Of Energy and Entropy: The Ineluctable Impact of Aging in Old Age Dementia




{kind=link}
Abstract
:1. Introduction
2. Peculiarity of AD in Old Age Subjects
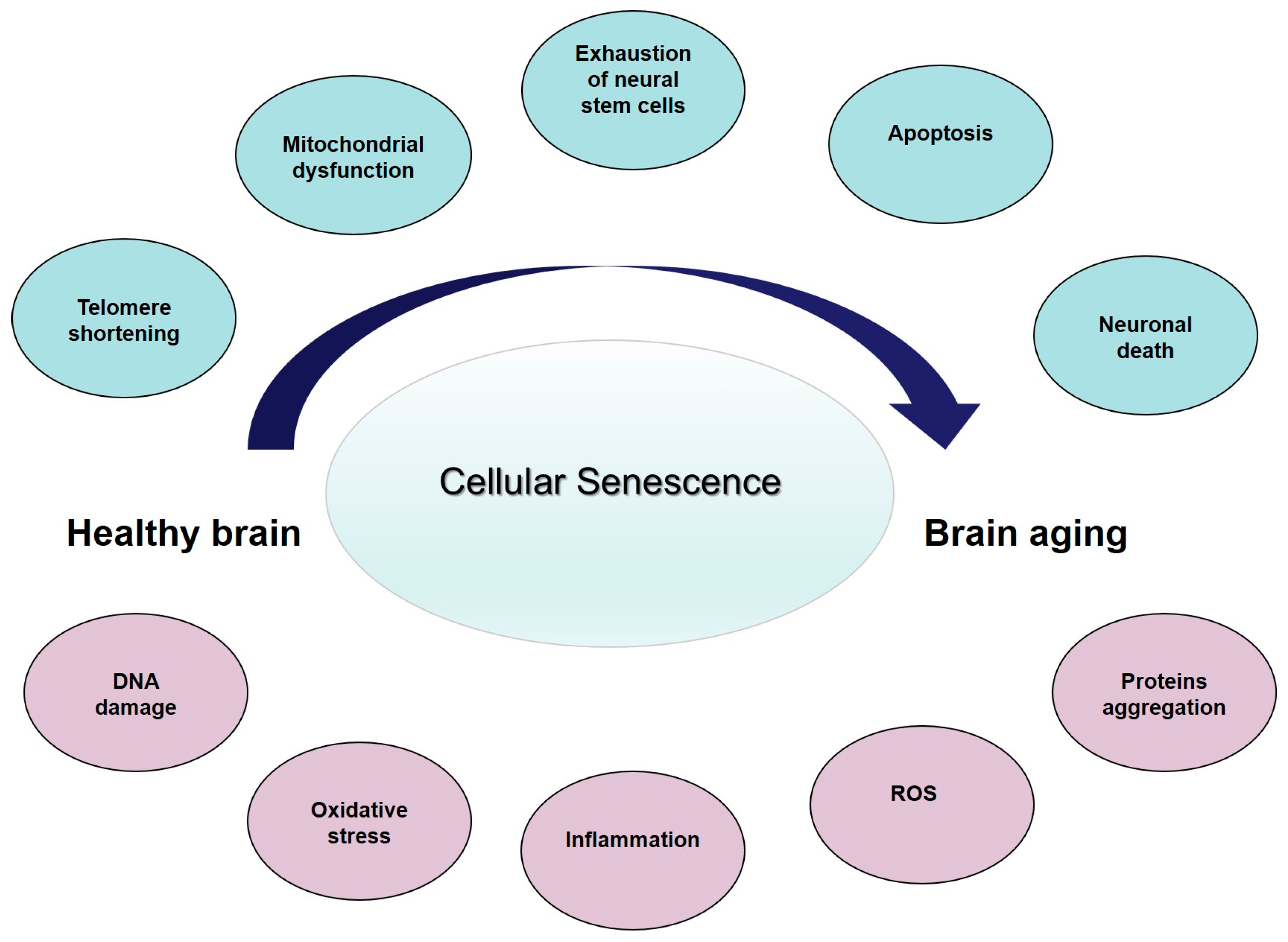
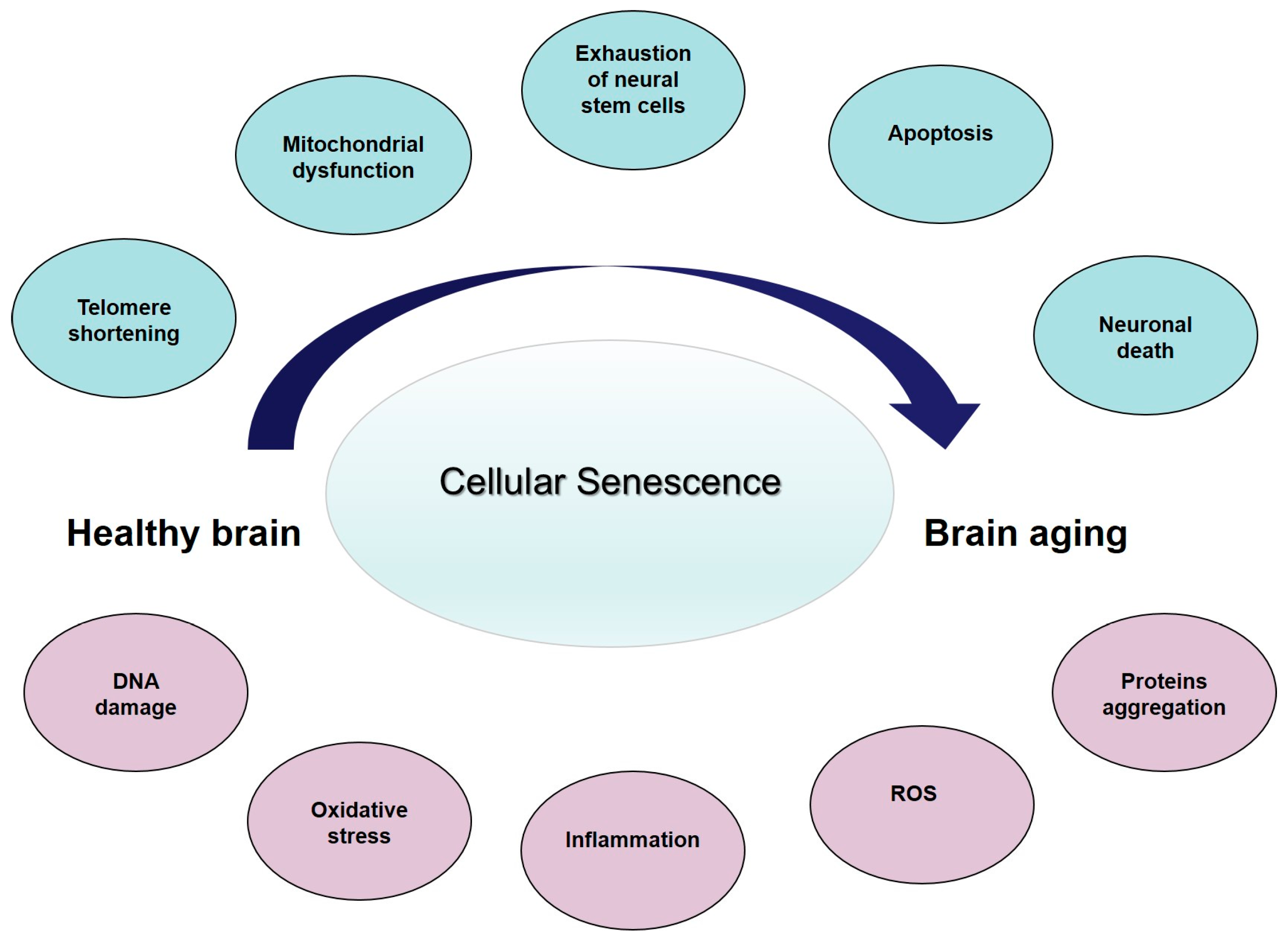
3. Cellular Senescence, Age-Related Changes, and Brain Aging
4. Aging and Dementia: An Entropic Point of View
5. Bioenergetics in Brain Aging and AD
6. Final Remarks and Perspective
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Flatt, T. A new definition of aging? Front. Genet. 2012, 3, 148. [Google Scholar] [CrossRef] [PubMed]
- Hedden, T.; Gabrieli, J.D.E. Insights into the aging mind: A view from cognitive neuroscience. Nat. Rev. Neurosci. 2004, 5, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Pierce, A.L.; Bullain, S.S.; Kawas, C.H. Late-Onset Alzheimer Disease. Neurol. Clin. 2017, 35, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Giulioli, C.; Amieva, H. Epidemiology of cognitive aging in the oldest old. Rev. Investig. Clin. 2016, 68, 33–39. [Google Scholar] [PubMed]
- Corrada, M.M.; Brookmeyer, R.; Paganini-Hill, A.; Berlau, D.; Kawas, C.H. Incidence continues to increase with age in the oldest old: The 90+ study. Ann. Neurol. 2010, 67, 114–121. [Google Scholar]
- National Institute of Aging. Available online: https://www.nia.nih.gov/sites/default/files/2017-06/WPAM (accessed on15 July 2017).
- National Population Projections. Available online: http://www.census.gov/population/www/projections/summarytables.html (accessed on 15 July 2017).
- Hebert, L.E.; Weuve, J.; Scherr, P.A. Alzheimer disease in the United States (2010–2050) estimated using the 2010 census. Neurology 2013, 80, 1778–1783. [Google Scholar] [PubMed]
- Drachman, D.A. If we live long enough, will we all be demented? Neurology 1994, 44, 1563–1565. [Google Scholar] [PubMed]
- Katzman, R. The prevalence and malignancy of Alzheimer’s disease: A major killer. Arch. Neurol. 1976, 3, 217–218. [Google Scholar]
- Scheuner, D.; Eckman, C.; Jensen, M.; Song, X.; Citron, M.; Suzuki, N.; Bird, T.D.; Hardy, J.; Hutton, M.; Kukull, W.; et al. Secreted amyloid β-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat. Med. 1996, 8, 864–870. [Google Scholar]
- Drachman, D.A. Aging of the brain, entropy, and Alzheimer disease. Neurology 2006, 67, 1340–1352. [Google Scholar] [PubMed]
- Bullain, S.S.; Corrada, M.M. Dementia in the oldest old. Continuum 2013, 19, 457–469. [Google Scholar] [CrossRef] [PubMed]
- Gardner, R.; Valcour, V.; Yaffe, K. Dementia in the oldest old: A multi-factorial and growing public health issue. Alzheimers Res. Ther. 2013, 5, 27. [Google Scholar] [CrossRef] [PubMed]
- Bullain, S.S.; Corrada, M.M.; Perry, S.M. Sound body sound mind? Physical performance and the risk of dementia in the oldest-old: The 901 Study. J. Am. Geriatr. Soc. 2016, 4, 1408–1415. [Google Scholar] [CrossRef] [PubMed]
- Farrer, L.A.; Cupples, L.A.; Haines, J.L. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease: A meta analysis. JAMA 1997, 278, 1349–1356. [Google Scholar] [CrossRef] [PubMed]
- Euser, S.M.; van Bemmel, T.; Schram, M.T. The effect of age on the association between blood pressure and cognitive function later in life. J. Am. Geriatr. Soc. 2009, 57, 1232–1237. [Google Scholar] [CrossRef] [PubMed]
- Boccardi, V.; Baroni, M.; Smirne, N.; Clodomiro, A.; Ercolani, S.; Longo, A.; Ruggiero, C.; Bruni, A.C.; Mecocci, P. Short-Term Response is not Predictive of Long-Term Response to Acetylcholinesterase Inhibitors in Old Age Subjects with Alzheimer’s Disease: A “Real World” Study. J. Alzheimers Dis. 2017, 56, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Paolacci, L.; Giannandrea, D.; Mecocci, P.; Parnetti, L. Biomarkers for Early Diagnosis of Alzheimer’s Disease in the Oldest Old: Yes or No? J. Alzheimers Dis. 2017, 58, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R.; Albert, M.S.; Knopman, D.S.; McKhann, G.M.; Sperling, R.A.; Carrillo, M.C.; Thies, B.; Phelps, C.H. Introduction to the recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Braak, E. Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol. Aging 1997, 18, 351–357. [Google Scholar] [CrossRef]
- Savva, G.M.; Wharton, S.B.; Ince, P.G.; Forster, G.; Matthews, F.E.; Brayne, C. Age, neuropathology, and dementia. N. Engl. J. Med. 2009, 360, 2302–2309. [Google Scholar] [CrossRef] [PubMed]
- Brumback-Peltz, C.; Balasubramanian, A.B.; Corrada, M.M.; Kawas, C.H. Diagnosing dementia in the oldest-old. Maturitas 2011, 70, 164–168. [Google Scholar] [CrossRef] [PubMed]
- Bennett, D.A.; Wilson, R.S.; Boyle, P.A.; Buchman, A.S.; Schneider, J.A. Relation of neuropathology to cognition in persons without cognitive impairment. Ann. Neurol. 2012, 72, 599–609. [Google Scholar] [CrossRef] [PubMed]
- The 90+ study. Curr. Alzheimer Res. 2012, 9, 709–717.
- Chui, H.C.; Ramirez-Gomez, L. Clinical and imaging features of mixed Alzheimer and vascular pathologies. Alzheimers Res. Ther. 2015, 7, 21. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Pozo, A.; Qian, J.; Monsel, L.S.E.; Blacker, D.; Gomez-Isla, T.; Betensky, R.A.; Growdon, J.H.; Johnson, K.A.; Frosch, M.P.; Sperling, R.A. Mild to moderate Alzheimer dementia with insufficient neuropathological changes. Ann. Neurol. 2014, 75, 597–601. [Google Scholar] [CrossRef] [PubMed]
- Delaere, P.; He, Y.; Fayet, G.; Duyckaerts, C.; Hau, J.J. Beta A4 deposits are constant in the brain of the oldest old: An immunocytochemical study of 20 French centenarians. Neurobiol. Aging 1993, 14, 191–194. [Google Scholar] [CrossRef]
- Giannakopoulos, P.; Hof, P.R.; Kovari, E.; Vallet, P.G.; Herrmann, F.R.; Bouras, C. Distinct patterns of neuronal loss and Alzheimer’s disease lesion distribution in elderly individuals older than 90 years. J. Neuropathol. Exp. Neurol. 1996, 55, 1210–1220. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A.; Attems, J. Prevalence of dementia disorders in the oldest-old: An autopsy study. Acta Neuropathol. 2010, 119, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Nelson, P.T.; Head, E.; Schmitt, F.A.; Davis, P.R.; Neltner, J.H.; Jicha, G.A.; Abner, E.L.; Smith, C.D.; Van Eldik, L.J.; Kryscio, R.J.; et al. Alzheimer’s disease is not “brain aging”: Neuropathological, genetic, and epidemiological human studies. Acta Neuropathol. 2011, 121, 571–587. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Slavin, M.J.; Perminder, S.S. Dementia in oldest old. Nat. Rev. Neurol. 2013, 9, 382–393. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, L.A.; White, L.R.; Ross, G.W.; Petrovitch, H.; Launer, L.J. Cerebral amyloid angiopathy and cognitive function: The HAAS autopsy study. Neurology 2002, 58, 1629–1634. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Thal, D.R.; Ghebremedhin, E.; Del Tredici, K. Stages of the pathologic process in Alzheimer disease: Age categories from 1 to100 years. J Neuropathol Exp Neurol. 2011, 70, 960–969. [Google Scholar] [CrossRef] [PubMed]
- White, L. Brain lesions at autopsy in older Japanese–American men as related to cognitive impairment and dementia in the final years of life: A summary report from the Honolulu–Asia Aging Study. J. Alzheimers Dis. 2009, 18, 713–725. [Google Scholar] [CrossRef] [PubMed]
- Elobeid, A.; Libard, S.; Leino, M.; Popova, S.N.; Alafuzoff, I. Altered proteins in the aging brain. J. Neuropathol. Exp. Neurol. 2016, 75, 316–325. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Velickaite, V.; Giedraitis, V.; Ström, K.; Alafuzoff, I.; Zetterberg, H.; Lannfelt, L.; Kilander, L.; Larsson, E.M.; Ingelsson, M. Cognitive function in very old men does not correlate to biomarkers of Alzheimer’s disease. BMC Geriatr. 2017, 17, 208. [Google Scholar] [CrossRef] [PubMed]
- Morrison, J.H.; Hof, P.R. Life and death of neurons in the aging brain. Science 1997, 278, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Whalley, L.J.; Deary, I.J.; Appleton, C.L.; Starr, J.M. Cognitive reserve and the neurobiology of cognitive aging. Ageing Res. Rev. 2004, 3, 369–382. [Google Scholar] [CrossRef] [PubMed]
- Hall, C.B.; Verghese, J.; Sliwinski, M. Dementia incidence may increase more slowly after age 90: Results from the Bronx Aging Study. Neurology 2005, 65, 882–886. [Google Scholar] [CrossRef] [PubMed]
- McShea, A.; Harris, P.L.; Webster, K.R.; Wahl, A.F.; Smith, M.A. Abnormal expression of the cell cycle regulators P16 and CDK4 in Alzheimer’s disease. Am. J. Pathol. 1997, 150, 1933–1939. [Google Scholar] [PubMed]
- Naylor, R.M.; Baker, D.J.; van Deursen, J.M. Senescent cells: A novel therapeutic target for aging and age-related diseases. Clin. Pharmacol. Ther. 2013, 93, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Tan, F.C.C.; Emmette, R.; Hutchison, E.E.; Mattson, M.P. Are there roles for brain cell senescence in aging and neurodegenerative disorders? Biogerontology 2014, 15, 643–660. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Ann. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [PubMed]
- Boccardi, V.; Pelini, L.; Ercolani, S.; Ruggiero, C.; Mecocci, P. From cellular senescence to Alzheimer’s disease: The role of telomere shortening. Ageing Res. Rev. 2015, 22, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Salama, R.; Sadaie, M.; Hoare, M.; Narita, M. Cellular senescence and its effector programs. Genes Dev. 2014, 28, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Carnero, A. Markers of cellular senescence. Methods Mol. Biol. 2013, 965, 63–81. [Google Scholar] [PubMed]
- Jurk, D.; Wang, C.; Miwa, S.; Maddick, M.; Korolchuk, V.; Tsolou, A.; Gonos, E.S.; Thrasivoulou, C.; Jill Saffrey, M.; Cameron, K.; et al. Postmitotic neurons develop a p21-dependent senescence-like phenotype driven by a DNA damage response. Aging Cell 2012, 11, 996–1004. [Google Scholar] [CrossRef] [PubMed]
- Streit, W.J. Microglia and the Response to Brain Injury. Neuroinflamm. Bench Bedside 2002, 39, 11–24. [Google Scholar]
- Pelvig, D.P.; Pakkenberg, H.; Stark, A.K.; Pakkenberg, B. Neocortical glial cell numbers in human brains. Neurobiol. Aging 2008, 29, 1754–1762. [Google Scholar] [CrossRef] [PubMed]
- Fabricius, K.; Jacobsen, J.S.; Pakkenberg, B. Effect of age on neocortical brain cells in 90+ year old human females, a cell counting study. Neurobiol. Aging 2013, 34, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Drachman, D.A. Aging and the brain: A new frontier. Ann. Neurol. 1997, 42, 819–828. [Google Scholar] [CrossRef] [PubMed]
- Scahill, R.; Frost, C.; Jenkins, R.; Whitwell, J.L.; Rossor, M.N.; Fox, N.C. A longitudinal study of brain volume changes in normal ageing using serial registered magnetic resonance imaging. Arch. Neurol. 2003, 60, 989–994. [Google Scholar] [CrossRef] [PubMed]
- Gunning-Dixon, F.; Raz, N. Neuroanatomical correlates of selected executive functions in middle aged and older adults: A prospective MRI study. Neuropsychologia 2003, 41, 1929–1941. [Google Scholar] [CrossRef]
- Peters, A. The effects of normal aging on myelin and nerve fibers: A review. J. Neurocytol. 2002, 31, 581–593. [Google Scholar] [CrossRef] [PubMed]
- Di Loreto, R.; Murphy, C.T. The cell biology of aging. Mol. Biol. Cell. 2015, 15, 4524–4531. [Google Scholar] [CrossRef] [PubMed]
- Toussaint, O.; Raes, M.; Remacle, J. Aging as multistep process characterized by lowering of entropy production leading the cell to a sequence of defined stages. Mech. Ageing Dev. 1991, 15, 45–64. [Google Scholar] [CrossRef]
- Toussaint, O.; Remacle, J.; Dierick, J.F.; Pascal, T.; Frippiat, C.; Royer, V.; Chainiaux, F. Approach of evolutionary theories of ageing, stress, senescence-like phenotypes, calorie restriction and hormesis from the view point of far-from-equilibrium thermodynamics. Mech. Ageing Dev. 2002, 30, 937–946. [Google Scholar] [CrossRef]
- Prigogine, I.; Wiame, J.M. Biologie et thermodynamique des phenomenes irréversibles. Experientia 1946, 2, 451–453. [Google Scholar] [CrossRef] [PubMed]
- Denbeigh, K.G. The Thermodynamics of the Steady State. J. Chem. Educ. 1951, 29, 322. [Google Scholar] [CrossRef]
- Navratil, V. Health, Ageing and Entropy. Available online: http://www.ped.muni.cz/z21/knihy/2011/39/texty/eng/34_navratil_eng.pdf (accessed on 15 July 2017).
- Aoki, I. Entropy principle for human development, growth and aging. J. Theor. Biol. 1991, 21, 215–223. [Google Scholar] [CrossRef]
- Alberts, B.; Johnson, A.; Lewis, J. Catalysis and the Use of Energy by Cells. In Molecular Biology of the Cell; Garland Science: New York, NY, USA, 2002. [Google Scholar]
- Stauch, K.L.; Purnell, P.R.; Villeneuve, L.M.; Fox, H.S. Proteomic analysis and functional characterization of mouse brain mitochondria during aging reveal alterations in energy metabolism. Proteomics 2015, 15, 1574–1586. [Google Scholar] [CrossRef] [PubMed]
- Holliday, M.A. Metabolic rate and organ size during growth from infancy to maturity and during late gastation and early infancy. Pediatrics 1971, 47, 169. [Google Scholar]
- Sokoloff, L. Energetics of functional activation in neural tissues. Neurochem. Res. 1999, 24, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Mooradian, A.D.; Morin, A.M.; Cipp, L.J.; Haspel, H.C. Glucose transport is reduced in the blood-brain barrier of aged rats. Brain Res. 1991, 14, 145–149. [Google Scholar] [CrossRef]
- Owen, O.E.; Morgan, A.P.; Kemp, H.G.; Sullivan, J.M.; Herrera, M.G.; Cahill, G.F. Brain Metabolism during Fasting. J. Clin. Investig. 1967, 46, 1589–1595. [Google Scholar] [CrossRef] [PubMed]
- Cunnane, S.; Nugent, S.; Roy, M.; Courchesne-Loyer, A.; Croteau, E.; Tremblay, S.; Castellano, A.; Pifferi, F.; Bocti, C.; Paquet, N.; et al. Brain fuel metabolism, aging, and Alzheimer’s disease. Nutrition 2011, 27, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Ingram, T.; Chakrabarti, L. Proteomic profiling of mitochondria: What does it tell us about the ageing brain? Aging 2016, 8, 3161–3179. [Google Scholar] [CrossRef] [PubMed]
- Kwong, L.K.; Sohal, R.S. Age-related changes in activities of mitochondrial electron transport complexes in various tissues of the mouse. Arch. Biochem. Biophys. 2000, 1, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Yin, F.; Sancheti, H.; Patil, I.; Cadenas, E. Energy metabolism and inflammation in brain aging and Alzheimer’s disease. Free Radic. Biol. Med. 2016, 100, 108–122. [Google Scholar] [CrossRef] [PubMed]
- Stauch, K.L.; Purnell, P.R.; Fox, H.S. Aging synaptic mitochondria exhibit dynamic proteomic changes while maintaining bioenergetic function. Aging 2014, 6, 320–334. [Google Scholar] [CrossRef] [PubMed]
- Mecocci, P.; MacGarvey, U.; Kaufman, A.E.; Koontz, D.; Shoffner, J.M.; Wallace, DC.; Beal, M.F. Oxidative damage to mitochondrial DNA shows marked age-dependent increases in human brain. Ann. Neurol. 1993, 34, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Mecocci, P.; MacGarvey, U.; Beal, M.F. Oxidative damage to mitochondrial DNA is increased in Alzheimer’s disease. Ann. Neurol. 1994, 36, 747–751. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H.; Khan, S.M. A “mitochondrial cascade hypothesis” for sporadic Alzheimer’s disease. Med. Hypotheses 2004, 63, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H. Brain aging, Alzheimer’s disease, and mitochondria. Biochim. Biophys. Acta 2011, 1812, 1630–1639. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boccardi, V.; Comanducci, C.; Baroni, M.; Mecocci, P. Of Energy and Entropy: The Ineluctable Impact of Aging in Old Age Dementia. Int. J. Mol. Sci. 2017, 18, 2672. https://doi.org/10.3390/ijms18122672
Boccardi V, Comanducci C, Baroni M, Mecocci P. Of Energy and Entropy: The Ineluctable Impact of Aging in Old Age Dementia. International Journal of Molecular Sciences. 2017; 18(12):2672. https://doi.org/10.3390/ijms18122672
Chicago/Turabian StyleBoccardi, Virginia, Chiara Comanducci, Marta Baroni, and Patrizia Mecocci. 2017. "Of Energy and Entropy: The Ineluctable Impact of Aging in Old Age Dementia" International Journal of Molecular Sciences 18, no. 12: 2672. https://doi.org/10.3390/ijms18122672
APA StyleBoccardi, V., Comanducci, C., Baroni, M., & Mecocci, P. (2017). Of Energy and Entropy: The Ineluctable Impact of Aging in Old Age Dementia. International Journal of Molecular Sciences, 18(12), 2672. https://doi.org/10.3390/ijms18122672