
- Moreth, K.; Iozzo, R.V.; Schaefer, L. Small leucine-rich proteoglycans orchestrate receptor crosstalk during inflammation. Cell Cycle 2012, 11, 2084–2091. [Google Scholar] [CrossRef] [PubMed]
- Frey, H.; Schroeder, N.; Manon-Jensen, T.; Iozzo, R.V.; Schaefer, L. Biological interplay between proteoglycans and their innate immune receptors in inflammation. FEBS J. 2013, 280, 2165–2179. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, L.; Babelova, A.; Kiss, E.; Hausser, H.J.; Baliova, M.; Krzyzankova, M.; Marsche, G.; Young, M.F.; Mihalik, D.; Gotte, M.; et al. The matrix component biglycan is proinflammatory and signals through toll-like receptors 4 and 2 in macrophages. J. Clin. Investig. 2005, 115, 2223–2233. [Google Scholar] [CrossRef] [PubMed]
- Zeng-Brouwers, J.; Beckmann, J.; Nastase, M.V.; Iozzo, R.V.; Schaefer, L. De novo expression of circulating biglycan evokes an innate inflammatory tissue response via Myd88/Trif pathways. Matrix Biol. 2014, 35, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, L.T.; Frey, H.; Nastase, M.V.; Tredup, C.; Hoffmann, A.; Poluzzi, C.; Zeng-Brouwers, J.; Manon-Jensen, T.; Schroder, K.; Brandes, R.P.; et al. Bimodal role of NADPH oxidases in the regulation of biglycan-triggered IL-1β synthesis. Matrix Biol. 2016, 49, 61–81. [Google Scholar] [CrossRef] [PubMed]
- Babelova, A.; Moreth, K.; Tsalastra-Greul, W.; Zeng-Brouwers, J.; Eickelberg, O.; Young, M.F.; Bruckner, P.; Pfeilschifter, J.; Schaefer, R.M.; Grone, H.J.; et al. Biglycan, a danger signal that activates the NLRP3 inflammasome via toll-like and P2X receptors. J. Biol. Chem. 2009, 284, 24035–24048. [Google Scholar] [CrossRef] [PubMed]
- Moreth, K.; Brodbeck, R.; Babelova, A.; Gretz, N.; Spieker, T.; Zeng-Brouwers, J.; Pfeilschifter, J.; Young, M.F.; Schaefer, R.M.; Schaefer, L. The proteoglycan biglycan regulates expression of the B cell chemoattractant CXCL13 and aggravates murine lupus nephritis. J. Clin. Investig. 2010, 120, 4251–4272. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Moreth, K.; Frey, H.; Hubo, M.; Zeng-Brouwers, J.; Nastase, M.V.; Hsieh, L.T.; Haceni, R.; Pfeilschifter, J.; Iozzo, R.V.; Schaefer, L. Biglycan-triggered TLR-2- and TLR-4-signaling exacerbates the pathophysiology of ischemic acute kidney injury. Matrix Biol. 2014, 35, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, L.; Tredup, C.; Gubbiotti, M.A.; Iozzo, R.V. Proteoglycan neofunctions: Regulation of inflammation and autophagy in cancer biology. FEBS J. 2017, 284, 10–26. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, L. Extracellular matrix molecules: Endogenous danger signals as new drug targets in kidney diseases. Curr. Opin. Pharmacol. 2010, 10, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Nastase, M.V.; Young, M.F.; Schaefer, L. Biglycan: A multivalent proteoglycan providing structure and signals. J. Histochem. Cytochem. 2012, 60, 963–975. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, L.T.; Nastase, M.V.; Zeng-Brouwers, J.; Iozzo, R.V.; Schaefer, L. Soluble biglycan as a biomarker of inflammatory renal diseases. Int. J. Biochem. Cell. Biol. 2014, 54, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Schwalm, S.; Pfeilschifter, J.; Huwiler, A. Targeting the Sphingosine Kinase/Sphingosine 1-Phosphate Pathway to Treat Chronic Inflammatory Kidney Diseases. Basic Clin. Pharmacol. Toxicol. 2014, 114, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Maceyka, M.; Spiegel, S. Sphingolipid metabolites in inflammatory disease. Nature 2014, 510, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.R.; Pyne, S.; Pyne, N.J. Sphingosine kinases: Emerging structure-function insights. Trends Biochem. Sci. 2016, 41, 395–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orr Gandy, K.A.; Obeid, L.M. Targeting the Sphingosine Kinase/Sphingosine 1-Phosphate Pathway in Disease: Review of Sphingosine Kinase Inhibitors. Biochim. Biophys. Acta 2013, 1831, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Pitson, S.M.; Moretti, P.A.; Zebol, J.R.; Lynn, H.E.; Xia, P.; Vadas, M.A.; Wattenberg, B.W. Activation of sphingosine kinase 1 by ERK1/2-mediated phosphorylation. EMBO J. 2003, 22, 5491–5500. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, S.E.; Harikumar, K.B.; Hait, N.C.; Allegood, J.; Strub, G.M.; Kim, E.Y.; Maceyka, M.; Jiang, H.; Luo, C.; Kordula, T.; et al. Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature 2010, 465, 1084–1088. [Google Scholar] [CrossRef] [PubMed]
- Harikumar, K.B.; Yester, J.W.; Surace, M.J.; Oyeniran, C.; Price, M.M.; Huang, W.C.; Hait, N.C.; Allegood, J.C.; Yamada, A.; Kong, X.; et al. K63-linked polyubiquitination of transcription factor IRF1 is essential for IL-1-induced production of chemokines CXCL10 and CCL5. Nat. Immunol. 2014, 15, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Pchejetski, D.; Nunes, J.; Coughlan, K.; Lall, H.; Pitson, S.M.; Waxman, J.; Sumbayev, V.V. The involvement of sphingosine kinase 1 in LPS-induced Toll-like receptor 4-mediated accumulation of HIF-1α protein, activation of ASK1 and production of the pro-inflammatory cytokine IL-6. Immunol. Cell Biol. 2011, 89, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Lai, W.Q.; Irwan, A.W.; Goh, H.H.; Melendez, A.J.; McInnes, I.B.; Leung, B.P. Distinct roles of sphingosine kinase 1 and 2 in murine collagen-induced arthritis. J. Immunol. 2009, 183, 2097–2103. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.A.; Barth, J.; Chang, R.; Obeid, L.M.; Gilkeson, G.S. Genetic Sphingosine Kinase 1 Deficiency Significantly Decreases Synovial Inflammation and Joint Erosions in Murine TNF-α-Induced Arthritis. J. Immunol. 2010, 185, 2570–2579. [Google Scholar] [CrossRef] [PubMed]
- Geng, T.; Sutter, A.; Harland, M.D.; Law, B.A.; Ross, J.S.; Lewin, D.; Palanisamy, A.; Russo, S.B.; Chavin, K.D.; Cowart, L.A. SphK1 mediates hepatic inflammation in a mouse model of NASH induced by high saturated fat feeding and initiates proinflammatory signaling in hepatocytes. J. Lipid Res. 2015, 56, 2359–2371. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Nagahashi, M.; Kim, E.Y.; Harikumar, K.B.; Yamada, A.; Huang, W.C.; Hait, N.C.; Allegood, J.C.; Price, M.M.; Avni, D.; et al. Sphingosine-1-Phosphate Links Persistent STAT3 Activation, Chronic Intestinal Inflammation, and Development of Colitis-Associated Cancer. Cancer Cell 2013, 23, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Schwalm, S.; Timcheva, T.M.; Filipenko, I.; Ebadi, M.; Hofmann, L.P.; Zangemeister-Wittke, U.; Pfeilschifter, J.; Huwiler, A. Sphingosine kinase 2 deficiency increases proliferation and migration of renal mouse mesangial cells and fibroblasts. Biol. Chem. 2015, 396, 813–825. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.L.; Grey, J.Y.; Thomas, S.; Qiu, F.H.; Medford, R.M.; Wasserman, M.A.; Kunsch, C. Sphingosine kinase-1 mediates TNF-α-induced MCP-1 gene expression in endothelial cells: Upregulation by oscillatory flow. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H1452–H1458. [Google Scholar] [CrossRef] [PubMed]
- Luheshi, N.M.; Giles, J.A.; Lopez-Castejon, G.; Brough, D. Sphingosine regulates the NLRP3-inflammasome and IL-1β release from macrophages. Eur. J. Immunol. 2012, 42, 716–725. [Google Scholar] [CrossRef] [PubMed]
- Schnute, M.E.; McReynolds, M.D.; Kasten, T.; Yates, M.; Jerome, G.; Rains, J.W.; Hall, T.; Chrencik, J.; Kraus, M.; Cronin, C.N.; et al. Modulation of cellular S1P levels with a novel, potent and specific inhibitor of sphingosine kinase-1. Biochem. J. 2012, 444, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.; Xin, C.; Pfeilschifter, J.; Huwiler, A. A Novel Mode of Action of the Putative Sphingosine Kinase Inhibitor 2-(p-hydroxyanilino)-4-(p-chlorophenyl) thiazole (SKI II): Induction of Lysosomal Sphingosine Kinase 1 Degradation. Cell. Physiol. Biochem. 2010, 26, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, S.; Milstien, S. The outs and the ins of sphingosine-1-phosphate in immunity. Nat. Rev. Immunol. 2011, 11, 403–415. [Google Scholar] [CrossRef] [PubMed]
- Kilpatrick, L.E.; Lee, J.Y.; Haines, K.M.; Campbell, D.E.; Sullivan, K.E.; Korchak, H.M. A role for PKC-δ and PI 3-kinase in TNF-α-mediated antiapoptotic signaling in the human neutrophil. Am. J. Physiol. Cell. Physiol. 2002, 283, C48–C57. [Google Scholar] [CrossRef] [PubMed]
- Mastrandrea, L.D.; Sessanna, S.M.; Laychock, S.G. Sphingosine kinase activity and sphingosine-1 phosphate production in rat pancreatic islets and INS-1 cells: Response to cytokines. Diabetes 2005, 54, 1429–1436. [Google Scholar] [CrossRef] [PubMed]
- Olivera, A.; Spiegel, S. Sphingosine-1-phosphate as second messenger in cell proliferation induced by PDGF and FCS mitogens. Nature 1993, 365, 557–560. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, M.; Shegogue, D.; Pei, H.; Bu, S.; Bielawska, A.; Bielawski, J.; Pettus, B.; Hannun, Y.A.; Obeid, L.; Trojanowska, M. Sphingosine kinase 1 (SPHK1) is induced by transforming growth factor-beta and mediates TIMP-1 up-regulation. J. Biol. Chem. 2004, 279, 53994–54001. [Google Scholar] [CrossRef] [PubMed]
- Stayrook, K.R.; Mack, J.K.; Cerabona, D.; Edwards, D.F.; Bui, H.H.; Niewolna, M.; Fournier, P.G.; Mohammad, K.S.; Waning, D.L.; Guise, T.A. TGFβ-Mediated induction of SphK1 as a potential determinant in human MDA-MB-231 breast cancer cell bone metastasis. Bonekey Rep. 2015, 4, 719. [Google Scholar] [CrossRef] [PubMed]
- Rius, R.A.; Edsall, L.C.; Spiegel, S. Activation of sphingosine kinase in pheochromocytoma PC12 neuronal cells in response to trophic factors. FEBS Lett. 1997, 417, 173–176. [Google Scholar] [CrossRef]
- Schonherr, E.; Jarvelainen, H.T.; Kinsella, M.G.; Sandell, L.J.; Wight, T.N. Platelet-derived growth factor and transforming growth factor-beta 1 differentially affect the synthesis of biglycan and decorin by monkey arterial smooth muscle cells. Arterioscler. Thromb. 1993, 13, 1026–1036. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, L.; Beck, K.F.; Raslik, I.; Walpen, S.; Mihalik, D.; Micegova, M.; Macakova, K.; Schonherr, E.; Seidler, D.G.; Varga, G.; et al. Biglycan, a nitric oxide-regulated gene, affects adhesion, growth, and survival of mesangial cells. J. Biol. Chem. 2003, 278, 26227–26237. [Google Scholar] [CrossRef] [PubMed]
- Schwiebs, A.; Friesen, O.; Katzy, E.; Ferreiros, N.; Pfeilschifter, J.M.; Radeke, H.H. Activation-induced cell death of dendritic cells is dependent on sphingosine kinase 1. Front. Pharmacol. 2016, 7, 94. [Google Scholar] [CrossRef] [PubMed]
- Prakash, H.; Luth, A.; Grinkina, N.; Holzer, D.; Wadgaonkar, R.; Gonzalez, A.P.; Anes, E.; Kleuser, B. Sphingosine Kinase-1 (SphK-1) Regulates Mycobacterium smegmatis Infection in Macrophages. PLoS ONE 2010, 5, e10657. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Pisonero, I.; Duenas, A.I.; Barreiro, O.; Montero, O.; Sanchez-Madrid, F.; Garcia-Rodriguez, C. Lipopolysaccharide and sphingosine-1-phosphate cooperate to induce inflammatory molecules and leukocyte adhesion in endothelial cells. J. Immunol. 2012, 189, 5402–5410. [Google Scholar] [CrossRef] [PubMed]
- Aoki, M.; Aoki, H.; Ramanathan, R.; Hait, N.C.; Takabe, K. Sphingosine-1-phosphate signaling in immune cells and inflammation: Roles and therapeutic potential. Mediat. Inflamm. 2016, 2016, 8606878. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Lee, H.J.; Mariko, B.; Lu, Y.C.; Dannenberg, A.J.; Haka, A.S.; Maxfield, F.R.; Camerer, E.; Proia, R.L.; Hla, T. Sphingosine kinases are not required for inflammatory responses in macrophages. J. Biol. Chem. 2013, 288, 32563–32573. [Google Scholar] [CrossRef] [PubMed]
- Jung, I.D.; Lee, J.S.; Kim, Y.J.; Jeong, Y.I.; Lee, C.M.; Baumruker, T.; Billlich, A.; Banno, Y.; Lee, M.G.; Ahn, S.C.; et al. Sphingosine kinase inhibitor suppresses a Th1 polarization via the inhibition of immunostimulatory activity in murine bone marrow-derived dendritic cells. Int. Immunol. 2007, 19, 411–426. [Google Scholar] [CrossRef] [PubMed]
- Elass-Rochard, E.; Rombouts, Y.; Coddeville, B.; Maes, E.; Blervaque, R.; Hot, D.; Kremer, L.; Guerardel, Y. Structural determination and Toll-like Receptor 2-dependent Proinflammatory Activity of Dimycolyl-diarabino-glycerol from Mycobacterium marinum. J. Biol. Chem. 2012, 287, 34432–34444. [Google Scholar] [CrossRef] [PubMed]
- Xia, P.; Wang, L.; Moretti, P.A.; Albanese, N.; Chai, F.; Pitson, S.M.; d'Andrea, R.J.; Gamble, J.R.; Vadas, M.A. Sphingosine Kinase Interacts with TRAF2 and Dissects Tumor Necrosis Factor-α Signaling. J. Biol. Chem. 2002, 277, 7996–8003. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Pu, G.; Tang, R.; Zhang, D.; Pan, W. Activation of Nuclear Factor Kappa B in the Hepatic Stellate Cells of Mice with Schistosomiasis Japonica. PLoS ONE 2014, 9, e104323. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Mosteller, R.D.; Broek, D. Sphingosine kinase protects lipopolysaccharide-activated macrophages from apoptosis. Mol. Cell. Biol. 2004, 24, 7359–7369. [Google Scholar] [CrossRef] [PubMed]
- Cummings, R.; Zhao, Y.; Jacoby, D.; Spannhake, E.W.; Ohba, M.; Garcia, J.G.; Watkins, T.; He, D.; Saatian, B.; Natarajan, V. Protein Kinase Cδ Mediates Lysophosphatidic Acid-induced NF-κB Activation and Interleukin-8 Secretion in Human Bronchial Epithelial Cells. J. Biol. Chem. 2004, 279, 41085–41094. [Google Scholar] [CrossRef] [PubMed]
- Adada, M.M.; Orr-Gandy, K.A.; Snider, A.J.; Canals, D.; Hannun, Y.A.; Obeid, L.M.; Clarke, C.J. Sphingosine Kinase 1 Regulates Tumor Necrosis Factor-mediated RANTES Induction through p38 Mitogen-activated Protein Kinase but Independently of Nuclear Factor κB Activation. J. Biol. Chem. 2013, 288, 27667–27679. [Google Scholar] [CrossRef] [PubMed]
- Mendez-Samperio, P.; Perez, A.; Trejo, A. Sphingosine kinase, phosphatidylinositol 3-kinase, Akt, NF-κB, and p300 are required for CCL5 production in Mycobacterium bovis Bacillus Calmette-Guérin (BCG)-infected epithelial cells. Cell. Immunol. 2007, 249, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Mendez-Samperio, P.; Trejo, A.; Perez, A. Mycobacterium bovis Bacillus Calmette-Guérin Induces CCL5 Secretion via the Toll-like receptor 2-NF-κB and -Jun N-Terminal Kinase Signaling Pathways. Clin. Vaccine Immunol. 2008, 15, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Anti-inflammatory agents: Present and future. Cell 2010, 140, 935–950. [Google Scholar] [CrossRef] [PubMed]
- Sedger, L.M.; McDermott, M.F. TNF and TNF-receptors: From mediators of cell death and inflammation to therapeutic giants: Past, present and future. Cytokine Growth Factor Rev. 2014, 25, 453–472. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, S.E.; Firestein, G.S. Mitogen activated protein kinase inhibitors: Where are we now and where are we going? Ann. Rheum. Dis. 2006, 65, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Wadgaonkar, R.; Patel, V.; Grinkina, N.; Romano, C.; Liu, J.; Zhao, Y.; Sammani, S.; Garcia, J.G.; Natarajan, V. Differential regulation of sphingosine kinases 1 and 2 in lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 296, 603–613. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Kalari, S.K.; Usatyuk, P.V.; Gorshkova, I.; He, D.; Watkins, T.; Brindley, D.N.; Sun, C.; Bittman, R.; Garcia, J.G.; et al. Intracellular generation of sphingosine 1-phosphate in human lung endothelial cells: Role of lipid phosphate phosphatase-1 and sphingosine kinase 1. J. Biol. Chem. 2007, 282, 14165–14177. [Google Scholar] [CrossRef] [PubMed]
- Oskeritzian, C.A.; Alvarez, S.E.; Hait, N.C.; Price, M.M.; Milstien, S.; Spiegel, S. Distinct roles of sphingosine kinases 1 and 2 in human mast-cell functions. Blood 2008, 111, 4193–4200. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.; Wilson, P.; Brandewie, K.; Taneja, D.; Schaefer, L.; Mitchell, B.; Tannock, L.R. Renal accumulation of biglycan and lipid retention accelerates diabetic nephropathy. Am. J. Pathol. 2011, 179, 1179–1187. [Google Scholar] [CrossRef] [PubMed]
- Gulyas, M.; Dobra, K.; Hjerpe, A. Expression of genes coding for proteoglycans and Wilms’ tumour susceptibility gene 1 (WT1) by variously differentiated benign human mesothelial cells. Differentiation 1999, 65, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Nastase, M.V.; Iozzo, R.V.; Schaefer, L. Key roles for the small leucine-rich proteoglycans in renal and pulmonary pathophysiology. Biochem. Biophys. Acta 2014, 1840, 2460–2470. [Google Scholar] [CrossRef]
- Yaghobian, D.; Don, A.S.; Yaghobian, S.; Chen, X.; Pollock, C.A.; Saad, S. Increased sphingosine 1-phosphate mediates inflammation and fibrosis in tubular injury in diabetic nephropathy. Clin. Exp. Pharmacol. Physiol. 2016, 43, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.; Babelova, A.; Moreth, K.; Xin, C.; Eberhardt, W.; Doller, A.; Pavenstadt, H.; Schaefer, L.; Pfeilschifter, J.; Huwiler, A. Transforming growth factor-β2 upregulates sphingosine kinase-1 activity, which in turn attenuates the fibrotic response to TGF-β2 by impeding CTGF expression. Kidney Int. 2009, 76, 857–867. [Google Scholar] [CrossRef] [PubMed]
- Koch, A.; Pfeilschifter, J.; Huwiler, A. Sphingosine 1-phosphate in renal diseases. Cell. Physiol. Biochem. 2013, 31, 745–760. [Google Scholar] [CrossRef] [PubMed]
- Schwalm, S.; Pfeilschifter, J.; Huwiler, A. Sphingosine-1-phosphate: A janus-faced mediator of fibrotic diseases. Biochim. Biophys. Acta 2013, 1831, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Bartels, K.; Grenz, A.; Eltzschig, H.K. Sphingosine-1-phosphate receptor signaling during acute kidney injury: The tissue is the issue. Kidney Int. 2014, 85, 733–735. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.K.; Bajwa, A.; Ye, H.; Vergis, A.L.; Awad, A.S.; Kharel, Y.; Lynch, K.R.; Okusa, M.D. Divergent roles of sphingosine kinases in kidney ischemia-reperfusion injury. Kidney Int. 2009, 75, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Park, S.W.; Kim, M.; Kim, M.; d’Agati, V.D.; Lee, H.T. Sphingosine kinase 1 protects against renal ischemia-reperfusion injury in mice by sphingosine-1-phosphate1 receptor activation. Kidney Int. 2011, 80, 1315–1327. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsieh, L.T.-H.; Nastase, M.-V.; Roedig, H.; Zeng-Brouwers, J.; Poluzzi, C.; Schwalm, S.; Fork, C.; Tredup, C.; Brandes, R.P.; Wygrecka, M.; et al. Biglycan- and Sphingosine Kinase-1 Signaling Crosstalk Regulates the Synthesis of Macrophage Chemoattractants. Int. J. Mol. Sci. 2017, 18, 595. https://doi.org/10.3390/ijms18030595
Hsieh LT-H, Nastase M-V, Roedig H, Zeng-Brouwers J, Poluzzi C, Schwalm S, Fork C, Tredup C, Brandes RP, Wygrecka M, et al. Biglycan- and Sphingosine Kinase-1 Signaling Crosstalk Regulates the Synthesis of Macrophage Chemoattractants. International Journal of Molecular Sciences. 2017; 18(3):595. https://doi.org/10.3390/ijms18030595
Chicago/Turabian StyleHsieh, Louise Tzung-Harn, Madalina-Viviana Nastase, Heiko Roedig, Jinyang Zeng-Brouwers, Chiara Poluzzi, Stephanie Schwalm, Christian Fork, Claudia Tredup, Ralf P. Brandes, Malgorzata Wygrecka, and et al. 2017. "Biglycan- and Sphingosine Kinase-1 Signaling Crosstalk Regulates the Synthesis of Macrophage Chemoattractants" International Journal of Molecular Sciences 18, no. 3: 595. https://doi.org/10.3390/ijms18030595
APA StyleHsieh, L. T.-H., Nastase, M.-V., Roedig, H., Zeng-Brouwers, J., Poluzzi, C., Schwalm, S., Fork, C., Tredup, C., Brandes, R. P., Wygrecka, M., Huwiler, A., Pfeilschifter, J., & Schaefer, L. (2017). Biglycan- and Sphingosine Kinase-1 Signaling Crosstalk Regulates the Synthesis of Macrophage Chemoattractants. International Journal of Molecular Sciences, 18(3), 595. https://doi.org/10.3390/ijms18030595