The Molecular and Phenotypic Basis of the Glioma Invasive Perivascular Niche

{kind=link}
{kind=link}
Abstract
1. Introduction
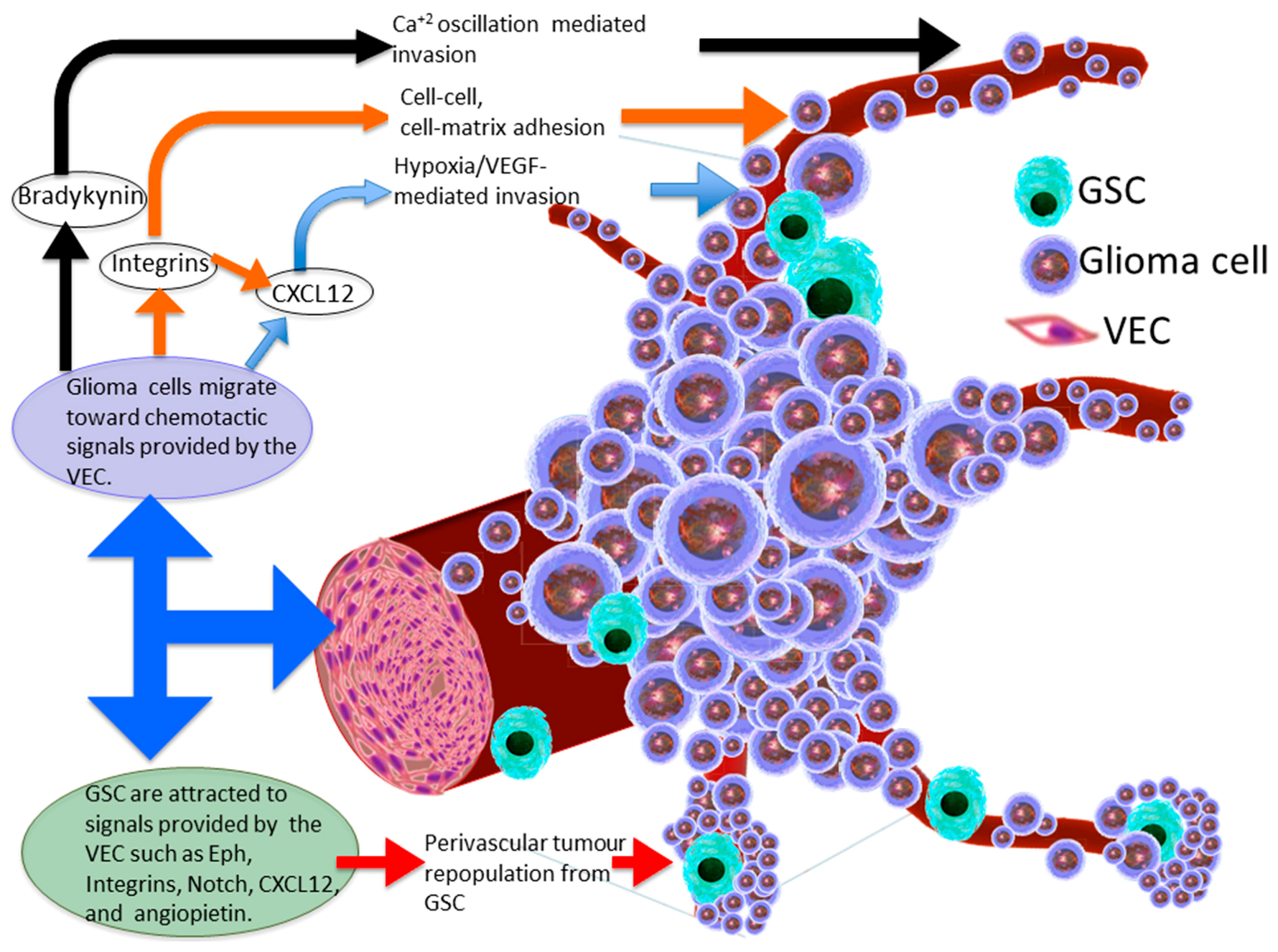
2. The Glioma Invasive Phenotype
3. Molecular Basis of Glioma Perivascular Invasion
4. Chemokine Signalling within the Perivascular Invasive Niche
5. Perivascular Niche Enhances Glioma Stem Cell Invasion
6. Vascular Endothelial Cell-Glioma Stem Cell Cross-Talk
7. Heterogeneous Location of Glioma Stem Cells
8. The Gliovascular Regulatory Unit
9. Conclusions
Conflicts of Interest
References
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Beauchesne, P. Extra-neural metastases of malignant gliomas: Myth or reality? Cancers 2011, 3, 461–477. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.E. Mutational analysis reveals the origin and therapy-driven evolution of reccurent glioma. Science 2014, 189, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Mazor, T.; Pankov, A.; Johnson, B.E.; Hong, C.; Hamilton, E.G.; Bell, R.J.A.; Smirnov, I.V.; Reis, G.F.; Phillips, J.J.; Barnes, M.J.; et al. DNA methylation and somatic mutations converge on the cell cycle and define similar evolutionary histories in brain tumors. Cancer Cell 2015, 28, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Suh, Y.; Lee, S.-J. Radiation treatment and cancer stem cells. Arch. Pharm. Res. 2015, 38, 408–413. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Clarke, I.D.; Hide, T.; Dirks, P.B. Cancer stem cells in nervous system tumors. Oncogene 2004, 23, 7267–7273. [Google Scholar] [CrossRef] [PubMed]
- Krusche, B.; Ottone, C.; Clements, M.P.; Johnstone, E.R.; Goetsch, K.; Lieven, H.; Mota, S.G.; Singh, P.; Khadayate, S.; Ashraf, A.; et al. EphrinB2 drives perivascular invasion and proliferation of glioblastoma stem-like cells. eLife 2016, 5, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Aoki, K.; Chiba, K.; Sato, Y.; Shiozawa, Y.; Shiraishi, Y.; Shimamura, T.; Niida, A.; Motomura, K.; Ohka, F.; et al. Mutational landscape and clonal architecture in grade II and III gliomas. Nat. Genet. 2015, 47, 458–468. [Google Scholar] [CrossRef] [PubMed]
- Sottoriva, A.; Spiteri, I.; Piccirillo, S.G.M.; Touloumis, A.; Collins, V.P.; Marioni, J.C.; Curtis, C.; Watts, C.; Tavaré, S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 4009–4014. [Google Scholar] [CrossRef] [PubMed]
- Ceccarelli, M.; Barthel, F.P.; Malta, T.M.; Sabedot, T.S.; Salama, S.R.; Murray, B.A.; Morozova, O.; Newton, Y.; Radenbaugh, A.; Pagnotta, S.M.; et al. Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell 2016, 164, 550–563. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Scherer, H.J. Structural development in gliomas. Am. J. Cancer 1938, 34, 333–351. [Google Scholar] [CrossRef]
- Farin, A.; Suzuki, S.O.; Weiker, M.; Goldman, J.E.; Bruce, J.N.; Canoll, P. Transplanted glioma cells migrate and proliferate on host brain vasculature: A dynamic analysis. Glia 2006, 53, 799–808. [Google Scholar] [CrossRef] [PubMed]
- Zagzag, D.; Esencay, M.; Mendez, O.; Yee, H.; Smirnova, I.; Huang, Y.; Chiriboga, L.; Lukyanov, E.; Liu, M.; Newcomb, E.W. Hypoxia- and Vascular Endothelial Growth Factor-Induced Stromal Cell-Derived Factor-1α/CXCR4 Expression in Glioblastomas. Am. J. Pathol. 2008, 173, 545–560. [Google Scholar] [CrossRef] [PubMed]
- Montana, V.; Sontheimer, H. Bradykinin Promotes the Chemotactic Invasion of Primary Brain Tumors. J. Neurosci. 2011, 31, 4858–4867. [Google Scholar] [CrossRef] [PubMed]
- Deryugina, E.I.; Bourdon, M.A.; Luo, G.X.; Reisfeld, R.A. Strongin, a Matrix metalloproteinase-2 activation modulates glioma cell migration. J. Cell Sci. 1997, 110 Pt 1, 2473–2482. [Google Scholar] [PubMed]
- Demuth, T.; Berens, M.E. Molecular mechanisms of glioma cell migration and invasion. J. Neurooncol. 2004, 70, 217–228. [Google Scholar] [CrossRef] [PubMed]
- D’Abaco, G.M.; Kaye, A.H. Integrins: Molecular determinants of glioma invasion. J. Clin. Neurosci. 2007, 14, 1041–1048. [Google Scholar] [CrossRef] [PubMed]
- Wolfenson, H.; Lavelin, I.; Geiger, B. Dynamic Regulation of the Structure and Functions of Integrin Adhesions. Dev. Cell 2013, 24, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowska, A.; Symons, M. Signaling determinants of glioma cell invasion. Adv. Exp. Med. Biol. 2013, 986, 121–141. [Google Scholar] [CrossRef] [PubMed]
- Rao, J.S. Molecular mechanisms of glioma invasiveness: The role of proteases. Nat. Rev. Cancer 2003, 3, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Lim, S.; Kim, Y. The Role of Myosin II in Glioma Invasion: A Mathematical Model. PLoS ONE 2017, 12, e0171312. [Google Scholar] [CrossRef] [PubMed]
- Beadle, C.; Assanah, M.C.; Monzo, P.; Vallee, R.; Rosenfeld, S.S.; Canoll, P. The Role of Myosin II in Glioma Invasion of the Brain. Mol. Biol. Cell 2008, 19, 3357–3368. [Google Scholar] [CrossRef] [PubMed]
- Watkins, S.; Sontheimer, H. Hydrodynamic Cellular Volume Changes Enable Glioma Cell Invasion. J. Neurosci. 2011, 31, 17250–17259. [Google Scholar] [CrossRef] [PubMed]
- Cuddapah, V.A.; Robel, S.; Watkins, S.; Sontheimer, H. A neurocentric perspective on glioma invasion. Nat. Rev. Neurosci. 2014, 15, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Watkins, S.; Robel, S.; Kimbrough, I.F.; Robert, S.M.; Ellis-Davies, G.; Sontheimer, H. Disruption of astrocyte–vascular coupling and the blood–brain barrier by invading glioma cells. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A Randomized Trial of Bevacizumab for Newly Diagnosed Glioblastoma. N. Engl. J. Med. 2014, 370, 699–708. [Google Scholar] [CrossRef] [PubMed]
- El Hallani, S.; Boisselier, B.; Peglion, F.; Rousseau, A.; Colin, C.; Idbaih, A.; Marie, Y.; Mokhtari, K.; Thomas, J.L.; Eichmann, A.; et al. A new alternative mechanism in glioblastoma vascularization: Tubular vasculogenic mimicry. Brain 2010, 133, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.J.; Ward, J.H.; Tan, C.; Grundy, R.G.; Rahman, R. Endothelial-like malignant glioma cells in dynamic three dimensional culture identifies a role for VEGF and FGFR in a tumor-derived angiogenic response. Oncotarget 2015, 6, 22191–22205. [Google Scholar] [CrossRef] [PubMed]
- Beenken, A.; Mohammadi, M. The FGF family: Biology, pathophysiology and therapy. Nat. Rev. Drug Discov. 2009, 8, 235–253. [Google Scholar] [CrossRef] [PubMed]
- Uriel, S.; Brey, E.M.; Greisler, H.P. Sustained low levels of fibroblast growth factor-1 promote persistent microvascular network formation. Am. J. Surg. 2006, 192, 604–609. [Google Scholar] [CrossRef] [PubMed]
- Presta, M.; Dell’Era, P.; Mitola, S.; Moroni, E.; Ronca, R.; Rusnati, M. Fibroblast growth factor/fibroblast growth factor receptor system in angiogenesis. Cytokine Growth Factor Rev. 2005, 16, 159–178. [Google Scholar] [CrossRef] [PubMed]
- Moreau, M.E.; Garbacki, N.; Molinaro, G.; Brown, N.J.; Marceau, F.; Adam, A. The kallikrein-kinin system: Current and future pharmacological targets. J. Pharmacol. Sci. 2005, 99, 6–38. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.S.; Han, K.S.; Ku, B.M.; Lee, Y.K.; Hong, J.; Shin, H.Y.; Almonte, A.G.; Woo, D.H.; Brat, D.J.; Hwang, E.M.; et al. Caffeine-mediated inhibition of calcium release channel inositol 1,4,5-trisphosphate receptor subtype 3 blocks glioblastoma invasion and extends survival. Cancer Res. 2010, 70, 1173–1183. [Google Scholar] [CrossRef] [PubMed]
- Higashida, H.; Yokoyama, S.; Hoshi, N.; Hashii, M.; Egorova, A.; Zhong, Z.G.; Noda, M.; Shahidullah, M.; Taketo, M.; Knijnik, R.; et al. Signal transduction from bradykinin, angiotensin, adrenergic and muscarinic receptors to effector enzymes, including ADP-ribosyl cyclase. Biol. Chem. 2001, 382, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.B.; Peng, C.; Liu, Y.H. Low dose of bradykinin selectively increases intracellular calcium in glioma cells. J. Neurol. Sci. 2007, 258, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Reetz, G.; Reiser, G. [Ca2+]i oscillations induced by bradykinin in rat glioma cells associated with Ca2+ store-dependent Ca2+ influx are controlled by cell volume and by membrane potential. Cell Calcium 1996, 19, 143–156. [Google Scholar] [CrossRef]
- Thompson, E.G.; Sontheimer, H. A role for ion channels in perivascular glioma invasion. Eur. Biophys. J. 2016, 45, 635–648. [Google Scholar] [CrossRef] [PubMed]
- Seifert, S.; Sontheimer, H. Bradykinin enhances invasion of malignant glioma into the brain parenchyma by inducing cells to undergo amoeboid migration. J. Physiol. 2014, 592, 5109–5127. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.Y.; Leung, Y.M.; Huang, S.M.; Wong, K.L. Bradykinin-induced cell migration and COX-2 production mediated by the bradykinin B1 receptor in glioma cells. J. Cell. Biochem. 2010, 110, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.S.; Thompson, C.B. Metabolic Reprogramming: A Cancer Hallmark Even Warburg Did Not Anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Parpura, V.; Basarsky, T.A.; Liu, F.; Jeftinija, K.; Jeftinija, S.; Haydon, P.G. Glutamate-mediated astrocyte–neuron signalling. Nature 1994, 369, 744–747. [Google Scholar] [CrossRef] [PubMed]
- Verderio, C.; Matteoli, M. Astrocytes and Microglial Cells: Modulation ATP Mediates Calcium Signaling Between ATP Mediates Calcium Signaling between Astrocytes and Microglial Cells: Modulation by IFN-γ Claudia Verderio 2 and Michela Matteoli. J. Immunol. 2001, 166, 6383–6391. [Google Scholar] [CrossRef] [PubMed]
- Montana, V. Vesicular Glutamate Transporter-Dependent Glutamate Release from Astrocytes. J. Neurosci. 2004, 24, 2633–2642. [Google Scholar] [CrossRef] [PubMed]
- Martineau, M.; Galli, T.; Baux, G.; Mothet, J.P. Confocal imaging and tracking of the exocytotic routes for D-serine-mediated gliotransmission. Glia 2008, 56, 1271–1284. [Google Scholar] [CrossRef] [PubMed]
- Hamadi, A.; Giannone, G.; Takeda, K.; Rondé, P. Glutamate involvement in calcium–dependent migration of astrocytoma cells. Cancer Cell Int. 2014, 14, 42. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, H.L.; Wu, C.Y.; Yang, C.M. Bradykinin induces matrix metalloproteinase-9 expression and cell migration through a PKC-δ-dependent ERK/Elk-1 pathway in astrocytes. Glia 2008, 56, 619–632. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Fink, K.L.; Mikkelsen, T.; Cloughesy, T.F.; O’Neill, A.; Plotkin, S.; Glantz, M.; Ravin, P.; Raizer, J.J.; Rich, K.M.; et al. Randomized phase II study of cilengitide, an integrin-targeting arginine-glycine-aspartic acid peptide, in recurrent glioblastoma multiforme. J. Clin. Oncol. 2008, 26, 5610–5617. [Google Scholar] [CrossRef] [PubMed]
- Würth, R.; Bajetto, A.; Harrison, J.K.; Barbieri, F.; Florio, T. CXCL12 modulation of CXCR4 and CXCR7 activity in human glioblastoma stem-like cells and regulation of the tumor microenvironment. Front. Cell. Neurosci. 2014, 8, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Carson-Walter, E.B.; Cooper, A.; Winans, B.N.; Johnson, M.D.; Walter, K.A. Vascular gene expression patterns are conserved in primary and metastatic brain tumors. J. Neurooncol. 2010, 99, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Zagzag, D.; Lukyanov, Y.; Lan, L.; Ali, M.A.; Esencay, M.; Mendez, O.; Yee, H.; Voura, E.B.; Newcomb, E.W. Hypoxia-inducible factor 1 and VEGF upregulate CXCR4 in glioblastoma: Implications for angiogenesis and glioma cell invasion. Lab. Investig. 2006, 86, 1221–1232. [Google Scholar] [CrossRef] [PubMed]
- Esencay, M.; Sarfraz, Y.; Zagzag, D. CXCR7 is induced by hypoxia and mediates glioma cell migration towards SDF-1alpha. BMC Cancer 2013, 13, 347. [Google Scholar] [CrossRef] [PubMed]
- Yadav, V.; Zamler, D.; Baker, G.J.; Kadiyala, P.; Erdreich-Epstein, A.; DeCarvalho, A.C.; Mikkelsen, T.; Castro, M.G.; Lowenstein, P.R. CXCR4 increases in vivo glioma perivascular invasion, and reduces radiation induced apoptosis: A genetic knockdown study. Oncotarget 2016, 7, 83701–83719. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Huang, Z.; Zhou, W.; Wu, Q.; Donnola, S.; Liu, J.K.; Fang, X.; Sloan, A.E.; Mao, Y.; Lathia, J.D.; et al. Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell 2013, 153, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, C.; Poppleton, H.; Kocak, M.; Hogg, T.L.; Fuller, C.; Hamner, B.; Oh, E.Y.; Gaber, M.W.; Finklestein, D.; Allen, M.; et al. A Perivascular Niche for Brain Tumor Stem Cells. Cancer Cell 2007, 11, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Hira, V.V.V.; Ploegmakers, K.J.; Grevers, F.; Verbovšek, U.; Silvestre-Roig, C.; Aronica, E.; Tigchelaar, W.; Turnšek, T.L.; Molenaar, R.J.; Van Noorden, C.J.F. CD133+ and Nestin+ Glioma Stem-Like Cells Reside Around CD31 + Arterioles in Niches that Express SDF-1α, CXCR4, Osteopontin and Cathepsin K. J. Histochem. Cytochem. 2015, 63, 481–493. [Google Scholar] [CrossRef] [PubMed]
- Cortina, C.; Palomo-Ponce, S.; Iglesias, M.; Fernández-Masip, J.L.; Vivancos, A.; Whissell, G.; Humà, M.; Peiró, N.; Gallego, L.; Jonkheer, S.; et al. EphB–ephrin-B interactions suppress colorectal cancer progression by compartmentalizing tumor cells. Nat. Genet. 2007, 39, 1376–1383. [Google Scholar] [CrossRef] [PubMed]
- Astin, J.W.; Batson, J.; Kadir, S.; Charlet, J.; Persad, R.A.; Gillatt, D.; Oxley, J.D.; Nobes, C.D. Competition amongst Eph receptors regulates contact inhibition of locomotion and invasiveness in prostate cancer cells. Nat. Cell Biol. 2010, 12, 1194–1204. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.S.; Costello, M.A.; Talsma, C.E.; Flack, C.G.; Crowley, J.G.; Hamm, L.L.; He, X.; Hervey-Jumper, S.L.; Heth, J.A.; Muraszko, K.M.; et al. Endothelial cells create a stem cell niche in glioblastoma by providing NOTCH ligands that nurture self-renewal of cancer stem-like cells. Cancer Res. 2011, 71, 6061–6072. [Google Scholar] [CrossRef] [PubMed]
- Charles, N.; Ozawa, T.; Squatrito, M.; Bleau, A.M.; Brennan, C.W.; Hambardzumyan, D.; Holland, E.C. Perivascular Nitric Oxide Activates Notch Signaling and Promotes Stem-like Character in PDGF-Induced Glioma Cells. Cell Stem Cell 2010, 6, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Martin, V.; Fueyo, J.; Lee, O.-H.; Xu, J.; Cortes-Santiago, N.; Alonso, M.M.; Aldape, K.; Colman, H.; Gomez-Manzano, C. Tie2/TEK modulates the interaction of glioma and brain tumor stem cells with endothelial cells and promotes an invasive phenotype. Oncotarget 2010, 1, 700–709. [Google Scholar] [CrossRef] [PubMed]
- Becher, O.J.; Hambardzumyan, D.; Fomchenko, E.I.; Momota, H.; Mainwaring, L.; Bleau, A.M.; Katz, A.M.; Edgar, M.; Kenney, A.M.; Cordon-Cardo, C.; et al. Gli activity correlates with tumor grade in platelet-derived growth factor-induced gliomas. Cancer Res. 2008, 68, 2241–2249. [Google Scholar] [CrossRef] [PubMed]
- Galan-Moya, E.M.; Le Guelte, A.; Fernandes, E.L.; Thirant, C.; Dwyer, J.; Bidere, N.; Couraud, P.-O.; Scott, M.G.H.; Junier, M.-P.; Chneiweiss, H.; et al. Secreted factors from brain endothelial cells maintain glioblastoma stem-like cell expansion through the mTOR pathway. EMBO Rep. 2011, 12, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Roos, A.; Ding, Z.; Loftus, J.C.; Tran, N.L. Molecular and Microenvironmental Determinants of Glioma Stem-Like Cell Survival and Invasion. Front. Oncol. 2017, 7, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Mathiisen, T.M.; Lehre, K.P.; Danbolt, N.C.; Ottersen, O.P. The perivascular astroglial sheath provides a complete covering of the brain microvessels: An electron microscopic 3D reconstruction. Glia 2010, 58, 1094–1103. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C.; Nedergaard, M. Glial regulation of the cerebral microvasculature. Nat. Neurosci. 2007, 10, 1369–1376. [Google Scholar] [CrossRef] [PubMed]
- Pellerin, L.; Pellegri, G.; Bittar, P.G.; Charnay, Y.; Bouras, C.; Martin, J.L.; Stella, N.; Magistretti, P.J. Evidence supporting the existence of an activity-dependent astrocyte-neuron lactate shuttle. Dev. Neurosci. 1998, 20, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Akella, N.S.; Twieg, D.B.; Mikkelsen, T.; Hochberg, F.H.; Grossman, S.; Cloud, G.A.; Nabors, L.B. Assessment of brain tumor angiogenesis inhibitors using perfusion magnetic resonance imaging: Quality and analysis results of a phase I trial. J. Magn. Reson. Imaging 2004, 20, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Scully, S.; Francescone, R.; Faibish, M.; Bentley, B.; Taylor, S.L.; Oh, D.; Schapiro, R.; Moral, L.; Yan, W.; Shao, R. Transdifferentiation of Glioblastoma Stem-Like Cells into Mural Cells Drives Vasculogenic Mimicry in Glioblastomas. J. Neurosci. 2012, 32, 12950–12960. [Google Scholar] [CrossRef] [PubMed]
- Castells, M.; Thibault, B.; Delord, J.-P.; Couderc, B. Implication of Tumor Microenvironment in Chemoresistance: Tumor-Associated Stromal Cells Protect Tumor Cells from Cell Death. Int. J. Mol. Sci. 2012, 13, 9545–9571. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wakeman, T.P.; Lathia, J.D.; Hjelmeland, A.B.; Wang, X.F.; White, R.R.; Rich, J.N.; Sullenger, B.A. Notch promotes radioresistance of glioma stem cells. Stem Cells 2010, 28, 17–28. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diksin, M.; Smith, S.J.; Rahman, R. The Molecular and Phenotypic Basis of the Glioma Invasive Perivascular Niche. Int. J. Mol. Sci. 2017, 18, 2342. https://doi.org/10.3390/ijms18112342
Diksin M, Smith SJ, Rahman R. The Molecular and Phenotypic Basis of the Glioma Invasive Perivascular Niche. International Journal of Molecular Sciences. 2017; 18(11):2342. https://doi.org/10.3390/ijms18112342
Chicago/Turabian StyleDiksin, Mohammed, Stuart J. Smith, and Ruman Rahman. 2017. "The Molecular and Phenotypic Basis of the Glioma Invasive Perivascular Niche" International Journal of Molecular Sciences 18, no. 11: 2342. https://doi.org/10.3390/ijms18112342
APA StyleDiksin, M., Smith, S. J., & Rahman, R. (2017). The Molecular and Phenotypic Basis of the Glioma Invasive Perivascular Niche. International Journal of Molecular Sciences, 18(11), 2342. https://doi.org/10.3390/ijms18112342