Immunogenomic Classification of Colorectal Cancer and Therapeutic Implications

 ,
,  ,
, 
 ,
,
Abstract
1. Introduction
2. Prognostic Signatures
2.1. Tumor-Infiltrating Lymphocytes (TILs)
2.2. Gene Signatures
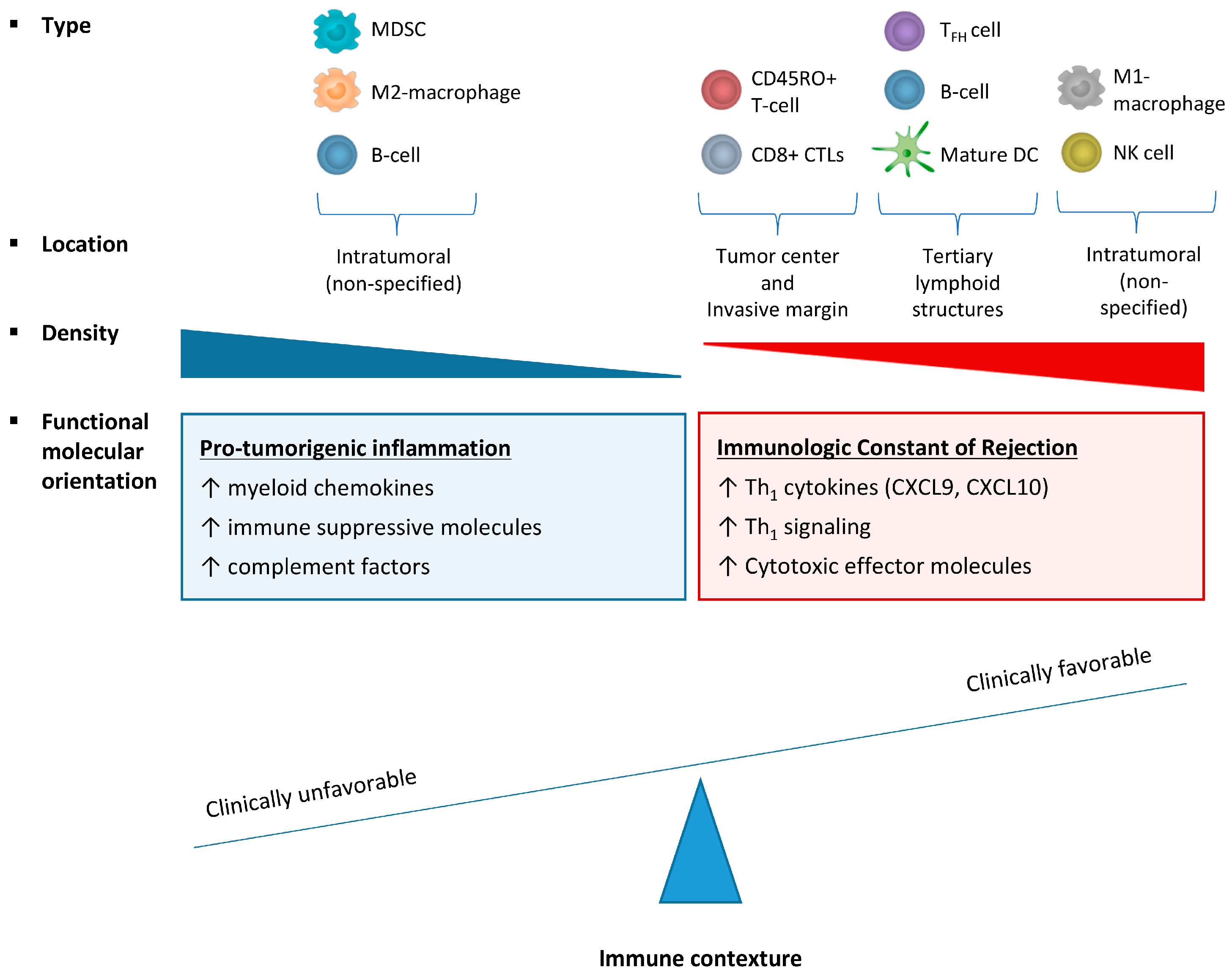
2.3. Effect of Immune Cell Infiltration Depends on the Tumor Microenvironment (TME)
2.4. Immune Signatures in CRC Metastasis
3. Predictive Signatures
3.1. Immune Signatures Predictive for Conventional Therapy
3.2. Immune Signatures Predictive for Immunotherapy
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| 5-FU | 5-fluorouracil |
| CAF | Cancer associated fibroblast |
| CMS | Consensus molecular subtype |
| CRC | Colorectal cancer |
| CT | Core of the tumor |
| CTL | Cytotoxic T lymphocyte |
| DC | Dendritic cell |
| DC-LAMP+ | Dendritic cell lysosome-associated membrane glycoprotein positive |
| EMT | Epithelial to mesenchymal transition |
| HLA | Human leukocyte antigen |
| ICR | Immunologic constant of rejection |
| IM | Invasive margin |
| MDSC | Myeloid derived suppressor cell |
| MEK | Mitogen-activated protein kinase (mapk) kinase |
| MHC | Major histocompatibility complex |
| MSI | Microsatellite instability |
| MSS | Microsatellite stable |
| NK | Natural killer cell |
| Th | T-helper cell |
| TIL | Tumor infiltrating lymphocyte |
| TLS | Tertiary lymphoid structure |
| TME | Tumor microenvironment |
| TNM | Tumor node metastasis |
| T-reg | Regulatory T cell |
References
- Puppa, G.; Sonzogni, A.; Colombari, R.; Pelosi, G. TNM staging system of colorectal carcinoma: A critical appraisal of challenging issues. Arch. Pathol. Lab. Med. 2010, 134, 837–852. [Google Scholar] [CrossRef] [PubMed]
- Moline, J.; Mahdi, H.; Yang, B.; Biscotti, C.; Roma, A.A.; Heald, B.; Rose, P.G.; Michener, C.; Eng, C. Implementation of tumor testing for lynch syndrome in endometrial cancers at a large academic medical center. Gynecol. Oncol. 2013, 130, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Abbas, O.; Mahalingam, M. Cutaneous sebaceous neoplasms as markers of Muir-Torre syndrome: A diagnostic algorithm. J. Cutan. Pathol. 2009, 36, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Gryfe, R.; Kim, H.; Hsieh, E.T.; Aronson, M.D.; Holowaty, E.J.; Bull, S.B.; Redston, M.; Gallinger, S. Tumor microsatellite instability and clinical outcome in young patients with colorectal cancer. N. Engl. J. Med. 2000, 342, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Fridman, W.H.; Pagès, F.; Sautès-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Pagès, F.; Marincola, F.M.; Angell, H.K.; Thurin, M.; Lugli, A.; Zlobec, I.; Berger, A.; Bifulco, C.; Botti, G.; et al. Cancer classification using the immunoscore: A worldwide task force. J. Transl. Med. 2012, 10, 205. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Angell, H.K.; Bedognetti, D.; Marincola, F.M. The Continuum of Cancer Immunosurveillance: Prognostic, Predictive, and Mechanistic Signatures. Immunity 2013, 39, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Harlin, H.; Meng, Y.; Peterson, A.C.; Zha, Y.; Tretiakova, M.; Slingluff, C.; McKee, M.; Gajewski, T.F. Chemokine expression in melanoma metastases associated with CD8+ T-Cell Recruitment. Cancer Res. 2009, 69, 3077–3085. [Google Scholar] [CrossRef] [PubMed]
- Clemente, C.G.; Mihm, M.C.; Bufalino, R.; Zurrida, S.; Collini, P.; Cascinelli, N. Prognostic value of tumor infiltrating lymphocytes in the vertical growth phase of primary cutaneous melanoma. Cancer 1996, 77, 1303–1310. [Google Scholar] [CrossRef]
- Fuertes, M.B.; Kacha, A.K.; Kline, J.; Woo, S.-R.; Kranz, D.M.; Murphy, K.M.; Gajewski, T.F. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8α+ dendritic cells. J. Exp. Med. 2011, 208, 2005–2016. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Andre, F.; Pusztai, L. Molecular classification of breast cancer: Implications for selection of adjuvant chemotherapy. Nat. Clin. Pract. Oncol. 2006, 3, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Formenti, S.C.; Demaria, S. Combining radiotherapy and cancer immunotherapy: A paradigm shift. J. Natl. Cancer Inst. 2013, 105, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Gibney, G.T.; Weiner, L.M.; Atkins, M.B. Predictive biomarkers for checkpoint inhibitor-based immunotherapy. Lancet Oncol. 2016, 17, e542–e551. [Google Scholar] [CrossRef]
- Pagès, F.; Galon, J.; Fridman, W.H. The essential role of the in situ immune reaction in human colorectal cancer. J. Leukoc. Biol. 2008, 84, 981–987. [Google Scholar] [CrossRef] [PubMed]
- Quante, M.; Varga, J.; Wang, T.C.; Greten, F.R. The gastrointestinal tumor microenvironment. Gastroenterology 2013, 145, 63–78. [Google Scholar] [CrossRef] [PubMed]
- Becht, E.; de Reyniès, A.; Giraldo, N.A.; Pilati, C.; Buttard, B.; Lacroix, L.; Selves, J.; Sautès-Fridman, C.; Laurent-Puig, P.; Fridman, W.H. Immune and stromal classification of colorectal cancer is associated with molecular subtypes and relevant for precision immunotherapy. Clin. Cancer Res. 2016, 22, 4057–4066. [Google Scholar] [CrossRef] [PubMed]
- Chifman, J.; Pullikuth, A.; Chou, J.W.; Bedognetti, D.; Miller, L.D. Conservation of immune gene signatures in solid tumors and prognostic implications. BMC Cancer 2016, 16, 911. [Google Scholar] [CrossRef] [PubMed]
- Boissière-Michot, F.; Lazennec, G.; Frugier, H.; Jarlier, M.; Roca, L.; Duffour, J.; du Paty, E.; Laune, D.; Blanchard, F.; Le Pessot, F.; et al. Characterization of an adaptive immune response in microsatellite-instable colorectal cancer. Oncoimmunology 2014, 3, e29256. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Chen, Z. The effect of immune microenvironment on the progression and prognosis of colorectal cancer. Med. Oncol. Northwood Lond. Engl. 2014, 31, 82. [Google Scholar] [CrossRef] [PubMed]
- Edin, S.; Wikberg, M.L.; Dahlin, A.M.; Rutegård, J.; Öberg, Å.; Oldenborg, P.-A.; Palmqvist, R. The distribution of macrophages with a M1 or M2 phenotype in relation to prognosis and the molecular characteristics of colorectal cancer. PLoS ONE 2012, 7, e47045. [Google Scholar] [CrossRef] [PubMed]
- Väyrynen, J.P.; Sajanti, S.A.; Klintrup, K.; Mäkelä, J.; Herzig, K.-H.; Karttunen, T.J.; Tuomisto, A.; Mäkinen, M.J. Characteristics and significance of colorectal cancer associated lymphoid reaction. Int. J. Cancer 2014, 134, 2126–2135. [Google Scholar] [CrossRef] [PubMed]
- Bindea, G.; Mlecnik, B.; Tosolini, M.; Kirilovsky, A.; Waldner, M.; Obenauf, A.C.; Angell, H.; Fredriksen, T.; Lafontaine, L.; Berger, A.; et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity 2013, 39, 782–795. [Google Scholar] [CrossRef] [PubMed]
- Herrera, M.; Herrera, A.; Domínguez, G.; Silva, J.; García, V.; García, J.M.; Gómez, I.; Soldevilla, B.; Muñoz, C.; Provencio, M.; et al. Cancer-associated fibroblast and M2 macrophage markers together predict outcome in colorectal cancer patients. Cancer Sci. 2013, 104, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Duan, Y.; Cheng, X.; Chen, X.; Xie, W.; Long, H.; Lin, Z.; Zhu, B. IL-17 is associated with poor prognosis and promotes angiogenesis via stimulating VEGF production of cancer cells in colorectal carcinoma. Biochem. Biophys. Res. Commun. 2011, 407, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Salama, P.; Phillips, M.; Grieu, F.; Morris, M.; Zeps, N.; Joseph, D.; Platell, C.; Iacopetta, B. Tumor-infiltrating FOXP3+ T regulatory cells show strong prognostic significance in colorectal cancer. J. Clin. Oncol. 2009, 27, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Barbera-Guillem, E.; Nelson, M.B.; Barr, B.; Nyhus, J.K.; May, K.F.; Feng, L.; Sampsel, J.W. B lymphocyte pathology in human colorectal cancer. Experimental and clinical therapeutic effects of partial B cell depletion. Cancer Immunol. Immunother. 2000, 48, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Dieu-Nosjean, M.-C.; Goc, J.; Giraldo, N.A.; Sautès-Fridman, C.; Fridman, W.H. Tertiary lymphoid structures in cancer and beyond. Trends Immunol. 2014, 35, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Di Caro, G.; Bergomas, F.; Grizzi, F.; Doni, A.; Bianchi, P.; Malesci, A.; Laghi, L.; Allavena, P.; Mantovani, A.; Marchesi, F. Occurrence of tertiary lymphoid tissue is associated with T-cell infiltration and predicts better prognosis in early-stage colorectal cancers. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 2147–2158. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Mlecnik, B.; Marliot, F.; Ou, F.-S.; Carlo Bruno, B.; Lugli, A.; Zlobec, I.; Rau, T.T.; Hartmann, A.; Masucci, G.V.; et al. Validation of the Immunoscore (IM) as a prognostic marker in stage I/II/III colon cancer: Results of a worldwide consortium-based analysis of 1336 patients. J. Clin. Oncol. 2016, 34, 3500. [Google Scholar] [CrossRef]
- Pagès, F.; Kirilovsky, A.; Mlecnik, B.; Asslaber, M.; Tosolini, M.; Bindea, G.; Lagorce, C.; Wind, P.; Marliot, F.; Bruneval, P.; et al. In situ cytotoxic and memory T cells predict outcome in patients with early-stage colorectal cancer. J. Clin. Oncol. 2009, 27, 5944–5951. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Fox, B.A.; Bifulco, C.B.; Masucci, G.; Rau, T.; Botti, G.; Marincola, F.M.; Ciliberto, G.; Pages, F.; Ascierto, P.A.; et al. Immunoscore and Immunoprofiling in cancer: An update from the melanoma and immunotherapy bridge 2015. J. Transl. Med. 2016, 14. [Google Scholar] [CrossRef] [PubMed]
- Mlecnik, B.; Bindea, G.; Angell, H.K.; Maby, P.; Angelova, M.; Tougeron, D.; Church, S.E.; Lafontaine, L.; Fischer, M.; Fredriksen, T.; et al. Integrative analyses of colorectal cancer show immunoscore is a stronger predictor of patient survival than microsatellite instability. Immunity 2016, 44, 698–711. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Worschech, A.; Marincola, F.M. The immunologic constant of rejection. Trends Immunol. 2008, 29, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Spivey, T.L.; Uccellini, L.; Ascierto, M.L.; Zoppoli, G.; de Giorgi, V.; Delogu, L.G.; Engle, A.M.; Thomas, J.M.; Wang, E.; Marincola, F.M.; et al. Gene expression profiling in acute allograft rejection: Challenging the immunologic constant of rejection hypothesis. J. Transl. Med. 2011, 9, 174. [Google Scholar] [CrossRef] [PubMed]
- Bedognetti, D.; Spivey, T.L.; Zhao, Y.; Uccellini, L.; Tomei, S.; Dudley, M.E.; Ascierto, M.L.; de Giorgi, V.; Liu, Q.; Delogu, L.G.; et al. CXCR3/CCR5 pathways in metastatic melanoma patients treated with adoptive therapy and interleukin-2. Br. J. Cancer 2013, 109, 2412–2423. [Google Scholar] [CrossRef] [PubMed]
- Murtas, D.; Maric, D.; de Giorgi, V.; Reinboth, J.; Worschech, A.; Fetsch, P.; Filie, A.; Ascierto, M.L.; Bedognetti, D.; Liu, Q.; et al. IRF-1 responsiveness to IFN-γ predicts different cancer immune phenotypes. Br. J. Cancer 2013. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Bedognetti, D.; Marincola, F.M. Prediction of response to anticancer immunotherapy using gene signatures. J. Clin. Oncol. 2013, 31, 2369–2371. [Google Scholar] [CrossRef] [PubMed]
- Bedognetti, D.; Tomei, S.; Hendrickx, W.; Marincola, F.M.; Wang, E. Toward the identification of genetic determinants of responsiveness to cancer immunotherapy. In Developments in T Cell Based Cancer Immunotherapies; Ascierto, P.A., Stroncek, D.F., Wang, E., Eds.; Cancer Drug Discovery and Development, Humana Press: New York, NY, USA, 2015; pp. 99–127. ISBN 978-3-319-21167-1. [Google Scholar]
- Bedognetti, D.; Hendrickx, W.; Ceccarelli, M.; Miller, L.D.; Seliger, B. Disentangling the relationship between tumor genetic programs and immune responsiveness. Curr. Opin. Immunol. 2016, 39, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Hendrickx, W.; Simeone, I.; Anjum, S.; Mokrab, Y.; Bertucci, F.; Finetti, P.; Curigliano, G.; Seliger, B.; Cerulo, L.; Tomei, S.; et al. Identification of genetic determinants of breast cancer immune phenotypes by integrative genome-scale analysis. OncoImmunology 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Miller, L.D.; Chou, J.A.; Black, M.A.; Print, C.; Chifman, J.; Alistar, A.; Putti, T.; Zhou, X.; Bedognetti, D.; Hendrickx, W.; et al. Immunogenic subtypes of breast cancer delineated by gene classifiers of immune responsiveness. Cancer Immunol. Res. 2016. [Google Scholar] [CrossRef] [PubMed]
- Barbie, D.A.; Tamayo, P.; Boehm, J.S.; Kim, S.Y.; Moody, S.E.; Dunn, I.F.; Schinzel, A.C.; Sandy, P.; Meylan, E.; Scholl, C.; et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 2009, 462, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Angelova, M.; Charoentong, P.; Hackl, H.; Fischer, M.L.; Snajder, R.; Krogsdam, A.M.; Waldner, M.J.; Bindea, G.; Mlecnik, B.; Galon, J.; et al. Characterization of the immunophenotypes and antigenomes of colorectal cancers reveals distinct tumor escape mechanisms and novel targets for immunotherapy. Genome Biol. 2015, 16, 64. [Google Scholar] [CrossRef] [PubMed]
- Nagalla, S.; Chou, J.W.; Willingham, M.C.; Ruiz, J.; Vaughn, J.P.; Dubey, P.; Lash, T.L.; Hamilton-Dutoit, S.J.; Bergh, J.; Sotiriou, C.; et al. Interactions between immunity, proliferation and molecular subtype in breast cancer prognosis. Genome Biol. 2013, 14, R34. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Becht, E.; Giraldo, N.A.; Lacroix, L.; Buttard, B.; Elarouci, N.; Petitprez, F.; Selves, J.; Laurent-Puig, P.; Sautès-Fridman, C.; Fridman, W.H.; et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 2016, 17, 218. [Google Scholar] [CrossRef] [PubMed]
- Sandel, M.H.; Dadabayev, A.R.; Menon, A.G.; Morreau, H.; Melief, C.J.M.; Offringa, R.; van der Burg, S.H.; Janssen-van Rhijn, C.M.; Ensink, N.G.; Tollenaar, R.A.E.M.; et al. Prognostic value of tumor-infiltrating dendritic cells in colorectal cancer: Role of maturation status and intratumoral localization. Clin. Cancer Res. 2005, 11, 2576–2582. [Google Scholar] [CrossRef] [PubMed]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef] [PubMed]
- Calon, A.; Lonardo, E.; Berenguer-Llergo, A.; Espinet, E.; Hernando-Momblona, X.; Iglesias, M.; Sevillano, M.; Palomo-Ponce, S.; Tauriello, D.V.F.; Byrom, D.; et al. Stromal gene expression defines poor-prognosis subtypes in colorectal cancer. Nat. Genet. 2015, 47, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.-L.; Pittet, M.J.; Gorelik, L.; Flavell, R.A.; Weissleder, R.; von Boehmer, H.; Khazaie, K. Regulatory T cells suppress tumor-specific CD8 T cell cytotoxicity through TGF-β signals in vivo. Proc. Natl. Acad. Sci. USA 2005, 102, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.A.; Massagué, J. TGF-β directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 2005, 8, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Pang, Y.; Moses, H.L. TGF-β and immune cells: An important regulatory axis in the tumor microenvironment and progression. Trends Immunol. 2010, 31, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Spranger, S.; Gajewski, T.F. Tumor-intrinsic oncogene pathways mediating immune avoidance. Oncoimmunology 2015, 5, e1086862. [Google Scholar] [CrossRef] [PubMed]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Aref, A.R.; Skoulidis, F.; Herter-Sprie, G.S.; Buczkowski, K.A.; Liu, Y.; Awad, M.M.; Denning, W.L.; et al. STK11/LKB1 deficiency promotes neutrophil recruitment and proinflammatory cytokine production to suppress T-cell activity in the lung tumor microenvironment. Cancer Res. 2016, 76, 999–1008. [Google Scholar] [CrossRef] [PubMed]
- Launonen, V. Mutations in the human LKB1/STK11 gene. Hum. Mutat. 2005, 26, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Watson, N.F.S.; Ramage, J.M.; Madjd, Z.; Spendlove, I.; Ellis, I.O.; Scholefield, J.H.; Durrant, L.G. Immunosurveillance is active in colorectal cancer as downregulation but not complete loss of MHC class I expression correlates with a poor prognosis. Int. J. Cancer 2006, 118, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Zeestraten, E.C.M.; Reimers, M.S.; Saadatmand, S.; Goossens-Beumer, I.J.; Dekker, J.-W.T.; Liefers, G.J.; van den Elsen, P.J.; van de Velde, C.J.H.; Kuppen, P.J.K. Combined analysis of HLA class I, HLA-E and HLA-G predicts prognosis in colon cancer patients. Br. J. Cancer 2014, 110, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, S.; Long, E.O. A Human Histocompatibility Leukocyte Antigen (HLA)-G–specific Receptor Expressed on All Natural Killer Cells. J. Exp. Med. 1999, 189, 1093–1100. [Google Scholar] [CrossRef] [PubMed]
- Braud, V.M.; Allan, D.S.J.; O’Callaghan, C.A.; Söderström, K.; D’Andrea, A.; Ogg, G.S.; Lazetic, S.; Young, N.T.; Bell, J.I.; Phillips, J.H.; et al. HLA-E binds to natural killer cell receptors CD94/NKG2A, B and C. Nature 1998, 391, 795–799. [Google Scholar] [CrossRef] [PubMed]
- Swets, M.; König, M.H.; Zaalberg, A.; Dekker-Ensink, N.G.; Gelderblom, H.; van de Velde, C.J.H.; van den Elsen, P.J.; Kuppen, P.J.K. HLA-G and classical HLA class I expression in primary colorectal cancer and associated liver metastases. Hum. Immunol. 2016, 77, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Wei, A.C.; Greig, P.D.; Grant, D.; Taylor, B.; Langer, B.; Gallinger, S. Survival after hepatic resection for colorectal metastases: A 10-year experience. Ann. Surg. Oncol. 2006, 13, 668–676. [Google Scholar] [CrossRef] [PubMed]
- Pathak, S.; Poston, G.J. Colorectal liver metastases; the current scenario. Indian J. Surg. Oncol. 2010, 1, 350–355. [Google Scholar] [CrossRef] [PubMed]
- Riihimäki, M.; Hemminki, A.; Sundquist, J.; Hemminki, K. Patterns of metastasis in colon and rectal cancer. Sci. Rep. 2016, 6, 29765. [Google Scholar] [CrossRef] [PubMed]
- Viadana, E.; Bross, I.D.J.; Pickren, J.W. The Metastatic Spread of Cancers of the Digestive System in Man. Oncology 1978, 35, 114–126. [Google Scholar] [CrossRef] [PubMed]
- Weiss, L.; Grundmann, E.; Torhorst, J.; Hartveit, F.; Moberg, I.; Eder, M.; Fenoglio-Preiser, C.M.; Napier, J.; Horne, C.H.; Lopez, M.J. Haematogenous metastatic patterns in colonic carcinoma: An analysis of 1541 necropsies. J. Pathol. 1986, 150, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Paget, S. The distribution of secondary growths in cancer of the breast. Cancer Metastasis Rev. 1989, 8, 98–101. [Google Scholar] [CrossRef]
- Fidler, I.J. The pathogenesis of cancer metastasis: The “seed and soil” hypothesis revisited. Nat. Rev. Cancer 2003, 3, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Langley, R.R.; Fidler, I.J. The seed and soil hypothesis revisited — The role of tumor-stroma interactions in metastasis to different organs. Int. J. Cancer J. Int. Cancer 2011, 128, 2527–2535. [Google Scholar] [CrossRef] [PubMed]
- Seretis, F.; Seretis, C.; Youssef, H.; Chapman, M. Colorectal cancer: Seed and soil hypothesis revisited. Anticancer Res. 2014, 34, 2087–2094. [Google Scholar] [PubMed]
- Chan, T.; Wiltrout, R.H.; Weiss, J.M. Immunotherapeutic modulation of the suppressive liver and tumor microenvironments. Int. Immunopharmacol. 2011, 11, 876–886. [Google Scholar] [CrossRef] [PubMed]
- Halama, N.; Michel, S.; Kloor, M.; Zoernig, I.; Benner, A.; Spille, A.; Pommerencke, T.; von Knebel, D.M.; Folprecht, G.; Luber, B.; et al. Localization and density of immune cells in the invasive margin of human colorectal cancer liver metastases are prognostic for response to chemotherapy. Cancer Res. 2011, 71, 5670–5677. [Google Scholar] [CrossRef] [PubMed]
- Van den Eynde, M.; Mlecnik, B.; Machiels, J.-P.H.; Debetancourt, D.; Bindea, G.; Jouret-Mourin, A.; Sempoux, C.; Carrasco, J.; Gigot, J.F.; Hubert, C.; et al. Characterization of the immune microenvironment of synchronous primary tumor and liver colorectal metastases. J. Clin. Oncol. 2015, 33, 3610. [Google Scholar] [CrossRef]
- Halama, N.; Spille, A.; Lerchl, T.; Brand, K.; Herpel, E.; Welte, S.; Keim, S.; Lahrmann, B.; Klupp, F.; Kahlert, C.; et al. Hepatic metastases of colorectal cancer are rather homogeneous but differ from primary lesions in terms of immune cell infiltration. Oncoimmunology 2013, 2, e24116. [Google Scholar] [CrossRef] [PubMed]
- Kwak, Y.; Koh, J.; Kim, D.-W.; Kang, S.-B.; Kim, W.H.; Lee, H.S. Immunoscore encompassing CD3+ and CD8+ T cell densities in distant metastasis is a robust prognostic marker for advanced colorectal cancer. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed]
- Remark, R.; Alifano, M.; Cremer, I.; Lupo, A.; Dieu-Nosjean, M.-C.; Riquet, M.; Crozet, L.; Ouakrim, H.; Goc, J.; Cazes, A.; et al. Characteristics and clinical impacts of the immune environments in colorectal and renal cell carcinoma lung metastases: Influence of tumor origin. Clin. Cancer Res. 2013, 19, 4079–4091. [Google Scholar] [CrossRef] [PubMed]
- Vignot, S.; Lefebvre, C.; Frampton, G.M.; Meurice, G.; Yelensky, R.; Palmer, G.; Capron, F.; Lazar, V.; Hannoun, L.; Miller, V.A.; et al. Comparative analysis of primary tumour and matched metastases in colorectal cancer patients: Evaluation of concordance between genomic and transcriptional profiles. Eur. J. Cancer 1990 2015, 51, 791–799. [Google Scholar] [CrossRef] [PubMed]
- Roessler, S.; Lin, G.; Forgues, M.; Budhu, A.; Hoover, S.; Simpson, R.M.; Wu, X.; He, P.; Qin, L.-X.; Tang, Z.-Y.; et al. Integrative genomic and transcriptomic characterization of matched primary and metastatic liver and colorectal carcinoma. Int. J. Biol. Sci. 2015, 11, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Koh, K.H.; Rhee, H.; Kang, H.J.; Yang, E.; You, K.T.; Lee, H.; Min, B.S.; Kim, N.K.; Nam, S.W.; Kim, H. Differential gene expression profiles of metastases in paired primary and metastatic colorectal carcinomas. Oncology 2008, 75, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Sayagués, J.M.; Corchete, L.A.; Gutiérrez, M.L.; Sarasquete, M.E.; del mar Abad, M.; Bengoechea, O.; Fermiñán, E.; Anduaga, M.F.; del Carmen, S.; Iglesias, M.; et al. Genomic characterization of liver metastases from colorectal cancer patients. Oncotarget 2016, 7, 72908–72922. [Google Scholar] [CrossRef]
- André, T.; Boni, C.; Navarro, M.; Tabernero, J.; Hickish, T.; Topham, C.; Bonetti, A.; Clingan, P.; Bridgewater, J.; Rivera, F.; et al. Improved overall survival with oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment in stage II or III colon cancer in the MOSAIC trial. J. Clin. Oncol. 2009, 27, 3109–3116. [Google Scholar] [CrossRef] [PubMed]
- Bosset, J.-F.; Collette, L.; Calais, G.; Mineur, L.; Maingon, P.; Radosevic-Jelic, L.; Daban, A.; Bardet, E.; Beny, A.; Ollier, J.-C.; et al. Chemotherapy with preoperative radiotherapy in rectal cancer. N. Engl. J. Med. 2006, 355, 1114–1123. [Google Scholar] [CrossRef] [PubMed]
- Ribic, C.M.; Sargent, D.J.; Moore, M.J.; Thibodeau, S.N.; French, A.J.; Goldberg, R.M.; Hamilton, S.R.; Laurent-Puig, P.; Gryfe, R.; Shepherd, L.E.; et al. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N. Engl. J. Med. 2003, 349, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Carethers, J.M.; Smith, E.J.; Behling, C.A.; Nguyen, L.; Tajima, A.; Doctolero, R.T.; Cabrera, B.L.; Goel, A.; Arnold, C.A.; Miyai, K.; et al. Use of 5-fluorouracil and survival in patients with microsatellite-unstable colorectal cancer. Gastroenterology 2004, 126, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Benatti, P.; Gafà, R.; Barana, D.; Marino, M.; Scarselli, A.; Pedroni, M.; Maestri, I.; Guerzoni, L.; Roncucci, L.; Menigatti, M.; et al. Microsatellite instability and colorectal cancer prognosis. Clin. Cancer Res. 2005, 11, 8332–8340. [Google Scholar] [CrossRef] [PubMed]
- Jover, R.; Zapater, P.; Castells, A.; Llor, X.; Andreu, M.; Cubiella, J.; Piñol, V.; Xicola, R.M.; Bujanda, L.; Reñé, J.M.; et al. Mismatch repair status in the prediction of benefit from adjuvant fluorouracil chemotherapy in colorectal cancer. Gut 2006, 55, 848–855. [Google Scholar] [CrossRef] [PubMed]
- Hsu, H.-C.; Thiam, T.K.; Lu, Y.-J.; Yeh, C.Y.; Tsai, W.-S.; You, J.F.; Hung, H.Y.; Tsai, C.-N.; Hsu, A.; Chen, H.-C.; et al. Mutations of KRAS/NRAS/BRAF predict cetuximab resistance in metastatic colorectal cancer patients. Oncotarget 2016, 7, 22257–22270. [Google Scholar] [CrossRef] [PubMed]
- Apetoh, L.; Ghiringhelli, F.; Tesniere, A.; Obeid, M.; Ortiz, C.; Criollo, A.; Mignot, G.; Maiuri, M.C.; Ullrich, E.; Saulnier, P.; et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat. Med. 2007, 13, 1050–1059. [Google Scholar] [CrossRef] [PubMed]
- Venereau, E.; Casalgrandi, M.; Schiraldi, M.; Antoine, D.J.; Cattaneo, A.; Marchis, F.D.; Liu, J.; Antonelli, A.; Preti, A.; Raeli, L.; et al. Mutually exclusive redox forms of HMGB1 promote cell recruitment or proinflammatory cytokine release. J. Exp. Med. 2012, 209, 1519–1528. [Google Scholar] [CrossRef] [PubMed]
- Cottone, L.; Capobianco, A.; Gualteroni, C.; Perrotta, C.; Bianchi, M.E.; Rovere-Querini, P.; Manfredi, A.A. 5-Fluorouracil causes leukocytes attraction in the peritoneal cavity by activating autophagy and HMGB1 release in colon carcinoma cells. Int. J. Cancer 2015, 136, 1381–1389. [Google Scholar] [CrossRef] [PubMed]
- Cottone, L.; Capobianco, A.; Gualteroni, C.; Monno, A.; Raccagni, I.; Valtorta, S.; Canu, T.; Tomaso, T.D.; Lombardo, A.; Esposito, A.; et al. Leukocytes recruited by tumor-derived HMGB1 sustain peritoneal carcinomatosis. Oncoimmunology 2016, 5, e1122860. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wu, K.; Zhao, E.; Shi, L.; Li, R.; Zhang, P.; Yin, Y.; Shuai, X.; Wang, G.; Tao, K. HMGB1 recruits myeloid derived suppressor cells to promote peritoneal dissemination of colon cancer after resection. Biochem. Biophys. Res. Commun. 2013, 436, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Kanterman, J.; Sade-Feldman, M.; Biton, M.; Ish-Shalom, E.; Lasry, A.; Goldshtein, A.; Hubert, A.; Baniyash, M. Adverse immunoregulatory effects of 5FU and CPT11 chemotherapy on myeloid-derived suppressor cells and colorectal cancer outcomes. Cancer Res. 2014, 74, 6022–6035. [Google Scholar] [CrossRef] [PubMed]
- Derer, A.; Frey, B.; Fietkau, R.; Gaipl, U.S. Immune-modulating properties of ionizing radiation: Rationale for the treatment of cancer by combination radiotherapy and immune checkpoint inhibitors. Cancer Immunol. Immunother. 2016, 65, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Frey, D.M.; Droeser, R.A.; Viehl, C.T.; Zlobec, I.; Lugli, A.; Zingg, U.; Oertli, D.; Kettelhack, C.; Terracciano, L.; Tornillo, L. High frequency of tumor-infiltrating FOXP3+ regulatory T cells predicts improved survival in mismatch repair-proficient colorectal cancer patients. Int. J. Cancer 2010, 126, 2635–2643. [Google Scholar] [CrossRef] [PubMed]
- Dunne, P.D.; McArt, D.G.; O’Reilly, P.G.; Coleman, H.G.; Allen, W.L.; Loughrey, M.; Schaeybroeck, S.V.; McDade, S.; Salto-Tellez, M.; Longley, D.B.; et al. Immune-derived PD-L1 gene expression defines a subgroup of stage ii/iii colorectal cancer patients with favorable prognosis who may be harmed by adjuvant chemotherapy. Cancer Immunol. Res. 2016, 4, 582–591. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, M.S.; DeConti, R.C. Immunosuppression by 5-fluorouracil. Cancer 1970, 26, 884–889. [Google Scholar] [CrossRef]
- Limagne, E.; Euvrard, R.; Thibaudin, M.; Rébé, C.; Derangère, V.; Chevriaux, A.; Boidot, R.; Végran, F.; Bonnefoy, N.; Vincent, J.; et al. Accumulation of MDSC and Th17 cells in patients with metastatic colorectal cancer predicts the efficacy of a FOLFOX-bevacizumab drug treatment regimen. Cancer Res. 2016, 76, 5241–5252. [Google Scholar] [CrossRef] [PubMed]
- Teng, F.; Mu, D.; Meng, X.; Kong, L.; Zhu, H.; Liu, S.; Zhang, J.; Yu, J. Tumor infiltrating lymphocytes (TILs) before and after neoadjuvant chemoradiotherapy and its clinical utility for rectal cancer. Am. J. Cancer Res. 2015, 5, 2064–2074. [Google Scholar] [PubMed]
- Agostini, M.; Janssen, K.-P.; Kim, I.-J.; D’Angelo, E.; Pizzini, S.; Zangrando, A.; Zanon, C.; Pastrello, C.; Maretto, I.; Digito, M.; et al. An integrative approach for the identification of prognostic and predictive biomarkers in rectal cancer. Oncotarget 2015, 6, 32561–32574. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrial.gov. Available online: https://clinicaltrials.gov/ct2/home (accessed on 3 May 2017).
- Mellman, I.; Coukos, G.; Dranoff, G. Cancer immunotherapy comes of age. Nature 2011, 480, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Marrone, K.A.; Brahmer, J.R. Using immune checkpoint inhibitors in lung cancer. Oncology 2016, 30, 713–721. [Google Scholar] [PubMed]
- Ott, P.A.; Hodi, F.S.; Robert, C. CTLA-4 and PD-1/PD-L1 blockade: New immunotherapeutic modalities with durable clinical benefit in melanoma patients. Clin. Cancer Res. 2013, 19, 5300–5309. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.T.; Siefker-Radtke, A.O.; Gao, J. The state of immune checkpoint inhibition in urothelial carcinoma: Current evidence and future areas of exploration. Cancer J. 2016, 22, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Jing, W.; Gershan, J.A.; Weber, J.; Tlomak, D.; McOlash, L.; Sabatos-Peyton, C.; Johnson, B.D. Combined immune checkpoint protein blockade and low dose whole body irradiation as immunotherapy for myeloma. J. Immunother. Cancer 2015, 3, 2. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Rini, B.I.; McDermott, D.F.; Redman, B.G.; Kuzel, T.M.; Harrison, M.R.; Vaishampayan, U.N.; Drabkin, H.A.; George, S.; Logan, T.F.; et al. Nivolumab for metastatic renal cell carcinoma: Results of a randomized phase ii trial. J. Clin. Oncol. 2015, 33, 1430–1437. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 blockade in tumors with mismatch-repair deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed]
- Overman, M.J.; Kopetz, S.; McDermott, R.S.; Leach, J.; Lonardi, S.; Lenz, H.-J.; Morse, M.A.; Desai, J.; Hill, A.; Axelson, M.D.; et al. Nivolumab ± ipilimumab in treatment (tx) of patients (pts) with metastatic colorectal cancer (mCRC) with and without high microsatellite instability (MSI-H): CheckMate-142 interim results. J. Clin. Oncol. 2016, 34, 3501. [Google Scholar]
- Xiao, Y.; Freeman, G.J. The Microsatellite Instable (MSI) Subset of Colorectal Cancer is a particularly good candidate for checkpoint blockade immunotherapy. Cancer Discov. 2015, 5, 16–18. [Google Scholar] [CrossRef] [PubMed]
- Parmiani, G.; Maccalli, C.; Maio, M. Integrating immune checkpoint blockade with anti-neo/mutated antigens reactivity to increase the clinical outcome of immunotherapy. Vaccines 2015, 3, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Wurz, G.T.; Kao, C.-J.; DeGregorio, M.W. Novel cancer antigens for personalized immunotherapies: Latest evidence and clinical potential. Ther. Adv. Med. Oncol. 2016, 8, 4–31. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Fang, W.; Zhan, J.; Hong, S.; Tang, Y.; Kang, S.; Zhang, Y.; He, X.; Zhou, T.; Qin, T.; et al. Upregulation of PD-L1 by EGFR activation mediates the immune escape in EGFR-driven NSCLC: Implication for optional immune targeted therapy for NSCLC patients with EGFR mutation. J. Thorac. Oncol. 2015, 10, 910–923. [Google Scholar] [CrossRef] [PubMed]
- Ebert, P.J.R.; Cheung, J.; Yang, Y.; McNamara, E.; Hong, R.; Moskalenko, M.; Gould, S.E.; Maecker, H.; Irving, B.A.; Kim, J.M.; et al. MAP kinase inhibition promotes T cell and anti-tumor activity in combination with PD-L1 checkpoint blockade. Immunity 2016, 44, 609–621. [Google Scholar] [CrossRef] [PubMed]
- Hatzivassiliou, G.; Haling, J.R.; Chen, H.; Song, K.; Price, S.; Heald, R.; Hewitt, J.F.M.; Zak, M.; Peck, A.; Orr, C.; et al. Mechanism of MEK inhibition determines efficacy in mutant KRAS-versus BRAF-driven cancers. Nature 2013, 501, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Bendell, J.C.; Kim, T.W.; Goh, B.C.; Wallin, J.; Oh, D.-Y.; Han, S.-W.; Lee, C.B.; Hellmann, M.D.; Desai, J.; Lewin, J.H.; et al. Clinical activity and safety of cobimetinib (cobi) and atezolizumab in colorectal cancer (CRC). J. Clin. Oncol. 2016, 34, 3502. [Google Scholar]
- Schaer, D.; Li, Y.; Castaneda, S.; Inigo, I.; Surguladze, D.; Xu, X.; Nugent, D.; Murphy, M.; Hall, G.; Benhadji, K.; et al. Targeting the TGFβ pathway with galunisertib, a TGFβRI SMI, promotes anti-tumor immunity leading to durable, complete responses, as monotherapy and in combination with checkpoint inhibition. J. Immunother. Cancer 2015, 3, P402. [Google Scholar] [CrossRef]
- Tauriello, D.V.F.; Batlle, E. Targeting the microenvironment in advanced colorectal cancer. Trends Cancer 2016, 2, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Zhao, W.; Yan, C.; Watson, C.C.; Massengill, M.; Xie, M.; Massengill, C.; Noyes, D.R.; Martinez, G.V.; Afzal, R.; et al. HDAC Inhibitors Enhance T-Cell Chemokine Expression and Augment Response to PD-1 Immunotherapy in Lung Adenocarcinoma. Clin. Cancer Res. 2016, 22, 4119–4132. [Google Scholar] [CrossRef] [PubMed]
- Ramjiawan, R.R.; Griffioen, A.W.; Duda, D.G. Anti-angiogenesis for cancer revisited: Is there a role for combinations with immunotherapy? Angiogenesis 2017, 20, 185–204. [Google Scholar] [CrossRef] [PubMed]
- Pfirschke, C.; Engblom, C.; Rickelt, S.; Cortez-Retamozo, V.; Garris, C.; Pucci, F.; Yamazaki, T.; Poirier-Colame, V.; Newton, A.; Redouane, Y.; et al. Immunogenic chemotherapy sensitizes tumors to checkpoint blockade therapy. Immunity 2016, 44, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Zhen, Y.-H.; Liu, X.-H.; Yang, Y.; Li, B.; Tang, J.-L.; Zeng, Q.-X.; Hu, J.; Zeng, X.-N.; Zhang, L.; Wang, Z.-J.; et al. Phase I/II study of adjuvant immunotherapy with sentinel lymph node T lymphocytes in patients with colorectal cancer. Cancer Immunol. Immunother. 2015, 64, 1083–1093. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.S.; Zea, A.H.; Rini, B.I.; Ireland, J.L.; Elson, P.; Cohen, P.; Golshayan, A.; Rayman, P.A.; Wood, L.; Garcia, J.; et al. Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin. Cancer Res. 2009, 15, 2148–2157. [Google Scholar] [CrossRef] [PubMed]
- Katoh, H.; Watanabe, M. Myeloid-derived suppressor cells and therapeutic strategies in cancer. Mediat. Inflamm. 2015, 2015, 159269. [Google Scholar] [CrossRef] [PubMed]
- Courau, T.; Nehar-Belaid, D.; Florez, L.; Levacher, B.; Vazquez, T.; Brimaud, F.; Bellier, B.; Klatzmann, D. TGF-β and VEGF cooperatively control the immunotolerant tumor environment and the efficacy of cancer immunotherapies. JCI Insight 2016, 1, e85974. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| CMS1 | CMS2 | CMS3 | CMS4 |
|---|---|---|---|
| Immune checkpoint inhibition (anti-PD-1/PD-L1, anti-CTLA-4, anti-IDO) [110,111,112] | Combined EGF pathway inhibition and immune checkpoint inhibition [115] | Combined MEK-inhibitor and immune checkpoint inhibition [59,116,117,118] | Combined TGF pathway inhibition and immune checkpoint inhibition [119,120] |
| Combined HDAC inhibitors and immune checkpoint inhibition [121] | Combined HDAC inhibitors and immune checkpoint inhibition [121] | Combined angiogenesis blockade and immune checkpoint inhibition [100,122] | |
| Combined neoantigen-based peptide vaccination and immune checkpoint inhibition [113,114] | Immuno-chemotherapy [123] | Immuno-chemotherapy [123] | |
| Passive immunotherapy (DCs vaccines, ACT) [124] | Passive immunotherapy (DCs vaccines, ACT) [124] | Anti-T-reg and/or anti-MDSCs treatment [125,126] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roelands, J.; Kuppen, P.J.K.; Vermeulen, L.; Maccalli, C.; Decock, J.; Wang, E.; Marincola, F.M.; Bedognetti, D.; Hendrickx, W. Immunogenomic Classification of Colorectal Cancer and Therapeutic Implications. Int. J. Mol. Sci. 2017, 18, 2229. https://doi.org/10.3390/ijms18102229
Roelands J, Kuppen PJK, Vermeulen L, Maccalli C, Decock J, Wang E, Marincola FM, Bedognetti D, Hendrickx W. Immunogenomic Classification of Colorectal Cancer and Therapeutic Implications. International Journal of Molecular Sciences. 2017; 18(10):2229. https://doi.org/10.3390/ijms18102229
Chicago/Turabian StyleRoelands, Jessica, Peter J. K. Kuppen, Louis Vermeulen, Cristina Maccalli, Julie Decock, Ena Wang, Francesco M. Marincola, Davide Bedognetti, and Wouter Hendrickx. 2017. "Immunogenomic Classification of Colorectal Cancer and Therapeutic Implications" International Journal of Molecular Sciences 18, no. 10: 2229. https://doi.org/10.3390/ijms18102229
APA StyleRoelands, J., Kuppen, P. J. K., Vermeulen, L., Maccalli, C., Decock, J., Wang, E., Marincola, F. M., Bedognetti, D., & Hendrickx, W. (2017). Immunogenomic Classification of Colorectal Cancer and Therapeutic Implications. International Journal of Molecular Sciences, 18(10), 2229. https://doi.org/10.3390/ijms18102229