Kinases of eIF2a Switch Translation of mRNA Subset during Neuronal Plasticity


Abstract
1. Neuron-Specific Translation Initiation Regulation and the Role of Kinases Phosphorylating α-Subunit of Eukaryotic Initiation Factor 2 (eIF2α) in This Process
1.1. Neuronal Plasticity and Translation
1.2. Eukaryotic Initiation Factor 2 (eIF2) and Four Kinases of Its α-Subunit
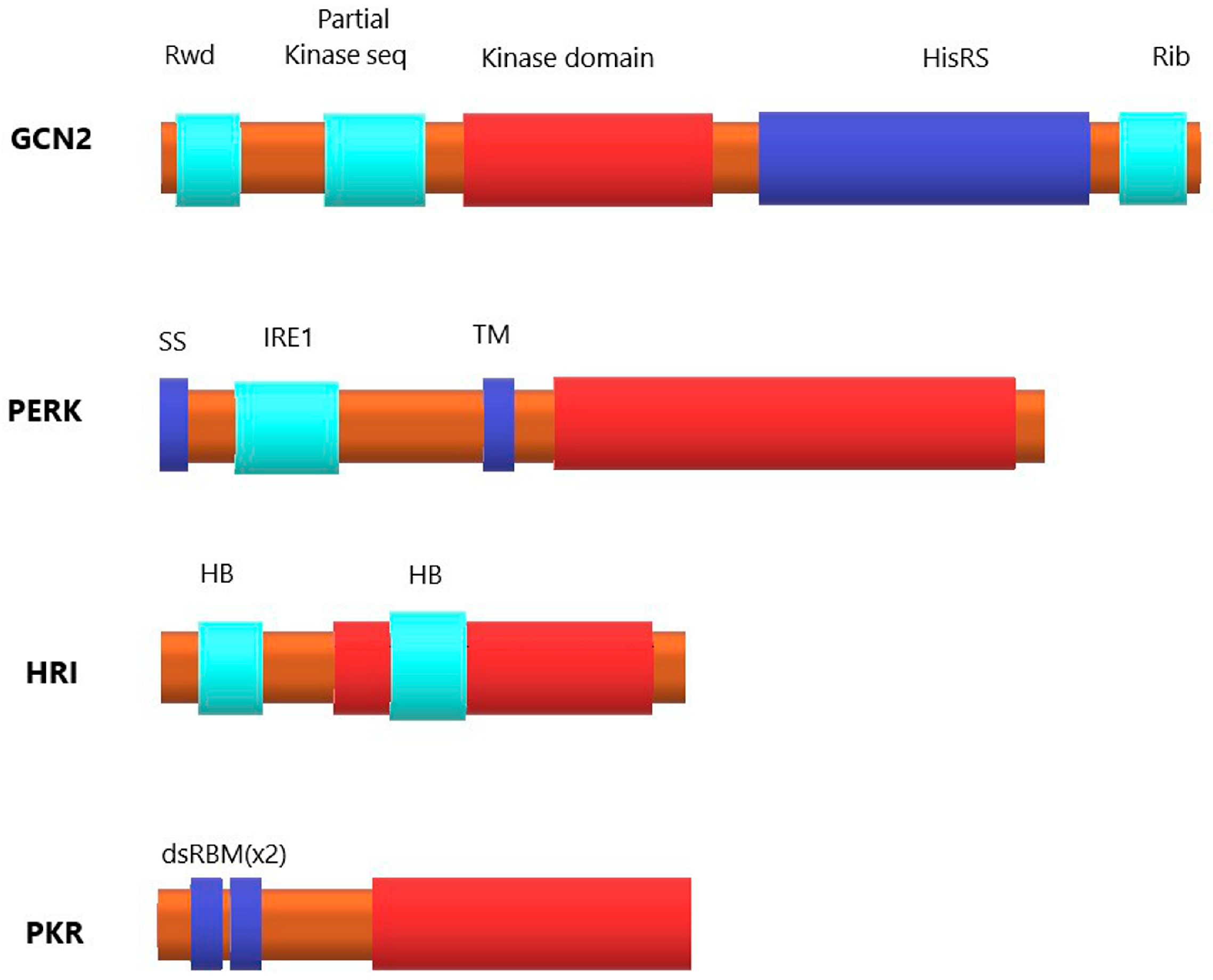
1.2.1. Kinase General Control Nonderepressible 2 (GCN2)
1.2.2. PKR-like Endoplasmic Reticulum Kinase (PERK)
1.2.3. Heme-regulated eIF2α kinase (HRI)
1.2.4. Protein Kinase R (PKR)
1.3. The Mechanism of Translation Shift Caused by eIF2α Phosphorylation and Its Role in Neurons
2. Neuron-Specific Proteins that May Be Regulated by eIF2α Phosphorylation
2.1. Activating Transcription Factor 4 (ATF4)
2.2. ER Stress-Related Proteins: Growth Arrest and DNA Damage-Inducible Protein (GADD34) and CCAAT-Enhancer-Binding Protein Homologous Protein (CHOP)
2.3. Beta-Site APP-Cleaving Enzyme 1 (BACE1), the Enzyme Connected with Alzheimer’s Disease
2.4. Glutamate Ionotropic Receptor NMDA Type Subunit 2B (GluN2B)
2.5. Oligophrenin-1
2.6. Postsynaptic Density Proteins: Synapse-Associated Protein 90/Postsynaptic Density Protein-95-Associated Protein 3 (SAPAP3) and SH3 and Multiple Ankyrin Repeat Domains 1 (Shank1)
2.7. Neuron-Specific BCL2-Antagonist/Killer (N-Bak), a Constitutively Repressed Pro-Apoptotic Protein
2.8. Protein Kinase Mζ, “the Memory Molecule”
3. Protein Kinase Mζ, Its Functions and Regulation of Its Translation
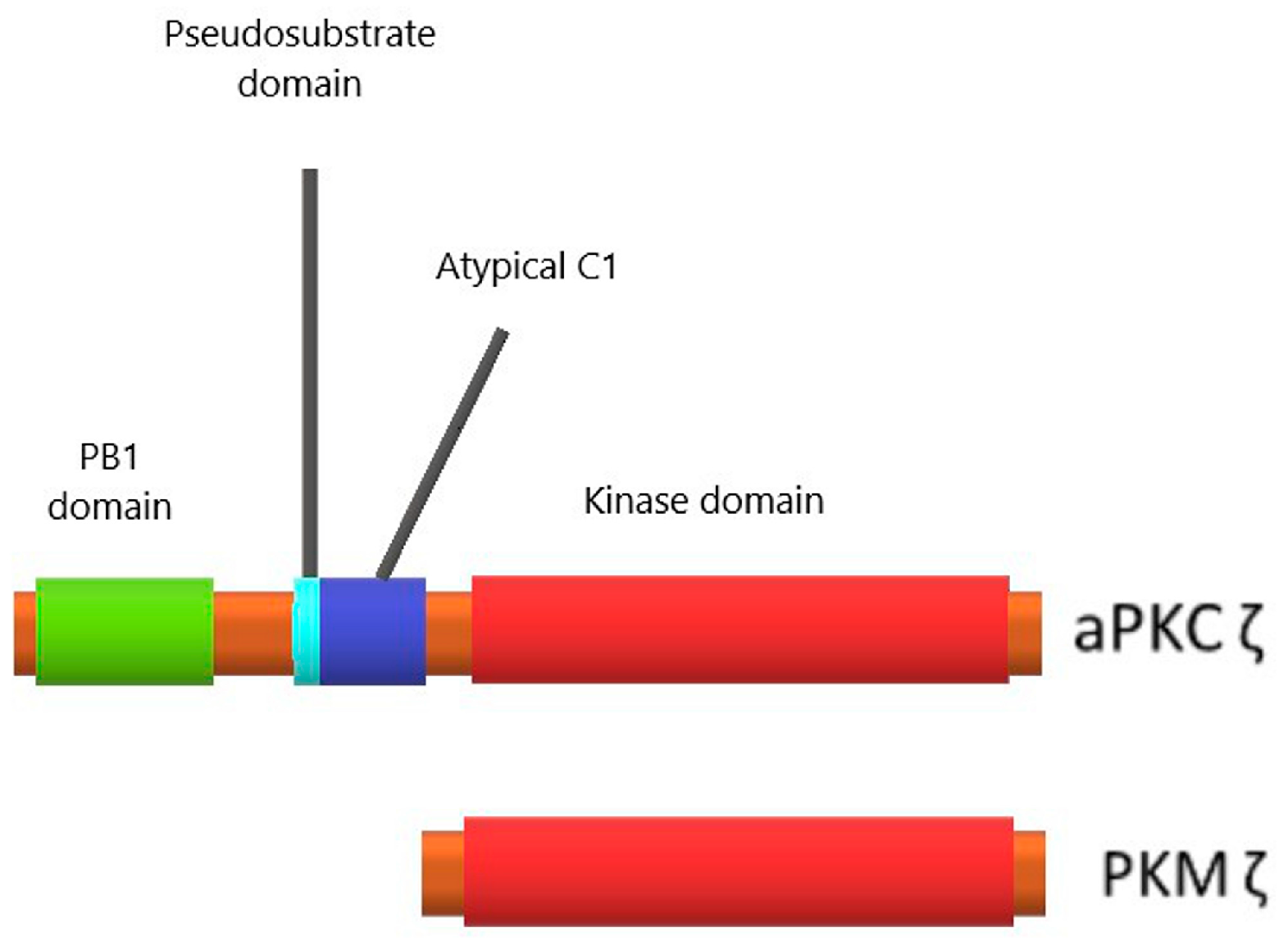
3.1. Protein Kinase Mζ Structure and Its Difference from Structures of Other Related Kinases
3.2. Protein Kinase Mζ Cellular Localization and Function
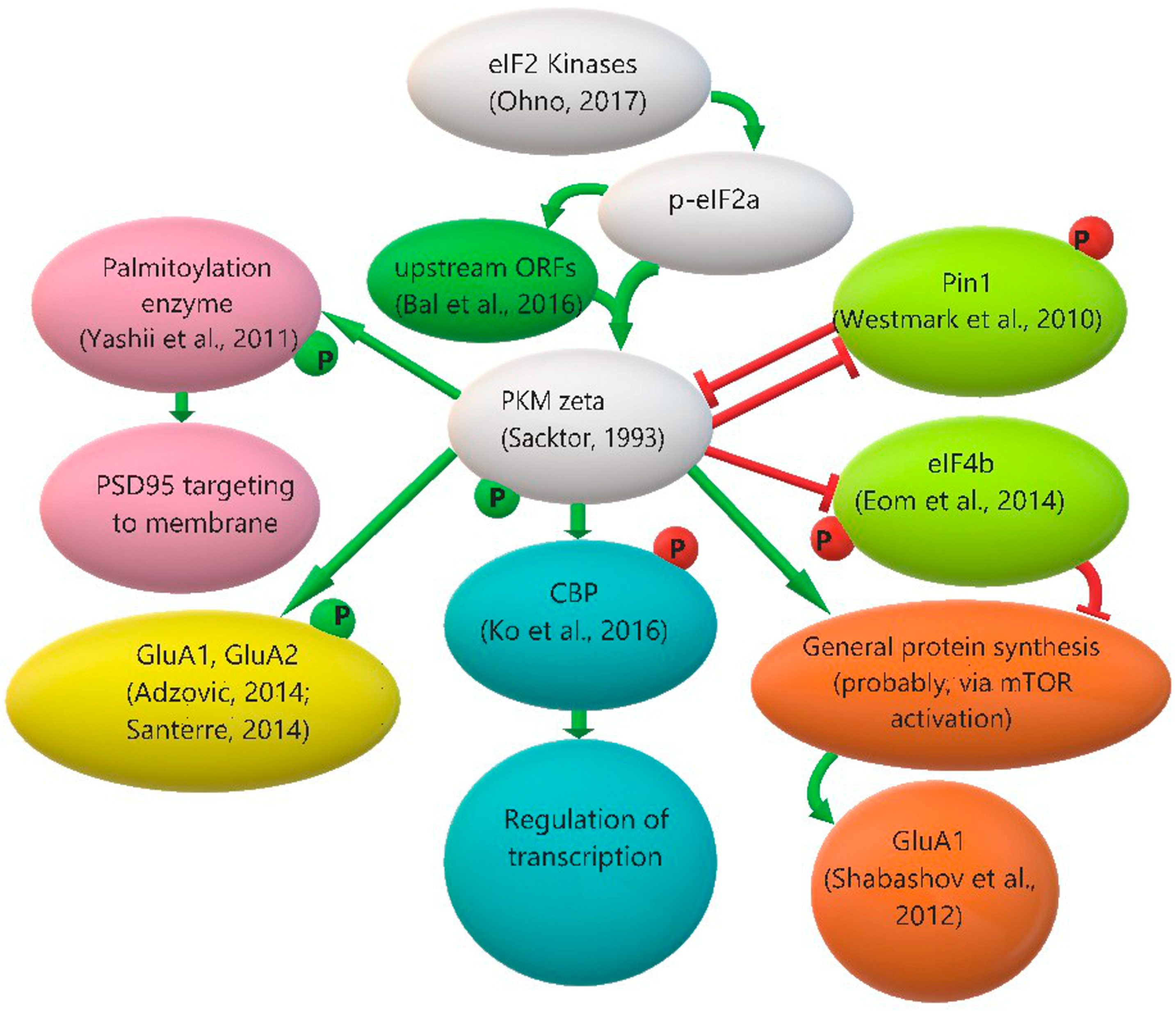
3.3. Protein Kinase Mζ Translation Regulation: The Role of eIF2α Phosphorylation and Other Possible Mechanisms
4. Summary
Acknowledgments
Conflicts of Interest
Abbreviations
| ∆eIF4G | Truncated eucaryotic translation initiation factor 4G |
| 3′-UTR | 3′-Untranslated region |
| 5′-UTR | 5′-Untranslated region |
| 7-NI | 7-Nitroindazole |
| AMPA | α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid |
| aPKC | Atypical protein kinase C |
| APP | Amyloid precursor protein |
| ATF4 | Activating transcription factor 4 |
| ATF5 | Activating transcription factor 5 |
| ATF6 | Activating transcription factor 6 |
| ATP | Adenosine triphosphate |
| BACE1 | Beta-site APP-cleaving enzyme 1 |
| BACE1-293 | HEK-293 cells stably overexpressing BACE1 |
| Bax | BCL-2-associated X protein |
| BC1/BC200 RNA | Brain cytoplasmic RNA |
| BDNF | Brain-derived neurotropic factor |
| BiP/GPR78 | Binding immunoglobulin protein (BiP), also known as 78 kDa glucose-regulated protein |
| C1 | Protein kinase C conserved region 1 |
| C2 | Protein kinase C conserved region 2 |
| CA1 | Cornu Ammonis region 1 |
| CaMKII | Ca2+/calmodulin-dependent protein kinase II |
| CBP | CREB-binding protein |
| CHO-K1 | Chinese hamster ovary K1 cells |
| CHOP | CCAAT-enhancer-binding protein homologous protein |
| CREB-2 | cAMP-response element binding protein 2 |
| C-terminal | Carboxyl terminal |
| DAG | Diacylglycerol |
| DDIT3 | DNA damage-inducible transcript 3 |
| DHPG | Dihydroxyphenylglycine |
| dsRBM | Double-stranded RNA binding motif |
| ds-RNA | Double-stranded RNA |
| eIF2 | Eucaryotic translation initiation factor 2 |
| eIF2α | α-subunit of eucaryotic translation initiation factor 2 |
| eIF3 | Eucaryotic translation initiation factor 3 |
| eIF4B | Eucaryotic translation initiation factor 4B |
| eIF4E | Eucaryotic translation initiation factor 4E |
| eIF4G | Eucaryotic translation initiation factor 4G |
| ER | Endoplasmic reticulum |
| G-actin | Globular actin |
| GADD34 | Growth arrest and DNA damage-inducible protein |
| GCN1 | General control of amino-acid synthesis 1-like protein 1 |
| GCN2 | General control nonderepressible 2 (kinase) |
| GCN2DN | Dominant-negative GCN2 allele |
| GFP | Green fluorescent protein |
| GluA1 | Glutamate ionotropic receptor AMPA type subunit 1 |
| GluA2 | Glutamate ionotropic receptor AMPA type subunit 2 |
| GluN2B | Glutamate ionotropic receptor NMDA type subunit 2B |
| GTP | Guanosine triphosphate |
| H2B | Histone 2B |
| H3 | Histone 3 |
| HEK | Human embryonic kidney cells |
| HeLa | Henrietta Lacks cells |
| HisRS | Histidyl-tRNA synthetase |
| HRI | Heme-reguated eIF2α kinase |
| IRE1 | Inositol-requiring enzyme 1 |
| ISRIB | Integrated stress response inhibitor |
| L-LTP | Late long-term potentiation |
| LTD | Long-term depression |
| LTP | Long-term potentiation |
| MAPK | Mitogen-activated protein kinase |
| Met-tRNAi | Initiator methionyl-tRNA |
| mGluR | Metabotropic glutamate receptor |
| mGluR1 | Metabotropic glutamate receptor 1 |
| mGluR5 | Metabotropic glutamate receptor 5 |
| mGluR-LTD | mGluR-Dependent long-term depression |
| mRNA | Messenger RNA |
| mTOR | Mammalian target of rapamycin |
| mTORC2 | Mammalian target of rapamycin complex 2 |
| N-Bak | Neuron-specific BCL2-antagonist/killer |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NMDA | N-Methyl-d-aspartate |
| NMDAR | N-Methyl-d-aspartate receptor |
| NMDAR-LTD | NMDAR-dependent long-term depression |
| nNOS | Neuronal NO-synthase |
| nt | Nucleotide |
| N-terminal | Amino-terminal |
| Ophn1 | Oligophrenin-1 |
| P58IPK | Protein kinase inhibitor P58 |
| PACT | Protein activator |
| PAR6 | Partitioning-defective 6 |
| PB1 | Phox and Bem1 |
| PDK1 | Phosphoinositide-dependent kinase-1 |
| p-eIF2α | Phosphorylated α-subunit of eucaryotic translation initiation factor 2 |
| PERK | Protein kinase R-like endoplasmic reticulum kinase |
| PERKDN | Dominant-negative PERK allele |
| PI3-kinase | Phosphoinositide 3-kinase |
| PICK1 | Protein interacting with C-kinase 1 |
| Pin1 | Peptidyl-prolyl cis-trans isomerase NIMA-interacting 1 |
| PKA | Protein kinase A |
| PKC | Protein kinase C |
| PKCζ | Protein kinase Cζ |
| PKMζ | Protein kinase Mζ |
| PKR | Protein kinase R |
| PP1c | Protein phosphatase 1c |
| PPP1R15A | Protein phosphatase regulatory subunit 15A |
| PS1 | Presenilin 1 |
| PSD | Postsynaptic density |
| PSD-95 | Postsynaptic density protein 95 |
| Rho | Ras homolog |
| RNA-seq | RNA sequencing |
| rRNA | Ribosomal RNA |
| RT-qPCR | Reverse transcription quantitative polymerase chain reaction |
| RWD | Domain in RING finger and WD repeat containing proteins and DEXDc-like helicases |
| Sal003 | Salubrinal analog 003 |
| SAPAP3 | Synapse-associated protein 90/postsynaptic density protein-95-associated protein 3 |
| Shank1 | SH3 and multiple ankyrin repeat domains 1 |
| shRNA | Small hairpin RNA |
| siHRI | Small interfering RNA that binds with HRI mRNA |
| siRNA | Small interfering RNA |
| SNP | Sodium nitroprusside |
| SS | Signal sequence |
| STAT | Signal transducer and activator of transcription |
| Tg2576 | AβPP-transgenic 2576 mice |
| TM | Transmembrane segment |
| tRNA | Transfer RNA |
| uORF | Upstream open reading frame |
| UPR | Unfolded protein response |
| UV | Ultraviolet |
| VEGF-A | Vascular endothelial growth factor A |
| XBP-1 | X-box binding protein 1 |
| ZDHHC8 | Zinc finger DHHC-type containing 8 |
| ZIP | Zeta inhibitory peptide |
References
- Bramham, C.R.; Wells, D.G. Dendritic mRNA: Transport, translation and function. Nat. Rev. Neurosci. 2007, 8, 776–789. [Google Scholar] [CrossRef] [PubMed]
- Eom, T.; Muslimov, I.A.; Tsokas, P.; Berardi, V.; Zhong, J.; Sacktor, T.C.; Tiedge, H. Neuronal BC RNAs cooperate with eIF4B to mediate activity-dependent translational control. J. Cell Biol. 2014, 207, 237–252. [Google Scholar] [CrossRef] [PubMed]
- Halliday, M.; Mallucci, G.R. Targeting the unfolded protein response in neurodegeneration: A new approach to therapy. Neuropharmacology 2014, 76 Pt A, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Wolozin, B. Physiological protein aggregation run amuck: Stress granules and the genesis of neurodegenerative disease. Discov. Med. 2014, 17, 47–52. [Google Scholar] [PubMed]
- Jackson, R.J.; Hellen, C.U.T.; Pestova, T.V. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell Biol. 2010, 11, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Ohno, M. PERK as a hub of multiple pathogenic pathways leading to memory deficits and neurodegeneration in Alzheimer’s disease. Brain Res. Bull. 2017. [Google Scholar] [CrossRef] [PubMed]
- Baird, T.D.; Wek, R.C. Eukaryotic initiation factor 2 phosphorylation and translational control in metabolism. Adv. Nutr. 2012, 3, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Taniuchi, S.; Miyake, M.; Tsugawa, K.; Oyadomari, M.; Oyadomari, S. Integrated stress response of vertebrates is regulated by four eIF2α kinases. Sci. Rep. 2016, 6, 32886. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, N.; Gorman, A.M.; Gupta, S.; Samali, A. The eIF2α kinases: Their structures and functions. Cell. Mol. Life Sci. 2013, 70, 3493–3511. [Google Scholar] [CrossRef] [PubMed]
- Raven, J.F.; Koromilas, A.E. PERK and PKR: Old kinases learn new tricks. Cell Cycle 2008, 7, 1146–1150. [Google Scholar] [CrossRef] [PubMed]
- Wek, R.C.; Jiang, H.Y.; Anthony, T.G. Coping with stress: EIF2 kinases and translational control. Biochem. Soc. Trans. 2006, 34 Pt 1, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Costa-Mattioli, M.; Gobert, D.; Harding, H.; Herdy, B.; Azzi, M.; Bruno, M.; Bidinosti, M.; Ben Mamou, C.; Marcinkiewicz, E.; Yoshida, M.; et al. Translational control of hippocampal synaptic plasticity and memory by the eIF2α kinase GCN2. Nature 2005, 436, 1166–1173. [Google Scholar] [CrossRef] [PubMed]
- Lenox, A.R.; Bhootada, Y.; Gorbatyuk, O.; Fullard, R.; Gorbatyuk, M. Unfolded protein response is activated in aged retinas. Neurosci. Lett. 2015, 609, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Ill-Raga, G.; Tajes, M.; Busquets-García, A.; Ramos-Fernández, E.; Vargas, L.M.; Bosch-Morató, M.; Guivernau, B.; Valls-Comamala, V.; Eraso-Pichot, A.; Guix, F.X.; et al. Physiological control of nitric oxide in neuronal BACE1 translation by heme-regulated eIF2α kinase HRI induces synaptogenesis. Antioxid. Redox Signal. 2015, 22, 1295–1307. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Fernández, E.; Tajes, M.; Ill-Raga, G.; Vargas, L.; Busquets-García, A.; Bosch-Morató, M.; Guivernau, B.; Valls-Comamala, V.; Gomis, M.; Grau, C.; et al. Glutamatergic stimulation induces GluN2B translation by the nitric oxide-heme-regulated eIF2α kinase in cortical neurons. Oncotarget 2016, 7, 58876–58892. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, E.; Naveau, M.; Mechulam, Y. Eukaryotic and archaeal translation initiation factor 2: A heterotrimeric tRNA carrier. FEBS Lett. 2010, 584, 405–412. [Google Scholar] [CrossRef] [PubMed]
- DuRose, J.B.; Scheuner, D.; Kaufman, R.J.; Rothblum, L.I.; Niwa, M. Phosphorylation of eukaryotic translation initiation factor 2α coordinates rRNA transcription and translation inhibition during endoplasmic reticulum stress. Mol. Cell. Biol. 2009, 29, 4295–4307. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.D.; Harding, H.P.; Ron, D. Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. J. Cell Biol. 2004, 167, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Vattem, K.M.; Wek, R.C. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc. Natl. Acad. Sci. USA 2004, 101, 11269–11274. [Google Scholar] [CrossRef] [PubMed]
- Novoa, I.; Zeng, H.; Harding, H.P.; Ron, D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2α. J. Cell Biol. 2001, 153, 1011–1022. [Google Scholar] [CrossRef] [PubMed]
- Dalet, A.; Argüello, R.J.; Combes, A.; Spinelli, L.; Jaeger, S.; Fallet, M.; Vu Manh, T.P.; Mendes, A.; Perego, J.; Reverendo, M.; et al. Protein synthesis inhibition and GADD34 control IFN-β heterogeneous expression in response to dsRNA. EMBO J. 2017, 36, 761–782. [Google Scholar] [CrossRef] [PubMed]
- Gentz, S.H.; Bertollo, C.M.; Souza-Fagundes, E.M.; da Silva, A.M. Implication of eIF2α kinase GCN2 in induction of apoptosis and endoplasmic reticulum stress-responsive genes by sodium salicylate. J. Pharm. Pharmacol. 2013, 65, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Bellato, H.M.; Hajj, G.N. Translational control by eIF2α in neurons: Beyond the stress response. Cytoskeleton 2016, 73, 551–565. [Google Scholar] [CrossRef] [PubMed]
- Trinh, M.A.; Klann, E. Translational control by eIF2α kinases in long-lasting synaptic plasticity and long-term memory. Neurobiol. Learn. Mem. 2013, 105, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Sidrauski, C.; Acosta-Alvear, D.; Khoutorsky, A.; Vedantham, P.; Hearn, B.R.; Li, H.; Gamache, K.; Gallagher, C.M.; Ang, K.K.; Wilson, C.; et al. Pharmacological brake-release of mRNA translation enhances cognitive memory. Elife 2013, 2, e00498. [Google Scholar] [CrossRef] [PubMed]
- Costa-Mattioli, M.; Gobert, D.; Stern, E.; Gamache, K.; Colina, R.; Cuello, C.; Sossin, W.; Kaufman, R.; Pelletier, J.; Rosenblum, K.; et al. eIF2α phosphorylation bidirectionally regulates the switch from short- to long-term synaptic plasticity and memory. Cell 2007, 129, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Costa-Mattioli, M.; Sossin, W.S.; Klann, E.; Sonenberg, N. Translational control of long-lasting synaptic plasticity and memory. Neuron 2009, 61, 10–26. [Google Scholar] [CrossRef] [PubMed]
- Di Prisco, G.V.; Huang, W.; Buffington, S.A.; Hsu, C.C.; Bonnen, P.E.; Placzek, A.N.; Sidrauski, C.; Krnjević, K.; Kaufman, R.J.; Walter, P.; et al. Translational control of mGluR-dependent long-term depression and object-place learning by eIF2α. Nat. Neurosci. 2014, 17, 1073–1082. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Belforte, J.E.; Lu, Y.; Yabe, Y.; Pickel, J.; Smith, C.B.; Je, H.S.; Lu, B.; Nakazawa, K. eIF2α phosphorylation-dependent translation in CA1 pyramidal cells impairs hippocampal memory consolidation without affecting general translation. J. Neurosci. 2010, 30, 2582–2594. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, T.; Sadleir, K.R.; Maus, E.; Velliquette, R.A.; Zhao, J.; Cole, S.L.; Eimer, W.A.; Hitt, B.; Bembinster, L.A.; Lammich, S.; et al. Phosphorylation of the translation initiation factor eIF2α increases BACE1 levels and promotes amyloidogenesis. Neuron 2008, 60, 988–1009. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Trinh, M.A.; Wexler, A.J.; Bourbon, C.; Gatti, E.; Pierre, P.; Cavener, D.R.; Klann, E. Suppression of eIF2α kinases alleviates Alzheimer’s disease-related plasticity and memory deficits. Nat. Neurosci. 2013, 16, 1299–1305. [Google Scholar] [CrossRef] [PubMed]
- Biever, A.; Boubaker-Vitre, J.; Cutando, L.; Gracia-Rubio, I.; Costa-Mattioli, M.; Puighermanal, E.; Valjent, E. Repeated exposure to D-amphetamine decreases global protein synthesis and regulates the translation of a subset of mRNAs in the striatum. Front. Mol. Neurosci. 2017, 9, 165. [Google Scholar] [CrossRef] [PubMed]
- Jousse, C.; Bruhat, A.; Carraro, V.; Urano, F.; Ferrara, M.; Ron, D.; Fafournoux, P. Inhibition of CHOP translation by a peptide encoded by an open reading frame localized in the chop 5′-UTR. Nucleic Acids Res. 2001, 29, 4341–4351. [Google Scholar] [CrossRef] [PubMed]
- Chua, J.J.; Schob, C.; Rehbein, M.; Gkogkas, C.G.; Richter, D.; Kindler, S. Synthesis of two SAPAP3 isoforms from a single mRNA is mediated via alternative translational initiation. Sci. Rep. 2012, 2, 484. [Google Scholar] [CrossRef] [PubMed]
- Studtmann, K.; Olschläger-Schütt, J.; Buck, F.; Richter, D.; Sala, C.; Bockmann, J.; Kindler, S.; Kreienkamp, H.J. A non-canonical initiation site is required for efficient translation of the dendritically localized Shank1 mRNA. PLoS ONE 2014, 9, e88518. [Google Scholar] [CrossRef] [PubMed]
- Jakobson, M.; Jakobson, M.; Llano, O.; Palgi, J.; Arumäe, U. Multiple mechanisms repress N-Bak mRNA translation in the healthy and apoptotic neurons. Cell Death Dis. 2013, 4, e777. [Google Scholar] [CrossRef] [PubMed]
- Bal, N.V.; Susorov, D.; Chesnokova, E.; Kasianov, A.; Mikhailova, T.; Alkalaeva, E.; Balaban, P.M.; Kolosov, P. Upstream open reading frames located in the leader of protein kinase Mζ mRNA regulate its translation. Front. Mol. Neurosci. 2016, 9, 103. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Hendershot, L.M. Delineation of a negative feedback regulatory loop that controls protein translation during endoplasmic reticulum stress. J. Biol. Chem. 2003, 278, 34864–34873. [Google Scholar] [CrossRef] [PubMed]
- Bartsch, D.; Ghirardi, M.; Skehel, P.A.; Karl, K.A.; Herder, S.P.; Chen, M.; Bailey, C.H.; Kandel, E.R. Aplysia CREB2 represses long-term facilitation: Relief of repression converts transient facilitation into long-term functional and structural change. Cell 1995, 83, 979–992. [Google Scholar] [CrossRef]
- Chen, A.; Muzzio, I.A.; Malleret, G.; Bartsch, D.; Verbitsky, M.; Pavlidis, P.; Yonan, A.L.; Vronskaya, S.; Grody, M.B.; Cepeda, I.; et al. Inducible enhancement of memory storage and synaptic plasticity in transgenic mice expressing an inhibitor of ATF4 (CREB-2) and C/EBP proteins. Neuron 2003, 39, 655–669. [Google Scholar] [CrossRef]
- Harding, H.P.; Novoa, I.; Zhang, Y.; Zeng, H.; Wek, R.; Schapira, M.; Ron, D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 2000, 6, 1099–1108. [Google Scholar] [CrossRef]
- Marciniak, S.J.; Yun, C.Y.; Oyadomari, S.; Novoa, I.; Zhang, Y.; Jungreis, R.; Nagata, K.; Harding, H.P.; Ron, D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004, 18, 3066–3077. [Google Scholar] [CrossRef] [PubMed]
- Sulzer, D.; Sonders, M.S.; Poulsen, N.W.; Galli, A. Mechanisms of neurotransmitter release by amphetamines: A review. Prog. Neurobiol. 2005, 75, 406–433. [Google Scholar] [CrossRef] [PubMed]
- Paschen, W.; Hayashi, T.; Saito, A.; Chan, P.H. GADD34 protein levels increase after transient ischemia in the cortex but not in the CA1 subfield: Implications for post-ischemic recovery of protein synthesis in ischemia-resistant cells. J. Neurochem. 2004, 90, 694–701. [Google Scholar] [CrossRef] [PubMed]
- Chambers, J.E.; Dalton, L.E.; Clarke, H.J.; Malzer, E.; Dominicus, C.S.; Patel, V.; Moorhead, G.; Ron, D.; Marciniak, S.J. Actin dynamics tune the integrated stress response by regulating eukaryotic initiation factor 2α dephosphorylation. Elife 2015, 4, e04872. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Rato, C.; Yan, Y.; Crespillo-Casado, A.; Clarke, H.J.; Harding, H.P.; Marciniak, S.J.; Read, R.J.; Ron, D. G-actin provides substrate-specificity to eukaryotic initiation factor 2α holophosphatases. Elife 2015, 4, e04871. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.Y.; Cevallos, R.C.; Jan, E. An upstream open reading frame regulates translation of GADD34 during cellular stresses that induce eIF2α phosphorylation. J. Biol. Chem. 2009, 284, 6661–6673. [Google Scholar] [CrossRef] [PubMed]
- Young, S.K.; Willy, J.A.; Wu, C.; Sachs, M.S.; Wek, R.C. Ribosome reinitiation directs gene-specific translation and regulates the integrated stress response. J. Biol. Chem. 2015, 290, 28257–28271. [Google Scholar] [CrossRef] [PubMed]
- Jin, K.; Mao, X.O.; Eshoo, M.W.; del Rio, G.; Rao, R.; Chen, D.; Simon, R.P.; Greenberg, D.A. cDNA microarray analysis of changes in gene expression induced by neuronal hypoxia in vitro. Neurochem. Res. 2002, 27, 1105–1112. [Google Scholar] [CrossRef] [PubMed]
- Méchaly, I.; Bourane, S.; Piquemal, D.; Al-Jumaily, M.; Ventéo, S.; Puech, S.; Scamps, F.; Valmier, J.; Carroll, P. Gene profiling during development and after a peripheral nerve traumatism reveals genes specifically induced by injury in dorsal root ganglia. Mol. Cell. Neurosci. 2006, 32, 217–229. [Google Scholar] [CrossRef] [PubMed]
- Oyadomari, S.; Mori, M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004, 11, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Yamada, M.; Tanaka, H.; Aida, K.; Tsuruma, K.; Shimazawa, M.; Hozumi, I.; Inuzuka, T.; Takahashi, H.; Hara, H. Involvement of CHOP, an ER-stress apoptotic mediator, in both human sporadic ALS and ALS model mice. Neurobiol. Dis. 2009, 36, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Papouin, T.; Ladépêche, L.; Ruel, J.; Sacchi, S.; Labasque, M.; Hanini, M.; Groc, L.; Pollegioni, L.; Mothet, J.P.; Oliet, S.H. Synaptic and extrasynaptic NMDA receptors are gated by different endogenous coagonists. Cell 2012, 150, 633–646. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Zhao, J.; Gu, Q.H.; Chen, R.Q.; Xu, Z.; Yan, J.Z.; Wang, S.H.; Liu, S.Y.; Chen, Z.; Lu, W. Distinct trafficking and expression mechanisms underlie LTP and LTD of NMDA receptor-mediated synaptic responses. Hippocampus 2010, 20, 646–658. [Google Scholar] [CrossRef] [PubMed]
- Neyman, S.; Manahan-Vaughan, D. Metabotropic glutamate receptor 1 (mGluR1) and 5 (mGluR5) regulate late phases of LTP and LTD in the hippocampal CA1 region in vitro. Eur. J. Neurosci. 2008, 27, 1345–1352. [Google Scholar] [CrossRef] [PubMed]
- Huber, K.M.; Kayser, M.S.; Bear, M.F. Role for rapid dendritic protein synthesis in hippocampal mGluR-dependent long-term depression. Science 2000, 288, 1254–1257. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.J.; Huang, W.; Kalikulov, D.; Yoo, J.W.; Placzek, A.N.; Stoica, L.; Zhou, H.; Bell, J.C.; Friedlander, M.J.; Krnjević, K.; et al. Suppression of PKR promotes network excitability and enhanced cognition by interferon-γ-mediated disinhibition. Cell 2011, 147, 1384–1396. [Google Scholar] [CrossRef] [PubMed]
- Nadif Kasri, N.; Nakano-Kobayashi, A.; Malinow, R.; Li, B.; Van Aelst, L. The Rho-linked mental retardation protein oligophrenin-1 controls synapse maturation and plasticity by stabilizing AMPA receptors. Genes Dev. 2009, 23, 1289–1302. [Google Scholar] [CrossRef] [PubMed]
- Welch, J.M.; Lu, J.; Rodriguiz, R.M.; Trotta, N.C.; Peca, J.; Ding, J.D.; Feliciano, C.; Chen, M.; Adams, J.P.; Luo, J.; et al. Cortico-striatal synaptic defects and OCD-like behaviours in Sapap3-mutant mice. Nature 2007, 448, 894–900. [Google Scholar] [CrossRef] [PubMed]
- Züchner, S.; Wendland, J.R.; Ashley-Koch, A.E.; Collins, A.L.; Tran-Viet, K.N.; Quinn, K.; Timpano, K.C.; Cuccaro, M.L.; Pericak-Vance, M.A.; Steffens, D.C.; et al. Multiple rare SAPAP3 missense variants in trichotillomania and OCD. Mol. Psychiatry 2009, 14, 6–9. [Google Scholar] [CrossRef] [PubMed]
- Jakobson, M.; Lintulahti, A.; Arumäe, U. mRNA for N-Bak, a neuron-specific BH3-only splice isoform of Bak, escapes nonsense-mediated decay and is translationally repressed in the neurons. Cell Death Dis. 2012, 3, e269. [Google Scholar] [CrossRef] [PubMed]
- Uo, T.; Kinoshita, Y.; Morrison, R.S. Neurons exclusively express N-Bak, a BH3 domain-only Bak isoform that promotes neuronal apoptosis. J. Biol. Chem. 2005, 280, 9065–9073. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C.; Oh, N.; Cho, H.; Choe, J.; Kim, Y.K. Nonsense-mediated translational repression involves exon junction complex downstream of premature translation termination codon. FEBS Lett. 2010, 584, 795–800. [Google Scholar] [CrossRef] [PubMed]
- Newton, A.C. Protein kinase C: Structural and spatial regulation by phosphorylation, cofactors, and macromolecular interactions. Chem. Rev. 2001, 101, 2353–2364. [Google Scholar] [CrossRef] [PubMed]
- Pu, Y.; Peach, M.L.; Garfield, S.H.; Wincovitch, S.; Marquez, V.E.; Blumberg, P.M. Effects on ligand interaction and membrane translocation of the positively charged arginine residues situated along the C1 domain binding cleft in the atypical protein kinase C isoforms. J. Biol. Chem. 2006, 281, 33773–33788. [Google Scholar] [CrossRef] [PubMed]
- Tobias, I.S.; Kaulich, M.; Kim, P.K.; Simon, N.; Jacinto, E.; Dowdy, S.F.; King, C.C.; Newton, A.C. Protein kinase Cζ exhibits constitutive phosphorylation and phosphatidylinositol-3,4,5-triphosphate-independent regulation. Biochem. J. 2016, 473, 509–523. [Google Scholar] [CrossRef] [PubMed]
- Graybill, C.; Wee, B.; Atwood, S.X.; Prehoda, K.E. Partitioning-defective protein 6 (Par-6) activates atypical protein kinase C (aPKC) by pseudosubstrate displacement. J. Biol. Chem. 2012, 287, 21003–21011. [Google Scholar] [CrossRef] [PubMed]
- Tsai, L.C.; Xie, L.; Dore, K.; Xie, L.; Del Rio, J.C.; King, C.C.; Martinez-Ariza, G.; Hulme, C.; Malinow, R.; Bourne, P.E.; et al. Zeta inhibitory peptide disrupts electrostatic interactions that maintain atypical protein kinase C in its active conformation on the scaffold p62. J. Biol. Chem. 2015, 290, 21845–21856. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, A.I.; Blace, N.; Crary, J.F.; Serrano, P.A.; Leitges, M.; Libien, J.M.; Weinstein, G.; Tcherapanov, A.; Sacktor, T.C. Protein kinase Mz synthesis from a brain mRNA encoding an independent protein kinase Cz catalytic domain. Implications for the molecular mechanism of memory. J. Biol. Chem. 2003, 278, 40305–40316. [Google Scholar] [CrossRef] [PubMed]
- Muslimov, I.A.; Nimmrich, V.; Hernandez, A.I.; Tcherepanov, A.; Sacktor, T.C.; Tiedge, H. Dendritic transport and localization of protein kinase Mzeta mRNA: Implications for molecular memory consolidation. J. Biol. Chem. 2004, 279, 52613–52622. [Google Scholar] [CrossRef] [PubMed]
- Hernández, A.I.; Oxberry, W.C.; Crary, J.F.; Mirra, S.S.; Sacktor, T.C. Cellular and subcellular localization of PKMζ. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2013, 369, 20130140. [Google Scholar] [CrossRef] [PubMed]
- Pastalkova, E.; Serrano, P.; Pinkhasova, D.; Wallace, E.; Fenton, A.A.; Sacktor, T.C. Storage of spatial information by the maintenance mechanism of LTP. Science 2006, 313, 1141–1144. [Google Scholar] [CrossRef] [PubMed]
- Shema, R.; Sacktor, T.C.; Dudai, Y. Rapid erasure of long-term memory associations in the cortex by an inhibitor of PKM zeta. Science 2007, 317, 951–953. [Google Scholar] [CrossRef] [PubMed]
- Serrano, P.; Friedman, E.L.; Kenney, J.; Taubenfeld, S.M.; Zimmerman, J.M.; Hanna, J.; Alberini, C.; Kelley, A.E.; Maren, S.; Rudy, J.W.; et al. PKMzeta maintains spatial, instrumental, and classically conditioned long-term memories. PLoS Biol. 2008, 6, e318. [Google Scholar] [CrossRef] [PubMed]
- Von Kraus, L.M.; Sacktor, T.C.; Francis, J.T. Erasing sensorimotor memories via PKMzeta inhibition. PLoS ONE 2010, 5, e11125. [Google Scholar] [CrossRef] [PubMed]
- Sacktor, T.C. Memory maintenance by PKMζ—An evolutionary perspective. Mol. Brain 2012, 5, 31. [Google Scholar] [CrossRef] [PubMed]
- Borodinova, A.A.; Zuzina, A.B.; Balaban, P.M. Role of atypical protein kinases in maintenance of long-term memory and synaptic plasticity. Biochemistry 2017, 82, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Balaban, P.M. Molecular mechanisms of memory modifications. Zhurnal Vysshei Nervnoi Deyatelnosti Imeni I.P. Pavlova 2017, 67, 131–140. [Google Scholar] [CrossRef]
- Sacktor, T.C.; Osten, P.; Valsamis, H.; Jiang, X.; Naik, M.U.; Sublette, E. Persistent activation of the zeta isoform of protein kinase C in the maintenance of long-term potentiation. Proc. Natl. Acad. Sci. USA 1993, 90, 8342–8346. [Google Scholar] [CrossRef] [PubMed]
- Osten, P.; Valsamis, L.; Harris, A.; Sacktor, T.C. Protein synthesis-dependent formation of protein kinase Mzeta in long-term potentiation. J. Neurosci. 1996, 16, 2444–2451. [Google Scholar] [PubMed]
- Kelly, M.T.; Crary, J.F.; Sacktor, T.C. Regulation of protein kinase Mz synthesis by multiple kinases in long-term potentiation. J. Neurosci. 2007, 27, 3439–3444. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.; Tsokas, P.; Serrano, P.; Hernández, A.I.; Tian, D.; Cottrell, J.E.; Shouval, H.Z.; Fenton, A.A.; Sacktor, T.C. Persistent increased PKMζ in long-term and remote spatial memory. Neurobiol. Learn. Mem. 2017, 138, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Balaban, P.M.; Roshchin, M.; Timoshenko, A.K.; Zuzina, A.B.; Lemak, M.; Ierusalimsky, V.N.; Aseyev, N.A.; Malyshev, A.Y. Homolog of protein kinase Mz maintains context aversive memory and underlying long-term facilitation in terrestrial snail Helix. Front. Cell. Neurosci. 2015, 9, 222. [Google Scholar] [CrossRef] [PubMed]
- Ling, D.S.; Benardo, L.S.; Serrano, P.A.; Blace, N.; Kelly, M.T.; Crary, J.F.; Sacktor, T.C. Protein kinase Mzeta is necessary and sufficient for LTP maintenance. Nat. Neurosci. 2002, 5, 295–296. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Han, H.; Li, H.; Bai, Y.; Wang, W.; Tu, M.; Peng, Y.; Zhou, L.; He, W.; Wu, X.; et al. Long-term potentiation decay and memory loss are mediated by AMPAR endocytosis. J. Clin. Investig. 2015, 125, 234–247. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Sheng, T.; Ren, S.; Tian, T.; Lu, W. Distinct roles of PKCι/λ and PKMζ in the initiation and maintenance of hippocampal long-term potentiation and memory. Cell Rep. 2016, 16, 1954–1961. [Google Scholar] [CrossRef] [PubMed]
- Drier, E.A.; Tello, M.K.; Cowan, M.; Wu, P.; Blace, N.; Sacktor, T.C.; Yin, J.C. Memory enhancement and formation by atypical PKM activity in Drosophila melanogaster. Nat. Neurosci. 2002, 5, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Shema, R.; Haramati, S.; Ron, S.; Hazvi, S.; Chen, A.; Sacktor, T.C.; Dudai, Y. Enhancement of consolidated long-term memory by overexpression of protein kinase Mzeta in the neocortex. Science 2011, 331, 1207–1210. [Google Scholar] [CrossRef] [PubMed]
- Schuette, S.R.; Fernández-Fernández, D.; Lamla, T.; Rosenbrock, H.; Hobson, S. Overexpression of protein kinase Mζ in the hippocampus enhances long-term potentiation and long-term contextual but not cued fear memory in rats. J. Neurosci. 2016, 36, 4313–4324. [Google Scholar] [CrossRef] [PubMed]
- Ko, H.G.; Kim, J.I.; Sim, S.E.; Kim, T.; Yoo, J.; Choi, S.L.; Baek, S.H.; Yu, W.J.; Yoon, J.B.; Sacktor, T.C.; et al. The role of nuclear PKMζ in memory maintenance. Neurobiol. Learn. Mem. 2016, 135, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.; Pestova, T.V.; Hellen, C.U.; Tiedge, H. Translational control by a small RNA: Dendritic BC1 RNA targets the eukaryotic initiation factor 4A helicase mechanism. Mol. Cell. Biol. 2008, 28, 3008–3019. [Google Scholar] [CrossRef] [PubMed]
- Westmark, P.R.; Westmark, C.J.; Wang, S.; Levenson, J.; O’Riordan, K.J.; Burger, C.; Malter, J.S. Pin1 and PKMzeta sequentially control dendritic protein synthesis. Sci. Signal. 2010, 3, ra18. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Kelly, M.T.; Sajikumar, S.; Serrano, P.; Tian, D.; Bergold, P.J.; Frey, J.U.; Sacktor, T.C. PKM zeta maintains late long-term potentiation by N-ethylmaleimide-sensitive factor/GluR2-dependent trafficking of postsynaptic AMPA receptors. J. Neurosci. 2008, 28, 7820–7827. [Google Scholar] [CrossRef] [PubMed]
- Adzovic, L.; Domenici, L. Insulin induces phosphorylation of the AMPA receptor subunit GluR1, reversed by ZIP, and over-expression of Protein Kinase M zeta, reversed by amyloid beta. J. Neurochem. 2014, 131, 582–587. [Google Scholar] [CrossRef]
- Santerre, J.L.; Rogow, J.A.; Kolitz, E.B.; Pal, R.; Landin, J.D.; Gigante, E.D.; Werner, D.F. Ethanol dose-dependently elicits opposing regulatory effects on hippocampal AMPA receptor GluA2 subunits through a zeta inhibitory peptide-sensitive kinase in adolescent and adult Sprague-Dawley rats. Neuroscience 2014, 280, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Shabashov, D.; Shohami, E.; Yaka, R. Inactivation of PKMζ in the NAc shell abolished cocaine-conditioned reward. J. Mol. Neurosci. 2012, 47, 546–553. [Google Scholar] [CrossRef] [PubMed]
- Yoshii, A.; Murata, Y.; Kim, J.; Zhang, C.; Shokat, K.M.; Constantine-Paton, M. TrkB and protein kinase Mζ regulate synaptic localization of PSD-95 in developing cortex. J. Neurosci. 2011, 31, 11894–11904. [Google Scholar] [CrossRef] [PubMed]
- Leseux, L.; Laurent, G.; Laurent, C.; Rigo, M.; Blanc, A.; Olive, D.; Bezombes, C. PKC zeta mTOR pathway: A new target for rituximab therapy in follicular lymphoma. Blood 2008, 111, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Velazquez-Garcia, S.; Valle, S.; Rosa, T.C.; Takane, K.K.; Demirci, C.; Alvarez-Perez, J.C.; Mellado-Gil, J.M.; Ernst, S.; Scott, D.K.; Vasavada, R.C.; et al. Activation of protein kinase C-ζ in pancreatic β-cells in vivo improves glucose tolerance and induces β-cell expansion via mTOR activation. Diabetes 2011, 60, 2546–2559. [Google Scholar] [CrossRef] [PubMed]
- Pöyry, T.A.A.; Kaminski, A.; Jackson, R.J. What determines whether mammalian ribosomes resume scanning after translation of a short upstream open reading frame? Genes Dev. 2004, 18, 62–75. [Google Scholar] [CrossRef] [PubMed]
- Carrara, M.; Sigurdardottir, A.; Bertolotti, A. Decoding the selectivity of eIF2α holophosphatases and PPP1R15A inhibitors. Nat. Struct. Mol. Biol. 2017, 24, 708–716. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Protein Name | Protein Function | The Number of uORFs and Presence of Other Translation-Impeding Features in mRNA 5′-UTR | eIF2α Kinases that May be Involved in mRNA Translation Regulation |
|---|---|---|---|
| Activating transcription factor 4 (ATF4), also known as cAMP-response element binding protein 2 (CREB-2) | transcription repressor | 2 uORFs, uORF2 overlaps with the main frame | General control nonderepressible 2 (GCN2) [12,31] PKR-like endoplasmic reticulum kinase (PERK) [29,31] |
| Growth arrest and DNA damage-inducible protein (GADD34), also known as protein phosphatase regulatory subunit 15A (PPP1R15A) | regulatory subunit of p-eIF2α phosphatase, activated by DNA lesion | 2 uORFs | GCN2 and PERK [20] *, Protein kinase R (PKR) [21] * |
| CCAAT-enhancer-binding protein homologous protein (CHOP), also known as DNA damage-inducible transcript 3 (DDIT3) | transcription repressor, activated by DNA lesion | 3 uORFs; the short peptide translated from uORF2 is important for the repression of the main frame translation [33] | Presumably not PERK [31] GCN2 [22] * |
| Beta-site APP-cleaving enzyme 1 (BACE1) | protease, most known for its role in β-amyloid production | 4 uORFs, complicated secondary structure | PERK [30] Heme-regulated eIF2α kinase (HRI) [14] |
| Glutamate ionotropic receptor NMDA type subunit 2B (GluN2B) | ionotropic glutamate receptor subunit | 3 uORFs | HRI [15] |
| Oligophrenin-1 | Rho-GTPase-activating protein | 2 uORFs | PKR [28] |
| Synapse-Associated Protein 90/Postsynaptic Density Protein-95-Associated Protein 3 (SAPAP3) | scaffolding protein of the postsynaptic density | 4 uORFs, high GC content; uORF2 is also important for translation start shift between 2 distinct start codons in protein-coding frame [34] | No data |
| SH3 and multiple ankyrin repeat domains 1 (Shank1) | scaffolding protein of the postsynaptic density | 3 conventional uORFs (uORF3 overlaps with the main frame) and the fourth unique uORF that starts with non-canonical start codon ACG and upregulates main frame translation | eIF2α phosphorylation is not important for regulation of this mRNA translation [35] |
| Neuron-specific BCL2-antagonist/killer (N-Bak) | pro-apoptotic factor | 1 or 2 uORFs in different species; also, 3′-UTR of N-Bak contains premature termination codon and exon–exon junction | eIF2α phosphorylation is not important for regulation of this mRNA translation; it was demonstrated that N-Bak mRNA translation is repressed consistently, even during apoptosis [36] |
| Protein kinase Mζ (PKMζ) | kinase, known for its importance in memory formation | 7 uORFs (unusually many, supposedly each of them contributes to the translation repression) [37] | PERK [31] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chesnokova, E.; Bal, N.; Kolosov, P. Kinases of eIF2a Switch Translation of mRNA Subset during Neuronal Plasticity. Int. J. Mol. Sci. 2017, 18, 2213. https://doi.org/10.3390/ijms18102213
Chesnokova E, Bal N, Kolosov P. Kinases of eIF2a Switch Translation of mRNA Subset during Neuronal Plasticity. International Journal of Molecular Sciences. 2017; 18(10):2213. https://doi.org/10.3390/ijms18102213
Chicago/Turabian StyleChesnokova, Ekaterina, Natalia Bal, and Peter Kolosov. 2017. "Kinases of eIF2a Switch Translation of mRNA Subset during Neuronal Plasticity" International Journal of Molecular Sciences 18, no. 10: 2213. https://doi.org/10.3390/ijms18102213
APA StyleChesnokova, E., Bal, N., & Kolosov, P. (2017). Kinases of eIF2a Switch Translation of mRNA Subset during Neuronal Plasticity. International Journal of Molecular Sciences, 18(10), 2213. https://doi.org/10.3390/ijms18102213