Stress and the HPA Axis: Balancing Homeostasis and Fertility


Abstract
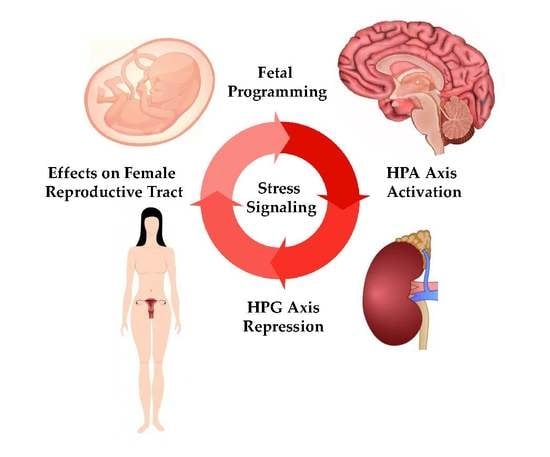
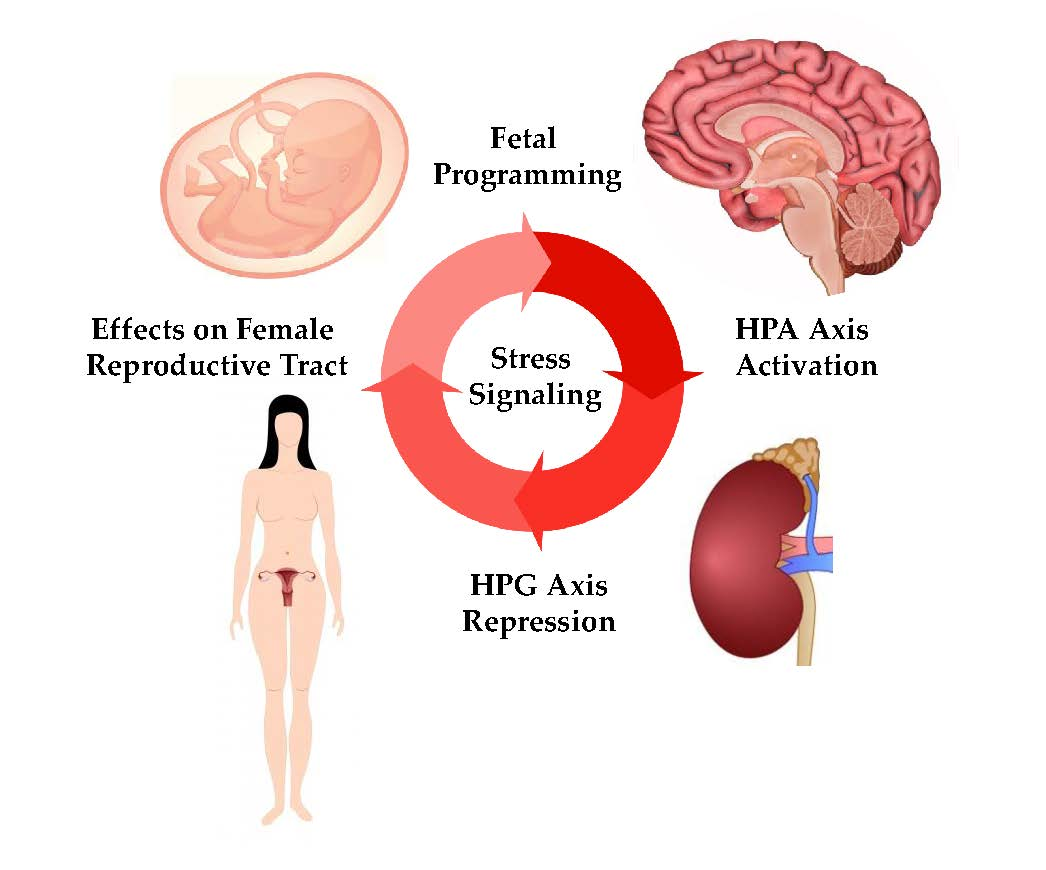{kind=link}
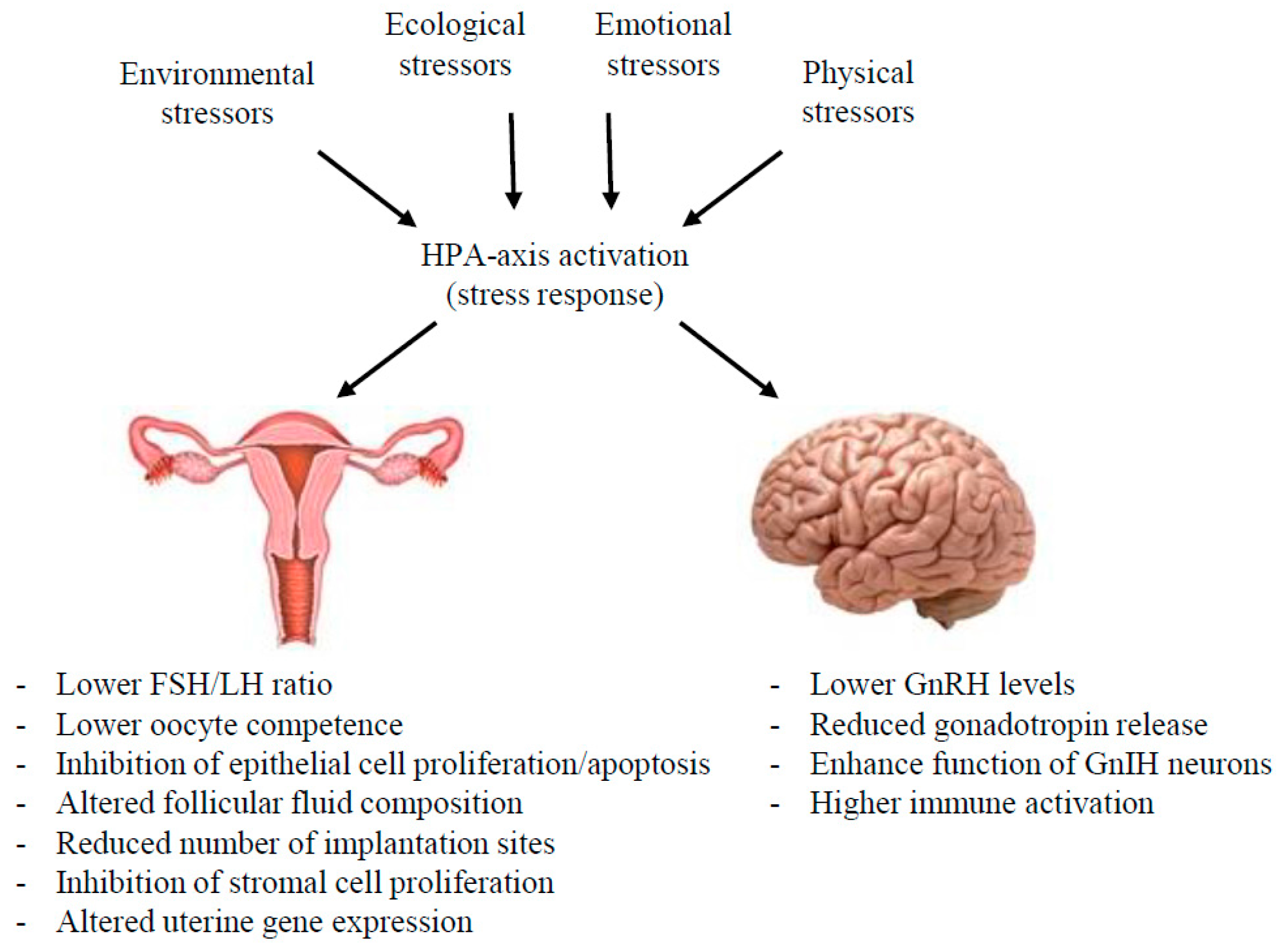{kind=link}
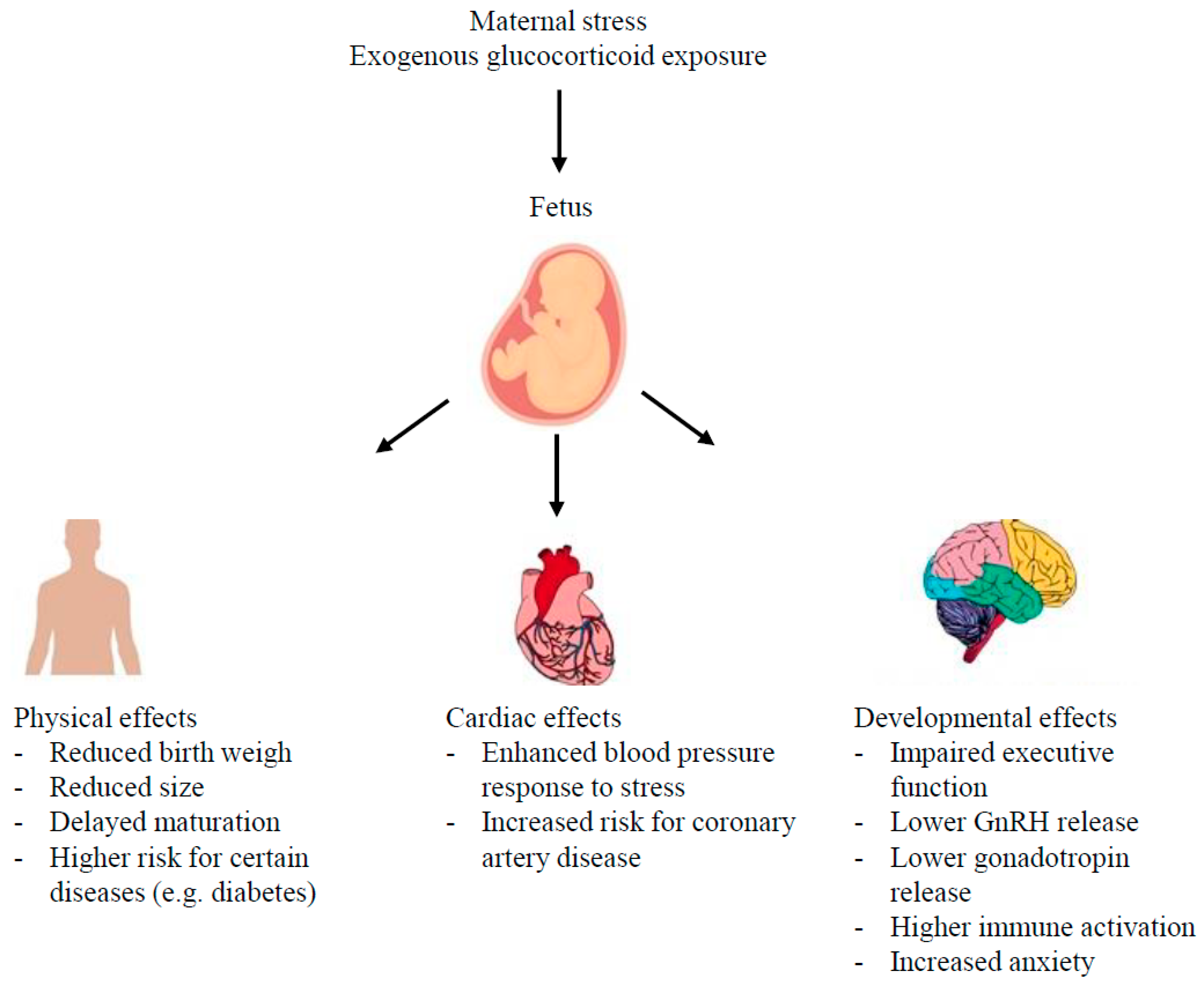{kind=link}
1. Introduction
2. Mechanisms of Hormone Signaling
3. The Impacts of Stress throughout the HPG Axis
3.1. Effects in Female Reproductive Tract Organs
3.2. The Placenta and Parturition
3.3. Other Markers of the Stress Response
4. Stress Impact on Programming
Sex Differences in the Response to Stress
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Smith, S.M.; Vale, W.W. The role of the hypothalamic-pituitary-adrenal axis in neuroendocrine responses to stress. Dialogues Clin. Neurosci. 2006, 8, 383–395. [Google Scholar] [PubMed]
- Carrasco, G.A.; van de Kar, L.D. Neuroendocrine pharmacology of stress. Eur. J. Pharmacol. 2003, 463, 235–272. [Google Scholar] [CrossRef]
- Stephens, M.A.; Wand, G. Stress and the HPA axis: Role of glucocorticoids in alcohol dependence. Alcohol. Res. 2012, 34, 468–483. [Google Scholar] [PubMed]
- Rhodes, M.E. Chapter 10-adrenocorticotropic hormone A2-ink, George. In Stress: Neuroendocrinology and Neurobiology; Academic Press: San Diego, CA, USA, 2017; pp. 109–116. [Google Scholar] [CrossRef]
- Richards, J.S.; Pangas, S.A. The ovary: Basic biology and clinical implications. J. Clin. Investig. 2010, 120, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Namwanje, M.; Brown, C.W. Activins and inhibins: Roles in development, physiology, and disease. Cold Spring Harbor Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Whirledge, S.; Cidlowski, J.A. Glucocorticoids and reproduction: Traffic control on the road to reproduction. Trends Endocrinol. Metab. TEM 2017, 28, 399–415. [Google Scholar] [CrossRef] [PubMed]
- Magiakou, M.A.; Mastorakos, G.; Webster, E.; Chrousos, G.P. The hypothalamic-pituitary-adrenal axis and the female reproductive system. Ann. N. Y. Acad. Sci. 1997, 816, 42–56. [Google Scholar] [CrossRef] [PubMed]
- Oakley, R.H.; Cidlowski, J.A. Cellular processing of the glucocorticoid receptor gene and protein: New mechanisms for generating tissue-specific actions of glucocorticoids. J. Biol. Chem. 2011, 286, 3177–3184. [Google Scholar] [CrossRef] [PubMed]
- Overman, R.A.; Yeh, J.Y.; Deal, C.L. Prevalence of oral glucocorticoid usage in the United States: A general population perspective. Arthritis Care Res. 2013, 65, 294–298. [Google Scholar] [CrossRef] [PubMed]
- Grad, I.; Picard, D. The glucocorticoid responses are shaped by molecular chaperones. Mol. Cell. Endocrinol. 2007, 275, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Pratt, W.B.; Toft, D.O. Steroid receptor interactions with heat shock protein and immunophilin chaperones. Endocr. Rev. 1997, 18, 306–360. [Google Scholar] [CrossRef] [PubMed]
- Luisi, B.F.; Xu, W.X.; Otwinowski, Z.; Freedman, L.P.; Yamamoto, K.R.; Sigler, P.B. Crystallographic analysis of the interaction of the glucocorticoid receptor with DNA. Nature 1991, 352, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Strahle, U.; Klock, G.; Schutz, G. A DNA sequence of 15 base pairs is sufficient to mediate both glucocorticoid and progesterone induction of gene expression. Proc. Natl. Acad. Sci. USA 1987, 84, 7871–7875. [Google Scholar] [CrossRef] [PubMed]
- Surjit, M.; Ganti, K.P.; Mukherji, A.; Ye, T.; Hua, G.; Metzger, D.; Li, M.; Chambon, P. Widespread negative response elements mediate direct repression by agonist-liganded glucocorticoid receptor. Cell 2011, 145, 224–241. [Google Scholar] [CrossRef] [PubMed]
- Weikum, E.R.; Knuesel, M.T.; Ortlund, E.A.; Yamamoto, K.R. Glucocorticoid receptor control of transcription: Precision and plasticity via allostery. Nat. Rev. Mol. Cell Biol. 2017, 18, 159–174. [Google Scholar] [CrossRef] [PubMed]
- Vedder, H.; Weiss, I.; Holsboer, F.; Reul, J.M. Glucocorticoid and mineralocorticoid receptors in rat neocortical and hippocampal brain cells in culture: Characterization and regulatory studies. Brain Res. 1993, 605, 18–24. [Google Scholar] [CrossRef]
- Fuller, P.J.; Yang, J.; Young, M.J. 30 years of the mineralocorticoid receptor: Coregulators as mediators of mineralocorticoid receptor signalling diversity. J. Endocrinol. 2017, 234, T23–T34. [Google Scholar] [CrossRef] [PubMed]
- De Kloet, E.R.; Joels, M. Brain mineralocorticoid receptor function in control of salt balance and stress-adaptation. Physiol. Behav. 2017, 178, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Shahjahan, M.; Kitahashi, T.; Ando, H. Temperature affects sexual maturation through the control of kisspeptin, kisspeptin receptor, GnRH and GTH subunit gene expression in the grass puffer during the spawning season. Gen. Comp. Endocrinol. 2017, 243, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Clarke, I.J.; Bartolini, D.; Conductier, G.; Henry, B.A. Stress Increases Gonadotropin Inhibitory Hormone Cell Activity and Input to GnRH Cells in Ewes. Endocrinology 2016, 157, 4339–4350. [Google Scholar] [CrossRef] [PubMed]
- Lopes, P.C.; Wingfield, J.C.; Bentley, G.E. Lipopolysaccharide injection induces rapid decrease of hypothalamic GnRH mRNA and peptide, but does not affect GnIH in zebra finches. Horm. Behav. 2012, 62, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Dobson, H.; Fergani, C.; Routly, J.E.; Smith, R.F. Effects of stress on reproduction in ewes. Anim. Reprod. Sci. 2012, 130, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Breen, K.M.; Karsch, F.J. New insights regarding glucocorticoids, stress and gonadotropin suppression. Front. Neuroendocrinol. 2006, 27, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Ciechanowska, M.; Lapot, M.; Antkowiak, B.; Mateusiak, K.; Paruszewska, E.; Malewski, T.; Paluch, M.; Przekop, F. Effect of short-term and prolonged stress on the biosynthesis of gonadotropin-releasing hormone (GnRH) and GnRH receptor (GnRHR) in the hypothalamus and GnRHR in the pituitary of ewes during various physiological states. Anim. Reprod. Sci. 2016, 174, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Kirby, E.D.; Geraghty, A.C.; Ubuka, T.; Bentley, G.E.; Kaufer, D. Stress increases putative gonadotropin inhibitory hormone and decreases luteinizing hormone in male rats. Proc. Natl. Acad. Sci. USA 2009, 106, 11324–11329. [Google Scholar] [CrossRef] [PubMed]
- Geraghty, A.C.; Muroy, S.E.; Zhao, S.; Bentley, G.E.; Kriegsfeld, L.J.; Kaufer, D. Knockdown of hypothalamic RFRP3 prevents chronic stress-induced infertility and embryo resorption. eLife 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Breen, K.M.; Thackray, V.G.; Hsu, T.; Mak-McCully, R.A.; Coss, D.; Mellon, P.L. Stress levels of glucocorticoids inhibit LHβ-subunit gene expression in gonadotrope cells. Mol. Endocrinol. 2012, 26, 1716–1731. [Google Scholar] [CrossRef] [PubMed]
- Wagenmaker, E.R.; Moenter, S.M. Exposure to Acute Psychosocial Stress Disrupts the Luteinizing Hormone Surge Independent of Estrous Cycle Alterations in Female Mice. Endocrinology 2017, 158, 2593–2602. [Google Scholar] [CrossRef] [PubMed]
- Warren, M.P.; Siris, E.S.; Petrovich, C. The Influence of Severe Illness on Gonadotropin Secretion in the Postmenopausal Female. J. Clin. Endocrinol. Metab. 1977, 45, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Ranganathan, P.; Kumar, R.G.; Davis, K.; McCullough, E.H.; Berga, S.L.; Wagner, A.K. Longitudinal sex and stress hormone profiles among reproductive age and post-menopausal women after severe TBI: A case series analysis. Brain Inj. 2016, 30, 452–461. [Google Scholar] [CrossRef] [PubMed]
- Suter, D.E.; Schwartz, N.B. Effects of glucocorticoids on secretion of luteinizing hormone and follicle-stimulating hormone by female rat pituitary cells in vitro. Endocrinology 1985, 117, 849–854. [Google Scholar] [CrossRef] [PubMed]
- Barroso, G.; Oehninger, S.; Monzo, A.; Kolm, P.; Gibbons, W.E.; Muasher, S.J. High FSH: LH ratio and low LH levels in basal cycle day 3: Impact on follicular development and IVF outcome. J. Assist. Reprod. Genet. 2001, 18, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Luo, E.; Stephens, S.B.; Chaing, S.; Munaganuru, N.; Kauffman, A.S.; Breen, K.M. Corticosterone Blocks Ovarian Cyclicity and the LH Surge via Decreased Kisspeptin Neuron Activation in Female Mice. Endocrinology 2016, 157, 1187–1199. [Google Scholar] [CrossRef] [PubMed]
- Rivier, C.; Vale, W. Influence of corticotropin-releasing factor on reproductive functions in the rat. Endocrinology 1984, 114, 914–921. [Google Scholar] [CrossRef] [PubMed]
- Traslavina, G.A.; Franci, C.R. Divergent roles of the CRH receptors in the control of gonadotropin secretion induced by acute restraint stress at proestrus. Endocrinology 2012, 153, 4838–4848. [Google Scholar] [CrossRef] [PubMed]
- Barbarino, A.; de Marinis, L.; Tofani, A.; Della Casa, S.; D’Amico, C.; Mancini, A.; Corsello, S.M.; Sciuto, R.; Barini, A. Corticotropin-releasing hormone inhibition of gonadotropin release and the effect of opioid blockade. J. Clin. Endocrinol. Metab. 1989, 68, 523–528. [Google Scholar] [CrossRef] [PubMed]
- Nteeba, J.; Sanz-Fernandez, M.V.; Rhoads, R.P.; Baumgard, L.H.; Ross, J.W.; Keating, A.F. Heat Stress Alters Ovarian Insulin-Mediated Phosphatidylinositol-3 Kinase and Steroidogenic Signaling in Gilt Ovaries. Biol. Reprod. 2015, 92, 148. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.W.; Hale, B.J.; Seibert, J.T.; Romoser, M.R.; Adur, M.K.; Keating, A.F.; Baumgard, L.H. Physiological mechanisms through which heat stress compromises reproduction in pigs. Mol. Reprod. Dev. 2017. [Google Scholar] [CrossRef] [PubMed]
- Gosden, R.G.; Hunter, R.H.; Telfer, E.; Torrance, C.; Brown, N. Physiological factors underlying the formation of ovarian follicular fluid. J. Reprod. Fertil. 1988, 82, 813–825. [Google Scholar] [CrossRef] [PubMed]
- Fortune, J.E. Ovarian follicular growth and development in mammals. Biol. Reprod. 1994, 50, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Chen, F.; Kong, Q.Q.; Ning, S.F.; Yuan, H.J.; Lian, H.Y.; Luo, M.J.; Tan, J.H. Stresses on Female Mice Impair Oocyte Developmental Potential: Effects of Stress Severity and Duration on Oocytes at the Growing Follicle Stage. Reprod. Sci. 2016, 23, 1148–1157. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.J.; Han, X.; He, N.; Wang, G.L.; Gong, S.; Lin, J.; Gao, M.; Tan, J.H. Glucocorticoids impair oocyte developmental potential by triggering apoptosis of ovarian cells via activating the Fas system. Sci. Rep. 2016, 6, 24036. [Google Scholar] [CrossRef] [PubMed]
- Jimena, P.; Castilla, J.A.; Peran, F.; Ramirez, J.P.; Vergara, F., Jr.; Molina, R.; Vergara, F.; Herruzo, A. Adrenal hormones in human follicular fluid. Acta Endocrinol. 1992, 127, 403–406. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.L.; Tan, X.W.; Cui, X.Z.; Yuan, H.J.; Li, H.; Jiao, G.Z.; Ji, C.L.; Tan, J.H. Preimplantation maternal stress impairs embryo development by inducing oviductal apoptosis with activation of the Fas system. Mol. Hum. Reprod. 2016, 22, 778–790. [Google Scholar] [CrossRef] [PubMed]
- Wiebold, J.L.; Stanfield, P.H.; Becker, W.C.; Hillers, J.K. The effect of restraint stress in early pregnancy in mice. J. Reprod. Fertil. 1986, 78, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Dong, Y.; Wang, Z.; Cao, J.; Chen, Y. Restraint stress delays endometrial adaptive remodeling during mouse embryo implantation. Stress 2015, 18, 699–709. [Google Scholar] [CrossRef] [PubMed]
- Jafari, Z.; Faraji, J.; Mirza Agha, B.; Metz, G.A.S.; Kolb, B.E.; Mohajerani, M.H. The Adverse Effects of Auditory Stress on Mouse Uterus Receptivity and Behaviour. Sci. Rep. 2017, 7, 4720. [Google Scholar] [CrossRef] [PubMed]
- Bitman, J.; Cecil, H.C. Differential Inhibition by Cortisol of Estrogen-Stimulated Uterine Responses. Endocrinology 1967, 80, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Wetendorf, M.; Wu, S.P.; Wang, X.; Creighton, C.J.; Wang, T.; Lanz, R.B.; Blok, L.; Tsai, S.Y.; Tsai, M.J.; Lydon, J.P.; et al. Decreased epithelial progesterone receptor A at the window of receptivity is required for preparation of the endometrium for embryo attachmentdagger. Biol. Reprod. 2017, 96, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Rhen, T.; Grissom, S.; Afshari, C.; Cidlowski, J.A. Dexamethasone blocks the rapid biological effects of 17beta-estradiol in the rat uterus without antagonizing its global genomic actions. FASEB J. 2003, 17, 1849–1870. [Google Scholar] [CrossRef] [PubMed]
- Whirledge, S.; Xu, X.; Cidlowski, J.A. Global gene expression analysis in human uterine epithelial cells defines new targets of glucocorticoid and estradiol antagonism. Biol. Reprod. 2013, 89, 66. [Google Scholar] [CrossRef] [PubMed]
- Whirledge, S.; Kisanga, E.P.; Taylor, R.N.; Cidlowski, J.A. Pioneer Factors FOXA1 and FOXA2 Assist Selective Glucocorticoid Receptor Signaling In Human Endometrial Cells. Endocrinology 2017. [Google Scholar] [CrossRef] [PubMed]
- Whirledge, S.; Cidlowski, J.A. Estradiol antagonism of glucocorticoid-induced GILZ expression in human uterine epithelial cells and murine uterus. Endocrinology 2013, 154, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Sahlin, L. Dexamethasone attenuates the estradiol-induced increase of IGF-I mRNA in the rat uterus. J. Steroid Biochem. Mol. Biol. 1995, 55, 9–15. [Google Scholar] [CrossRef]
- Zhu, L.; Pollard, J.W. Estradiol-17β regulates mouse uterine epithelial cell proliferation through insulin-like growth factor 1 signaling. Proc. Natl. Acad. Sci. USA 2007, 104, 15847–15851. [Google Scholar] [CrossRef] [PubMed]
- Hantak, A.M.; Bagchi, I.C.; Bagchi, M.K. Role of uterine stromal-epithelial crosstalk in embryo implantation. Int. J. Dev. Biol. 2014, 58, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, K.; Venkatakrishnan, R.; Salker, M.S.; Lucas, E.S.; Shaheen, F.; Kuroda, M.; Blanks, A.; Christian, M.; Quenby, S.; Brosens, J.J. Induction of 11β-HSD 1 and activation of distinct mineralocorticoid receptor- and glucocorticoid receptor-dependent gene networks in decidualizing human endometrial stromal cells. Mol. Endocrinol. 2013, 27, 192–202. [Google Scholar] [CrossRef] [PubMed]
- Gangestad, S.W.; Caldwell Hooper, A.E.; Eaton, M.A. On the function of placental corticotropin-releasing hormone: A role in maternal-fetal conflicts over blood glucose concentrations. Biol. Rev. Camb. Philos. Soc. 2012, 87, 856–873. [Google Scholar] [CrossRef] [PubMed]
- Thomson, M. The physiological roles of placental corticotropin releasing hormone in pregnancy and childbirth. J. Physiol. Biochem. 2013, 69, 559–573. [Google Scholar] [CrossRef] [PubMed]
- Linton, E.A.; Perkins, A.V.; Woods, R.J.; Eben, F.; Wolfe, C.D.; Behan, D.P.; Potter, E.; Vale, W.W.; Lowry, P.J. Corticotropin releasing hormone-binding protein (CRH-BP): Plasma levels decrease during the third trimester of normal human pregnancy. J. Clin. Endocrinol. Metab. 1993, 76, 260–262. [Google Scholar] [CrossRef] [PubMed]
- Hillhouse, E.W.; Grammatopoulos, D.; Milton, N.G.; Quartero, H.W. The identification of a human myometrial corticotropin-releasing hormone receptor that increases in affinity during pregnancy. J. Clin. Endocrinol. Metab. 1993, 76, 736–741. [Google Scholar] [CrossRef] [PubMed]
- Vrachnis, N.; Malamas, F.M.; Sifakis, S.; Tsikouras, P.; Iliodromiti, Z. Immune aspects and myometrial actions of progesterone and CRH in labor. Clin. Dev. Immunol. 2012, 2012, 937618. [Google Scholar] [CrossRef] [PubMed]
- Cong, B.; Zhang, L.; Gao, L.; Ni, X. Reduced expression of CRH receptor type 1 in upper segment human myometrium during labour. Reprod. Biol. Endocrinol. 2009, 7, 43. [Google Scholar] [CrossRef] [PubMed]
- Bukowski, R.; Hankins, G.D.; Saade, G.R.; Anderson, G.D.; Thornton, S. Labor-associated gene expression in the human uterine fundus, lower segment, and cervix. PLoS Med. 2006, 3, e169. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Shen, H.; Yu, L.; Peng, M.; Lai, W.S.; Ding, Y.L. Corticotropin-releasing hormone activates connexin 43 via activator protein-1 transcription factor in human myometrial smooth muscle cells. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E1789–E1794. [Google Scholar] [CrossRef] [PubMed]
- You, X.; Liu, J.; Xu, C.; Liu, W.; Zhu, X.; Li, Y.; Sun, Q.; Gu, H.; Ni, X. Corticotropin-releasing hormone (CRH) promotes inflammation in human pregnant myometrium: The evidence of CRH initiating parturition? J. Clin. Endocrinol. Metab. 2014, 99, E199–E208. [Google Scholar] [CrossRef] [PubMed]
- Christiaens, I.; Zaragoza, D.B.; Guilbert, L.; Robertson, S.A.; Mitchell, B.F.; Olson, D.M. Inflammatory processes in preterm and term parturition. J. Reprod. Immunol. 2008, 79, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.S.; Liu, C.; Li, W.J.; Zhu, P.; Li, J.N.; Sun, K. Involvement of CRH and hCG in the induction of aromatase by cortisol in human placental syncytiotrophoblasts. Placenta 2014, 35, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Robinson, B.G.; Emanuel, R.L.; Frim, D.M.; Majzoub, J.A. Glucocorticoid stimulates expression of corticotropin-releasing hormone gene in human placenta. Proc. Natl. Acad. Sci. USA 1988, 85, 5244–5248. [Google Scholar] [CrossRef] [PubMed]
- Heron, M. Deaths: Leading causes for 2008. Natl. Vital Stat. Rep. 2012, 60, 1–94. [Google Scholar] [PubMed]
- Goldenberg, R.L.; Culhane, J.F.; Iams, J.D.; Romero, R. Epidemiology and causes of preterm birth. Lancet 2008, 371, 75–84. [Google Scholar] [CrossRef]
- Witt, W.P.; Cheng, E.R.; Wisk, L.E.; Litzelman, K.; Chatterjee, D.; Mandell, K.; Wakeel, F. Preterm birth in the United States: The impact of stressful life events prior to conception and maternal age. Am. J. Public Health 2014, 104, S73–S80. [Google Scholar] [CrossRef] [PubMed]
- Khashan, A.S.; McNamee, R.; Abel, K.M.; Mortensen, P.B.; Kenny, L.C.; Pedersen, M.G.; Webb, R.T.; Baker, P.N. Rates of preterm birth following antenatal maternal exposure to severe life events: A population-based cohort study. Hum. Reprod. 2009, 24, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, S.L.; Christian, L.M.; Alston, A.D.; Salsberry, P.J. Childhood stress and birth timing among African American women: Cortisol as biological mediator. Psychoneuroendocrinology 2017, 84, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Wadhwa, P.D.; Porto, M.; Garite, T.J.; Chicz-DeMet, A.; Sandman, C.A. Maternal corticotropin-releasing hormone levels in the early third trimester predict length of gestation in human pregnancy. Am. J. Obstet. Gynecol. 1998, 179, 1079–1085. [Google Scholar] [CrossRef]
- Wadhwa, P.D.; Garite, T.J.; Porto, M.; Glynn, L.; Chicz-DeMet, A.; Dunkel-Schetter, C.; Sandman, C.A. Placental corticotropin-releasing hormone (CRH), spontaneous preterm birth, and fetal growth restriction: A prospective investigation. Am. J. Obstet. Gynecol. 2004, 191, 1063–1069. [Google Scholar] [CrossRef] [PubMed]
- Stewart, C.P.; Oaks, B.M.; Laugero, K.D.; Ashorn, U.; Harjunmaa, U.; Kumwenda, C.; Chaima, D.; Maleta, K.; Ashorn, P.; Dewey, K.G. Maternal cortisol and stress are associated with birth outcomes, but are not affected by lipid-based nutrient supplements during pregnancy: An analysis of data from a randomized controlled trial in rural Malawi. BMC Pregnancy Childbirth 2015, 15, 346. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Blanco, A.; Diago, V.; Serrano de la Cruz, V.; Hervas, D.; Chafer-Pericas, C.; Vento, M. Can stress biomarkers predict preterm birth in women with threatened preterm labor? Psychoneuroendocrinology 2017, 83, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Liu, C.; Sun, K. Induction of Amnion Epithelial Apoptosis by Cortisol via tPA/Plasmin System. Endocrinology 2016, 157, 4487–4498. [Google Scholar] [CrossRef] [PubMed]
- Mi, Y.; Wang, W.; Zhang, C.; Liu, C.; Lu, J.; Li, W.; Zuo, R.; Myatt, L.; Sun, K. Autophagic Degradation of Collagen 1A1 by Cortisol in Human Amnion Fibroblasts. Endocrinology 2017, 158, 1005–1014. [Google Scholar] [CrossRef] [PubMed]
- Hampson, V.; Liu, D.; Billett, E.; Kirk, S. Amniotic membrane collagen content and type distribution in women with preterm premature rupture of the membranes in pregnancy. Br. J. Obstet. Gynaecol. 1997, 104, 1087–1091. [Google Scholar] [CrossRef] [PubMed]
- Potestio, F.A.; Zakar, T.; Olson, D.M. Glucocorticoids stimulate prostaglandin synthesis in human amnion cells by a receptor-mediated mechanism. J. Clin. Endocrinol. Metab. 1988, 67, 1205–1210. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Q.; Zhu, P.; Myatt, L.; Sun, K. Roles of glucocorticoids in human parturition: A controversial fact? Placenta 2014, 35, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Craft, I.; Brummer, V.; Horwell, D.; Morgan, H. Betamethazone induction of labour. Proc. R. Soc. Med. 1976, 69, 827–828. [Google Scholar] [CrossRef] [PubMed]
- Mati, J.K.; Horrobin, D.F.; Bramley, P.S. Induction of labour in sheep and in humans by single doses of corticosteroids. Br. Med. J. 1973, 2, 149–151. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, Y.; Kawano, A.; Okamoto, S.; Ando, T.; Ishitobi, Y.; Tanaka, Y.; Inoue, A.; Imanaga, J.; Kanehisa, M.; Higuma, H.; et al. Differences in salivary alpha-amylase and cortisol responsiveness following exposure to electrical stimulation versus the Trier Social Stress Tests. PLoS ONE 2012, 7, e39375. [Google Scholar] [CrossRef] [PubMed]
- Lynch, C.D.; Sundaram, R.; Maisog, J.M.; Sweeney, A.M.; Buck Louis, G.M. Preconception stress increases the risk of infertility: Results from a couple-based prospective cohort study—The LIFE study. Hum. Reprod. 2014, 29, 1067–1075. [Google Scholar] [CrossRef] [PubMed]
- Louis, G.M.B.; Lum, K.J.; Sundaram, R.; Chen, Z.; Kim, S.; Lynch, C.D.; Schisterman, E.F.; Pyper, C. Stress reduces conception probabilities across the fertile window: Evidence in support of relaxation. Fertil. Steril. 2011, 95, 2184–2189. [Google Scholar] [CrossRef] [PubMed]
- Liggins, G.C. Premature delivery of foetal lambs infused with glucocorticoids. J. Endocrinol. 1969, 45, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Liggins, G.C.; Howie, R.N. A controlled trial of antepartum glucocorticoid treatment for prevention of the respiratory distress syndrome in premature infants. Pediatrics 1972, 50, 515–525. [Google Scholar] [PubMed]
- Matsuzaki, Y.; Xu, Y.; Ikegami, M.; Besnard, V.; Park, K.S.; Hull, W.M.; Wert, S.E.; Whitsett, J.A. Stat3 is required for cytoprotection of the respiratory epithelium during adenoviral infection. J. Immunol. 2006, 177, 527–537. [Google Scholar] [CrossRef] [PubMed]
- Gilstrap, L.C.; Christensen, R.; Clewell, W.H.; D’Alton, M.E.; Davidson, E.C.; Escobedo, M.B.; Gjerdingen, D.K.; Finegold, J.G.; Goldenberg, R.L.; Grimes, D.A.; et al. Effect of corticosteroids for fetal maturation on perinatal outcomes. NIH Consensus Development Panel on the Effect of Corticosteroids for Fetal Maturation on Perinatal Outcomes. JAMA 1995, 273, 413–418. [Google Scholar] [CrossRef]
- Cottrell, E.C.; Seckl, J.R. Prenatal stress, glucocorticoids and the programming of adult disease. Front. Behav. Neurosci. 2009, 3, 19. [Google Scholar] [CrossRef] [PubMed]
- Seckl, J.R. Prenatal glucocorticoids and long-term programming. Eur. J. Endocrinol. 2004, 151, 49–62. [Google Scholar] [CrossRef]
- Moisiadis, V.G.; Constantinof, A.; Kostaki, A.; Szyf, M.; Matthews, S.G. Prenatal Glucocorticoid Exposure Modifies Endocrine Function and Behaviour for 3 Generations Following Maternal and Paternal Transmission. Sci. Rep. 2017, 7, 11814. [Google Scholar] [CrossRef] [PubMed]
- Painter, R.C.; Roseboom, T.J.; Bleker, O.P. Prenatal exposure to the Dutch famine and disease in later life: An overview. Reprod. Toxicol. 2005, 20, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Roseboom, T.J.; Painter, R.C.; van Abeelen, A.F.; Veenendaal, M.V.; de Rooij, S.R. Hungry in the womb: What are the consequences? Lessons from the Dutch famine. Maturitas 2011, 70, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Tobi, E.W.; Goeman, J.J.; Monajemi, R.; Gu, H.; Putter, H.; Zhang, Y.; Slieker, R.C.; Stok, A.P.; Thijssen, P.E.; Muller, F.; et al. DNA methylation signatures link prenatal famine exposure to growth and metabolism. Nat. Commun. 2014, 5, 5592. [Google Scholar] [CrossRef] [PubMed]
- Montoya-Williams, D.; Quinlan, J.; Clukay, C.; Rodney, N.C.; Kertes, D.A.; Mulligan, C.J. Associations between maternal prenatal stress, methylation changes in IGF1 and IGF2, and birth weight. J. Dev. Orig. Health Dis. 2017, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Yehuda, R.; Engel, S.M.; Brand, S.R.; Seckl, J.; Marcus, S.M.; Berkowitz, G.S. Transgenerational effects of posttraumatic stress disorder in babies of mothers exposed to the World Trade Center attacks during pregnancy. J. Clin. Endocrinol. Metab. 2005, 90, 4115–4118. [Google Scholar] [CrossRef] [PubMed]
- Lipkind, H.S.; Curry, A.E.; Huynh, M.; Thorpe, L.E.; Matte, T. Birth outcomes among offspring of women exposed to the September 11, 2001, terrorist attacks. Obstet. Gynecol. 2010, 116, 917–925. [Google Scholar] [CrossRef] [PubMed]
- Fuller, S.C. The effect of prenatal natural disaster exposure on school outcomes. Demography 2014, 51, 1501–1525. [Google Scholar] [CrossRef] [PubMed]
- Laplante, D.P.; Brunet, A.; Schmitz, N.; Ciampi, A.; King, S. Project Ice Storm: Prenatal maternal stress affects cognitive and linguistic functioning in 5 1/2-year-old children. J. Am. Acad. Child Adolesc. Psychiatry 2008, 47, 1063–1072. [Google Scholar] [CrossRef] [PubMed]
- Hampshire, A.; Highfield, R.R.; Parkin, B.L.; Owen, A.M. Fractionating human intelligence. Neuron 2012, 76, 1225–1237. [Google Scholar] [CrossRef] [PubMed]
- Simcock, G.; Kildea, S.; Elgbeili, G.; Laplante, D.P.; Cobham, V.; King, S. Prenatal maternal stress shapes children’s theory of mind: The QF2011 Queensland Flood Study. J. Dev. Orig. Health Dis. 2017, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Van den Bergh, B.R.H.; van den Heuvel, M.I.; Lahti, M.; Braeken, M.; de Rooij, S.R.; Entringer, S.; Hoyer, D.; Roseboom, T.; Raikkonen, K.; King, S.; et al. Prenatal developmental origins of behavior and mental health: The influence of maternal stress in pregnancy. Neurosci. Biobehav. Rev. 2017. [Google Scholar] [CrossRef] [PubMed]
- Blair, M.M.; Glynn, L.M.; Sandman, C.A.; Davis, E.P. Prenatal maternal anxiety and early childhood temperament. Stress 2011, 14, 644–651. [Google Scholar] [CrossRef] [PubMed]
- Davis, E.P.; Sandman, C.A. The timing of prenatal exposure to maternal cortisol and psychosocial stress is associated with human infant cognitive development. Child. Dev. 2010, 81, 131–148. [Google Scholar] [CrossRef] [PubMed]
- Boddicker, R.L.; Koltes, J.E.; Fritz-Waters, E.R.; Koesterke, L.; Weeks, N.; Yin, T.; Mani, V.; Nettleton, D.; Reecy, J.M.; Baumgard, L.H.; et al. Genome-wide methylation profile following prenatal and postnatal dietary omega-3 fatty acid supplementation in pigs. Anim. Genet. 2016, 47, 658–671. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.S.; Boddicker, R.L.; Sanz-Fernandez, M.V.; Ross, J.W.; Selsby, J.T.; Lucy, M.C.; Safranski, T.J.; Rhoads, R.P.; Baumgard, L.H. Effects of mammalian in utero heat stress on adolescent body temperature. Int. J. Hyperth. 2013, 29, 696–702. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.S.; Sanz Fernandez, M.V.; Seibert, J.T.; Ross, J.W.; Lucy, M.C.; Safranski, T.J.; Elsasser, T.H.; Kahl, S.; Rhoads, R.P.; Baumgard, L.H. In utero heat stress increases postnatal core body temperature in pigs. J. Anim. Sci. 2015, 93, 4312–4322. [Google Scholar] [CrossRef] [PubMed]
- Grant, K.-A.; Sandman, C.A.; Wing, D.A.; Dmitrieva, J.; Davis, E.P. Prenatal Programming of Postnatal Susceptibility to Memory Impairments: A Developmental Double Jeopardy. Psychol. Sci. 2015, 26, 1054–1062. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.; Doyle, W.J.; Baum, A. Socioeconomic status is associated with stress hormones. Psychosom. Med. 2006, 68, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Miller, G.E.; Borders, A.E.; Crockett, A.H.; Ross, K.M.; Qadir, S.; Keenan-Devlin, L.; Leigh, A.K.; Ham, P.; Ma, J.; Arevalo, J.M.G.; et al. Maternal socioeconomic disadvantage is associated with transcriptional indications of greater immune activation and slower tissue maturation in placental biopsies and newborn cord blood. Brain Behav. Immun. 2017, 64, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Tyrrell, J.; Melzer, D.; Henley, W.; Galloway, T.S.; Osborne, N.J. Associations between socioeconomic status and environmental toxicant concentrations in adults in the USA: NHANES 2001–2010. Environ. Int. 2013, 59, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, T.; Grecian, S.M.; Reynolds, R.M. Sex differences in early-life programming of the hypothalamic-pituitary-adrenal axis in humans suggest increased vulnerability in females: A systematic review. J. Dev. Orig. Health Dis. 2017, 8, 244–255. [Google Scholar] [CrossRef] [PubMed]
- Clifton, V.L. Review: Sex and the human placenta: Mediating differential strategies of fetal growth and survival. Placenta 2010, 31, S33–S39. [Google Scholar] [CrossRef] [PubMed]
- Mericq, V.; Medina, P.; Kakarieka, E.; Marquez, L.; Johnson, M.C.; Iniguez, G. Differences in expression and activity of 11β-hydroxysteroid dehydrogenase type 1 and 2 in human placentas of term pregnancies according to birth weight and gender. Eur. J. Endocrinol. 2009, 161, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Cuffe, J.S.M.; Saif, Z.; Perkins, A.V.; Moritz, K.M.; Clifton, V.L. Dexamethasone and sex regulate placental glucocorticoid receptor isoforms in mice. J. Endocrinol. 2017, 234, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Lu, N.Z.; Cidlowski, J.A. Translational regulatory mechanisms generate N-terminal glucocorticoid receptor isoforms with unique transcriptional target genes. Mol. Cell 2005, 18, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Yun, H.J.; Kim, C.Y.; Cho, Y.W.; Lee, Y.; Kim, M.H. Prenatal exposure to dexamethasone in the mouse induces sex-specific differences in placental gene expression. Dev. Growth Differ. 2017, 59, 515–525. [Google Scholar] [CrossRef] [PubMed]
- Sandman, C.A.; Glynn, L.M.; Davis, E.P. Is there a viability-vulnerability tradeoff? Sex differences in fetal programming. J. Psychosom. Res. 2013, 75, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Braithwaite, E.C.; Murphy, S.E.; Ramchandani, P.G.; Hill, J. Associations between biological markers of prenatal stress and infant negative emotionality are specific to sex. Psychoneuroendocrinology 2017, 86, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Negro-Vilar, A. Stress and other environmental factors affecting fertility in men and women: Overview. Environ. Health Perspect. 1993, 101, S59–S64. [Google Scholar] [CrossRef]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Joseph, D.N.; Whirledge, S. Stress and the HPA Axis: Balancing Homeostasis and Fertility. Int. J. Mol. Sci. 2017, 18, 2224. https://doi.org/10.3390/ijms18102224
Joseph DN, Whirledge S. Stress and the HPA Axis: Balancing Homeostasis and Fertility. International Journal of Molecular Sciences. 2017; 18(10):2224. https://doi.org/10.3390/ijms18102224
Chicago/Turabian StyleJoseph, Dana N., and Shannon Whirledge. 2017. "Stress and the HPA Axis: Balancing Homeostasis and Fertility" International Journal of Molecular Sciences 18, no. 10: 2224. https://doi.org/10.3390/ijms18102224
APA StyleJoseph, D. N., & Whirledge, S. (2017). Stress and the HPA Axis: Balancing Homeostasis and Fertility. International Journal of Molecular Sciences, 18(10), 2224. https://doi.org/10.3390/ijms18102224