Catalytic Oxidation of Lignins into the Aromatic Aldehydes: General Process Trends and Development Prospects


Abstract
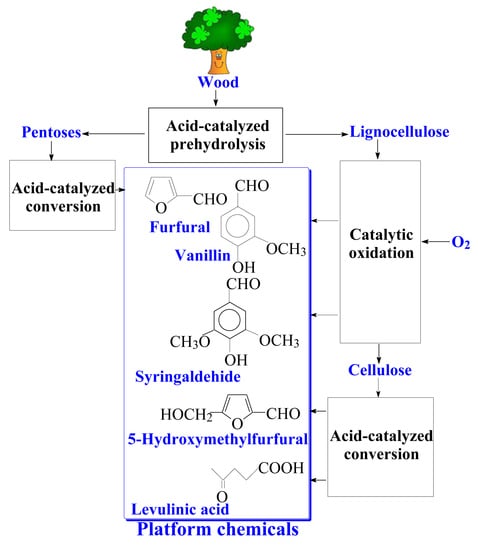
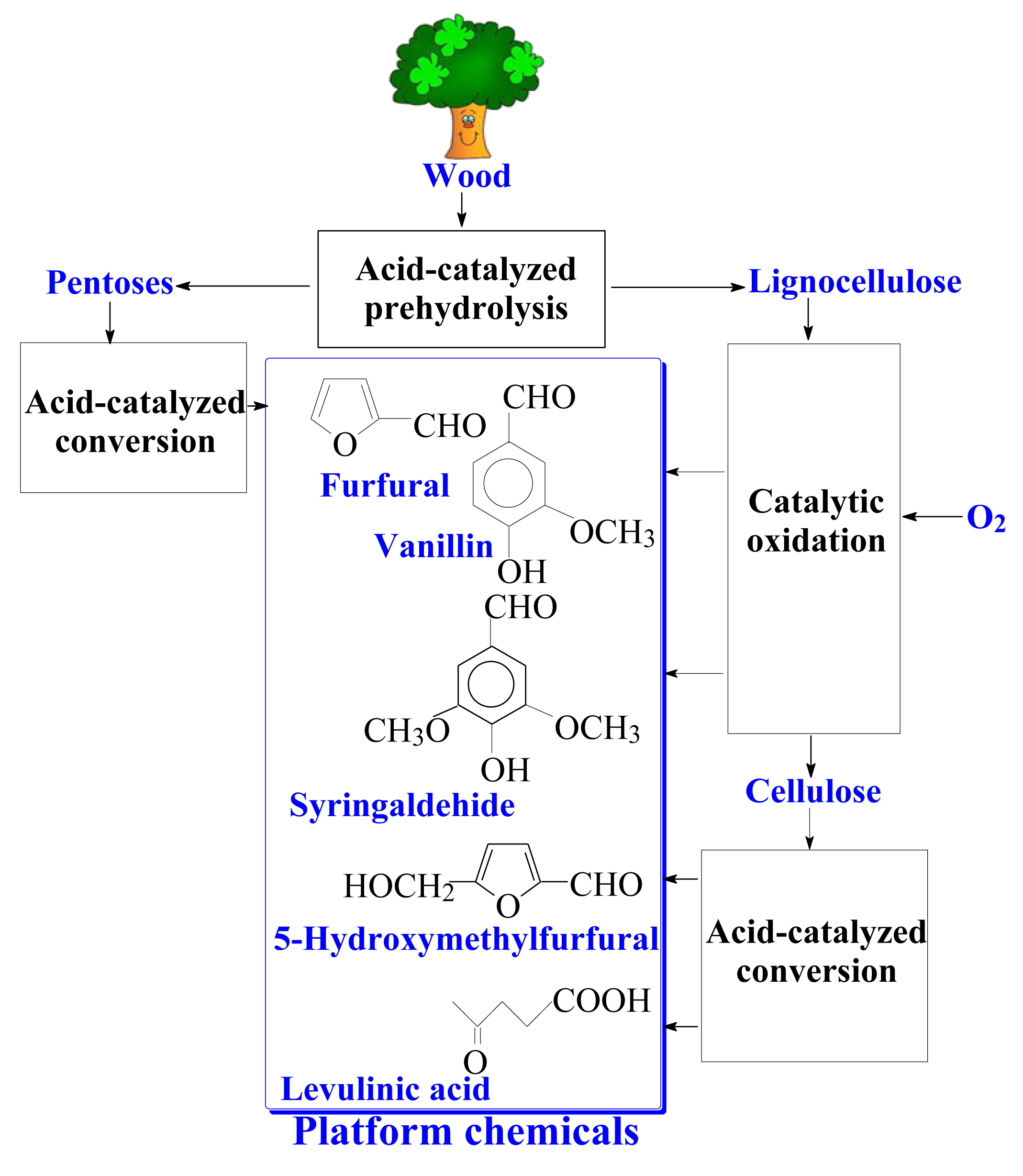
1. Introduction
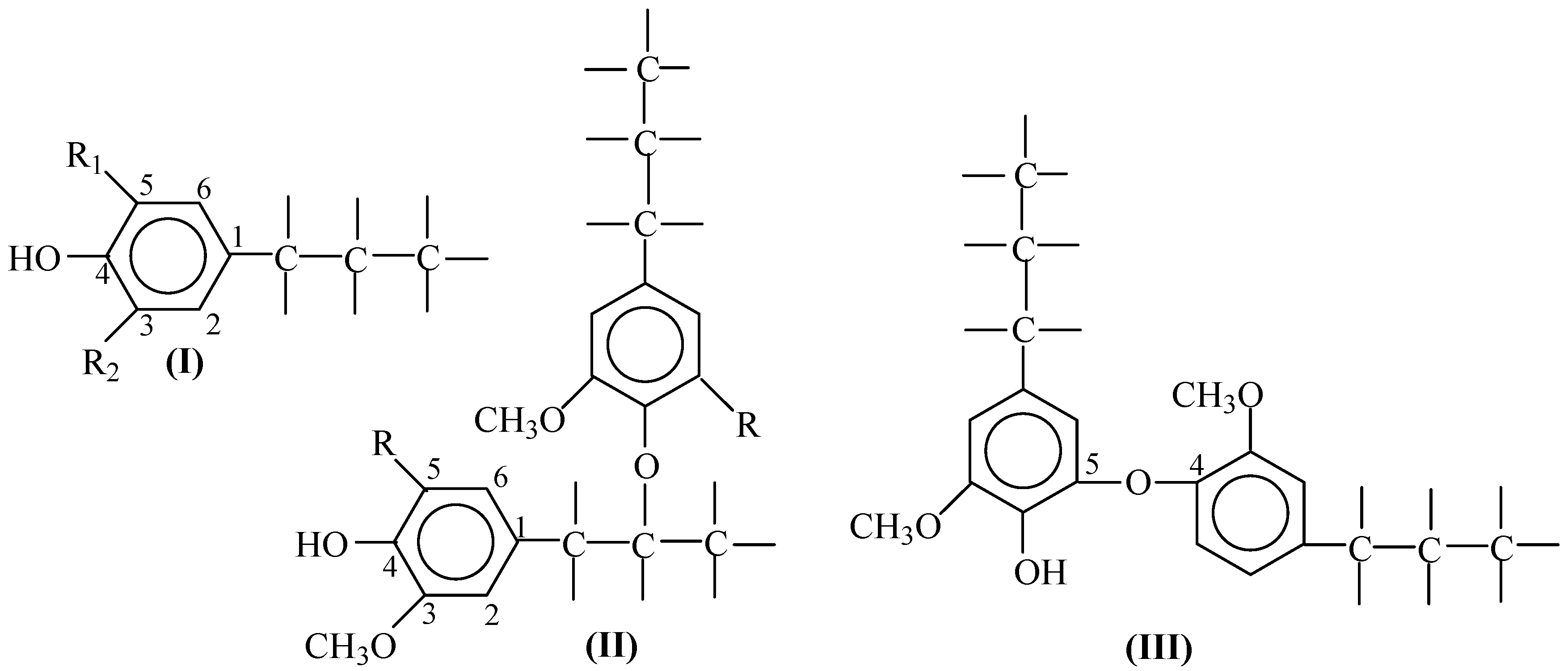
2. Lignin Structure
3. Oxidation of Lignins into the Aromatic Aldehydes by Nitrobenzene


4. Catalytic Oxidation of Lignins by Oxygen
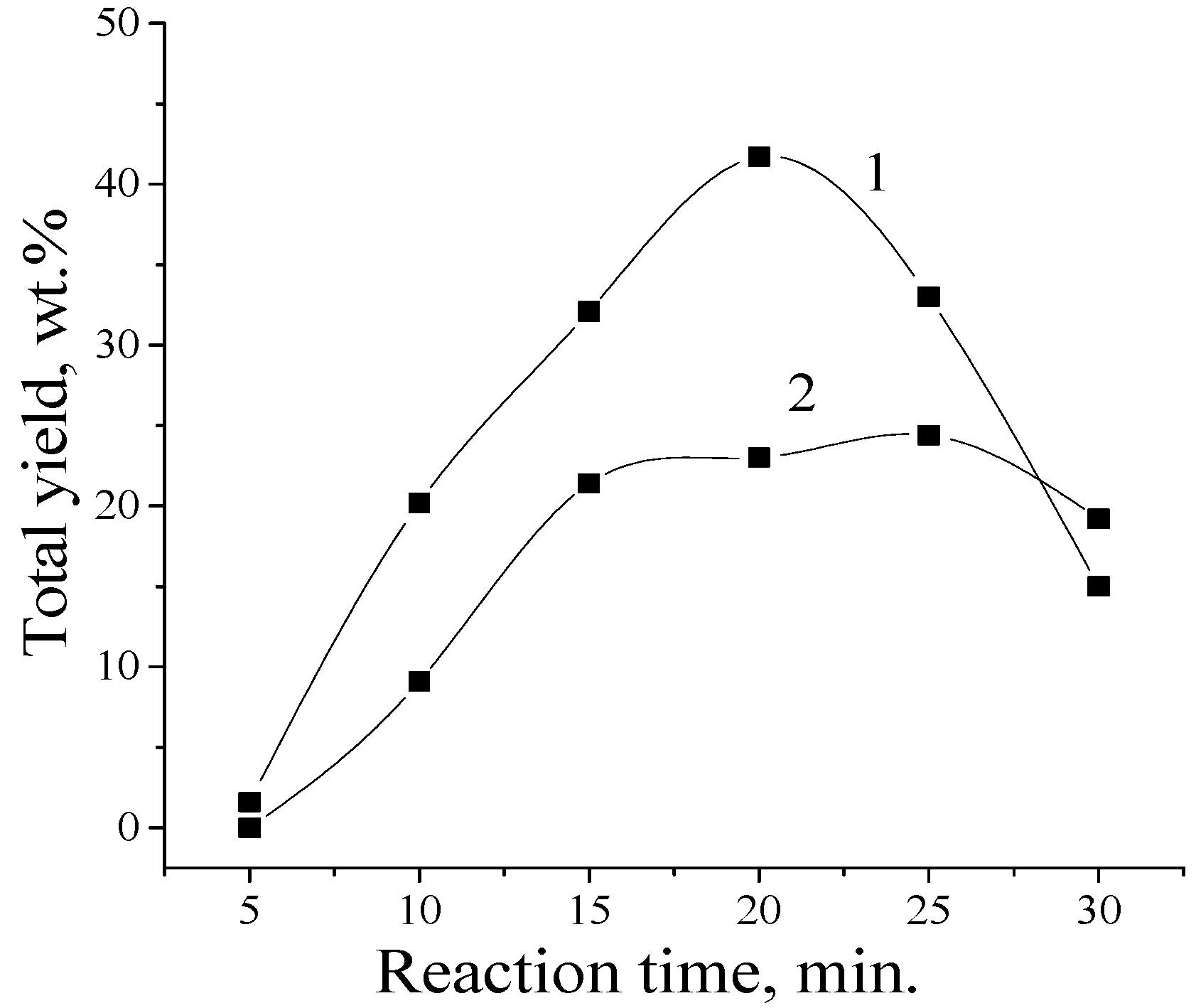
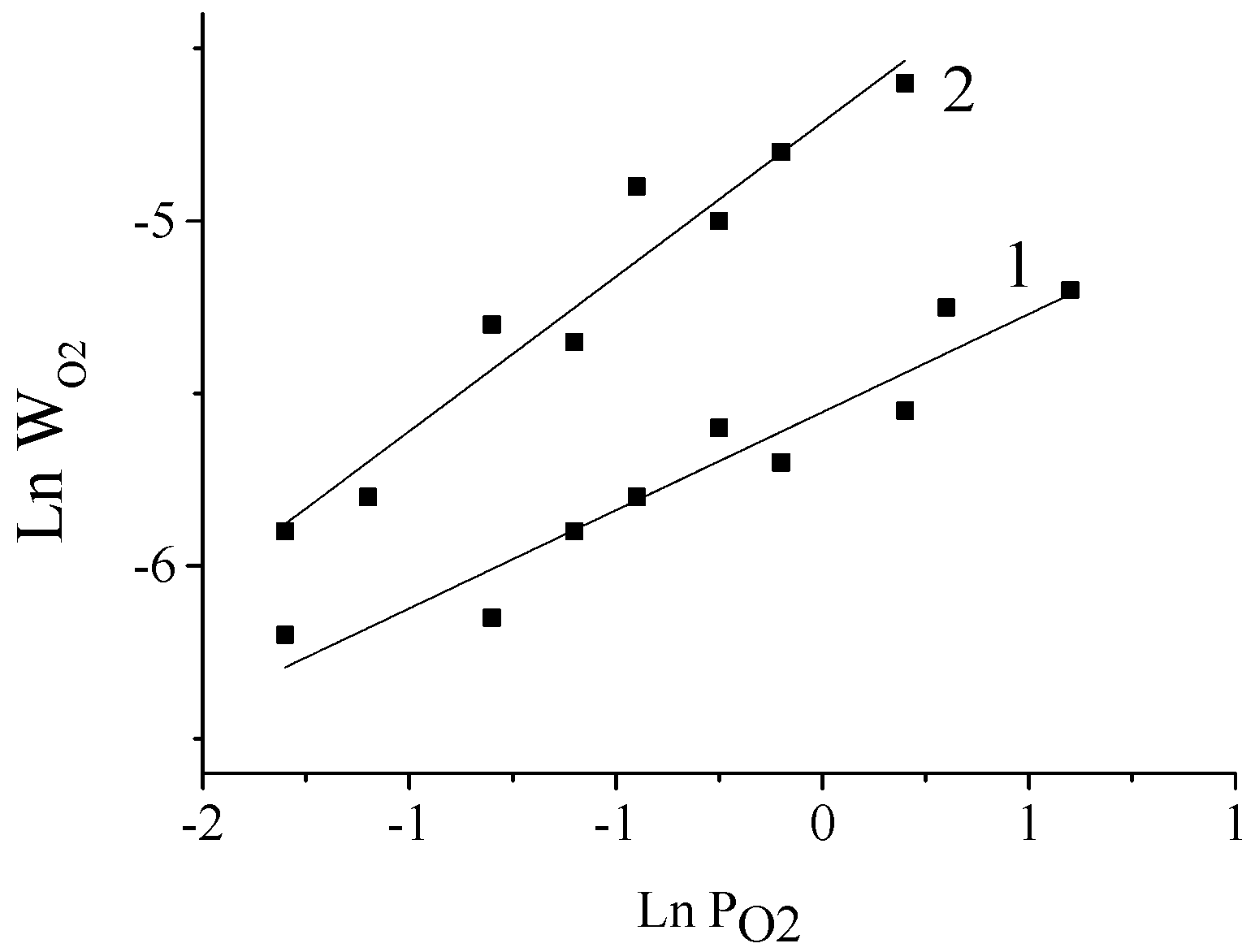
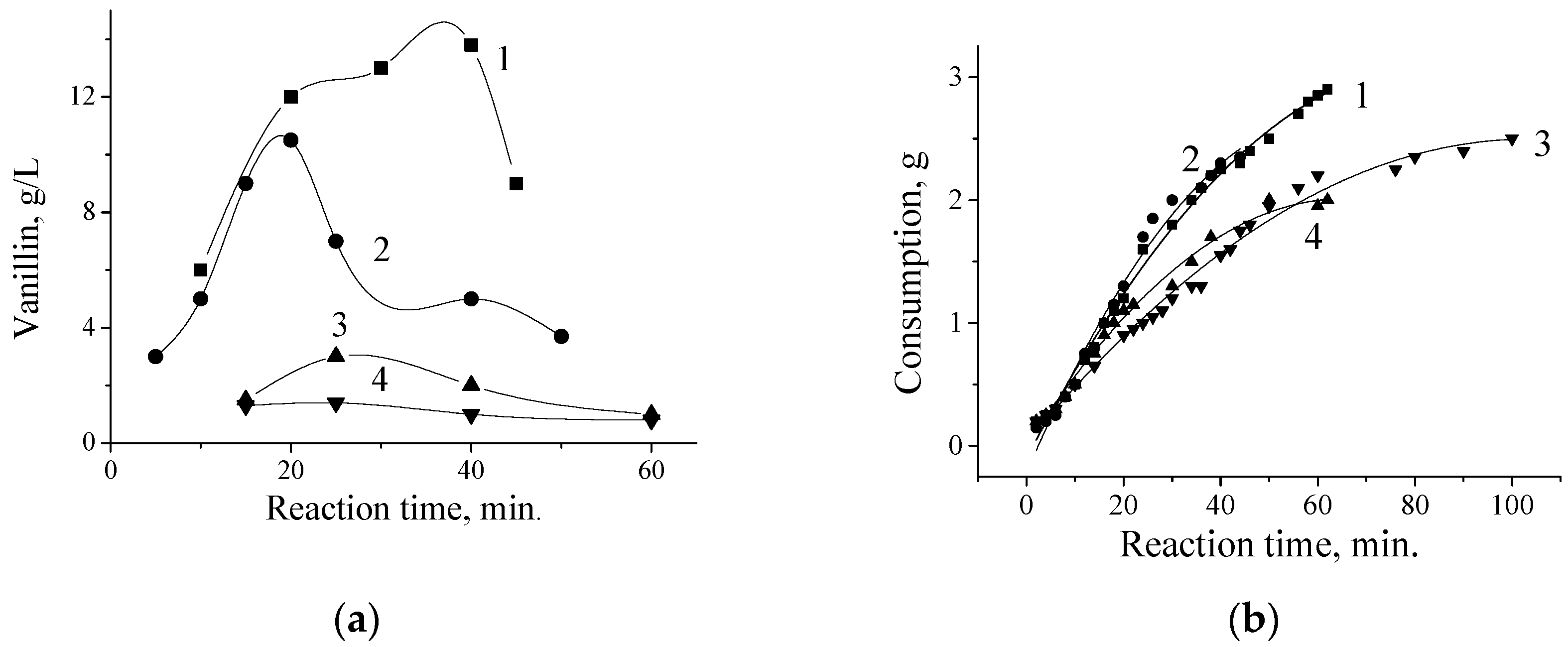
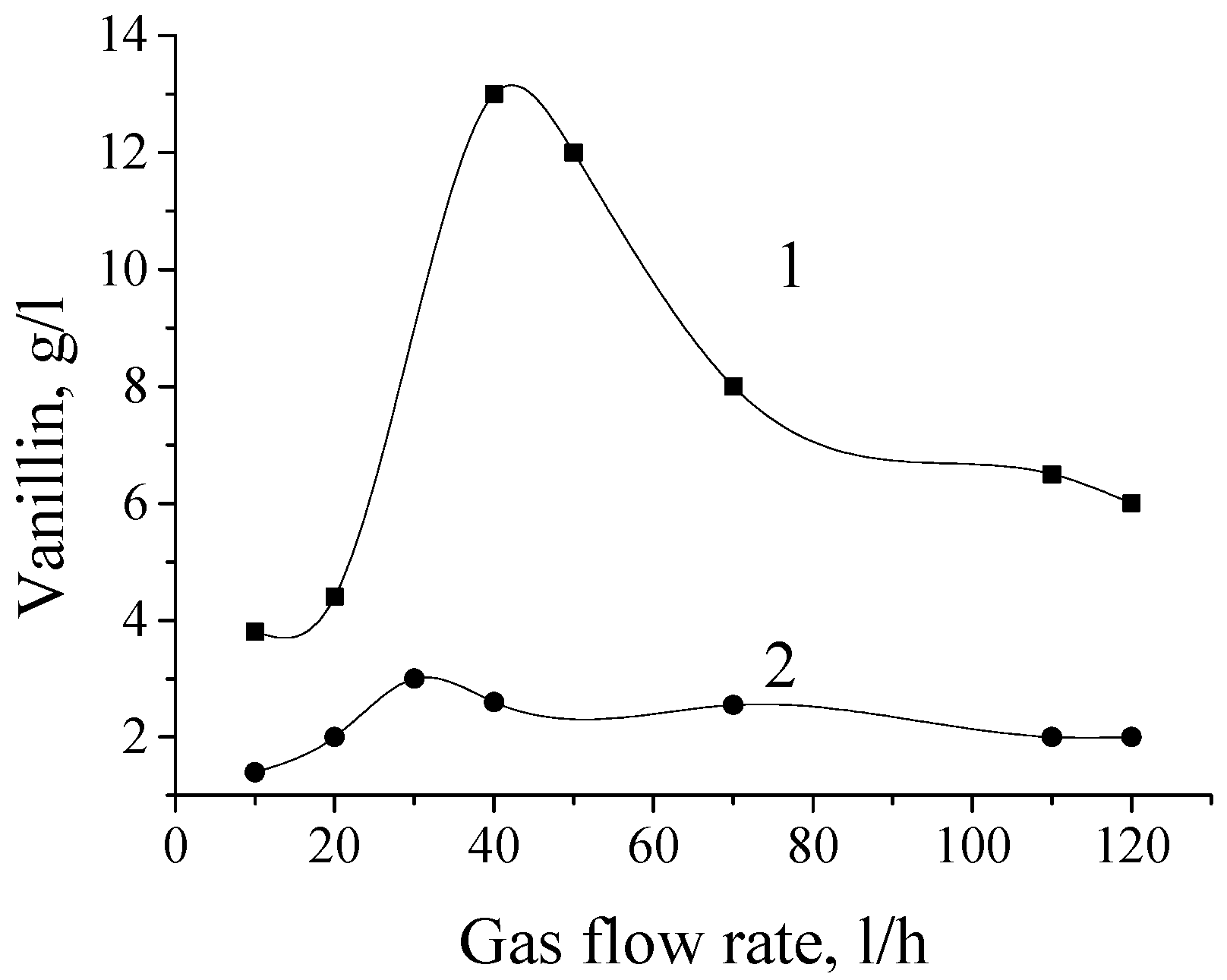
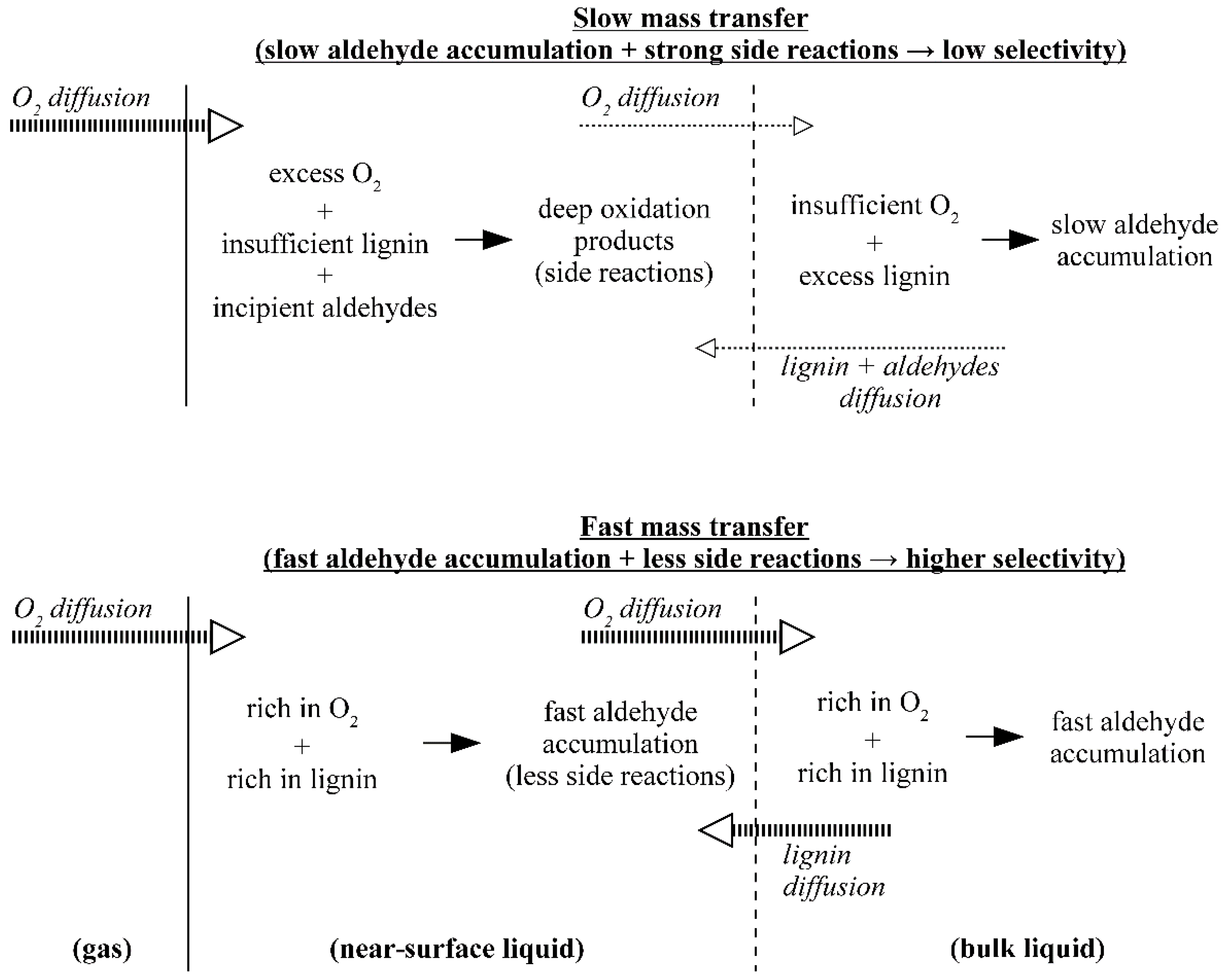
5. The Effects of Diffusion in the Processes of Lignin Oxidation
6. The Influence of Temperature on Yield of the Aromatic Aldehydes in Oxidation by Oxygen
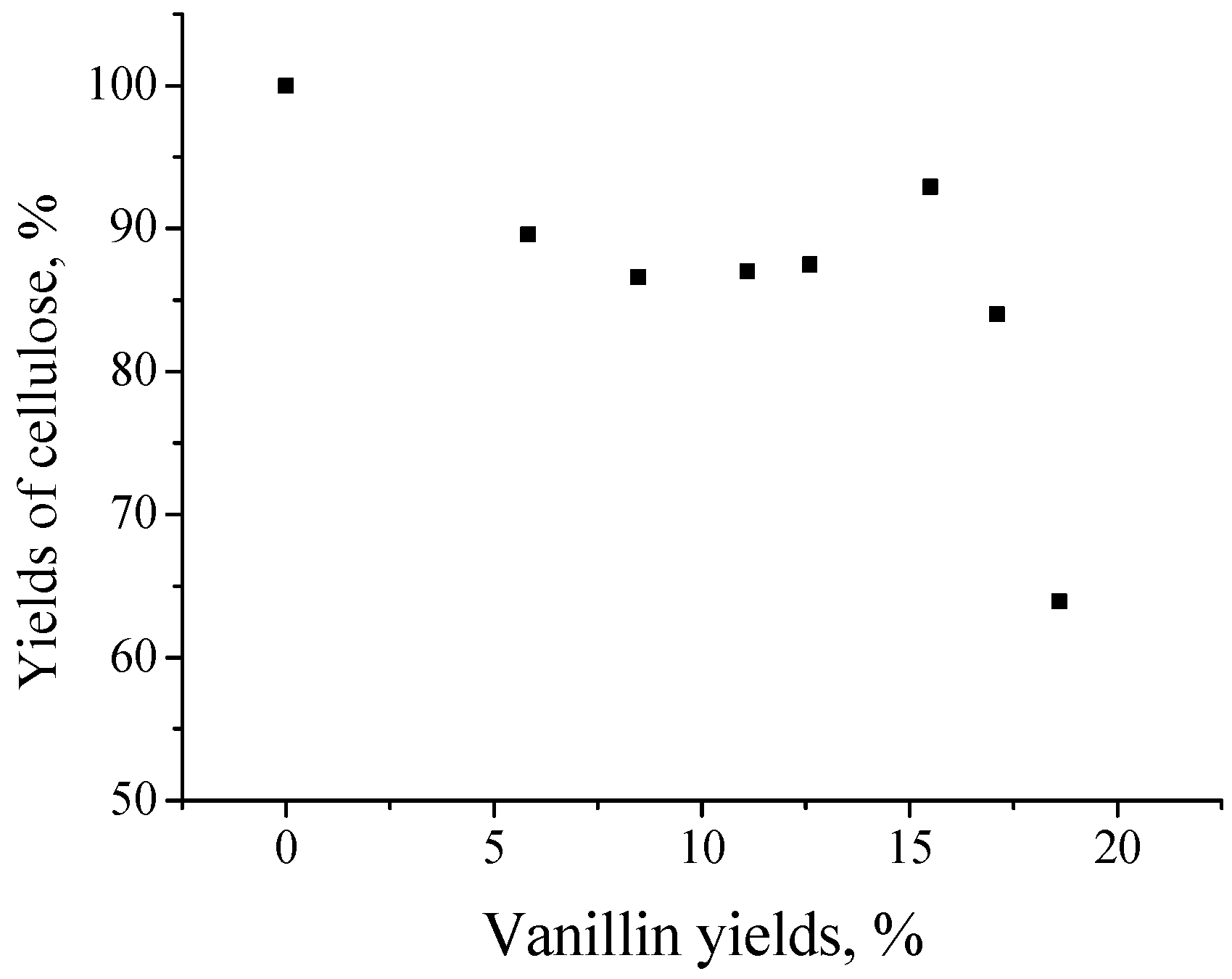
7. Kinetic Trends of Oxidation of Lignins
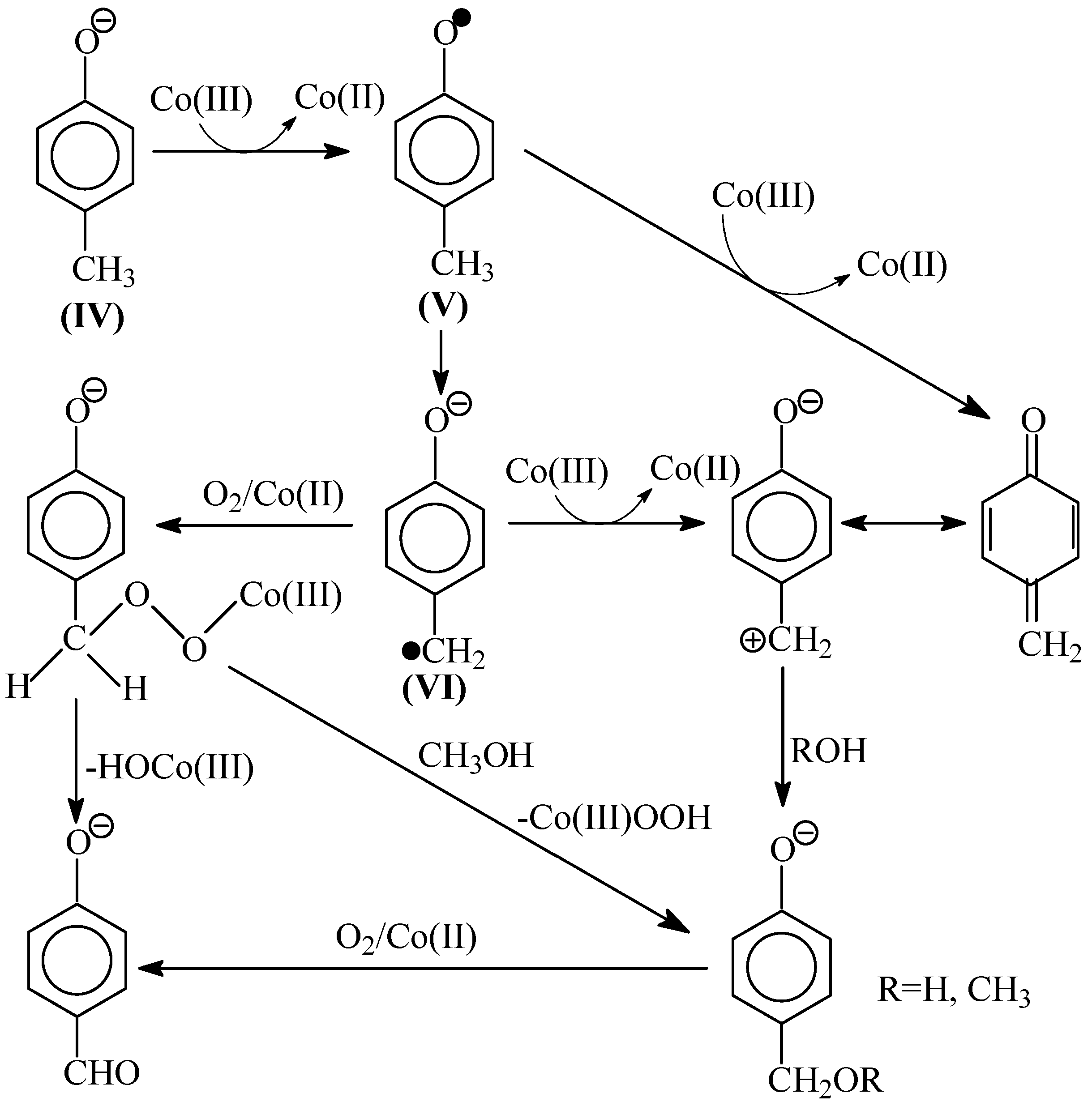
8. The Mechanism of Oxidation of Lignins

- Oxidation of lignins into vanillin and syringaldehyde proceeds with oxidants of a different nature (nitrobenzene, copper oxide [30], oxygen with and without [71] catalysts) with high and similar selectivities (over 40 wt. % of aldehydes). It appears unlikely that the same outcome can transpire through different oxidant-dependent mechanisms in such a chemically complicated system. Therefore, a mechanism hypothesis should be universal with respect to oxidant nature.
- Production of vanillin and syringaldehyde from lignins is accompanied by the formation of the corresponding aceto-derivatives as side products.
- Vanillin, syringaldehyde, and their aceto-derivatives are produced in smaller amounts in lignin alkaline hydrolysis without oxidants. This indicates that lignin oxidation and lignin alkaline hydrolysis may have common stages, and these should probably be the final stages.














9. Development Prospects for the Technology of the Aromatic Aldehydes Production from Lignins
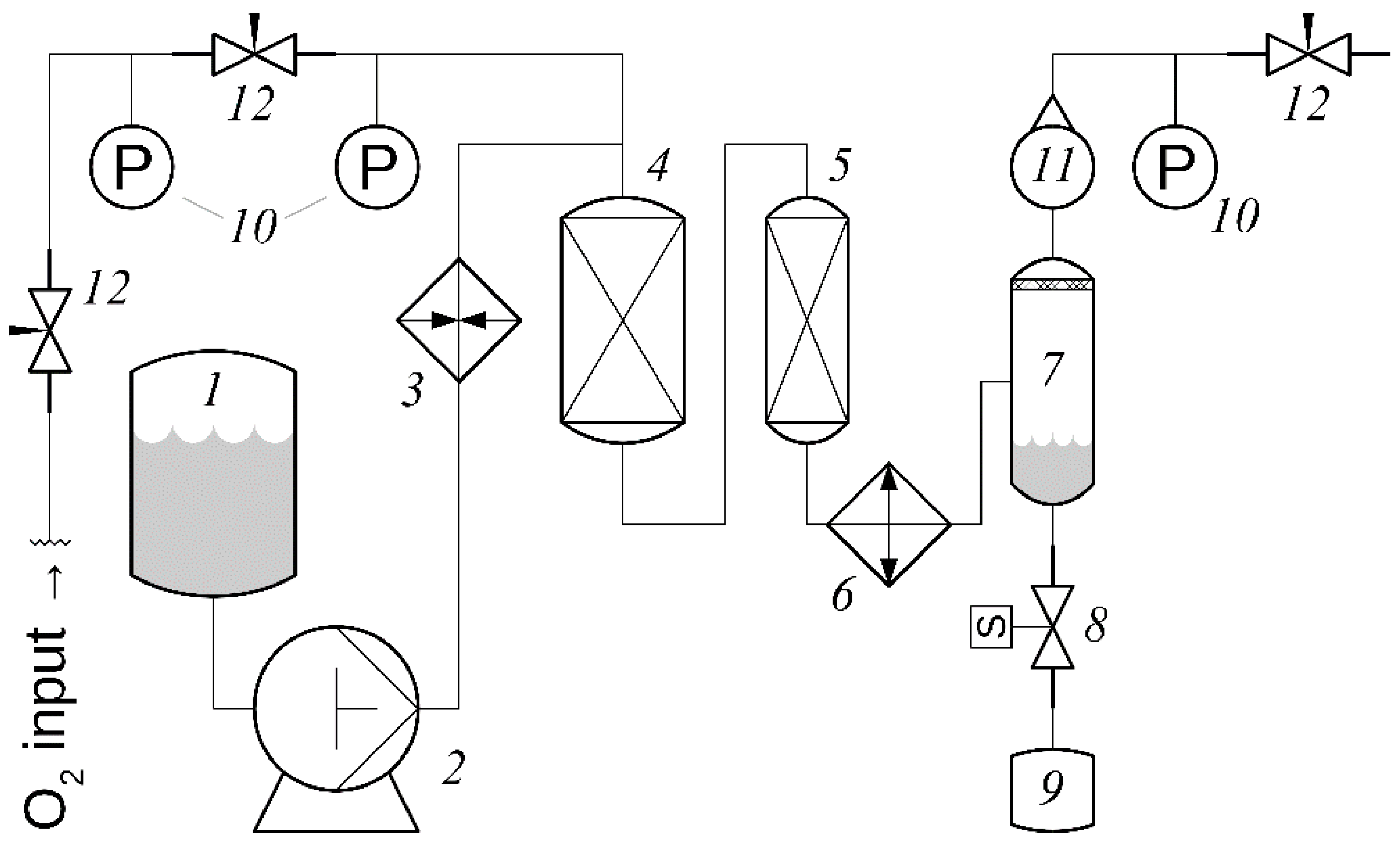
9.1. Oxidation of a Lignosulfonate Solution or Aspen Wood Slurry in a Flow Reactor System
9.2. The Prospects of Comprehensive Wood Processing into the Aromatic Aldehydes and Valuable Carbohydrate-Derived Products
10. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| PPU | Phenylpropane units |
| NBO | Nitrobenzene oxidation |
References
- Brauns, F.E.; Brauns, D.A. The Chemistry of Lignin; Lesnaya Promyshlennost: Moskow, Russia, 1964; p. 583. [Google Scholar]
- Zakzeski, J.; Bruijnincx, P.C.A.; Jongerius, A.L.; Weckhuysen, B.M. The Catalytic valorization of lignin for the production of renewable chemicals. Chem. Rev. 2010, 110, 3552–3599. [Google Scholar] [CrossRef] [PubMed]
- Pandey, M.P.; Kim, C.S. Lignin depolymerization and conversion: A Review of thermochemical methods. Chem. Eng. Technol. 2011, 34, 29–41. [Google Scholar] [CrossRef]
- Li, C.Z.; Zhao, X.C.; Wang, A.Q.; Huber, G.W.; Zhang, T. Catalytic transformation of lignin for the production of chemicals and fuels. Chem. Rev. 2015, 115, 11559–11624. [Google Scholar] [CrossRef] [PubMed]
- Lange, H.; Decina, S.; Crestini, C. Oxidative upgrade of lignin—Recent routes reviewed. Eur. Polym. J. 2013, 49, 1151–1173. [Google Scholar] [CrossRef]
- Ma, R.; Xu, Y.; Zhang, X. Catalytic oxidation of biorefinery lignin to value-added chemicals to support sustainable biofuel production. ChemSusChem 2015, 8, 24–51. [Google Scholar] [CrossRef] [PubMed]
- Behling, R.; Valange, S.; Chatel, G. Heterogeneous catalytic oxidation for lignin valorization into valuable chemicals: What results? What limitations? What trends? Green Chem. 2016, 18, 1839–1854. [Google Scholar] [CrossRef]
- Mark, H.V. (Ed.) Encyclopedia of Chemical Technology, 2nd ed.; Interscience Publishers: New York, NY, USA; John Wiley & Sons: London, UK, 1972; Volume 21, pp. 80–196. [Google Scholar]
- Dardelet, S.; Froment, P.; Lacoste, N.; Robert, A. Aldehyde syringique. Possibilites de production a partir de bois feuillus. Rev. ATIP 1985, 39, 267–274. [Google Scholar]
- Gogotov, A.F. Analysis of raw material base for obtaining of aromatic aldehides. Khimiya Rastitel’nogo Syr’ya 1999, 2, 73–79. [Google Scholar]
- Emsley, J. Plant a tree for chemistry. New Sci. 1987, 116, 39–42. [Google Scholar]
- Gogotov, A.F.; Babkin, V.A. Lignin—Potential source of value low-molecular substances. Khimiya Rastitel’nogo Syr’ya 1994, 2, 597–604. [Google Scholar]
- Mashkovsky, M.D. Medicinal Products; Novaya Volna: Moskow, Russia, 2014. [Google Scholar]
- Szallasi, A.; Blumberg, P.M. Mechanisms and terapeutic potentials of vanilloids (capsaicin-like molecules). Adv. Pharmacol. 1993, 24, 123–155. [Google Scholar] [PubMed]
- MacRae, W.D.; Towers, G.H.N. Biological activities of lignans. Phytochemistry 1984, 23, 1207–1220. [Google Scholar] [CrossRef]
- Erofeev, Y.V.; Afanas’eva, V.L.; Glushkov, R.V. Synthetic routes to 3,4,5-trimethoxybenzaldehyde. Pharm. Chem. J. 1990, 24, 501–510. [Google Scholar] [CrossRef]
- Heitner, C.; Dimmel, D.R.; Schmidt, J.A. (Eds.) Lignin and Lignans: Advances in Chemistry; CRC Press: London, UK; New York, NY, USA, 2010; p. 651. [Google Scholar]
- Nguyen, H.T.H.; Reis, M.H.; Qi, P.X.; Miller, S.A. Polyethylene ferulate (PEF) and congeners: Polystyrene mimics derived from biorenewable aromatics. Green Chem. 2015, 17, 4512–4517. [Google Scholar] [CrossRef]
- Fache, M.; Boutevin, B.; Caillol, S. Vanillin production from lignin and its use as a renewable chemical. ACS Sustain. Chem. Eng. 2016, 4, 35–46. [Google Scholar] [CrossRef]
- Zhao, S.; Abu-Omar, M.M. Renewable thermoplastics based on lignin-derived polyphenols. Macromolecules 2017, 50, 3573–3581. [Google Scholar] [CrossRef]
- Zhou, J.; Zhang, H.; Deng, J.; Wu, Y. High glass-transition temperature acrylate polymers derived from biomasses, syringaldehyde, and vanillin. Macromol. Chem. Phys. 2016, 217, 2402–2408. [Google Scholar] [CrossRef]
- Fengel, D.; Wegener, G. Wood (Chemistry, Ultrastructure, Reactions); Walter de Gruyter: Berlin, Germany; New York, NY, USA, 1984. [Google Scholar]
- Leopold, B. Studies on lignin. III. Oxidation of wood from Norway spruce with nitrobenzene and alkali. Acta Chem. Scand. 1952, 6, 38–48. [Google Scholar] [CrossRef]
- Kurshner, K. The difficulties in the production of vanillin from sulphite liquors. Rus. J. Appl. Chem. 1955, 28, 957–968. [Google Scholar]
- Bots, R.H. Process of Manufacturing Vanillin. U.S. Patent 1,643,805, 27 September 1927. [Google Scholar]
- Tomlinson, G.H.; Hibbert, H. Studies on lignin and related compounds. XXV. Mechanism of vanillin formation from spruce lignin sulfonic acids in relation to lignin structure. J. Am. Chem. Soc. 1936, 58, 348–353. [Google Scholar] [CrossRef]
- Min, D.; Xiang, Z.; Liu, J.; Jameel, H.; Chiang, V.; Jin, Y.; Chang, H. Improved protocol for alkaline nitrobenzene oxidation of woody and non-woody biomass. J. Wood Chem. Technol. 2015, 35, 52–61. [Google Scholar] [CrossRef]
- Adaci, S.; Tanimoto, M.; Tanaka, M.; Matsuno, R. Kinetics of the alkaline nitrobenzene oxidation of lignin in rice straw. Chem. Eng. J. 1992, 49, 17–21. [Google Scholar] [CrossRef]
- Leopold, B.; Malmstrom, I.L. Studies on lignin. IV. Investigation on nitrobenzene oxidation products of lignin from different woods by paper partition chromatography. Acta Chem. Scand. 1952, 6, 49–54. [Google Scholar] [CrossRef]
- Pepper, J.M.; Casselman, B.W.; Karapally, J.C. Lignin oxidation. Preferential use of cupric oxide. Can. J. Chem. 1967, 45, 3009–3012. [Google Scholar] [CrossRef]
- Gogotov, A.F.; Makovskaya, T.I.; Babkin, V.A. The effect of various additives on the yield of aromatic aldehydes in the aspen nitrobenzene oxidation. Chem. Sustain. Dev. 1996, 4, 187–192. [Google Scholar]
- Simpson, W.G.; Sondhimer, Е. Silica acid column chromatography in the alkaline nitrobenzene oxidation of wood. TAPPI 1960, 43, 1025–1026. [Google Scholar]
- Ikeda, T.; Holtman, K.; Kadla, J.F.; Chang, H.M.; Jameel, H. Studies on the effect of ball milling on lignin structure using a modified DFRC method. J. Agric. Food Chem. 2002, 50, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Leopold, B. Studies on lignin. Part VIII. Nitrobenzene oxidation and sulphonation of wood decayed by brown—Rotting fungi. Svensk Kemisk Tidskrift 1951, 63, 260–271. [Google Scholar]
- Ikeda, T.; Magara, K. Chemical properties of softwood soda-anthraquinone lignin. J. Wood Chem. Technol. 2015, 35, 167–177. [Google Scholar] [CrossRef]
- Moskovtsev, N.G.; Strel’skaya, S.A. Composition and properties of the sediment formed during the conversion of aqueous of wood prehydrolysates. Wood Chem. 1986, 1, 63–67. [Google Scholar]
- Jin, Z.; Jin, G.; Shao, S.; Katsumata, K.S. Lignin characteristics of bast fiber and core in kenaf, bark and wood of paper mulberry and mulberry. J. Wood Sci. 2016, 58, 144–152. [Google Scholar] [CrossRef]
- Gogotov, A.F. A new hypothesis of the interaction of lignin and an oxidizer in the process of nitrobenzene oxidation. Khimiya Rastitel’nogo Syr’ya 1999, 2, 65–72. [Google Scholar]
- Tarabanko, V.E.; Kaigorodov, K.L.; Koropachinskaya, N.V.; Chelbina, Y.V. Preparation of aromatic aldehydes from biobutanol production wastes. Chem. Sustain. 2012, 20, 471–476. [Google Scholar]
- Chirkin, G.A.; Tishenko, D.V. Redox-oxidative reactions during alkaline boiling of wood. Rus. J. Appl. Chem. 1962, 35, 153–159. [Google Scholar]
- Kleinert, T.N. Effect of temperature on the ratio of the contributions of fast and slowly soluble lignin. TAPPI 1964, 49, 53–57. [Google Scholar]
- Tarabanko, V.E.; Koropachinskaya, N.V.; Kudryashov, A.V.; Kuznetsov, B.N. Influence of lignin origin on the efficiency of the catalytic oxidation of lignin into vanillin and syringaldehyde. Russ. Chem. Bull. 1995, 2, 367–371. [Google Scholar] [CrossRef]
- Tarabanko, V.E.; Fomova, N.A.; Kuznetsov, B.N.; Kudryashev, A.V.; Ivanchenko, N.M. On the mechanism of vanillin formation in catalytic oxidation of lignin with oxygen. React. Kinet. Catal. Lett. 1995, 55, 161–170. [Google Scholar] [CrossRef]
- Tarabanko, V.E.; Pervishina, E.P.; Hendogina, Y.V. Kinetics of aspen wood oxidation by oxygen in alkaline media. React. Kinet. Catal. Lett. 2001, 72, 153–162. [Google Scholar] [CrossRef]
- Kamaldina, O.D.; Massov, Y.A. Preparation of Vanillin from Lignosulfonates; TsBTI TsINIS: Moscow, Russia, 1959. [Google Scholar]
- Epshtein, R.B. Preparation of vanillin from wood. Tr. Ukrainian NII Pischevoy Promishlennosty 1959, 2, 201–213. [Google Scholar]
- Villar, J.C.; Caperos, A.; Garcia Ochoa, F. Oxidation of hardwood kraft-lignin to phenolic derivatives with oxygen as oxidant. J. Wood Chem. Technol. 2001, 35, 245–255. [Google Scholar] [CrossRef]
- Wu, G.; Heits, M.; Chornet, E. Improved alkaline oxidation process for the production of aldehydes from steam-explosion lignin. Ind. Eng. Chem. Res. 1994, 33, 718–723. [Google Scholar] [CrossRef]
- Mathias, A.L.; Rodrigues, A.E. Production of vanillin by oxidation of pine kraft-lignins with oxygen. Holzforschung 1995, 49, 273–278. [Google Scholar] [CrossRef]
- Tarabanko, V.E.; Gulbis, G.R.; Ivanchenko, N.M.; Kuznetsov, B.N.; Kudryashev, A.V. Study of wood and lignosulfonates processing into fine products. Chem. Sustain. Dev. 1996, 4, 405–417. [Google Scholar]
- Koropachinskaya, N.V.; Tarabanko, V.E.; Chernyak, M.Y. Catalytic oxidation of birch wood (Betula Pendula Roth.) with oxygen in syringaldehyde and vanillin. Khimiya Rastitel’nogo Syr’ya 2003, 2, 9–14. [Google Scholar]
- Santos, S.G.; Marcues, A.P.; Lima, D.L.D.; Evtuguin, D.V.; Esteves, V.I. Kinetics of Eucaliptus lignosulfonate oxidation to aromatic aldehydes by oxygen in alkaline media. Ind. Eng. Chem. Res. 2011, 50, 291–298. [Google Scholar] [CrossRef]
- Villar, J.C.; Caperos, A.; Garcia Ochoa, F. Oxidation of hardwood kraft-lignin to phenolic derivatives. Nitrobenzene and copper oxide as oxidation. J. Wood Chem. Technol. 1997, 17, 259–285. [Google Scholar]
- Pinto, P.C.R.; Borges da Silva, E.A.; Rodrigues, A.E. Insights into oxidative conversion of lignin to high-added-value phenolic aldehydes. Ind. Eng. Chem. Res. 2011, 50, 741–748. [Google Scholar] [CrossRef]
- Xiang, Q.; Lee, Y.Y. Production of oxychemicals from precipitated hardwood lignin. Appl. Biochem. Biotechnol. 2001, 91–93, 71–80. [Google Scholar] [CrossRef]
- Zhang, J.H.; Deng, H.B.; Lin, L. Wet aerobic oxidation of lignin into aromatic aldehydes catalysed by a perovskite-type oxide: LaFe1−xCuxO3 (x = 0, 0.1, 0.2). Molecules 2009, 14, 2747–2757. [Google Scholar] [CrossRef] [PubMed]
- Sales, F.G.; Maranhao, L.C.A.; Filho, N.M.L.; Abreu, C.A.M. Kinetic evaluation and modeling of lignin catalytic wet oxidation to selective production of aromatic aldehydes. Ind. Eng. Chem. Res. 2006, 45, 6627–6631. [Google Scholar] [CrossRef]
- Goncalves, A.R.; Schuchardt, U. Oxidation of organosolv Lignins in Acetic Acid. Influence of oxygen pressure. Appl. Biochem. Biotechnol. 1999, 77, 127–132. [Google Scholar] [CrossRef]
- Werhan, H.; Mir, J.M.; Voitl, T.; von Rohr, P.R. Acidic oxidation of kraft lignin into aromatic monomers catalyzed by transition metal salts. Holzforschung 2011, 65, 703–709. [Google Scholar] [CrossRef]
- Werhan, H.; Assmann, N.; von Rohr, P.R. Lignin oxidation studies in a continuous two-phase flow microreactor. Chem. Eng. Process. 2013, 73, 29–37. [Google Scholar] [CrossRef]
- Deng, W.P.; Zhang, H.X.; Wu, X.J.; Li, R.S.; Zhang, Q.H.; Wang, Y. Oxidative conversion of lignin and lignin model compounds catalyzed by CeO2-supported Pd nanoparticles. Green Chem. 2015, 17, 5009–5018. [Google Scholar] [CrossRef]
- Partenheimer, W. The Aerobic oxidative cleavage of lignin to produce hydroxyaromatic benzaldehydes and carboxylic acids via metal/bromide catalysts in acetic acid/water mixtures. Adv. Synth. Catal. 2009, 351, 456–466. [Google Scholar] [CrossRef]
- Das, L.; Kolar, P.; Osborne, J.A.; Sharma-Shivappa, R.R.; Classen, J.J. Selective oxidation of lignin into aromatic aldehydes using niobium oxalate. Trans. ASABE 2016, 59, 727–735. [Google Scholar]
- Schmitt, D.; Regenbrecht, С.; Hartmer, M.; Stecker, F.; Waldvogel, S.R. Highly selective generation of vanillin by anodic degradation of lignin: A combined approach of electrochemistry and product isolation by adsorption. Beilstein J. Org. Chem. 2015, 11, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.Y.; Imbault, A.; Farnood, R. The promoting role of bismuth for the enhanced photocatalytic oxidation of lignin on Pt-TiO2 under solar light illumination. Appl. Catal. B 2017, 204, 296–303. [Google Scholar] [CrossRef]
- Chatel, G.; Rogers, R.D. Review: Oxidation of lignin using ionic liquids-an innovative strategy to produce renewable chemicals. ACS Sustain. Chem. Eng. 2014, 2, 322–339. [Google Scholar] [CrossRef]
- Tarabanko, V.E.; Pervishina, E.P.; Nevkrytova, T.A.; Pen, R.Z. Investigation of the oxidation kinetics of aspen wood by oxygen in an alkaline medium. Khimiya Rastitel’nogo Syr’ya 1999, 4, 53–59. [Google Scholar]
- Tarabanko, V.E.; Pervishina, E.P.; Tarabanko, N.V.; Chernyak, M.Y.; Kaygorodov, K.L.; Chelbina, Y.V.; Boyarchuk, D.V. Kinetics of fir wood oxidation by oxygen in aqueous alkaline media. Khimiya Rastitel’nogo Syr’ya 2016, 4, 57–63. [Google Scholar]
- Denisov, E.T. The Kinetics of Homogeneous Chemical Reactions; Vysshaya Shkola: Moskow, Russia, 1988; p. 391. [Google Scholar]
- Tarabanko, V.E.; Koropachinskaya, N.V.; Kydryashov, A.V.; Kyznetsov, B.N.; Polyakov, S.V.; Zolotukhin, V.N. Investigation of the process of processing wheat straw into aromatic aldehydes and levulinic acid. Khimiya Rastitel’nogo Syr’ya 1998, 3, 59–64. [Google Scholar]
- Schoefel, E.W. Manufacture of Vanillin. U.S. Patent 2,598,311, 27 May 1952. [Google Scholar]
- Tarabanko, V.E.; Pervishina, E.P.; Kuznetsov, B.N. Method of Processing of Small-Leaved Wood. Russian Patent 2,178,405, 20 January 2002. [Google Scholar]
- Kaigorodov, K.L.; Tarabanko, V.E.; Chernyak, M.Y.; Chelbina, Y.V.; Tarabanko, N.V.; Smirnova, M.A. Kinetics of low-temperature oxidation of enzime pine lignin (Pinus silvestris) in water-alkaline. Khimiya Rastitel’nogo Syr’ya 2017, 3, 63–70. [Google Scholar]
- Nonhebel, D.C.; Walton, J.C. Free-Radical Chemistry: Structure and Mechanism; Cambridge University Press: New York, NY, USA, 1974. [Google Scholar]
- Denisov, E.T. The Rate Constants of Homolytic Liquid-Phase Reactions; Khimia: Moskow, Russia, 1971; p. 712. [Google Scholar]
- Kleinert, T.N. Mechanisms of Alkaline Delignification: II. Free Radical Reactions. TAPPI 1966, 49, 126–130. [Google Scholar]
- Fargues, C.; Mathias, A.; Rodrigues, A. Kinetics of vanillin production from kraft lignin oxidation. Ind. Eng. Chem. Res. 1996, 35, 28–36. [Google Scholar] [CrossRef]
- Wong, Z.J.; Chen, K.F.; Li, J. Formation of vanillin and syringaldehyde in an oxygen delignification process. BioResources 2010, 5, 1509–1516. [Google Scholar]
- Hermer, E.I. Intensification of oxygen-alkaline delignification of lignocellulosic materials by the O-phenanthroline. Wood Chem. 1992, 4–5, 46–55. [Google Scholar]
- Denisov, E.T.; Mitzkevich, N.I.; Agabekov, V.E. The Mechanisms of Liquid-Phase Oxidation of Oxygen-Containing Compounds; Nauka i Tekhnika: Minsk, Belarus, 1975; p. 240. [Google Scholar]
- Wojnarovits, L.; Foldiak, G.; D’Angelantonio, M.; Emmi, S.S. Mechanism of OH radical-induced oxidation of p-cresol to p-methylphenoxyl radical. Res. Chem. Intermed. 2002, 28, 373–386. [Google Scholar] [CrossRef]
- Maa, Q.; Lianga, C.; Chenb, K.; Liuc, K.; Maod, J.; Chena, Z.; Li, H. Role of alkali in catalytic oxidation of p-cresols. J. Mol. Catal. A 2016, 420, 45–49. [Google Scholar] [CrossRef]
- Sheldon, R.A.; de Heij, N. Aromatic aldehydes via catalytic oxidation. In Role of Oxygen in Chemistry and Biochemistry; Ando, W., Moro-Oka, Y., Eds.; Elsevier: Amsterdam, The Netherlands, 1988; pp. 243–256. [Google Scholar]
- Barton, B.; Logie, G.C.; Schoonees, B.M.; Zeelie, B. Practical process for the air oxidation of cresols: Part A. mechanistic investigations. Org. Proc. Res. Dev. 2005, 9, 62–69. [Google Scholar] [CrossRef]
- Shevchenko, S.M.; Apushkinsky, A.G. Quinone methides in the chemistry of wood. Russ. Chem. Rev. 1992, 61, 105–131. [Google Scholar] [CrossRef]
- Tarabanko, V.E.; Petukhov, D.V.; Selutin, G.E. New Mechanism for the Catalytic Oxidation of Lignin to Vanillin. Kinet. Catal. 2004, 45, 569–577. [Google Scholar] [CrossRef]
- Tarabanko, V.E.; Il’ina, I.I.; Petukhov, D.V.; Pervishina, E.P. On the mechanism of oxidative cleavage of the carbon-carbon bond of lignins at the alkaline medium. Khimiya Rastitel’nogo Syr’ya 1997, 3, 51–58. [Google Scholar]
- Tarabanko, V.E.; Hendogina, Y.V.; Petukhov, D.V.; Pervishina, E.P. On the role of retroaldol reaction in the process of lignin oxidation into vanillin. Kinetics of the vanillylidene acetone cleavage in the alkaline media. React. Kinet. Catal. Lett. 2000, 69, 361–368. [Google Scholar] [CrossRef]
- Guthrie, J.P.; Cooper, K.J.; Cossar, J.; Dawson, B.A.; Taylor, K.F. The retroaldol reaction of cinnamaldehyde. Can. J. Chem. 1984, 62, 1441–1445. [Google Scholar] [CrossRef]
- Palm, V.A. (Ed.) Tables of Rate and Equilibrium Constants of Heterolytic Organic Reactions; VINITI: Moskow, Russia, 1975; Volume 1, p. 82. [Google Scholar]
- Ermakova, M.I.; Kirushina, M.F.; Zarubin, M.Y. Comparison of the OH-acidity of lignin-related phenols in water, alcohols and aqueous-alcoholic media. Wood Chem. 1984, 5, 23–29. [Google Scholar]
- Zakis, G.F. Functional Analysis of Lignins and Their Derivatives; Zinatne: Riga, Latvia, 1987; p. 230. [Google Scholar]
- Schultz, T.P.; Templeton, M.C. Proposed mechanism for nitrobenzene oxidation of lignin. Holzforschung 1986, 40, 93–97. [Google Scholar] [CrossRef]
- Schultz, T.P.; Hubbard, T.V.; Fisher, T.H. Oxidation of β-O-4 lignin models: Reaction difference between Ce(IV) ammonium nitrate and lignin peroxidase. Holzforschung 1995, 49, 528–532. [Google Scholar] [CrossRef]
- Gierer, J. The chemistry of delignification. A general concept. Part II. Holzforschung 1982, 36, 55–64. [Google Scholar] [CrossRef]
- Gierer, J.; Nilvebrant, N.O. Studies on the degradation by oxygen in aciditic media. Holzforschung 1994, 48, 51–58. [Google Scholar] [CrossRef]
- Nilvebrant, N.O. Stilbenes and Their Role in Bleaching. Synthesis and Behaviour in Autoxidative Reactions. Ph.D. Thesis, The Royal Institute of Technology, Stokholm, Sweden, 1993. [Google Scholar]
- Tarabanko, V.E.; Petukhov, D.V. Investigation of the mechanism and improvement of the process of oxidative cleavage of lignins into aromatic aldehydes. Chem. Sustain. Dev. 2003, 11, 645–657. [Google Scholar]
- Gierer, J.; Nelson, C.R. Mechanisms of aryl group migration in the formation of stilbens from 1.1-bis (p-hydroxyaryl)-ethane 2-Oaryl ethers. J. Org. Chem. 1978, 43, 4028–4032. [Google Scholar] [CrossRef]
- Gierer, J.; Imsgard, F. The reactions of lignins with oxygen and hydrogen peroxide in alkaline media. Svensk Paperstidn 1977, 80, 510–518. [Google Scholar]
- Wong, D.F.; Leary, G.; Arct, G. The reaction of lignin model stilbenes with alkali & oxygen & its relevance to mechanical pulp bleaching. In Proceedings of the 8th International Symposium on Wood and Pulping Chemistry, Helsinki, Finland, 6–9 June 1995; Volume 1, pp. 209–216. [Google Scholar]
- Tarabanko, V.E.; Petukhov, D.V.; Avramov, P.V. Study of the acidity of radical intermediates of lignin oxidation by quantum chemical methods. Khimiya Rastitel’nogo Syr’ya 1998, 3, 101–109. [Google Scholar]
- Koropachinskaya, N.V.; Tarabanko, V.E.; Levdanskyi, V.A. Catalytic oxidation of birch bark with oxygen into syringaldehyde and vanillin. Khimiya Rastitel’nogo Syr’ya 2004, 1, 27–30. [Google Scholar]
- Khudyakov, I.V.; Levin, P.P.; Kuz’min, V.A. Reversible Recombination of Radicals. Russ. Chem. Rev. 1980, 49, 982–1002. [Google Scholar] [CrossRef]
- Caldwell, E.S.; Steelink, C. Phenoxy radical intermediates in the enzimatic degradation of lignin model compounds. Biochim. Biophys. Acta 1969, 184, 420–431. [Google Scholar] [CrossRef]
- Hapiot, P.; Pinson, J. Oxidative dimerization of phenolic aldehydes related to lignin formation. J. Phys. Chem. 1994, 98, 2641–2645. [Google Scholar] [CrossRef]
- Ragnar, M.; Lindgren, C.T.; Nilvebrant, N.O. pKa-values of guaiacyl and syringyl phenols related to lignins. J. Wood Chem. Technol. 2000, 20, 277–305. [Google Scholar] [CrossRef]
- Dobos, D. Electrochemical Data. A Handbook for Electrochemists in Industry and Universities; Akademiai Kiado: Budapest, Hungary, 1978; p. 320. [Google Scholar]
- Shibaeva, L.V.; Metelitsa, D.I.; Denisoiv, E.T. Oxidation of phenol by molecular oxygen in aqueous solutions. 1. Kinetics of oxidation of phenol with oxygen. Kinet. Catal. 1969, 10, 1020–1025. [Google Scholar]
- Tarabanko, V.E.; Kudryashev, A.V.; Kuznetsov, B.N.; Gulbis, G.R.; Koropachinskaya, N.V.; Ivanchenko, N.M. Investigation of the process of catalytic oxidation of lignosulfonates to vanillin in a flow reactor. J. Appl. Chem. 1996, 69, 620–624. [Google Scholar]
- Ramachadran, P.A.; Chaudhari, R.V. Three-Phase Catalytic Reactors; Nauka: Novosibirsk, Russia, 1992; Volume 2, p. 396. [Google Scholar]
- Ioffe, I.I.; Reshetov, V.A.; Dobrotvorsyi, A.M. Heteroheneous Catalysis; Khimia: Leningrad, Russia, 1985; p. 224. [Google Scholar]
- Dardelet, S.; Froment, P.; Lacoste, N.; Robert, A. Vanilline et aldehyde syringique. Stabilite a l’oxydation en milieu alcalin par l’oxygene. Rev. ATIP 1985, 39, 369–376. [Google Scholar]
- ITS 1–2015. Manufacture of Pulp, Paper and Board; Information and Technical Reference Book; Rosstandart, NDT Bureau: Moscow, Russia, 2016; p. 216. [Google Scholar]
- Bjørsvik, H.-R. Fine Chemicals from Lignosulfonates. 1. Synthesis of Vanillin by Oxidation of Lignosulfonates. Org. Process Res. Dev. 1999, 3, 330–340. [Google Scholar] [CrossRef]
- Kalunyants, K.A.; Shanenko, E.F.; Zai’tseva, L.V. Modern methods of enzymatic hydrolysis of cellulosic materials. In Itogi Nauki e Tehkniki. Kmimia e Tehknologiya Pish’evyh Productov; VINITI: Moskow, Russia, 1988; Volume 1, p. 187. [Google Scholar]
- Ripachek, V. Biology of Wood-Destroing Fungi; Lesnaya Promyshlennost: Moskow, Russia, 1976; p. 245. [Google Scholar]
- Garcia, V.; Pakkila, J.; Ojamo, H.; Muurinen, E.; Keiski, R.L. Challenges in biobutanol production: How to improve the efficiency? Renew. Sustain. Energy Rev. 2011, 15, 964–980. [Google Scholar] [CrossRef]
- Tarabanko, V.E.; Pervishina, E.P.; Kuznetsov, B.N. Method of Wood Processing into Products of Fine Organic Synthesis. Russian Patent 2,119,427, 27 September 1998. [Google Scholar]
- Tarabanko, V.E.; Kozlov, I.A.; Pervishina, E.P.; Chernyak, M.Y.; Kuznetsov, B.N. Chemical Wood Processing Method. Russian Patent 2,158,192, 27 October 2000. [Google Scholar]
- Tarabanko, V.E.; Kaygorodov, K.L.; Skiba, E.A.; Tarabanko, N.; Chelbina, Y.V.; Baybakova, O.V.; Kuznetsov, B.N.; Djakovitch, L. Processing pine wood into vanillin and glucose by sequential catalytic oxidation and enzymatic hydrolysis. J. Wood Chem. Technol. 2017, 37, 43–51. [Google Scholar] [CrossRef]
- Tarabanko, V.E.; Shambazov, V.K.; Kuznetsova, S.A.; Kuznetsov, B.N. Method of Hardwood Processing into Valuable Organic Products. Russian Patent 2,219,048, 20 December 2003. [Google Scholar]
- Sharkov, V.I.; Kyibina, N.I. Chemistry of Hemicelluloses; Lesnaya Promyshlennost: Moskow, Russia, 1972; p. 440. [Google Scholar]
- Tarabanko, V.E.; Kudryashov, A.V.; Pervishina, E.P.; Kuznetsov, B.N.; Petukhov, D.V.; Hendogina, Y.V. New methods of separation of vanillin and syringcaldehyde. Khimiya Rastitel’nogo Syr’ya 1998, 3, 95–100. [Google Scholar]
- Tarabanko, V.E.; Kudryashov, A.V.; Gulbis, G.R.; Kuznetsov, B.N. Method of Separation of Vanillin and Syringcaldehyde. Russian Patent 2,059,600, 10 May 1996. [Google Scholar]
- Tarabanko, V.E.; Chelbina, Y.V.; Sokolenko, V.A.; Tarabanko, N.V. The study of vanillin extraction by octylamine. Solvent Extr. Ion Exch. 2007, 25, 99–107. [Google Scholar] [CrossRef]
- Tarabanko, V.E.; Chelbina, Y.V.; Kudryashev, A.V.; Tarabanko, N.V. Separation of vanillin and syringaldehyde produced from lignins. Sep. Sci. Technol. 2013, 48, 127–132. [Google Scholar] [CrossRef]
- Mota, M.I.F.; Rodrigue, P.P.C.; Loureiro, J.M.; Rodrigues, A.E. Recovery of vanillin and syringaldehyde from lignin oxidation: A review of separation and purification processes. Sep. Purif. Rev. 2016, 45, 227–259. [Google Scholar] [CrossRef]
- Nepenin, Y.A. Сellulose Technology; Lesnaya Promyshlennost: Moskow, Russia, 1990; Volume 2, p. 597. [Google Scholar]
- Chudakov, M.I. Industrial Use of Lignin; Lesnaya Promyshlennost: Moskow, Russia, 1983; p. 42. [Google Scholar]
- Study into the Establishment of An Aroma and Fragrance Fine Chemicals Value Chain in South Africa (Tender Number T79/07/03). Available online: https://www.thedti.gov.za/industrial_development/docs/fridge/Aroma_Part3.pdf (accessed on 15 October 2017).
- Corma, A.; Iborra, S.; Velty, A. Chemical routes for the transformation of biomass into chemicals. Chem. Rev. 2007, 107, 2411–2502. [Google Scholar] [CrossRef] [PubMed]
- Pileidis, F.D.; Titirici, M.M.; Filoklis, D. Levulinic acid biorefineries: New challenges for efficient utilization of biomass. ChemSusChem 2016, 9, 562–582. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lignin Type | Yield, wt. % of the Lignin | Reference No. | |
|---|---|---|---|
| Vanillin | Syringaldehyde | ||
| Silver fir wood (Abies alba) | 24.3 | - | [29] |
| Norway spruce wood (Picea abies) | 27.5 | 0.06 | [23] |
| White spruce wood (Picea glauca Voss) | 20.1 | - | [22,30] |
| Quaking aspen wood (Populus tremuloides Michx) | 12. | 30. | [30] |
| Quaking aspen wood (Populus tremuloides Michx) | 11 * | 53 * | [31] |
| Birch wood (Betula pendula Roth.) | 12 | 35 | [29] |
| Norway maple wood (Acer platanoides) | 13 | 37 | [32] |
| Milled wood lignin of Loblolly pine (Pinus taeda L.) | 26.6 | - | [33] |
| Organosolv ethanol lignin ** | 6.7 | 17.0 | [32] |
| Kraft softwood lignin | 13.1 | 0.6 | [32] |
| Kraft hardwood lignin | 5.3 | 7.9 | [32] |
| Sulfite softwood lignin | 16.5 | trace | [32] |
| Sulfite hardwood lignin | 6.1 | 10.1 | [32] |
| Brown rotted spruce wood | 13.9 | - | [34] |
| Pine soda lignin | 5.2 | - | [10] |
| Japanese cedar soda-anthraquinone lignin (Cryptomeria japonica) | 8–11.2 | - | [35] |
| Indulin AT commercial pine kraft lignin | 10.9 | - | [35] |
| Pine prehydrolysis liquor precipitate | 8.00 *** | - | [36] |
| Klason hydrolysis lignin | 1.5 | - | [24] |
| Lignin Type | Oxidant, Catalyst, Other Remarks | Yield, wt. % of Lignin | Reference No. |
|---|---|---|---|
| Spruce wood (28% of lignin) | Air, no catalyst | 11.4 | [46] |
| Spruce wood | Air, Cu(OH)2 10% of wood weight | 18.9 | [46] |
| Spruce wood | Air, MnO2 10% of wood weight | 18.2 | [46] |
| Spruce wood | Nitrobenzene | 20–27 | Table 1 |
| Pine wood | Oxygen, no catalyst | 12.9 | [42] |
| Pine wood | Oxygen, Cu(OH)2 | 23.1 | [42] |
| Brown rotted Pine wood | Oxygen, Cu(OH)2 | 19.8 | [42] |
| Aspen wood | Oxygen, CuO, flow reactor | 36 * | [50] |
| Aspen wood | Nitrobenzene | 43.6 * | Table 1 |
| Birch wood | Oxygen, Cu(OH)2 | 43 * | [51] |
| Birch wood | Nitrobenzene | 47 * | Table 1 |
| Softwood lignosulfonates | Air, no catalyst | 5–7 | [1,45] |
| Softwood lignosulfonates | Oxygen, Cu(OH)2 | 12 | [43] |
| Softwood lignosulfonates | Nitrobenzene | 16 | Table 1 |
| Softwood lignosulfonates | Oxygen, Co(OH)3, Mn3O4 | 10–15.5 | [1,24] |
| Eucalyptus lignosulfonates | Oxygen, Cu(OH)2 | 13.4 * | [52] |
| Kraft lignin | Oxygen, no catalyst | 4.5–10.8 | [49,53,54] |
| Kraft lignin | Nitrobenzene | 13.1 | Table 1 |
| Poplar precipitated hydrolysis lignin | Oxygen, Cu(OH)2, Fe(OH)3 | 15 * | 55 |
| Temperature, °C | Yields, wt. % Based on Lignin | ||
|---|---|---|---|
| Vanillin | Syringaldehyde | Total Yields | |
| 170 | 5.5 | 11.5 | 17.0 |
| 180 | 7.4 | 15.6 | 23.0 |
| 190 | 7.8 | 23.4 | 31.2 |
| 200 | 7.8 | 19.7 | 27.5 |
| No. | Catalyst, g/L | Base, g/L | T, °C | Initial pH | Initial O2 Consumption Rate, g/min | Peak Vanillin Concentration Parameters | ||
|---|---|---|---|---|---|---|---|---|
| [V] *, g/L | Time, min | Attained pH | ||||||
| 1 | - | NaOH, 120 | 160 | >13 | 0.094 | 6.5 | >60 | 9.7 |
| 2 | Cu(OH)2, 3.24 | NaOH, 120 | 160 | >13 | 0.110 | 8.7 | >60 | 10.4 |
| 3 | Cu(OH)2, 6.51 | NaOH, 120 | 160 | >13 | 0.136 | 11.3 | 40 | 9.8 |
| 4 | Cu(OH)2, 9.75 | NaOH, 120 | 160 | >13 | 0.080 | 13.9 | 40 | 9.9 |
| 5 | Cu(OH)2, 16.3 | NaOH, 120 | 160 | >13 | 0.103 | 14.7 | 40 | 10.9 |
| 6 | Cu(OH)2, 26.0 | NaOH, 120 | 160 | >13 | 0.137 | 11.4 | 20 | 10.6 |
| 7 | Cu(OH)2, 9.75 | NaOH, 80 | 160 | >13 | 0.115 | 10.7 | 20 | 11.0 |
| 8 ** | Cu(OH)2, 9.75 | NaOH, 40 + K2CO3, 300 | 160 | 10.8 | 0.056 | 3.3 | 25 | 10.5 |
| 9 ** | Cu(OH)2, 9.75 | K2CO3, 300 | 160 | 10.3 | 0.071 | 0.9 | 20 | 10.0 |
| 10 | - | NaOH, 120 | 110 | >13 | 0.018 | 5.4 | 70 | - |
| 11 | Cu(OH)2, 9.75 | NaOH, 120 | 110 | >13 | 0.050 | 7.7 | 70 | 9.9 |
| 12 | AgOH, 12.5 | NaOH, 120 | 110 | >13 | 0.042 | 5.7 | 70 | 9.5 |
| 13 | Cu(OH)2, 9.75 + AgOH, 12.5 | NaOH, 120 | 110 | >13 | 0.092 | 7.0 | >90 | 10.0 |
| Substance | Price, 103 USD/t | Global Production, 103 t/year | Yield from One Tonne of Wood, kg | Value of the Product from One Tonne of Wood, USD |
|---|---|---|---|---|
| Vanillin | 10–15 | 15–20 | 20 | 200–300 |
| 3,4,5-trimethoxybenzaldehyde from syringaldehyde | 25–30 | 10–20 | 50 | 1250–1500 |
| Levulinic acid | 3 | 2–5 | 100–120 | 300–360 |
| Furfural from pentosanes | 1–2 | 90 | 100–120 | 100–240 |
| Xylitol from xylose (in pentosanes) | 6–8 | 20–30 | 130–180 | 800–1200 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tarabanko, V.E.; Tarabanko, N. Catalytic Oxidation of Lignins into the Aromatic Aldehydes: General Process Trends and Development Prospects. Int. J. Mol. Sci. 2017, 18, 2421. https://doi.org/10.3390/ijms18112421
Tarabanko VE, Tarabanko N. Catalytic Oxidation of Lignins into the Aromatic Aldehydes: General Process Trends and Development Prospects. International Journal of Molecular Sciences. 2017; 18(11):2421. https://doi.org/10.3390/ijms18112421
Chicago/Turabian StyleTarabanko, Valery E., and Nikolay Tarabanko. 2017. "Catalytic Oxidation of Lignins into the Aromatic Aldehydes: General Process Trends and Development Prospects" International Journal of Molecular Sciences 18, no. 11: 2421. https://doi.org/10.3390/ijms18112421
APA StyleTarabanko, V. E., & Tarabanko, N. (2017). Catalytic Oxidation of Lignins into the Aromatic Aldehydes: General Process Trends and Development Prospects. International Journal of Molecular Sciences, 18(11), 2421. https://doi.org/10.3390/ijms18112421