Advances in Proteomic Techniques for Cytokine Analysis: Focus on Melanoma Research


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction to Melanoma and Cytokines
1.1. Melanoma
1.2. Spontaneous Regression
1.3. Melanoma Treatment and Immunotherapies
1.4. Tumour Microenvironment
1.5. Cytokines
2. Cytokine Detection Techniques
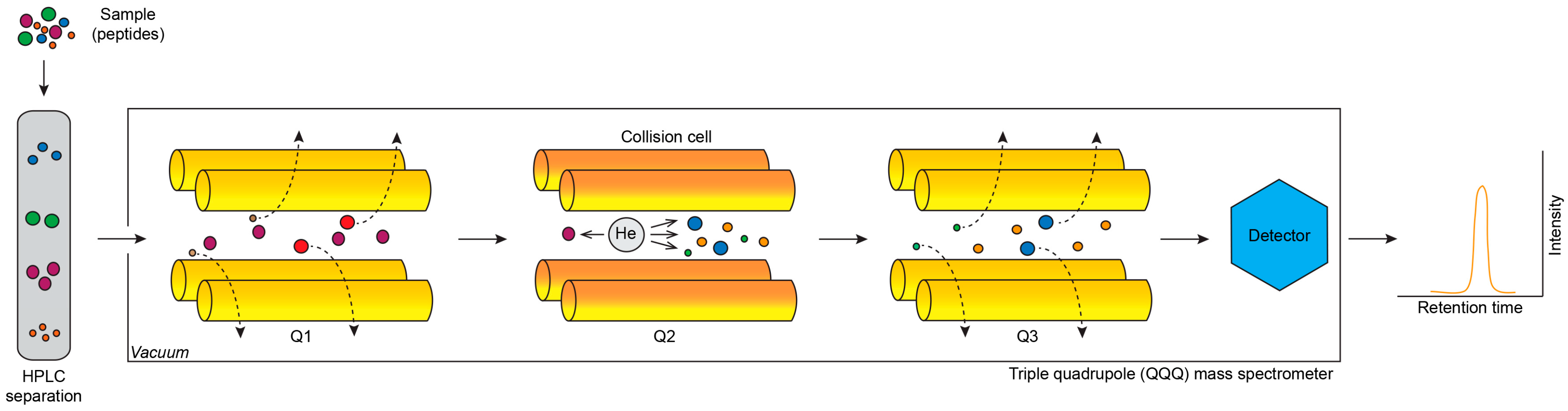
2.1. Mass Spectrometry
2.1.1. Data Dependent Acquisition (DDA)—Shotgun Proteomics
2.1.2. Data Independent Acquisition
2.1.3. Selected Reaction Monitoring and Multiple Reaction Monitoring
2.1.4. Immunoassays Coupled with MS Analyses
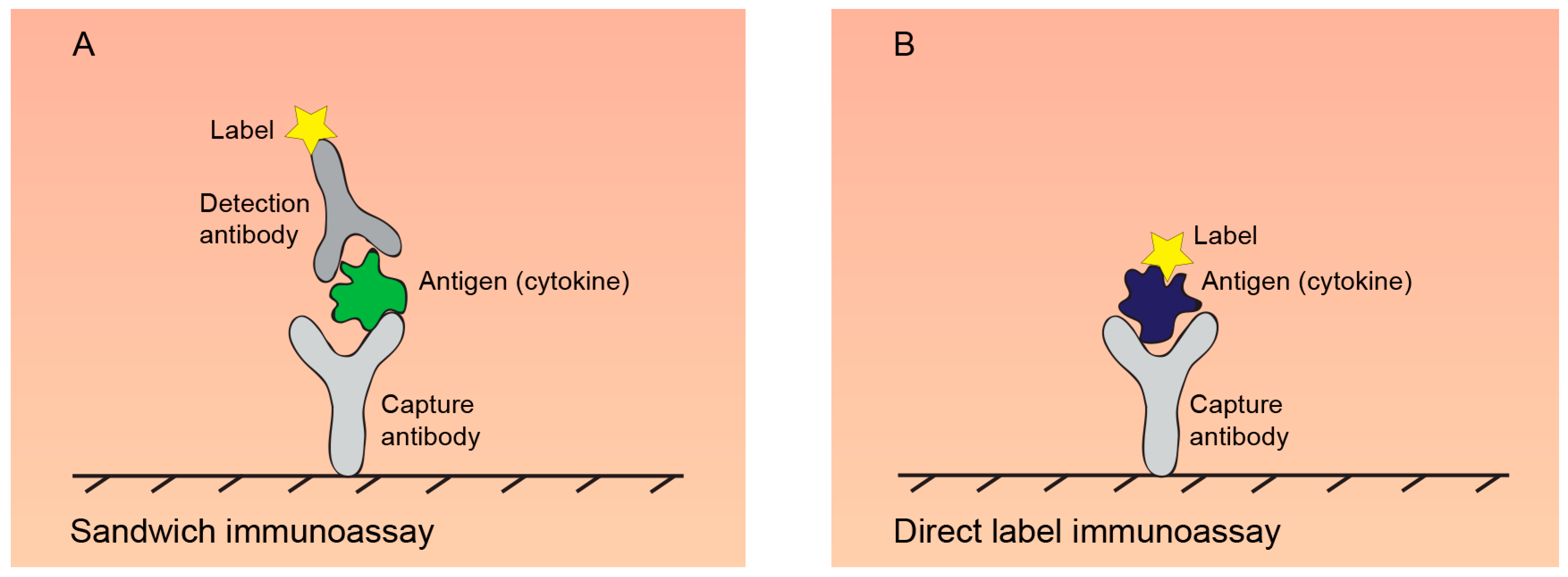
2.2. Immunoassays for Quantification of Secreted Cytokines
2.2.1. ELISA
2.2.2. Western Blot
2.2.3. Electrochemiluminescence Immunoassays
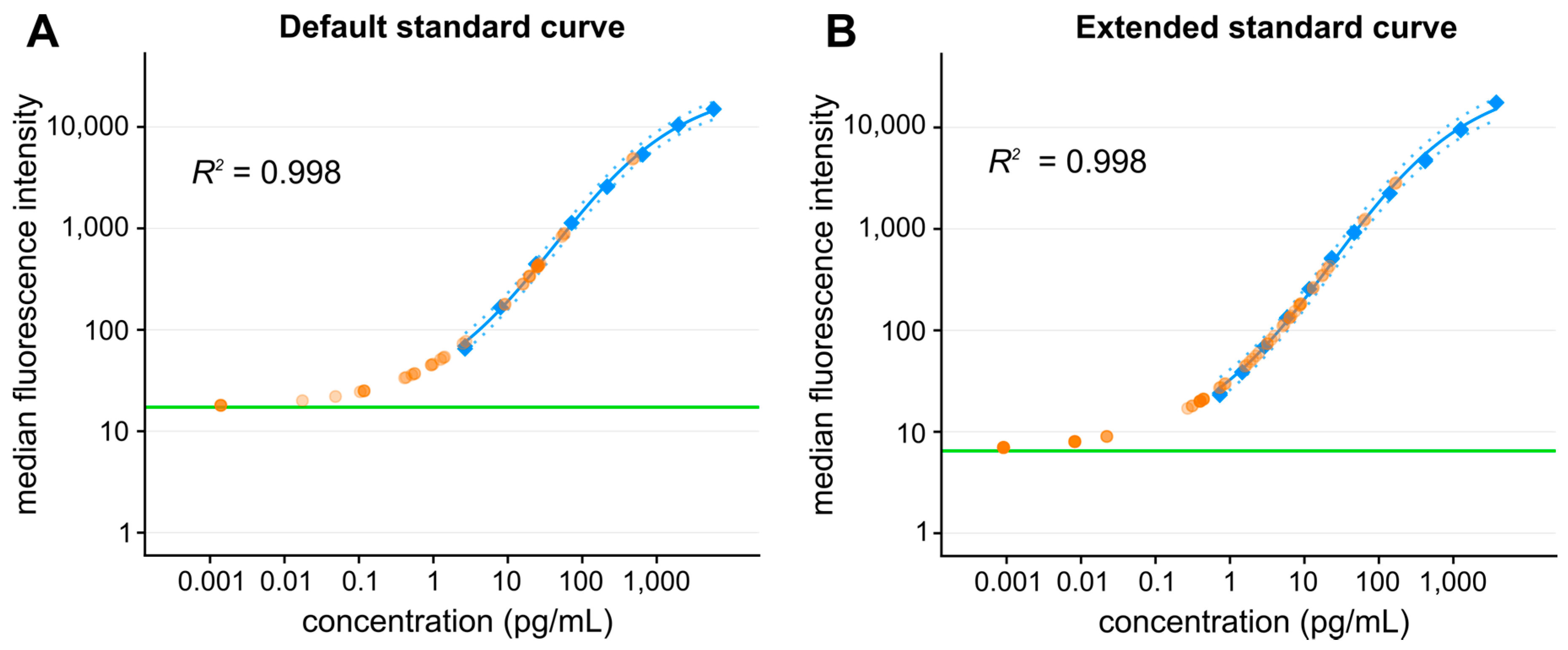
2.2.4. Antibody Arrays
Bead-Based Arrays
Planar Antibody Arrays
2.3. Emerging Techniques for Ultrasensitive Detection of Secreted Cytokines
2.3.1. Single Molecule Counting (Singulex)
2.3.2. Single Molecule Array (Simoa)
2.3.3. Immuno-PCR
2.3.4. Proximity Ligation Assay
2.3.5. Proximity Extension Assay
2.3.6. ImmunoMagnetic Reduction Assay
2.4. Single-Cell Analyses
2.4.1. ELISpot
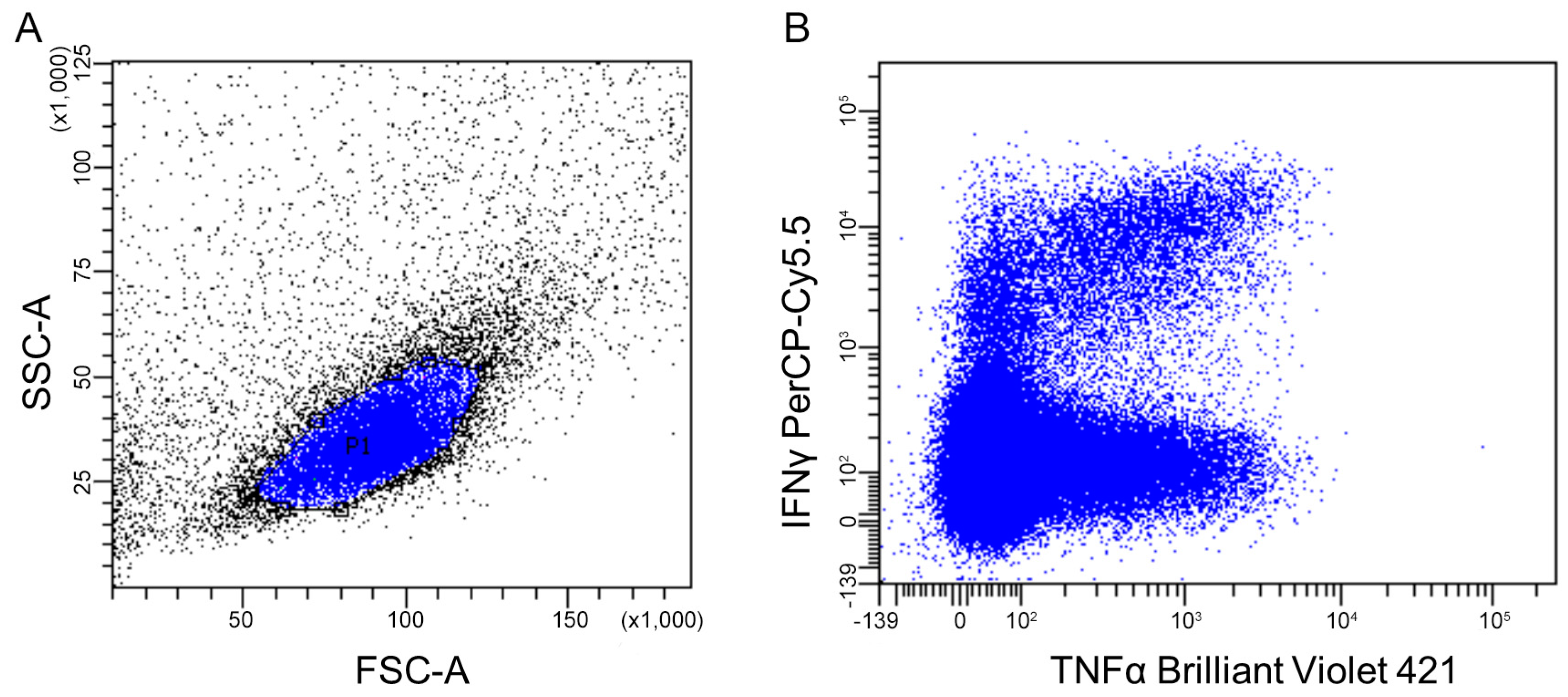
2.4.2. Flow Cytometry
2.4.3. Mass Cytometry
2.4.4. Single Cell Arrays
2.5. Other Techniques for Cytokine Detection
3. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| bFGF | basic fibroblast growth factor |
| CAFs | cancer-associated fibroblasts |
| CCL | C-C motif chemokine |
| CTLA-4 | T-lymphocyte-associated antigen 4 |
| CXCL1 | chemokine (C-X-C motif) ligand 1 (GROα) |
| DDA | data dependent acquisition |
| DIA | data independent acquisition |
| DIADI | direct immunoaffinity desorption/ionization |
| ECL | electrochemiluminescence |
| ELISpot | enzyme-linked immunospot assay |
| EVs | extracellular vesicles |
| G-CSF | granulocyte-colony stimulating factor |
| GDF15 | growth/differentiation factor 15 |
| GM-CSF | granulocyte-macrophage colony-stimulating factor |
| GROα | GRO1 oncogene |
| HGF | hepatocyte growth factor |
| HPLC | high-performance liquid chromatography |
| IFN | interferon |
| IGF | insulin-like growth factor |
| IL | interleukin |
| IL-10 | interferon gamma-induced protein 10 |
| IL1-ra | interleukin-1 receptor antagonist |
| IMR | immunomagnetic reduction |
| KGF | keratinocyte growth factor |
| MALDI | matrix-assisted laser desorption ionisation |
| MART-1 | melanoma-associated antigen recognized by T cells 1 |
| MCP-1 | monocyte chemoattractant protein 1 |
| M-CSF | macrophage colony-stimulating factor |
| MIF | macrophage migration inhibitory factor |
| MIP-1β | macrophage inflammatory protein-1β |
| MRM | multiple reaction monitoring |
| MS | mass spectrometry |
| MS/MS | tandem mass spectrometry |
| NK | natural killer |
| NRG-1 | neuregulin-1 |
| PBMCs | peripheral blood mononuclear cells |
| PD-1 | programmed cell-death protein 1 |
| PDGF | platelet-derived growth factor |
| PD-ligand | ligand of PD-1 receptor |
| PEA | proximity extension assay |
| PLA | proximity ligation assay |
| SCF | stem cell factor |
| Simoa | single molecule array |
| SISCAPA | stable isotope standards and capture by anti-peptide antibodies |
| SMC | single molecule counting |
| SRM | selected reaction monitoring |
| SWATH-MS | sequential windowed acquisition of all theoretical mass spectra |
| TAMs | tumour-associated macrophages |
| TARC | thymus and activation regulated chemokine (CCL17) |
| TGFβ | transforming growth factor-β |
| TNFα | tumour necrosis factor-α |
| Tregs | regulatory T-cells |
| VEGF-A | vascular endothelial growth factor A |
References
- Ferlay, J.; Steliarova-Foucher, E.; Lortet-Tieulent, J.; Rosso, S.; Coebergh, J.W.W.; Comber, H.; Forman, D.; Bray, F. Cancer incidence and mortality patterns in Europe: Estimates for 40 countries in 2012. Eur. J. Cancer 2013, 49, 1374–1403. [Google Scholar] [CrossRef] [PubMed]
- Guy, G.P., Jr.; Thomas, C.C.; Thompson, T.; Watson, M.; Massetti, G.M.; Richardson, L.C. Vital signs: Melanoma incidence and mortality trends and projections—United States, 1982–2030. Morb. Mortal. Wkly. Rep. 2015, 64, 591–596. [Google Scholar] [PubMed]
- Dunki-Jacobs, E.M.; Callender, G.G.; McMasters, K.M. Current management of melanoma. Curr. Probl. Surg. 2013, 50, 351–382. [Google Scholar] [CrossRef] [PubMed]
- Lo, J.A.; Fisher, D.E. The melanoma revolution: From UV carcinogenesis to a new era in therapeutics. Science 2014, 346, 945–949. [Google Scholar] [CrossRef] [PubMed]
- Ali, Z.; Yousaf, N.; Larkin, J. Melanoma epidemiology, biology and prognosis. EJC Suppl. EJC Off. J. EORTC Eur. Organ. Res. Treat. Cancer Al 2013, 11, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Gilchrest, B.A.; Eller, M.S.; Geller, A.C.; Yaar, M. The pathogenesis of melanoma induced by ultraviolet radiation. N. Engl. J. Med. 1999, 340, 1341–1348. [Google Scholar] [CrossRef] [PubMed]
- Lea, C.S.; Scotto, J.A.; Buffler, P.A.; Fine, J.; Barnhill, R.L.; Berwick, M. Ambient UVB and melanoma risk in the United States: A case-control analysis. Ann. Epidemiol. 2007, 17, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Rivers, J.K. Melanoma. Lancet 1996, 347, 803–806. [Google Scholar] [CrossRef]
- Beaumont, K.A.; Mohana-Kumaran, N.; Haass, N.K. Modeling Melanoma In Vitro and In Vivo. Healthcare 2013, 2, 27–46. [Google Scholar] [CrossRef] [PubMed]
- Kuzu, O.F.; Nguyen, F.D.; Noory, M.A.; Sharma, A. Current State of Animal (Mouse) Modeling in Melanoma Research. Cancer Growth Metastasis 2015, 8, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Van der Weyden, L.; Patton, E.E.; Wood, G.A.; Foote, A.K.; Brenn, T.; Arends, M.J.; Adams, D.J. Cross-species models of human melanoma. J. Pathol. 2016, 238, 152–165. [Google Scholar] [CrossRef] [PubMed]
- Bourneuf, E. The MeLiM Minipig: An Original Spontaneous Model to Explore Cutaneous Melanoma Genetic Basis. Front. Genet. 2017, 8, 146. [Google Scholar] [CrossRef] [PubMed]
- Cole, W.H.; Everson, T.C. Spontaneous regression of cancer: Preliminary report. Ann. Surg. 1956, 144, 366–383. [Google Scholar] [PubMed]
- High, W.A.; Stewart, D.; Wilbers, C.R.H.; Cockerell, C.J.; Hoang, M.P.; Fitzpatrick, J.E. Completely regressed primary cutaneous malignant melanoma with nodal and/or visceral metastases: A report of 5 cases and assessment of the literature and diagnostic criteria. J. Am. Acad. Dermatol. 2005, 53, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Blessing, K.; McLaren, K.M. Histological regression in primary cutaneous melanoma: Recognition, prevalence and significance. Histopathology 1992, 20, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Haanen, J.B.A.G. Immunotherapy of melanoma. EJC Suppl. EJC Off. J. EORTC Eur. Organ. Res. Treat. Cancer Al 2013, 11, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Kalialis, L.V.; Drzewiecki, K.T.; Klyver, H. Spontaneous regression of metastases from melanoma: Review of the literature. Melanoma Res. 2009, 19, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Aung, P.P.; Nagarajan, P.; Prieto, V.G. Regression in primary cutaneous melanoma: Etiopathogenesis and clinical significance. Lab. Investig. J. Tech. Methods Pathol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Maio, M. Melanoma as a model tumour for immuno-oncology. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2012, 23 (Suppl. 8), viii10–viii14. [Google Scholar] [CrossRef] [PubMed]
- Martín, J.M.; Pinazo, I.; Mateo, J.F.; Escandell, I.; Jordá, E.; Monteagudo, C. Assessment of regression in successive primary melanomas. Actas Dermosifiliogr. 2014, 105, 768–773. [Google Scholar] [CrossRef] [PubMed]
- Creagan, E.T.; Ahmann, D.L.; Green, S.J.; Long, H.J.; Frytak, S.; O’Fallon, J.R.; Itri, L.M. Phase II study of low-dose recombinant leukocyte A interferon in disseminated malignant melanoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 1984, 2, 1002–1005. [Google Scholar] [CrossRef] [PubMed]
- Robinson, W.A.; Mughal, T.I.; Thomas, M.R.; Johnson, M.; Spiegel, R.J. Treatment of metastatic malignant melanoma with recombinant interferon alpha 2. Immunobiology 1986, 172, 275–282. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Lotze, M.T.; Muul, L.M.; Chang, A.E.; Avis, F.P.; Leitman, S.; Linehan, W.M.; Robertson, C.N.; Lee, R.E.; Rubin, J.T. A progress report on the treatment of 157 patients with advanced cancer using lymphokine-activated killer cells and interleukin-2 or high-dose interleukin-2 alone. N. Engl. J. Med. 1987, 316, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Dutcher, J.P.; Creekmore, S.; Weiss, G.R.; Margolin, K.; Markowitz, A.B.; Roper, M.; Parkinson, D.; Ciobanu, N.; Fisher, R.I.; Boldt, D.H. A phase II study of interleukin-2 and lymphokine-activated killer cells in patients with metastatic malignant melanoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 1989, 7, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Maker, A.V.; Phan, G.Q.; Attia, P.; Yang, J.C.; Sherry, R.M.; Topalian, S.L.; Kammula, U.S.; Royal, R.E.; Haworth, L.R.; Levy, C.; et al. Tumor regression and autoimmunity in patients treated with cytotoxic T lymphocyte-associated antigen 4 blockade and interleukin 2: A phase I/II study. Ann. Surg. Oncol. 2005, 12, 1005–1016. [Google Scholar] [CrossRef] [PubMed]
- Aris, M.; Mordoh, J.; Barrio, M.M. Immunomodulatory Monoclonal Antibodies in Combined Immunotherapy Trials for Cutaneous Melanoma. Front. Immunol. 2017, 8, 1024. [Google Scholar] [CrossRef] [PubMed]
- Ryu, S.; Youn, C.; Moon, A.R.; Howland, A.; Armstrong, C.A.; Song, P.I. Therapeutic Inhibitors against Mutated BRAF and MEK for the Treatment of Metastatic Melanoma. Chonnam Med. J. 2017, 53, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Dudley, M.E.; Wunderlich, J.R.; Robbins, P.F.; Yang, J.C.; Hwu, P.; Schwartzentruber, D.J.; Topalian, S.L.; Sherry, R.; Restifo, N.P.; Hubicki, A.M.; et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science 2002, 298, 850–854. [Google Scholar] [CrossRef] [PubMed]
- Atkins, M.B.; Hsu, J.; Lee, S.; Cohen, G.I.; Flaherty, L.E.; Sosman, J.A.; Sondak, V.K.; Kirkwood, J.M.; Eastern Cooperative Oncology Group. Phase III trial comparing concurrent biochemotherapy with cisplatin, vinblastine, dacarbazine, interleukin-2 and interferon alfa-2b with cisplatin, vinblastine and dacarbazine alone in patients with metastatic malignant melanoma (E3695): A trial coordinated by the Eastern Cooperative Oncology Group. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2008, 26, 5748–5754. [Google Scholar] [CrossRef]
- Seung, S.K.; Curti, B.D.; Crittenden, M.; Walker, E.; Coffey, T.; Siebert, J.C.; Miller, W.; Payne, R.; Glenn, L.; Bageac, A.; et al. Phase 1 study of stereotactic body radiotherapy and interleukin-2—Tumor and immunological responses. Sci. Transl. Med. 2012, 4, 137ra74. [Google Scholar] [CrossRef] [PubMed]
- Sosman, J.A.; Carrillo, C.; Urba, W.J.; Flaherty, L.; Atkins, M.B.; Clark, J.I.; Dutcher, J.; Margolin, K.A.; Mier, J.; Gollob, J.; et al. Three phase II cytokine working group trials of gp100 (210M) peptide plus high-dose interleukin-2 in patients with HLA-A2-positive advanced melanoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2008, 26, 2292–2298. [Google Scholar] [CrossRef] [PubMed]
- Schwartzentruber, D.J.; Lawson, D.H.; Richards, J.M.; Conry, R.M.; Miller, D.M.; Treisman, J.; Gailani, F.; Riley, L.; Conlon, K.; Pockaj, B.; et al. gp100 peptide vaccine and interleukin-2 in patients with advanced melanoma. N. Engl. J. Med. 2011, 364, 2119–2127. [Google Scholar] [CrossRef] [PubMed]
- Luke, J.J.; Flaherty, K.T.; Ribas, A.; Long, G.V. Targeted agents and immunotherapies: Optimizing outcomes in melanoma. Nat. Rev. Clin. Oncol. 2017, 14, 463–482. [Google Scholar] [CrossRef] [PubMed]
- Dvořánková, B.; Szabo, P.; Kodet, O.; Strnad, H.; Kolář, M.; Lacina, L.; Krejčí, E.; Naňka, O.; Šedo, A.; Smetana, K. Intercellular crosstalk in human malignant melanoma. Protoplasma 2017, 254, 1143–1150. [Google Scholar] [CrossRef] [PubMed]
- Lacina, L.; Kodet, O.; Dvořánková, B.; Szabo, P.; Smetana, K. Ecology of melanoma cell. Histol. Histopathol. 2018, 247–254. [Google Scholar] [CrossRef]
- Lacina, L.; Plzak, J.; Kodet, O.; Szabo, P.; Chovanec, M.; Dvorankova, B.; Smetana, K. Cancer Microenvironment: What Can We Learn from the Stem Cell Niche. Int. J. Mol. Sci. 2015, 16, 24094–24110. [Google Scholar] [CrossRef] [PubMed]
- Paulitschke, V.; Kunstfeld, R.; Mohr, T.; Slany, A.; Micksche, M.; Drach, J.; Zielinski, C.; Pehamberger, H.; Gerner, C. Entering a new era of rational biomarker discovery for early detection of melanoma metastases: Secretome analysis of associated stroma cells. J. Proteome Res. 2009, 8, 2501–2510. [Google Scholar] [CrossRef] [PubMed]
- D’Orazio, J.; Jarrett, S.; Amaro-Ortiz, A.; Scott, T. UV radiation and the skin. Int. J. Mol. Sci. 2013, 14, 12222–12248. [Google Scholar] [CrossRef] [PubMed]
- Kodet, O.; Lacina, L.; Krejčí, E.; Dvořánková, B.; Grim, M.; Štork, J.; Kodetová, D.; Vlček, Č.; Šáchová, J.; Kolář, M.; et al. Melanoma cells influence the differentiation pattern of human epidermal keratinocytes. Mol. Cancer 2015, 14, 1. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Viennet, C.; Robin, S.; Berthon, J.-Y.; He, L.; Humbert, P. Precise role of dermal fibroblasts on melanocyte pigmentation. J. Dermatol. Sci. 2017, 88, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Kolář, M.; Szabo, P.; Dvořánková, B.; Lacina, L.; Gabius, H.-J.; Strnad, H.; Sáchová, J.; Vlček, C.; Plzák, J.; Chovanec, M.; et al. Upregulation of IL-6, IL-8 and CXCL-1 production in dermal fibroblasts by normal/malignant epithelial cells in vitro: Immunohistochemical and transcriptomic analyses. Biol. Cell 2012, 104, 738–751. [Google Scholar] [CrossRef] [PubMed]
- Jobe, N.P.; Rösel, D.; Dvořánková, B.; Kodet, O.; Lacina, L.; Mateu, R.; Smetana, K.; Brábek, J. Simultaneous blocking of IL-6 and IL-8 is sufficient to fully inhibit CAF-induced human melanoma cell invasiveness. Histochem. Cell Biol. 2016, 146, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Hoejberg, L.; Bastholt, L.; Schmidt, H. Interleukin-6 and melanoma. Melanoma Res. 2012, 22, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Singh, A.P.; Sharma, B.; Owen, L.B.; Singh, R.K. CXCL8 and its cognate receptors in melanoma progression and metastasis. Future Oncol. 2010, 6, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Kučera, J.; Dvořánková, B.; Smetana, K.; Szabo, P.; Kodet, O. Fibroblasts isolated from the malignant melanoma influence phenotype of normal human keratinocytes. J. Appl. Biomed. 2015, 13, 195–198. [Google Scholar] [CrossRef]
- Gasser, S.; Lim, L.H.K.; Cheung, F.S.G. The role of the tumour microenvironment in immunotherapy. Endocr. Relat. Cancer 2017, 24, T283–T295. [Google Scholar] [CrossRef] [PubMed]
- Fløe, A.; Løppke, C.; Hilberg, O.; Wejse, C.; Brix, L.; Jacobsen, K. Development of an epitope panel for consistent identification of antigen-specific T-cells in humans. Immunology 2017, 152, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Zikich, D.; Schachter, J.; Besser, M.J. Predictors of tumor-infiltrating lymphocyte efficacy in melanoma. Immunotherapy 2016, 8, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, Z.; Wu, H.; Li, L.; Luo, Y.; Li, X.; Huang, G. Regulatory T cells in the immunotherapy of melanoma. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2016, 37, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Fujimura, T.; Kakizaki, A.; Furudate, S.; Kambayashi, Y.; Aiba, S. Tumor-associated macrophages in skin: How to treat their heterogeneity and plasticity. J. Dermatol. Sci. 2016, 83, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Mignogna, C.; Scali, E.; Camastra, C.; Presta, I.; Zeppa, P.; Barni, T.; Donato, G.; Bottoni, U.; Di Vito, A. Innate immunity in cutaneous melanoma. Clin. Exp. Dermatol. 2017, 42, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Tarazona, R.; Duran, E.; Solana, R. Natural Killer Cell Recognition of Melanoma: New Clues for a More Effective Immunotherapy. Front. Immunol. 2015, 6, 649. [Google Scholar] [CrossRef] [PubMed]
- Saadeh, D.; Kurban, M.; Abbas, O. Plasmacytoid dendritic cell role in cutaneous malignancies. J. Dermatol. Sci. 2016, 83, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Chiaruttini, G.; Mele, S.; Opzoomer, J.; Crescioli, S.; Ilieva, K.M.; Lacy, K.E.; Karagiannis, S.N. B cells and the humoral response in melanoma: The overlooked players of the tumor microenvironment. Oncoimmunology 2017, 6, e1294296. [Google Scholar] [CrossRef] [PubMed]
- Weidle, U.H.; Birzele, F.; Kollmorgen, G.; Rüger, R. The Multiple Roles of Exosomes in Metastasis. Cancer Genom. Proteom. 2017, 14, 1–15. [Google Scholar] [CrossRef] [PubMed]
- O’Loghlen, A. Role for extracellular vesicles in the tumour microenvironment. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018, 373. [Google Scholar] [CrossRef] [PubMed]
- Romano, G.; Kwong, L.N. miRNAs, Melanoma and Microenvironment: An Intricate Network. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Ratnikov, B.I.; Scott, D.A.; Osterman, A.L.; Smith, J.W.; Ronai, Z.A. Metabolic rewiring in melanoma. Oncogene 2017, 36, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Stenken, J.A.; Poschenrieder, A.J. Bioanalytical chemistry of cytokines—A review. Anal. Chim. Acta 2015, 853, 95–115. [Google Scholar] [CrossRef] [PubMed]
- Yao, M.; Brummer, G.; Acevedo, D.; Cheng, N. Cytokine Regulation of Metastasis and Tumorigenicity. Adv. Cancer Res. 2016, 132, 265–367. [Google Scholar] [CrossRef] [PubMed]
- Atretkhany, K.-S.N.; Drutskaya, M.S.; Nedospasov, S.A.; Grivennikov, S.I.; Kuprash, D.V. Chemokines, cytokines and exosomes help tumors to shape inflammatory microenvironment. Pharmacol. Ther. 2016, 168, 98–112. [Google Scholar] [CrossRef] [PubMed]
- Herraiz, C.; Jiménez-Cervantes, C.; Sánchez-Laorden, B.; García-Borrón, J.C. Functional interplay between secreted ligands and receptors in melanoma. Semin. Cell Dev. Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Li, A.; Tian, Y.; Wu, J.D.; Liu, Y.; Li, T.; Chen, Y.; Han, X.; Wu, K. The CXCL8-CXCR1/2 pathways in cancer. Cytokine Growth Factor Rev. 2016, 31, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Sanmamed, M.F.; Carranza-Rua, O.; Alfaro, C.; Oñate, C.; Martín-Algarra, S.; Perez, G.; Landazuri, S.F.; Gonzalez, A.; Gross, S.; Rodriguez, I.; et al. Serum interleukin-8 reflects tumor burden and treatment response across malignancies of multiple tissue origins. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 5697–5707. [Google Scholar] [CrossRef] [PubMed]
- Alegre, E.; Sammamed, M.; Fernandez-Landazuri, S.; Zubiri, L.; Gonzalez, A. Circulating Biomarkers in Malignant Melanoma. In Advances in Clinical Chemistry; Elsevier: Amsterdam, The Netherlands, 2015; Volume 69, pp. 47–89. ISBN 978-0-12-802265-8. [Google Scholar]
- Filitis, D.C.; Rauh, J.; Mahalingam, M. The HGF-cMET signaling pathway in conferring stromal-induced BRAF-inhibitor resistance in melanoma. Melanoma Res. 2015, 25, 470–478. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Umitsu, M.; De Silva, D.M.; Roy, A.; Bottaro, D.P. Hepatocyte growth factor/MET in cancer progression and biomarker discovery. Cancer Sci. 2017, 108, 296–307. [Google Scholar] [CrossRef] [PubMed]
- Lok, E.; Chung, A.S.; Swanson, K.D.; Wong, E.T. Melanoma brain metastasis globally reconfigures chemokine and cytokine profiles in patient cerebrospinal fluid. Melanoma Res. 2014, 24, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Najjar, Y.G.; Ding, F.; Lin, Y.; VanderWeele, R.; Butterfield, L.H.; Tarhini, A.A. Melanoma antigen-specific effector T cell cytokine secretion patterns in patients treated with ipilimumab. J. Transl. Med. 2017, 15, 39. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.H.; Zhu, Z.; Xiao, H.; Wakefield, M.R.; Bai, Q.; Nicholl, M.B.; Ding, V.A.; Fang, Y. Unveil the mysterious mask of cytokine-based immunotherapy for melanoma. Cancer Lett. 2017, 394, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Zhou, C.; Ren, S. Role of IL-2 in cancer immunotherapy. Oncoimmunology 2016, 5, e1163462. [Google Scholar] [CrossRef] [PubMed]
- Ives, N.J.; Suciu, S.; Eggermont, A.M.M.; Kirkwood, J.; Lorigan, P.; Markovic, S.N.; Garbe, C.; Wheatley, K.; International Melanoma Meta-Analysis Collaborative Group (IMMCG). Adjuvant interferon-α for the treatment of high-risk melanoma: An individual patient data meta-analysis. Eur. J. Cancer 2017, 82, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Hoeller, C.; Michielin, O.; Ascierto, P.A.; Szabo, Z.; Blank, C.U. Systematic review of the use of granulocyte-macrophage colony-stimulating factor in patients with advanced melanoma. Cancer Immunol. Immunother. 2016, 65, 1015–1034. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Qi, M.; Hutchinson, M.R.; Yang, G.; Goldys, E.M. Recent advances in cytokine detection by immunosensing. Biosens. Bioelectron. 2016, 79, 810–821. [Google Scholar] [CrossRef] [PubMed]
- Kulbe, H.; Chakravarty, P.; Leinster, D.A.; Charles, K.A.; Kwong, J.; Thompson, R.G.; Coward, J.I.; Schioppa, T.; Robinson, S.C.; Gallagher, W.M.; et al. A dynamic inflammatory cytokine network in the human ovarian cancer microenvironment. Cancer Res. 2012, 72, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, T.; Mann, M.; Aebersold, R.; Yates, J.R.; Bairoch, A.; Bergeron, J.J.M. Mass spectrometry in high-throughput proteomics: Ready for the big time. Nat. Methods 2010, 7, 681–685. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.L.; Anderson, N.G. The human plasma proteome: History, character and diagnostic prospects. Mol. Cell. Proteom. 2002, 1, 845–867. [Google Scholar] [CrossRef] [PubMed]
- Rocco, M.; Malorni, L.; Cozzolino, R.; Palmieri, G.; Rozzo, C.; Manca, A.; Parente, A.; Chambery, A. Proteomic profiling of human melanoma metastatic cell line secretomes. J. Proteome Res. 2011, 10, 4703–4714. [Google Scholar] [CrossRef] [PubMed]
- Alečković, M.; Wei, Y.; LeRoy, G.; Sidoli, S.; Liu, D.D.; Garcia, B.A.; Kang, Y. Identification of Nidogen 1 as a lung metastasis protein through secretome analysis. Genes Dev. 2017, 31, 1439–1455. [Google Scholar] [CrossRef] [PubMed]
- Boyle, G.M.; Pedley, J.; Martyn, A.C.; Banducci, K.J.; Strutton, G.M.; Brown, D.A.; Breit, S.N.; Parsons, P.G. Macrophage inhibitory cytokine-1 is overexpressed in malignant melanoma and is associated with tumorigenicity. J. Investig. Dermatol. 2009, 129, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Rosenberger, G.; Koh, C.C.; Guo, T.; Röst, H.L.; Kouvonen, P.; Collins, B.C.; Heusel, M.; Liu, Y.; Caron, E.; Vichalkovski, A.; et al. A repository of assays to quantify 10,000 human proteins by SWATH-MS. Sci. Data 2014, 1, 140031. [Google Scholar] [CrossRef] [PubMed]
- Collins, B.C.; Hunter, C.L.; Liu, Y.; Schilling, B.; Rosenberger, G.; Bader, S.L.; Chan, D.W.; Gibson, B.W.; Gingras, A.-C.; Held, J.M.; et al. Multi-laboratory assessment of reproducibility, qualitative and quantitative performance of SWATH-mass spectrometry. Nat. Commun. 2017, 8, 291. [Google Scholar] [CrossRef] [PubMed]
- Anjo, S.I.; Santa, C.; Manadas, B. SWATH-MS as a tool for biomarker discovery: From basic research to clinical applications. Proteomics 2017, 17. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Lim, H.S.R.; Lin, H.L.; Tan, H.T.; Lim, T.K.; Cheong, W.K.; Cheah, P.Y.; Tang, C.L.; Chow, P.K.H.; Chung, M.C.M. Analysis of colorectal cancer glyco-secretome identifies laminin β-1 (LAMB1) as a potential serological biomarker for colorectal cancer. Proteomics 2015, 15, 3905–3920. [Google Scholar] [CrossRef] [PubMed]
- Manfredi, M.; Martinotti, S.; Gosetti, F.; Ranzato, E.; Marengo, E. The secretome signature of malignant mesothelioma cell lines. J. Proteom. 2016, 145, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Addona, T.A.; Abbatiello, S.E.; Schilling, B.; Skates, S.J.; Mani, D.R.; Bunk, D.M.; Spiegelman, C.H.; Zimmerman, L.J.; Ham, A.-J.L.; Keshishian, H.; et al. Multi-site assessment of the precision and reproducibility of multiple reaction monitoring-based measurements of proteins in plasma. Nat. Biotechnol. 2009, 27, 633–641. [Google Scholar] [CrossRef] [PubMed]
- Percy, A.J.; Chambers, A.G.; Yang, J.; Hardie, D.B.; Borchers, C.H. Advances in multiplexed MRM-based protein biomarker quantitation toward clinical utility. Biochim. Biophys. Acta 2014, 1844, 917–926. [Google Scholar] [CrossRef] [PubMed]
- Parker, C.E.; Borchers, C.H. Mass spectrometry based biomarker discovery, verification and validation—Quality assurance and control of protein biomarker assays. Mol. Oncol. 2014, 8, 840–858. [Google Scholar] [CrossRef] [PubMed]
- Bredehöft, M.; Schänzer, W.; Thevis, M. Quantification of human insulin-like growth factor-1 and qualitative detection of its analogues in plasma using liquid chromatography/electrospray ionisation tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2008, 22, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.L.; Anderson, N.G.; Haines, L.R.; Hardie, D.B.; Olafson, R.W.; Pearson, T.W. Mass spectrometric quantitation of peptides and proteins using Stable Isotope Standards and Capture by Anti-Peptide Antibodies (SISCAPA). J. Proteome Res. 2004, 3, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, E.; Whiteaker, J.R.; Mani, D.R.; Jackson, A.M.; Zhao, L.; Pope, M.E.; Smith, D.; Rivera, K.D.; Anderson, N.L.; Skates, S.J.; et al. Interlaboratory evaluation of automated, multiplexed peptide immunoaffinity enrichment coupled to multiple reaction monitoring mass spectrometry for quantifying proteins in plasma. Mol. Cell. Proteom. 2012, 11, M111.013854. [Google Scholar] [CrossRef] [PubMed]
- Sherma, N.D.; Borges, C.R.; Trenchevska, O.; Jarvis, J.W.; Rehder, D.S.; Oran, P.E.; Nelson, R.W.; Nedelkov, D. Mass Spectrometric Immunoassay for the qualitative and quantitative analysis of the cytokine Macrophage Migration Inhibitory Factor (MIF). Proteome Sci. 2014, 12, 52. [Google Scholar] [CrossRef] [PubMed]
- Pompach, P.; Benada, O.; Rosůlek, M.; Darebná, P.; Hausner, J.; Růžička, V.; Volný, M.; Novák, P. Protein Chips Compatible with MALDI Mass Spectrometry Prepared by Ambient Ion Landing. Anal. Chem. 2016, 88, 8526–8534. [Google Scholar] [CrossRef] [PubMed]
- Pompach, P.; Nováková, J.; Kavan, D.; Benada, O.; Růžička, V.; Volný, M.; Novák, P. Planar Functionalized Surfaces for Direct Immunoaffinity Desorption/Ionization Mass Spectrometry. Clin. Chem. 2016, 62, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Engvall, E.; Jonsson, K.; Perlmann, P. Enzyme-linked immunosorbent assay. II. Quantitative assay of protein antigen, immunoglobulin G, by means of enzyme-labelled antigen and antibody-coated tubes. Biochim. Biophys. Acta 1971, 251, 427–434. [Google Scholar] [CrossRef]
- Van Weemen, B.K.; Schuurs, A.H.W.M. Immunoassay using antigen-enzyme conjugates. FEBS Lett. 1971, 15, 232–236. [Google Scholar] [CrossRef]
- Shah, K.; Maghsoudlou, P. Enzyme-linked immunosorbent assay (ELISA): The basics. Br. J. Hosp. Med. 2016, 77, C98–C101. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. Cytokine assays. BioTechniques 2002, 10, S4–S15. [Google Scholar]
- Towbin, H.; Staehelin, T.; Gordon, J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: Procedure and some applications. Proc. Natl. Acad. Sci. USA 1979, 76, 4350–4354. [Google Scholar] [CrossRef] [PubMed]
- Apte, R.N.; Dotan, S.; Elkabets, M.; White, M.R.; Reich, E.; Carmi, Y.; Song, X.; Dvozkin, T.; Krelin, Y.; Voronov, E. The involvement of IL-1 in tumorigenesis, tumor invasiveness, metastasis and tumor-host interactions. Cancer Metastasis Rev. 2006, 25, 387–408. [Google Scholar] [CrossRef] [PubMed]
- Schneider, K.S.; Thomas, C.J.; Groß, O. Inflammasome activation and inhibition in primary murine bone marrow-derived cells and assays for IL-1α, IL-1β and caspase-1. Methods Mol. Biol. 2013, 1040, 117–135. [Google Scholar] [CrossRef] [PubMed]
- Guey, B.; Petrilli, V. Assessing Caspase-1 Activation. Methods Mol. Biol. 2016, 1417, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Logan, P.; Burnier, J.; Burnier, M.N. Vascular endothelial growth factor expression and inhibition in uveal melanoma cell lines. Ecancermedicalscience 2013, 7, 336. [Google Scholar] [CrossRef] [PubMed]
- Gatla, H.R.; Singha, B.; Persaud, V.; Vancurova, I. Evaluating cytoplasmic and nuclear levels of inflammatory cytokines in cancer cells by western blotting. Methods Mol. Biol. 2014, 1172, 271–283. [Google Scholar] [CrossRef] [PubMed]
- Miskolci, V.; Hodgson, L.; Cox, D.; Vancurova, I. Western analysis of intracellular interleukin-8 in human mononuclear leukocytes. Methods Mol. Biol. 2014, 1172, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Wessendorf, J.H.; Garfinkel, S.; Zhan, X.; Brown, S.; Maciag, T. Identification of a nuclear localization sequence within the structure of the human interleukin-1 alpha precursor. J. Biol. Chem. 1993, 268, 22100–22104. [Google Scholar] [PubMed]
- Boraschi, D.; Lucchesi, D.; Hainzl, S.; Leitner, M.; Maier, E.; Mangelberger, D.; Oostingh, G.J.; Pfaller, T.; Pixner, C.; Posselt, G.; et al. IL-37: A new anti-inflammatory cytokine of the IL-1 family. Eur. Cytokine Netw. 2011, 22, 127–147. [Google Scholar] [CrossRef] [PubMed]
- Ross, R.; Grimmel, J.; Goedicke, S.; Möbus, A.M.; Bulau, A.-M.; Bufler, P.; Ali, S.; Martin, M.U. Analysis of nuclear localization of interleukin-1 family cytokines by flow cytometry. J. Immunol. Methods 2013, 387, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Bertheloot, D.; Latz, E. HMGB1, IL-1α, IL-33 and S100 proteins: Dual-function alarmins. Cell. Mol. Immunol. 2017, 14, 43–64. [Google Scholar] [CrossRef] [PubMed]
- Miskolci, V.; Ghosh, C.C.; Rollins, J.; Romero, C.; Vu, H.-Y.; Robinson, S.; Davidson, D.; Vancurova, I. TNFalpha release from peripheral blood leukocytes depends on a CRM1-mediated nuclear export. Biochem. Biophys. Res. Commun. 2006, 351, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Richter, M.M. Electrochemiluminescence (ECL). Chem. Rev. 2004, 104, 3003–3036. [Google Scholar] [CrossRef] [PubMed]
- Rhyne, P.W.; Wong, O.T.; Zhang, Y.J.; Weiner, R.S. Electrochemiluminescence in bioanalysis. Bioanalysis 2009, 1, 919–935. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Wang, E. Electrochemiluminescence of tris(2,2′-bipyridyl)ruthenium and its applications in bioanalysis: A review. Lumin. J. Biol. Chem. Lumin. 2011, 26, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Obenauer-Kutner, L.J.; Jacobs, S.J.; Kolz, K.; Tobias, L.M.; Bordens, R.W. A highly sensitive electrochemiluminescence immunoassay for interferon alfa-2b in human serum. J. Immunol. Methods 1997, 206, 25–33. [Google Scholar] [CrossRef]
- Hercules, D.M.; Lytle, F.E. Chemiluminescence from Reduction Reactions. J. Am. Chem. Soc. 1966, 88, 4745–4746. [Google Scholar] [CrossRef]
- Chowdhury, F.; Williams, A.; Johnson, P. Validation and comparison of two multiplex technologies, Luminex and Mesoscale Discovery, for human cytokine profiling. J. Immunol. Methods 2009, 340, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Zhu, J.; Van Eyk, J.E. Comparison of multiplex immunoassay platforms. Clin. Chem. 2010, 56, 314–318. [Google Scholar] [CrossRef] [PubMed]
- Dabitao, D.; Margolick, J.B.; Lopez, J.; Bream, J.H. Multiplex measurement of proinflammatory cytokines in human serum: Comparison of the Meso Scale Discovery electrochemiluminescence assay and the Cytometric Bead Array. J. Immunol. Methods 2011, 372, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Ryan, B.M.; Pine, S.R.; Chaturvedi, A.K.; Caporaso, N.; Harris, C.C. A combined prognostic serum interleukin-8 and interleukin-6 classifier for stage 1 lung cancer in the prostate, lung, colorectal and ovarian cancer screening trial. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2014, 9, 1494–1503. [Google Scholar] [CrossRef] [PubMed]
- Block, M.S.; Maurer, M.J.; Goergen, K.; Kalli, K.R.; Erskine, C.L.; Behrens, M.D.; Oberg, A.L.; Knutson, K.L. Plasma immune analytes in patients with epithelial ovarian cancer. Cytokine 2015, 73, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.W.; Zhou, Z.G.; Wang, M.; Dong, J.Q.; Du, K.P.; Li, S.; Liu, Y.L.; Lv, P.J.; Gao, J.B. Combination of IL-6, IL-10 and MCP-1 with traditional serum tumor markers in lung cancer diagnosis and prognosis. Genet. Mol. Res. 2016, 15. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, Y.; Furuya, H.; Bryant Greenwood, P.; Chan, O.; Dai, Y.; Thornquist, M.D.; Goodison, S.; Rosser, C.J. A multiplex immunoassay for the non-invasive detection of bladder cancer. J. Transl. Med. 2016, 14, 31. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.J.; Burgess, R.; Mao, Y.-Q.; Luo, S.; Tang, H.; Jones, V.S.; Weisheng, B.; Huang, R.-Y.; Chen, X.; Huang, R.-P. Antibody arrays in biomarker discovery. Adv. Clin. Chem. 2015, 69, 255–324. [Google Scholar] [CrossRef] [PubMed]
- Valekova, I.; Skalnikova, H.K.; Jarkovska, K.; Motlik, J.; Kovarova, H. Multiplex immunoassays for quantification of cytokines, growth factors and other proteins in stem cell communication. Methods Mol. Biol. 2015, 1212, 39–63. [Google Scholar] [CrossRef] [PubMed]
- Faresjö, M. A useful guide for analysis of immune markers by fluorochrome (Luminex) technique. Methods Mol. Biol. 2014, 1172, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Valekova, I.; Jarkovska, K.; Kotrcova, E.; Bucci, J.; Ellederova, Z.; Juhas, S.; Motlik, J.; Gadher, S.J.; Kovarova, H. Revelation of the IFNα, IL-10, IL-8 and IL-1β as promising biomarkers reflecting immuno-pathological mechanisms in porcine Huntington’s disease model. J. Neuroimmunol. 2016, 293, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg-Hasson, Y.; Hansmann, L.; Liedtke, M.; Herschmann, I.; Maecker, H.T. Effects of serum and plasma matrices on multiplex immunoassays. Immunol. Res. 2014, 58, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Tarhini, A.A.; Lin, Y.; Zahoor, H.; Shuai, Y.; Butterfield, L.H.; Ringquist, S.; Gogas, H.; Sander, C.; Lee, S.; Agarwala, S.S.; et al. Pro-Inflammatory Cytokines Predict Relapse-Free Survival after One Month of Interferon-α but Not Observation in Intermediate Risk Melanoma Patients. PLoS ONE 2015, 10, e0132745. [Google Scholar] [CrossRef] [PubMed]
- Shetty, G.; Beasley, G.M.; Sparks, S.; Barfield, M.; Masoud, M.; Mosca, P.J.; Pruitt, S.K.; Salama, A.K.S.; Chan, C.; Tyler, D.S.; et al. Plasma cytokine analysis in patients with advanced extremity melanoma undergoing isolated limb infusion. Ann. Surg. Oncol. 2013, 20, 1128–1135. [Google Scholar] [CrossRef] [PubMed]
- Triozzi, P.L.; Aldrich, W.; Crabb, J.W.; Singh, A.D. Spontaneous cellular and humoral tumor antigen responses in patients with uveal melanoma. Melanoma Res. 2015, 25, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Ly, L.V.; Bronkhorst, I.H.G.; van Beelen, E.; Vrolijk, J.; Taylor, A.W.; Versluis, M.; Luyten, G.P.M.; Jager, M.J. Inflammatory cytokines in eyes with uveal melanoma and relation with macrophage infiltration. Investig. Ophthalmol. Vis. Sci. 2010, 51, 5445–5451. [Google Scholar] [CrossRef] [PubMed]
- Sanz, H.; Aponte, J.J.; Harezlak, J.; Dong, Y.; Ayestaran, A.; Nhabomba, A.; Mpina, M.; Maurin, O.R.; Díez-Padrisa, N.; Aguilar, R.; et al. drLumi: An open-source package to manage data, calibrate and conduct quality control of multiplex bead-based immunoassays data analysis. PLoS ONE 2017, 12, e0187901. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.W. Binding of cells to matrixes of distinct antibodies coated on solid surface. J. Immunol. Methods 1983, 65, 217–223. [Google Scholar] [CrossRef]
- Antibody Arrays for Protein Detection. Available online: https://www.raybiotech.com/antibody-array (accessed on 27 November 2017).
- Kopf, E.; Zharhary, D. Antibody arrays—An emerging tool in cancer proteomics. Int. J. Biochem. Cell Biol. 2007, 39, 1305–1317. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Carbayo, M. Antibody array-based technologies for cancer protein profiling and functional proteomic analyses using serum and tissue specimens. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2010, 31, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Gál, P.; Varinská, L.; Fáber, L.; Novák, Š.; Szabo, P.; Mitrengová, P.; Mirossay, A.; Mučaji, P.; Smetana, K. How Signaling Molecules Regulate Tumor Microenvironment: Parallels to Wound Repair. Molecules 2017, 22. [Google Scholar] [CrossRef] [PubMed]
- Rissin, D.M.; Kan, C.W.; Campbell, T.G.; Howes, S.C.; Fournier, D.R.; Song, L.; Piech, T.; Patel, P.P.; Chang, L.; Rivnak, A.J.; et al. Single-molecule enzyme-linked immunosorbent assay detects serum proteins at subfemtomolar concentrations. Nat. Biotechnol. 2010, 28, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Fischer, S.K.; Joyce, A.; Spengler, M.; Yang, T.-Y.; Zhuang, Y.; Fjording, M.S.; Mikulskis, A. Emerging technologies to increase ligand binding assay sensitivity. AAPS J. 2015, 17, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Andreasson, U.; Blennow, K.; Zetterberg, H. Update on ultrasensitive technologies to facilitate research on blood biomarkers for central nervous system disorders. Alzheimers Dement. Amst. Neth. 2016, 3, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.G.; Gerszten, R.E. Emerging Affinity-Based Proteomic Technologies for Large-Scale Plasma Profiling in Cardiovascular Disease. Circulation 2017, 135, 1651–1664. [Google Scholar] [CrossRef] [PubMed]
- Simon, S.; Ezan, E. Ultrasensitive bioanalysis: Current status and future trends. Bioanalysis 2017, 9, 753–764. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Truong, J.; Reeves, W.B.; Hahm, J.-I. Emerging Cytokine Biosensors with Optical Detection Modalities and Nanomaterial-Enabled Signal Enhancement. Sensors 2017, 17. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Frade, J.M.; Martínez-Muñoz, L.; Villares, R.; Cascio, G.; Lucas, P.; Gomariz, R.P.; Mellado, M. Chemokine Detection Using Receptors Immobilized on an SPR Sensor Surface. Methods Enzymol. 2016, 570, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Son, K.; Liu, Y.; Revzin, A. Biosensors for Cell Analysis. Annu. Rev. Biomed. Eng. 2015, 17, 165–190. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Huang, N.-T.; Chung, M.-T.; Cornell, T.T.; Kurabayashi, K. Label-free cytokine micro- and nano-biosensing towards personalized medicine of systemic inflammatory disorders. Adv. Drug Deliv. Rev. 2015, 95, 90–103. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Tang, Y.; Alt, R.R.; Xie, X.; Li, F. Emerging techniques for ultrasensitive protein analysis. Analyst 2016, 141, 3473–3481. [Google Scholar] [CrossRef] [PubMed]
- Cretich, M.; Daaboul, G.G.; Sola, L.; Ünlü, M.S.; Chiari, M. Digital detection of biomarkers assisted by nanoparticles: Application to diagnostics. Trends Biotechnol. 2015, 33, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Noji, H. Digital Bioassays: Theory, Applications and Perspectives. Anal. Chem. 2017, 89, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.; Zhang, P.; Yu, H.; Lee, S.; Kang, S.H. Ultrasensitive Detection of α-Fetoprotein by Total Internal Reflection Scattering-Based Super-Resolution Microscopy for Superlocalization of Nano-Immunoplasmonics. Anal. Chem. 2016, 88, 11070–11076. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.H.B.; Fukushima, N.; Puskas, R.; Todd, J.; Goix, P. Development and preliminary clinical validation of a high sensitivity assay for cardiac troponin using a capillary flow (single molecule) fluorescence detector. Clin. Chem. 2006, 52, 2157–2159. [Google Scholar] [CrossRef] [PubMed]
- Todd, J.; Freese, B.; Lu, A.; Held, D.; Morey, J.; Livingston, R.; Goix, P. Ultrasensitive flow-based immunoassays using single-molecule counting. Clin. Chem. 2007, 53, 1990–1995. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, M.; Livingston, R.; Felberg, J.; Bishop, J.J. Multiplex single molecule counting technology used to generate interleukin 4, interleukin 6 and interleukin 10 reference limits. Anal. Biochem. 2016, 503, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Milutinovic, M.D.; Walt, D.R. Single molecule array (Simoa) assay with optimal antibody pairs for cytokine detection in human serum samples. Analyst 2015, 140, 6277–6282. [Google Scholar] [CrossRef] [PubMed]
- Rissin, D.M.; Kan, C.W.; Song, L.; Rivnak, A.J.; Fishburn, M.W.; Shao, Q.; Piech, T.; Ferrell, E.P.; Meyer, R.E.; Campbell, T.G.; et al. Multiplexed single molecule immunoassays. Lab. Chip 2013, 13, 2902–2911. [Google Scholar] [CrossRef] [PubMed]
- Rivnak, A.J.; Rissin, D.M.; Kan, C.W.; Song, L.; Fishburn, M.W.; Piech, T.; Campbell, T.G.; DuPont, D.R.; Gardel, M.; Sullivan, S.; et al. A fully-automated, six-plex single molecule immunoassay for measuring cytokines in blood. J. Immunol. Methods 2015, 424, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Sano, T.; Smith, C.L.; Cantor, C.R. Immuno-PCR: Very sensitive antigen detection by means of specific antibody-DNA conjugates. Science 1992, 258, 120–122. [Google Scholar] [CrossRef] [PubMed]
- Adler, M.; Spengler, M. Novel Strategies and Tools for Enhanced Sensitivity in Routine Biomolecule Analytics. Curr. Pharm. Anal. 2009, 5, 390–407. [Google Scholar] [CrossRef]
- Ryazantsev, D.Y.; Voronina, D.V.; Zavriev, S.K. Immuno-PCR: Achievements and Perspectives. Biochem. Biokhimiia 2016, 81, 1754–1770. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Li, J.; Wang, L. Immuno-PCR: An ultrasensitive immunoassay for biomolecular detection. Anal. Chim. Acta 2016, 910, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Niemeyer, C.M.; Adler, M.; Wacker, R. Detecting antigens by quantitative immuno-PCR. Nat. Protoc. 2007, 2, 1918–1930. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.H.; Sadroddiny, E. Application of immuno-PCR for the detection of early stage cancer. Mol. Cell. Probes 2016, 30, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Assumpção, A.L.F.V.; da Silva, R.C. Immuno-PCR in cancer and non-cancer related diseases: A review. Vet. Q. 2016, 36, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Fredriksson, S.; Gullberg, M.; Jarvius, J.; Olsson, C.; Pietras, K.; Gústafsdóttir, S.M.; Ostman, A.; Landegren, U. Protein detection using proximity-dependent DNA ligation assays. Nat. Biotechnol. 2002, 20, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Greenwood, C.; Ruff, D.; Kirvell, S.; Johnson, G.; Dhillon, H.S.; Bustin, S.A. Proximity assays for sensitive quantification of proteins. Biomol. Detect. Quantif. 2015, 4, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.-Y.; Wu, C.C.; Chiu, Y.C.; Yang, S.Y.; Horng, H.E.; Yang, H.C. Magnetic susceptibility reduction method for magnetically labeled immunoassay. Appl. Phys. Lett. 2006, 88, 212512. [Google Scholar] [CrossRef]
- Yang, S.-Y.; Chiu, M.-J.; Chen, T.-F.; Horng, H.-E. Detection of Plasma Biomarkers Using Immunomagnetic Reduction: A Promising Method for the Early Diagnosis of Alzheimer’s Disease. Neurol. Ther. 2017, 6, 37–56. [Google Scholar] [CrossRef] [PubMed]
- Lue, L.-F.; Sabbagh, M.N.; Chiu, M.-J.; Jing, N.; Snyder, N.L.; Schmitz, C.; Guerra, A.; Belden, C.M.; Chen, T.-F.; Yang, C.-C.; et al. Plasma Levels of Aβ42 and Tau Identified Probable Alzheimer’s Dementia: Findings in Two Cohorts. Front. Aging Neurosci. 2017, 9, 226. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.W.; Yang, S.Y.; Yu, C.Y.; Chieh, J.J.; Yang, C.-C.; Horng, H.-E.; Hong, C.-Y.; Yang, H.-C.; Wu, C.-C. Exploration of the relationship between the tumor burden and the concentration of vascular endothelial growth factor in liver-cancer-bearing animals using immunomagnetic reduction assay. J. Biomed. Nanotechnol. 2011, 7, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.-C.; Yang, S.-Y.; Ho, C.-S.; Chang, J.-F.; Liu, B.-H.; Huang, K.-W. Development of antibody functionalized magnetic nanoparticles for the immunoassay of carcinoembryonic antigen: A feasibility study for clinical use. J. Nanobiotechnol. 2014, 12, 44. [Google Scholar] [CrossRef] [PubMed]
- Chieh, J.-J.; Huang, K.W.; Chuang, C.P.; Wei, W.C.; Dong, J.J.; Lee, Y.Y. Immunomagnetic Reduction Assay on Des-Gamma-Carboxy Prothrombin for Screening of Hepatocellular Carcinoma. IEEE Trans. Biomed. Eng. 2016, 63, 1681–1686. [Google Scholar] [CrossRef] [PubMed]
- Product-IMR Reagent | MagQu. Available online: http://www.magqu.com/product/IMR%20Reagent?shs_term_node_tid_depth=39 (accessed on 27 November 2017).
- Yeung, D.; Ciotti, S.; Purushothama, S.; Gharakhani, E.; Kuesters, G.; Schlain, B.; Shen, C.; Donaldson, D.; Mikulskis, A. Evaluation of highly sensitive immunoassay technologies for quantitative measurements of sub-pg/mL levels of cytokines in human serum. J. Immunol. Methods 2016, 437, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Fichorova, R.N.; Richardson-Harman, N.; Alfano, M.; Belec, L.; Carbonneil, C.; Chen, S.; Cosentino, L.; Curtis, K.; Dezzutti, C.S.; Donoval, B.; et al. Biological and technical variables affecting immunoassay recovery of cytokines from human serum and simulated vaginal fluid: A multicenter study. Anal. Chem. 2008, 80, 4741–4751. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, P.K.; Gierahn, T.M.; Roederer, M.; Love, J.C. Single-cell technologies for monitoring immune systems. Nat. Immunol. 2014, 15, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Czerkinsky, C.C.; Nilsson, L.A.; Nygren, H.; Ouchterlony, O.; Tarkowski, A. A solid-phase enzyme-linked immunospot (ELISPOT) assay for enumeration of specific antibody-secreting cells. J. Immunol. Methods 1983, 65, 109–121. [Google Scholar] [CrossRef]
- Czerkinsky, C.; Andersson, G.; Ekre, H.P.; Nilsson, L.A.; Klareskog, L.; Ouchterlony, O. Reverse ELISPOT assay for clonal analysis of cytokine production. I. Enumeration of gamma-interferon-secreting cells. J. Immunol. Methods 1988, 110, 29–36. [Google Scholar] [CrossRef]
- Slota, M.; Lim, J.-B.; Dang, Y.; Disis, M.L. ELISpot for measuring human immune responses to vaccines. Expert Rev. Vaccines 2011, 10, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Hauer, A.C.; Bajaj-Elliott, M. Elispot Technique for Assaying Interleukins. In Interleukin Protocols; Methods in Molecular MedicineTM; Springer: Totowa, NJ, USA, 2001; pp. 17–28. ISBN 978-1-59259-146-6. [Google Scholar]
- Faresjö, M. The challenge of measuring elusive immune markers by enzyme-linked immuno-spot (ELISPOT) technique. Methods Mol. Biol. 2014, 1172, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Morse, M.A.; Osada, T.; Hobeika, A.; Patel, S.; Lyerly, H.K. Biomarkers and correlative endpoints for immunotherapy trials. Am. Soc. Clin. Oncol. Educ. Book Am. Soc. Clin. Oncol. Meet. 2013. [Google Scholar] [CrossRef] [PubMed]
- Kamentsky, L.A.; Melamed, M.R.; Derman, H. Spectrophotometer: New instrument for ultrarapid cell analysis. Science 1965, 150, 630–631. [Google Scholar] [CrossRef] [PubMed]
- Fulwyler, M.J. Electronic separation of biological cells by volume. Science 1965, 150, 910–911. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Mitson-Salazar, A.; Prussin, C. Detection of Intracellular Cytokines by Flow Cytometry. Curr. Protoc. Immunol. 2015, 110, 6.24.1–6.24.18. [Google Scholar] [CrossRef] [PubMed]
- Freer, G. Intracellular staining and detection of cytokines by fluorescence-activated flow cytometry. Methods Mol. Biol. 2014, 1172, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Schuerwegh, A.J.; Stevens, W.J.; Bridts, C.H.; De Clerck, L.S. Evaluation of monensin and brefeldin A for flow cytometric determination of interleukin-1 beta, interleukin-6 and tumor necrosis factor-alpha in monocytes. Cytometry 2001, 46, 172–176. [Google Scholar] [CrossRef] [PubMed]
- Sander, B.; Andersson, J.; Andersson, U. Assessment of cytokines by immunofluorescence and the paraformaldehyde-saponin procedure. Immunol. Rev. 1991, 119, 65–93. [Google Scholar] [CrossRef] [PubMed]
- Jung, T.; Schauer, U.; Heusser, C.; Neumann, C.; Rieger, C. Detection of intracellular cytokines by flow cytometry. J. Immunol. Methods 1993, 159, 197–207. [Google Scholar] [CrossRef]
- Prussin, C.; Metcalfe, D.D. Detection of intracytoplasmic cytokine using flow cytometry and directly conjugated anti-cytokine antibodies. J. Immunol. Methods 1995, 188, 117–128. [Google Scholar] [CrossRef]
- Foster, B.; Prussin, C.; Liu, F.; Whitmire, J.K.; Whitton, J.L. Detection of intracellular cytokines by flow cytometry. Curr. Protoc. Immunol. 2007. [Google Scholar] [CrossRef]
- Mukai, K.; Gaudenzio, N.; Gupta, S.; Vivanco, N.; Bendall, S.C.; Maecker, H.T.; Chinthrajah, R.S.; Tsai, M.; Nadeau, K.C.; Galli, S.J. Assessing basophil activation by using flow cytometry and mass cytometry in blood stored 24 hours before analysis. J. Allergy Clin. Immunol. 2017, 139, 889–899. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, C.S.; Aranda Lopez, P.; Dopheide, J.F.; Schmidt, F.; Theobald, M.; Schild, H.; Lauinger-Lörsch, E.; Nolte, F.; Radsak, M.P. Phenotypic and functional characterization of neutrophils and monocytes from patients with myelodysplastic syndrome by flow cytometry. Cell. Immunol. 2016, 308, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Manfredi, A.A.; Rovere-Querini, P.; D’Angelo, A.; Maugeri, N. Low molecular weight heparins prevent the induction of autophagy of activated neutrophils and the formation of neutrophil extracellular traps. Pharmacol. Res. 2017, 123, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Misale, M.S.; Witek Janusek, L.; Tell, D.; Mathews, H.L. Chromatin organization as an indicator of glucocorticoid induced natural killer cell dysfunction. Brain. Behav. Immun. 2018, 67, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Bai, Y.; Olivera, A.; Desai, A.; Metcalfe, D.D. An optimized protocol for the generation and functional analysis of human mast cells from CD34(+) enriched cell populations. J. Immunol. Methods 2017, 448, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Daud, A.I.; Loo, K.; Pauli, M.L.; Sanchez-Rodriguez, R.; Sandoval, P.M.; Taravati, K.; Tsai, K.; Nosrati, A.; Nardo, L.; Alvarado, M.D.; et al. Tumor immune profiling predicts response to anti-PD-1 therapy in human melanoma. J. Clin. Investig. 2016, 126, 3447–3452. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Shin, D.S.; Zaretsky, J.; Frederiksen, J.; Cornish, A.; Avramis, E.; Seja, E.; Kivork, C.; Siebert, J.; Kaplan-Lefko, P.; et al. PD-1 Blockade Expands Intratumoral Memory T Cells. Cancer Immunol. Res. 2016, 4, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Tietze, J.K.; Angelova, D.; Heppt, M.V.; Reinholz, M.; Murphy, W.J.; Spannagl, M.; Ruzicka, T.; Berking, C. The proportion of circulating CD45RO(+)CD8(+) memory T cells is correlated with clinical response in melanoma patients treated with ipilimumab. Eur. J. Cancer 2017, 75, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Kitano, S.; Tsuji, T.; Liu, C.; Hirschhorn-Cymerman, D.; Kyi, C.; Mu, Z.; Allison, J.P.; Gnjatic, S.; Yuan, J.D.; Wolchok, J.D. Enhancement of tumor-reactive cytotoxic CD4+ T cell responses after ipilimumab treatment in four advanced melanoma patients. Cancer Immunol. Res. 2013, 1, 235–244. [Google Scholar] [CrossRef] [PubMed]
- De Coaña, Y.P.; Wolodarski, M.; Poschke, I.; Yoshimoto, Y.; Yang, Y.; Nyström, M.; Edbäck, U.; Brage, S.E.; Lundqvist, A.; Masucci, G.V.; et al. Ipilimumab treatment decreases monocytic MDSCs and increases CD8 effector memory T cells in long-term survivors with advanced melanoma. Oncotarget 2017, 8, 21539–21553. [Google Scholar] [CrossRef] [PubMed]
- Wistuba-Hamprecht, K.; Martens, A.; Heubach, F.; Romano, E.; Geukes Foppen, M.; Yuan, J.; Postow, M.; Wong, P.; Mallardo, D.; Schilling, B.; et al. Peripheral CD8 effector-memory type 1 T-cells correlate with outcome in ipilimumab-treated stage IV melanoma patients. Eur. J. Cancer 2017, 73, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Diller, M.L.; Kudchadkar, R.R.; Delman, K.A.; Lawson, D.H.; Ford, M.L. Complete response to high-dose IL-2 and enhanced IFNγ+Th17 : TREG ratio in a melanoma patient. Melanoma Res. 2016, 26, 535–539. [Google Scholar] [CrossRef] [PubMed]
- Diller, M.L.; Kudchadkar, R.R.; Delman, K.A.; Lawson, D.H.; Ford, M.L. Exogenous IL-2 Induces FoxP3+ Th17 Cells In Vivo in Melanoma Patients. J. Immunother. 2016, 39, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Zelba, H.; Weide, B.; Martens, A.; Derhovanessian, E.; Bailur, J.K.; Kyzirakos, C.; Pflugfelder, A.; Eigentler, T.K.; Di Giacomo, A.M.; Maio, M.; et al. Circulating CD4+ T cells that produce IL4 or IL17 when stimulated by melan-A but not by NY-ESO-1 have negative impacts on survival of patients with stage IV melanoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 4390–4399. [Google Scholar] [CrossRef] [PubMed]
- Zelba, H.; Weide, B.; Martens, A.; Bailur, J.K.; Garbe, C.; Pawelec, G. The prognostic impact of specific CD4 T-cell responses is critically dependent on the target antigen in melanoma. Oncoimmunology 2015, 4, e955683. [Google Scholar] [CrossRef] [PubMed]
- Borchers, S.; Maβlo, C.; Müller, C.A.; Tahedl, A.; Volkind, J.; Nowak, Y.; Umansky, V.; Esterlechner, J.; Frank, M.H.; Ganss, C.; et al. Detection of ABCB5 tumour antigen-specific CD8(+) T cells in melanoma patients and implications for immunotherapy. Clin. Exp. Immunol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Bandura, D.R.; Baranov, V.I.; Ornatsky, O.I.; Antonov, A.; Kinach, R.; Lou, X.; Pavlov, S.; Vorobiev, S.; Dick, J.E.; Tanner, S.D. Mass cytometry: Technique for real time single cell multitarget immunoassay based on inductively coupled plasma time-of-flight mass spectrometry. Anal. Chem. 2009, 81, 6813–6822. [Google Scholar] [CrossRef] [PubMed]
- Cosma, A.; Nolan, G.; Gaudilliere, B. Mass cytometry: The time to settle down. Cytom. Part J. Int. Soc. Anal. Cytol. 2017, 91, 12–13. [Google Scholar] [CrossRef] [PubMed]
- Bendall, S.C.; Simonds, E.F.; Qiu, P.; Amir, E.D.; Krutzik, P.O.; Finck, R.; Bruggner, R.V.; Melamed, R.; Trejo, A.; Ornatsky, O.I.; et al. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science 2011, 332, 687–696. [Google Scholar] [CrossRef] [PubMed]
- O’Gorman, W.E.; Kong, D.S.; Balboni, I.M.; Rudra, P.; Bolen, C.R.; Ghosh, D.; Davis, M.M.; Nolan, G.P.; Hsieh, E.W.Y. Mass cytometry identifies a distinct monocyte cytokine signature shared by clinically heterogeneous pediatric SLE patients. J. Autoimmun. 2017. [Google Scholar] [CrossRef] [PubMed]
- Fisher, D.A.C.; Miner, C.A.; Engle, E.K.; Brost, T.M.; Malkova, O.; Oh, S.T. Mass Cytometry Analysis of Dysregulated Cytokine Production and Intracellular Signaling in Myelofibrosis. Blood 2016, 128, 4277. [Google Scholar]
- Newell, E.W.; Lin, W. High-dimensional analysis of human CD8(+) T cell phenotype, function and antigen specificity. Curr. Top. Microbiol. Immunol. 2014, 377, 61–84. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, E.M.; Kent, S.C.; Tripuraneni, V.; Orban, T.; Ploegh, H.L.; Hafler, D.A.; Love, J.C. Concurrent detection of secreted products from human lymphocytes by microengraving: Cytokines and antigen-reactive antibodies. Clin. Immunol. 2008, 129, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Stybayeva, G.; Silangcruz, J.; Yan, J.; Ramanculov, E.; Dandekar, S.; George, M.D.; Revzin, A. Detecting cytokine release from single T-cells. Anal. Chem. 2009, 81, 8150–8156. [Google Scholar] [CrossRef] [PubMed]
- Han, Q.; Bagheri, N.; Bradshaw, E.M.; Hafler, D.A.; Lauffenburger, D.A.; Love, J.C. Polyfunctional responses by human T cells result from sequential release of cytokines. Proc. Natl. Acad. Sci. USA 2012, 109, 1607–1612. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Chen, J.J.; Mu, L.; Xue, Q.; Wu, Y.; Wu, P.-H.; Li, J.; Vortmeyer, A.O.; Miller-Jensen, K.; Wirtz, D.; et al. High-throughput secretomic analysis of single cells to assess functional cellular heterogeneity. Anal. Chem. 2013, 85, 2548–2556. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Xue, Q.; Eisele, M.R.; Sulistijo, E.S.; Brower, K.; Han, L.; Amir, E.-A.D.; Pe’er, D.; Miller-Jensen, K.; Fan, R. Highly multiplexed profiling of single-cell effector functions reveals deep functional heterogeneity in response to pathogenic ligands. Proc. Natl. Acad. Sci. USA 2015, 112, E607–E615. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Fan, R.; Ahmad, H.; Shi, Q.; Comin-Anduix, B.; Chodon, T.; Koya, R.C.; Liu, C.-C.; Kwong, G.A.; Radu, C.G.; et al. A clinical microchip for evaluation of single immune cells reveals high functional heterogeneity in phenotypically similar T cells. Nat. Med. 2011, 17, 738–743. [Google Scholar] [CrossRef] [PubMed]
- McWhorter, F.Y.; Smith, T.D.; Luu, T.U.; Rahim, M.K.; Haun, J.B.; Liu, W.F. Macrophage secretion heterogeneity in engineered microenvironments revealed using a microwell platform. Integr. Biol. Quant. Biosci. Nano Macro 2016, 8, 751–760. [Google Scholar] [CrossRef] [PubMed]
- An, X.; Sendra, V.G.; Liadi, I.; Ramesh, B.; Romain, G.; Haymaker, C.; Martinez-Paniagua, M.; Lu, Y.; Radvanyi, L.G.; Roysam, B.; et al. Single-cell profiling of dynamic cytokine secretion and the phenotype of immune cells. PLoS ONE 2017, 12, e0181904. [Google Scholar] [CrossRef] [PubMed]
- Chalaris, A.; Garbers, C.; Rabe, B.; Rose-John, S.; Scheller, J. The soluble Interleukin 6 receptor: Generation and role in inflammation and cancer. Eur. J. Cell Biol. 2011, 90, 484–494. [Google Scholar] [CrossRef] [PubMed]
- Meager, A. Measurement of cytokines by bioassays: Theory and application. Methods 2006, 38, 237–252. [Google Scholar] [CrossRef] [PubMed]
- Kovarik, P.; Ebner, F.; Sedlyarov, V. Posttranscriptional regulation of cytokine expression. Cytokine 2017, 89, 21–26. [Google Scholar] [CrossRef] [PubMed]





© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kupcova Skalnikova, H.; Cizkova, J.; Cervenka, J.; Vodicka, P. Advances in Proteomic Techniques for Cytokine Analysis: Focus on Melanoma Research. Int. J. Mol. Sci. 2017, 18, 2697. https://doi.org/10.3390/ijms18122697
Kupcova Skalnikova H, Cizkova J, Cervenka J, Vodicka P. Advances in Proteomic Techniques for Cytokine Analysis: Focus on Melanoma Research. International Journal of Molecular Sciences. 2017; 18(12):2697. https://doi.org/10.3390/ijms18122697
Chicago/Turabian StyleKupcova Skalnikova, Helena, Jana Cizkova, Jakub Cervenka, and Petr Vodicka. 2017. "Advances in Proteomic Techniques for Cytokine Analysis: Focus on Melanoma Research" International Journal of Molecular Sciences 18, no. 12: 2697. https://doi.org/10.3390/ijms18122697
APA StyleKupcova Skalnikova, H., Cizkova, J., Cervenka, J., & Vodicka, P. (2017). Advances in Proteomic Techniques for Cytokine Analysis: Focus on Melanoma Research. International Journal of Molecular Sciences, 18(12), 2697. https://doi.org/10.3390/ijms18122697