Diabetes-Induced Dysfunction of Mitochondria and Stem Cells in Skeletal Muscle and the Nervous System

{kind=link}
{kind=link}
Abstract
1. Introduction
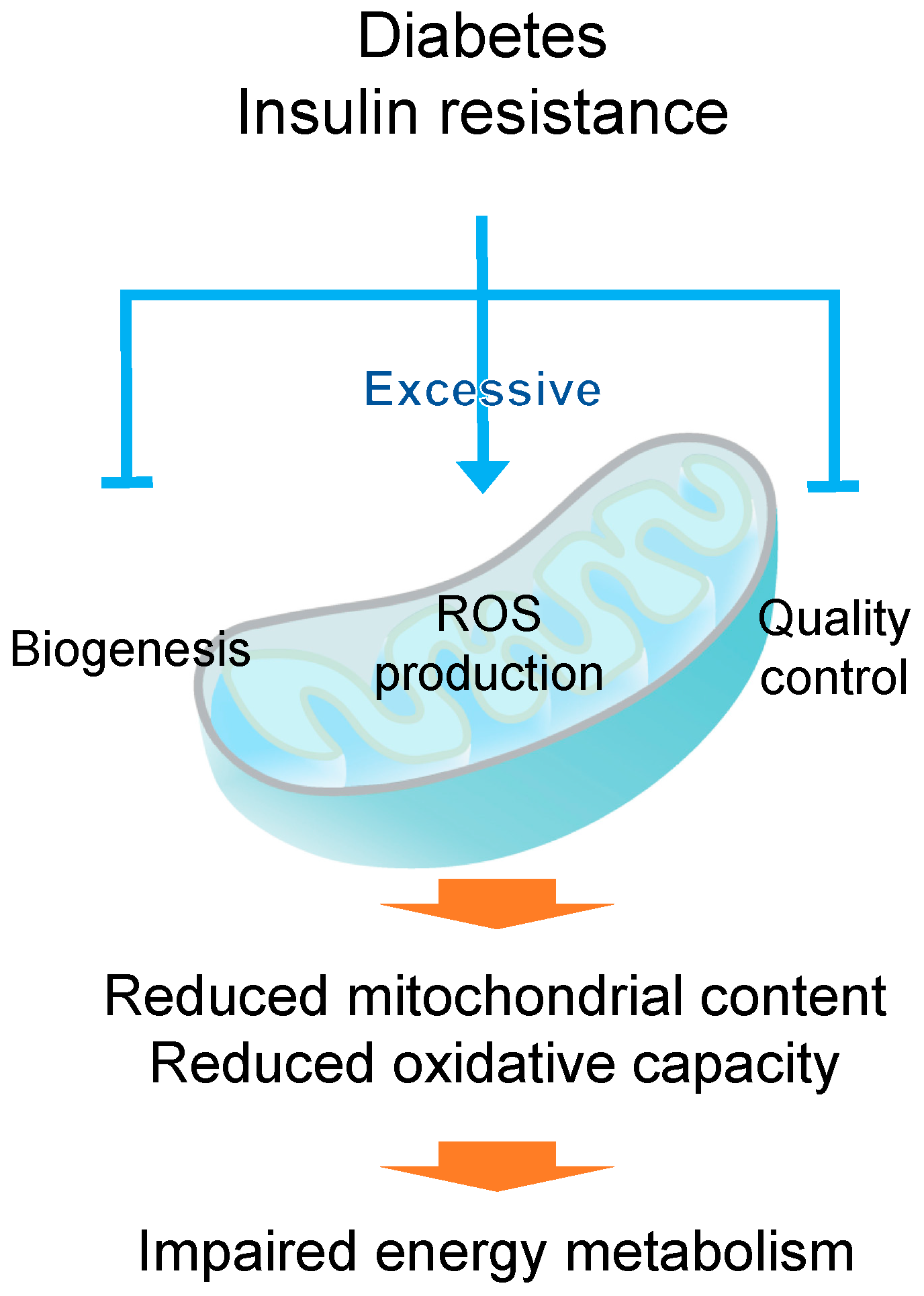
2. Mitochondrial Dysfunction in Diabetes
2.1. Mitochondrial Content and Dynamics in Diabetic Muscle
2.2. Mitochondrial Reactive Oxygen Species (ROS) Production in Diabetic Muscle
2.3. Alteration of Mitochondria in Neural Tissues
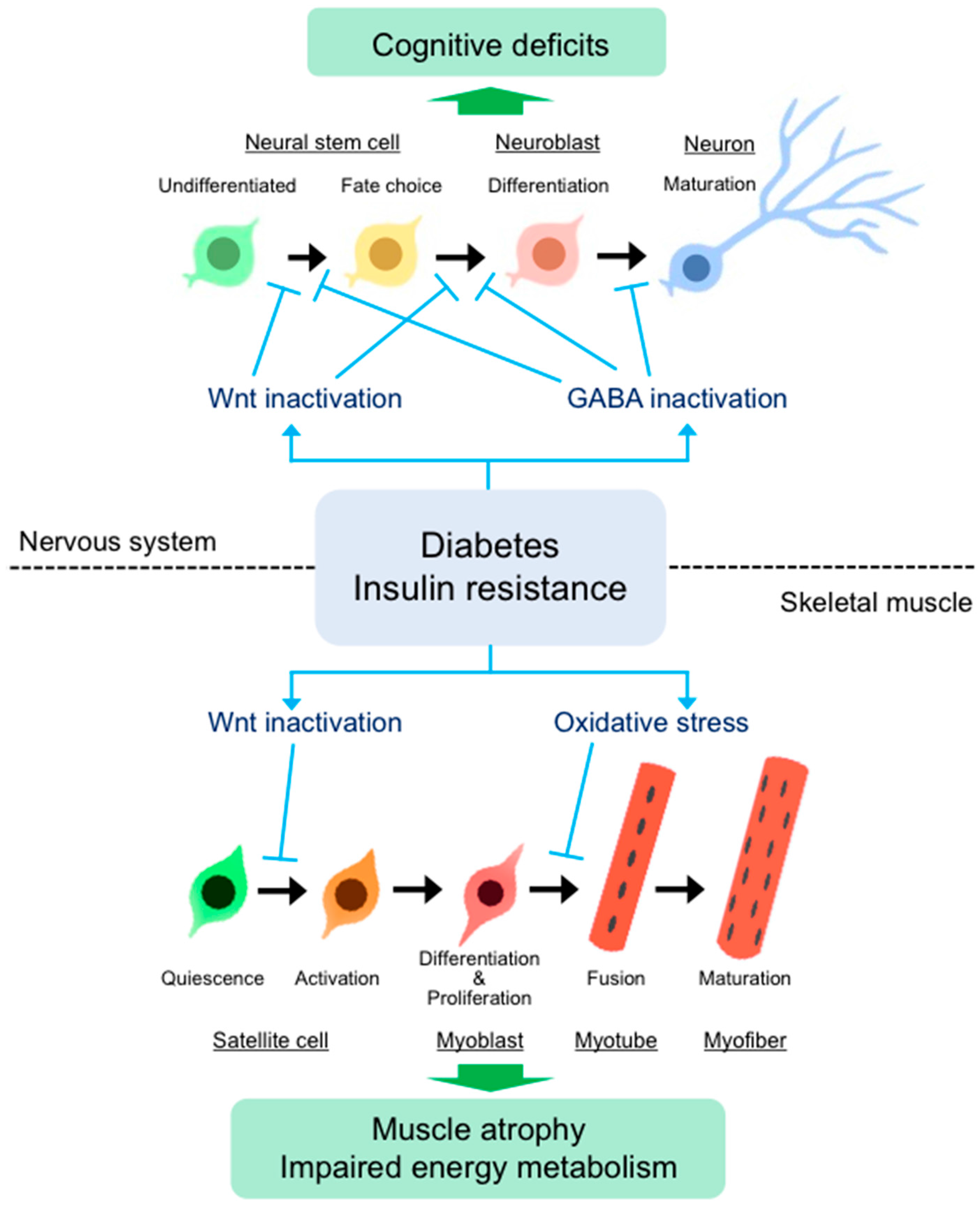
3. Alteration of Stem Cell Function in Diabetes
3.1. Impairment of Muscle Stem Cell Function in Diabetes
3.2. Impairment of Neural Stem Cell Function in Diabetes
4. Mitochondrial Function in Stem Cell Differentiation
5. Preventive Effects of Physical Exercise on Diabetic Alterations
5.1. Response of Mitochondria in Diabetic Muscle to Exercise
5.2. Effects of Exercise on Muscle Stem Cell Function
5.3. Effects of Exercise on Adult Neurogenesis
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care 2011, 34, S62–S69. [Google Scholar]
- Jiang, A.T.; Rowe, N.; Sener, A.; Luke, P. Simultaneous pancreas-kidney transplantation: The role in the treatment of type 1 diabetes and end-stage renal disease. Can. Urol. Assoc. J. 2014, 8, 135–138. [Google Scholar] [CrossRef] [PubMed]
- Groop, L.C.; Eriksson, J.G. The etiology and pathogenesis of non-insulin-dependent diabetes. Ann. Med. 1992, 24, 483–489. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, D.M.; Al-Sajee, D.; Hawke, T.J. Diabetic myopathy: Impact of diabetes mellitus on skeletal muscle progenitor cells. Front. Physiol. 2013, 4, 379. [Google Scholar] [CrossRef] [PubMed]
- Gispen, W.H.; Biessels, G.J. Cognition and synaptic plasticity in diabetes mellitus. Trends Neurosci. 2000, 23, 542–549. [Google Scholar] [CrossRef]
- Sexton, W.L.; Poole, D.C.; Mathieu-Costello, O. Microcirculatory structure-function relationships in skeletal muscle of diabetic rats. Am. J. Physiol. 1994, 266, H1502–H1511. [Google Scholar] [PubMed]
- Andersen, H.; Gadeberg, P.C.; Brock, B.; Jakobsen, J. Muscular atrophy in diabetic neuropathy: A stereological magnetic resonance imaging study. Diabetologia 1997, 40, 1062–1069. [Google Scholar] [CrossRef] [PubMed]
- Kamei, Y.; Miura, S.; Suzuki, M.; Kai, Y.; Mizukami, J.; Taniguchi, T.; Mochida, K.; Hata, T.; Matsuda, J.; Aburatani, H.; et al. Skeletal muscle foxo1 (FKHR) transgenic mice have less skeletal muscle mass, down-regulated type I (slow twitch/red muscle) fiber genes, and impaired glycemic control. J. Biol. Chem. 2004, 279, 41114–41123. [Google Scholar] [CrossRef] [PubMed]
- Hickey, M.S.; Carey, J.O.; Azevedo, J.L.; Houmard, J.A.; Pories, W.J.; Israel, R.G.; Dohm, G.L. Skeletal muscle fiber composition is related to adiposity and in vitro glucose transport rate in humans. Am. J. Phys. 1995, 268, E453–E457. [Google Scholar]
- Oberbach, A.; Bossenz, Y.; Lehmann, S.; Niebauer, J.; Adams, V.; Paschke, R.; Schon, M.R.; Bluher, M.; Punkt, K. Altered fiber distribution and fiber-specific glycolytic and oxidative enzyme activity in skeletal muscle of patients with type 2 diabetes. Diabetes Care 2006, 29, 895–900. [Google Scholar] [CrossRef] [PubMed]
- Greco, A.V.; Tataranni, P.A.; Mingrone, G.; de Gaetano, A.; Manto, A.; Cotroneo, P.; Ghirlanda, G. Daily energy metabolism in patients with type 1 diabetes mellitus. J. Am. Coll. Nutr. 1995, 14, 286–291. [Google Scholar] [CrossRef] [PubMed]
- Brandenburg, S.L.; Reusch, J.E.; Bauer, T.A.; Jeffers, B.W.; Hiatt, W.R.; Regensteiner, J.G. Effects of exercise training on oxygen uptake kinetic responses in women with type 2 diabetes. Diabetes Care 1999, 22, 1640–1646. [Google Scholar] [CrossRef] [PubMed]
- Regensteiner, J.G.; Sippel, J.; McFarling, E.T.; Wolfel, E.E.; Hiatt, W.R. Effects of non-insulin-dependent diabetes on oxygen consumption during treadmill exercise. Med. Sci. Sports Exerc. 1995, 27, 875–881. [Google Scholar] [CrossRef] [PubMed]
- Biessels, G.J.; Gispen, W.H. The impact of diabetes on cognition: What can be learned from rodent models? Neurobiol. Aging 2005, 26 (Suppl. 1), 36–41. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.J.; Freedland, K.E.; Clouse, R.E.; Lustman, P.J. The prevalence of comorbid depression in adults with diabetes: A meta-analysis. Diabetes Care 2001, 24, 1069–1078. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.D.; Katon, W.J.; Lovato, L.C.; Miller, M.E.; Murray, A.M.; Horowitz, K.R.; Bryan, R.N.; Gerstein, H.C.; Marcovina, S.; Akpunonu, B.E.; et al. Association of depression with accelerated cognitive decline among patients with type 2 diabetes in the accord-mind trial. JAMA Psychiatry 2013, 70, 1041–1047. [Google Scholar] [CrossRef] [PubMed]
- Leibson, C.L.; Rocca, W.A.; Hanson, V.A.; Cha, R.; Kokmen, E.; O’Brien, P.C.; Palumbo, P.J. The risk of dementia among persons with diabetes mellitus: A population-based cohort study. Ann. N. Y. Acad. Sci. 1997, 826, 422–427. [Google Scholar] [CrossRef] [PubMed]
- Ott, A.; Stolk, R.P.; van Harskamp, F.; Pols, H.A.; Hofman, A.; Breteler, M.M. Diabetes mellitus and the risk of dementia: The rotterdam study. Neurology 1999, 53, 1937–1942. [Google Scholar] [CrossRef] [PubMed]
- Gasparini, L.; Netzer, W.J.; Greengard, P.; Xu, H. Does insulin dysfunction play a role in Alzheimer’s disease? Trends Pharmacol. Sci. 2002, 23, 288–293. [Google Scholar] [CrossRef]
- Araki, Y.; Nomura, M.; Tanaka, H.; Yamamoto, H.; Yamamoto, T.; Tsukaguchi, I.; Nakamura, H. MRI of the brain in diabetes mellitus. Neuroradiology 1994, 36, 101–103. [Google Scholar] [CrossRef] [PubMed]
- Den Heijer, T.; Vermeer, S.E.; van Dijk, E.J.; Prins, N.D.; Koudstaal, P.J.; Hofman, A.; Breteler, M.M. Type 2 diabetes and atrophy of medial temporal lobe structures on brain MRI. Diabetologia 2003, 46, 1604–1610. [Google Scholar] [CrossRef] [PubMed]
- Boushel, R.; Gnaiger, E.; Schjerling, P.; Skovbro, M.; Kraunsoe, R.; Dela, F. Patients with type 2 diabetes have normal mitochondrial function in skeletal muscle. Diabetologia 2007, 50, 790–796. [Google Scholar] [CrossRef] [PubMed]
- Patti, M.E.; Butte, A.J.; Crunkhorn, S.; Cusi, K.; Berria, R.; Kashyap, S.; Miyazaki, Y.; Kohane, I.; Costello, M.; Saccone, R.; et al. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc. Natl. Acad. Sci. USA 2003, 100, 8466–8471. [Google Scholar] [CrossRef] [PubMed]
- Davidsen, P.K.; Gallagher, I.J.; Hartman, J.W.; Tarnopolsky, M.A.; Dela, F.; Helge, J.W.; Timmons, J.A.; Phillips, S.M. High responders to resistance exercise training demonstrate differential regulation of skeletal muscle microrna expression. J. Appl. Physiol. 2011, 110, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Baur, J.A.; Pearson, K.J.; Price, N.L.; Jamieson, H.A.; Lerin, C.; Kalra, A.; Prabhu, V.V.; Allard, J.S.; Lopez-Lluch, G.; Lewis, K.; et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature 2006, 444, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, T.; Edelstein, D.; Du, X.L.; Yamagishi, S.; Matsumura, T.; Kaneda, Y.; Yorek, M.A.; Beebe, D.; Oates, P.J.; Hammes, H.P.; et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 2000, 404, 787–790. [Google Scholar] [PubMed]
- Nishikawa, T.; Kukidome, D.; Sonoda, K.; Fujisawa, K.; Matsuhisa, T.; Motoshima, H.; Matsumura, T.; Araki, E. Impact of mitochondrial ros production in the pathogenesis of insulin resistance. Diabetes Res. Clin. Pract. 2007, 77, S161–S164. [Google Scholar] [CrossRef] [PubMed]
- Aragno, M.; Mastrocola, R.; Catalano, M.G.; Brignardello, E.; Danni, O.; Boccuzzi, G. Oxidative stress impairs skeletal muscle repair in diabetic rats. Diabetes 2004, 53, 1082–1088. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.H.; Cheng, M.; Koh, T.J. Impaired muscle regeneration in ob/ob and db/db mice. Sci. World J. 2011, 11, 1525–1535. [Google Scholar] [CrossRef] [PubMed]
- Fujimaki, S.; Wakabayashi, T.; Asashima, M.; Takemasa, T.; Kuwabara, T. Treadmill running induces satellite cell activation in diabetic mice. Biochem. Biophys. Rep. 2016, 8, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Beauquis, J.; Roig, P.; De Nicola, A.F.; Saravia, F. Short-term environmental enrichment enhances adult neurogenesis, vascular network and dendritic complexity in the hippocampus of type 1 diabetic mice. PLoS ONE 2010, 5, e13993. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.J.; Tan, Y.F.; Yue, J.T.; Vranic, M.; Wojtowicz, J.M. Impairment of hippocampal neurogenesis in streptozotocin-treated diabetic rats. Acta Neurol. Scand. 2008, 117, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Hidaka, R.; Machida, M.; Fujimaki, S.; Terashima, K.; Asashima, M.; Kuwabara, T. Monitoring neurodegeneration in diabetes using adult neural stem cells derived from the olfactory bulb. Stem Cell Res. Ther. 2013, 4, 51. [Google Scholar] [CrossRef] [PubMed]
- Wagatsuma, A.; Kotake, N.; Yamada, S. Muscle regeneration occurs to coincide with mitochondrial biogenesis. Mol. Cell. Biochem. 2011, 349, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Nichenko, A.S.; Southern, W.M.; Atuan, M.; Luan, J.; Peissig, K.B.; Foltz, S.J.; Beedle, A.M.; Warren, G.L.; Call, J.A. Mitochondrial maintenance via autophagy contributes to functional skeletal muscle regeneration and remodeling. Am. J. Physiol. Cell Physiol. 2016, 311, C190–C200. [Google Scholar] [CrossRef] [PubMed]
- Sandiford, S.D.; Kennedy, K.A.; Xie, X.; Pickering, J.G.; Li, S.S. Dual oxidase maturation factor 1 (DUOXA1) overexpression increases reactive oxygen species production and inhibits murine muscle satellite cell differentiation. Cell Commun. Signal. 2014, 12, 5. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Yadav, A.; Tiwari, S.K.; Seth, B.; Chauhan, L.K.; Khare, P.; Ray, R.S.; Chaturvedi, R.K. Dynamin-related protein 1 inhibition mitigates bisphenol a-mediated alterations in mitochondrial dynamics and neural stem cell proliferation and differentiation. J. Biol. Chem. 2016, 291, 15923–15939. [Google Scholar] [CrossRef] [PubMed]
- Kelley, D.E.; Williams, K.V.; Price, J.C. Insulin regulation of glucose transport and phosphorylation in skeletal muscle assessed by PET. Am. J. Physiol. 1999, 277, E361–E369. [Google Scholar] [PubMed]
- Goodpaster, B.H.; Theriault, R.; Watkins, S.C.; Kelley, D.E. Intramuscular lipid content is increased in obesity and decreased by weight loss. Metabolism 2000, 49, 467–472. [Google Scholar] [CrossRef]
- Petersen, K.F.; Befroy, D.; Dufour, S.; Dziura, J.; Ariyan, C.; Rothman, D.L.; DiPietro, L.; Cline, G.W.; Shulman, G.I. Mitochondrial dysfunction in the elderly: Possible role in insulin resistance. Science 2003, 300, 1140–1142. [Google Scholar] [CrossRef] [PubMed]
- Gaster, M.; Rustan, A.C.; Aas, V.; Beck-Nielsen, H. Reduced lipid oxidation in skeletal muscle from type 2 diabetic subjects may be of genetic origin: Evidence from cultured myotubes. Diabetes 2004, 53, 542–548. [Google Scholar] [CrossRef] [PubMed]
- Hood, D.A.; Tryon, L.D.; Carter, H.N.; Kim, Y.; Chen, C.C. Unravelling the mechanisms regulating muscle mitochondrial biogenesis. Biochem. J. 2016, 473, 2295–2314. [Google Scholar] [CrossRef] [PubMed]
- Erlich, A.T.; Tryon, L.D.; Crilly, M.J.; Memme, J.M.; Moosavi, Z.S.M.; Oliveira, A.N.; Beyfuss, K.; Hood, D.A. Function of specialized regulatory proteins and signaling pathways in exercise-induced muscle mitochondrial biogenesis. Integr. Med. Res. 2016, 5, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Ploumi, C.; Daskalaki, I.; Tavernarakis, N. Mitochondrial biogenesis and clearance: A balancing act. FEBS J. 2017, 284, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Handschin, C.; Chin, S.; Li, P.; Liu, F.; Maratos-Flier, E.; Lebrasseur, N.K.; Yan, Z.; Spiegelman, B.M. Skeletal muscle fiber-type switching, exercise intolerance, and myopathy in PGC-1α muscle-specific knock-out animals. J. Biol. Chem. 2007, 282, 30014–30021. [Google Scholar] [CrossRef] [PubMed]
- Benton, C.R.; Holloway, G.P.; Han, X.X.; Yoshida, Y.; Snook, L.A.; Lally, J.; Glatz, J.F.; Luiken, J.J.; Chabowski, A.; Bonen, A. Increased levels of peroxisome proliferator-activated receptor gamma, coactivator 1 α (PGC-1α) improve lipid utilisation, insulin signalling and glucose transport in skeletal muscle of lean and insulin-resistant obese zucker rats. Diabetologia 2010, 53, 2008–2019. [Google Scholar] [CrossRef] [PubMed]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstrale, M.; Laurila, E.; et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Evans, R. PPARs and ERRs: Molecular mediators of mitochondrial metabolism. Curr. Opin. Cell Biol. 2015, 33, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, S.N.; Emter, R.; Hock, M.B.; Knutti, D.; Cardenas, J.; Podvinec, M.; Oakeley, E.J.; Kralli, A. The estrogen-related receptor α (ERRα) functions in PPARγ coactivator 1α (PGC-1α)-induced mitochondrial biogenesis. Proc. Natl. Acad. Sci. USA 2004, 101, 6472–6477. [Google Scholar] [CrossRef] [PubMed]
- Huss, J.M.; Torra, I.P.; Staels, B.; Giguere, V.; Kelly, D.P. Estrogen-related receptor α directs peroxisome proliferator-activated receptor α signaling in the transcriptional control of energy metabolism in cardiac and skeletal muscle. Mol. Cell. Biol. 2004, 24, 9079–9091. [Google Scholar] [CrossRef] [PubMed]
- LaBarge, S.; McDonald, M.; Smith-Powell, L.; Auwerx, J.; Huss, J.M. Estrogen-related receptor-α (ERRα) deficiency in skeletal muscle impairs regeneration in response to injury. FASEB J. 2014, 28, 1082–1097. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.X.; Zhang, C.L.; Yu, R.T.; Cho, H.K.; Nelson, M.C.; Bayuga-Ocampo, C.R.; Ham, J.; Kang, H.; Evans, R.M. Regulation of muscle fiber type and running endurance by PPARδ. PLoS Biol. 2004, 2, e294. [Google Scholar] [CrossRef] [PubMed]
- Schuler, M.; Ali, F.; Chambon, C.; Duteil, D.; Bornert, J.M.; Tardivel, A.; Desvergne, B.; Wahli, W.; Chambon, P.; Metzger, D. PGC1α expression is controlled in skeletal muscles by PPARβ, whose ablation results in fiber-type switching, obesity, and type 2 diabetes. Cell Metab. 2006, 4, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Mensink, M.; Hesselink, M.K.; Russell, A.P.; Schaart, G.; Sels, J.P.; Schrauwen, P. Improved skeletal muscle oxidative enzyme activity and restoration of PGC-1α and PPARβ/δ gene expression upon rosiglitazone treatment in obese patients with type 2 diabetes mellitus. Int. J. Obes. 2007, 31, 1302–1310. [Google Scholar] [CrossRef] [PubMed]
- Seo, A.Y.; Joseph, A.M.; Dutta, D.; Hwang, J.C.; Aris, J.P.; Leeuwenburgh, C. New insights into the role of mitochondria in aging: Mitochondrial dynamics and more. J. Cell Sci. 2010, 123, 2533–2542. [Google Scholar] [CrossRef] [PubMed]
- Malka, F.; Guillery, O.; Cifuentes-Diaz, C.; Guillou, E.; Belenguer, P.; Lombes, A.; Rojo, M. Separate fusion of outer and inner mitochondrial membranes. EMBO Rep. 2005, 6, 853–859. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Vermulst, M.; Wang, Y.E.; Chomyn, A.; Prolla, T.A.; McCaffery, J.M.; Chan, D.C. Mitochondrial fusion is required for mtdna stability in skeletal muscle and tolerance of mtDNA mutations. Cell 2010, 141, 280–289. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Chomyn, A.; Chan, D.C. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J. Biol. Chem. 2005, 280, 26185–26192. [Google Scholar] [CrossRef] [PubMed]
- Parone, P.A.; Da Cruz, S.; Tondera, D.; Mattenberger, Y.; James, D.I.; Maechler, P.; Barja, F.; Martinou, J.C. Preventing mitochondrial fission impairs mitochondrial function and leads to loss of mitochondrial DNA. PLoS ONE 2008, 3, e3257. [Google Scholar] [CrossRef] [PubMed]
- Malena, A.; Loro, E.; Di Re, M.; Holt, I.J.; Vergani, L. Inhibition of mitochondrial fission favours mutant over wild-type mitochondrial DNA. Hum. Mol. Genet. 2009, 18, 3407–3416. [Google Scholar] [CrossRef] [PubMed]
- Bach, D.; Naon, D.; Pich, S.; Soriano, F.X.; Vega, N.; Rieusset, J.; Laville, M.; Guillet, C.; Boirie, Y.; Wallberg-Henriksson, H.; et al. Expression of Mfn2, the charcot-marie-tooth neuropathy type 2A gene, in human skeletal muscle: Effects of type 2 diabetes, obesity, weight loss, and the regulatory role of tumor necrosis factor α and interleukin-6. Diabetes 2005, 54, 2685–2693. [Google Scholar] [CrossRef] [PubMed]
- Joseph, A.M.; Joanisse, D.R.; Baillot, R.G.; Hood, D.A. Mitochondrial dysregulation in the pathogenesis of diabetes: Potential for mitochondrial biogenesis-mediated interventions. Exp. Diabetes Res. 2012, 2012, 642038. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Jin, P.; Yu, L.; Wang, Y.; Han, L.; Shi, T.; Li, X. Impaired mitochondrial dynamics and bioenergetics in diabetic skeletal muscle. PLoS ONE 2014, 9, e92810. [Google Scholar] [CrossRef] [PubMed]
- Pereira, R.O.; Tadinada, S.M.; Zasadny, F.M.; Oliveira, K.J.; Pires, K.M.P.; Olvera, A.; Jeffers, J.; Souvenir, R.; McGlauflin, R.; Seei, A.; et al. OPA1 deficiency promotes secretion of FGF21 from muscle that prevents obesity and insulin resistance. EMBO J. 2017. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [PubMed]
- Lahera, V.; de Las Heras, N.; Lopez-Farre, A.; Manucha, W.; Ferder, L. Role of mitochondrial dysfunction in hypertension and obesity. Curr. Hypertens. Rep. 2017, 19, 11. [Google Scholar] [CrossRef] [PubMed]
- Scheele, C.; Nielsen, A.R.; Walden, T.B.; Sewell, D.A.; Fischer, C.P.; Brogan, R.J.; Petrovic, N.; Larsson, O.; Tesch, P.A.; Wennmalm, K.; et al. Altered regulation of the PINK1 locus: A link between type 2 diabetes and neurodegeneration? FASEB J. 2007, 21, 3653–3665. [Google Scholar] [CrossRef] [PubMed]
- Song, F.; Jia, W.; Yao, Y.; Hu, Y.; Lei, L.; Lin, J.; Sun, X.; Liu, L. Oxidative stress, antioxidant status and DNA damage in patients with impaired glucose regulation and newly diagnosed type 2 diabetes. Clin. Sci. 2007, 112, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Bonnard, C.; Durand, A.; Peyrol, S.; Chanseaume, E.; Chauvin, M.A.; Morio, B.; Vidal, H.; Rieusset, J. Mitochondrial dysfunction results from oxidative stress in the skeletal muscle of diet-induced insulin-resistant mice. J. Clin. Investig. 2008, 118, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Anderson, E.J.; Lustig, M.E.; Boyle, K.E.; Woodlief, T.L.; Kane, D.A.; Lin, C.T.; Price, J.W., 3rd; Kang, L.; Rabinovitch, P.S.; Szeto, H.H.; et al. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J. Clin. Investig. 2009, 119, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Hoehn, K.L.; Salmon, A.B.; Hohnen-Behrens, C.; Turner, N.; Hoy, A.J.; Maghzal, G.J.; Stocker, R.; Van Remmen, H.; Kraegen, E.W.; Cooney, G.J.; et al. Insulin resistance is a cellular antioxidant defense mechanism. Proc. Natl. Acad. Sci. USA 2009, 106, 17787–17792. [Google Scholar] [CrossRef] [PubMed]
- Moreira, P.I.; Santos, M.S.; Sena, C.; Seica, R.; Oliveira, C.R. Insulin protects against amyloid β-peptide toxicity in brain mitochondria of diabetic rats. Neurobiol. Dis. 2005, 18, 628–637. [Google Scholar] [CrossRef] [PubMed]
- Mastrocola, R.; Restivo, F.; Vercellinatto, I.; Danni, O.; Brignardello, E.; Aragno, M.; Boccuzzi, G. Oxidative and nitrosative stress in brain mitochondria of diabetic rats. J. Endocrinol. 2005, 187, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Moreira, P.I.; Santos, M.S.; Moreno, A.M.; Seica, R.; Oliveira, C.R. Increased vulnerability of brain mitochondria in diabetic (Goto-kakizaki) rats with aging and amyloid-β exposure. Diabetes 2003, 52, 1449–1456. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, C.; Santos, M.S.; Oliveira, C.R.; Moreira, P.I. Alzheimer’s disease and type 2 diabetes-related alterations in brain mitochondria, autophagy and synaptic markers. Biochim. Biophys. Acta 2015, 1852, 1665–1675. [Google Scholar] [CrossRef] [PubMed]
- Moreira, P.I.; Cardoso, S.M.; Pereira, C.M.; Santos, M.S.; Oliveira, C.R. Mitochondria as a therapeutic target in Alzheimer’s disease and diabetes. CNS Neurol. Disord. Drug Targets 2009, 8, 492–511. [Google Scholar] [CrossRef] [PubMed]
- Correia, S.C.; Santos, R.X.; Carvalho, C.; Cardoso, S.; Candeias, E.; Santos, M.S.; Oliveira, C.R.; Moreira, P.I. Insulin signaling, glucose metabolism and mitochondria: Major players in alzheimer’s disease and diabetes interrelation. Brain Res. 2012, 1441, 64–78. [Google Scholar] [CrossRef] [PubMed]
- Sims-Robinson, C.; Kim, B.; Rosko, A.; Feldman, E.L. How does diabetes accelerate Alzheimer disease pathology? Nat. Rev. Neurol. 2010, 6, 551–559. [Google Scholar] [CrossRef] [PubMed]
- Kuang, S.; Rudnicki, M.A. The emerging biology of satellite cells and their therapeutic potential. Trends Mol. Med. 2008, 14, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Charge, S.B.; Rudnicki, M.A. Cellular and molecular regulation of muscle regeneration. Physiol. Rev. 2004, 84, 209–238. [Google Scholar] [CrossRef] [PubMed]
- Zammit, P.S.; Golding, J.P.; Nagata, Y.; Hudon, V.; Partridge, T.A.; Beauchamp, J.R. Muscle satellite cells adopt divergent fates: A mechanism for self-renewal? J. Cell Biol. 2004, 166, 347–357. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, J.J.; Mula, J.; Miyazaki, M.; Erfani, R.; Garrison, K.; Farooqui, A.B.; Srikuea, R.; Lawson, B.A.; Grimes, B.; Keller, C.; et al. Effective fiber hypertrophy in satellite cell-depleted skeletal muscle. Development 2011, 138, 3657–3666. [Google Scholar] [CrossRef] [PubMed]
- Zammit, P.S.; Relaix, F.; Nagata, Y.; Ruiz, A.P.; Collins, C.A.; Partridge, T.A.; Beauchamp, J.R. Pax7 and myogenic progression in skeletal muscle satellite cells. J. Cell Sci. 2006, 119, 1824–1832. [Google Scholar] [CrossRef] [PubMed]
- Fujimaki, S.; Machida, M.; Hidaka, R.; Asashima, M.; Takemasa, T.; Kuwabara, T. Intrinsic ability of adult stem cell in skeletal muscle: An effective and replenishable resource to the establishment of pluripotent stem cells. Stem Cells Int. 2013, 2013, 420164. [Google Scholar] [CrossRef] [PubMed]
- Rampalli, S.; Li, L.; Mak, E.; Ge, K.; Brand, M.; Tapscott, S.J.; Dilworth, F.J. P38 mapk signaling regulates recruitment of Ash2l-containing methyltransferase complexes to specific genes during differentiation. Nat. Struct. Mol. Biol. 2007, 14, 1150–1156. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.; Conboy, M.J.; Conboy, I.M. Pharmacological inhibition of myostatin/TGF-β receptor/pSmad3 signaling rescues muscle regenerative responses in mouse model of type 1 diabetes. Acta Pharmacol. Sin. 2013, 34, 1052–1060. [Google Scholar] [CrossRef] [PubMed]
- Krause, M.P.; Al-Sajee, D.; D’Souza, D.M.; Rebalka, I.A.; Moradi, J.; Riddell, M.C.; Hawke, T.J. Impaired macrophage and satellite cell infiltration occurs in a muscle-specific fashion following injury in diabetic skeletal muscle. PLoS ONE 2013, 8, e70971. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, D.M.; Zhou, S.; Rebalka, I.A.; MacDonald, B.; Moradi, J.; Krause, M.P.; Al-Sajee, D.; Punthakee, Z.; Tarnopolsky, M.A.; Hawke, T.J. Decreased satellite cell number and function in humans and mice with type 1 diabetes is the result of altered notch signaling. Diabetes 2016, 65, 3053–3061. [Google Scholar] [CrossRef] [PubMed]
- Peterson, J.M.; Bryner, R.W.; Alway, S.E. Satellite cell proliferation is reduced in muscles of obese zucker rats but restored with loading. Am. J. Physiol. Cell Physiol. 2008, 295, C521–C528. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Wang, H.; Lee, I.H.; Modi, S.; Wang, X.; Du, J.; Mitch, W.E. PTEN inhibition improves muscle regeneration in mice fed a high-fat diet. Diabetes 2010, 59, 1312–1320. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Zhu, M.; Zhang, S.; Foretz, M.; Viollet, B.; Du, M. Obesity impairs skeletal muscle regeneration through inhibition of ampk. Diabetes 2016, 65, 188–200. [Google Scholar] [PubMed]
- Woo, M.; Isganaitis, E.; Cerletti, M.; Fitzpatrick, C.; Wagers, A.J.; Jimenez-Chillaron, J.; Patti, M.E. Early life nutrition modulates muscle stem cell number: Implications for muscle mass and repair. Stem Cells Dev. 2011, 20, 1763–1769. [Google Scholar] [CrossRef] [PubMed]
- Levak-Frank, S.; Radner, H.; Walsh, A.; Stollberger, R.; Knipping, G.; Hoefler, G.; Sattler, W.; Weinstock, P.H.; Breslow, J.L.; Zechner, R. Muscle-specific overexpression of lipoprotein lipase causes a severe myopathy characterized by proliferation of mitochondria and peroxisomes in transgenic mice. J. Clin. Investig. 1995, 96, 976–986. [Google Scholar] [CrossRef] [PubMed]
- Tamilarasan, K.P.; Temmel, H.; Das, S.K.; Al Zoughbi, W.; Schauer, S.; Vesely, P.W.; Hoefler, G. Skeletal muscle damage and impaired regeneration due to lpl-mediated lipotoxicity. Cell Death Dis. 2012, 3, e354. [Google Scholar] [CrossRef] [PubMed]
- Green, C.J.; Pedersen, M.; Pedersen, B.K.; Scheele, C. Elevated NF-κB activation is conserved in human myocytes cultured from obese type 2 diabetic patients and attenuated by amp-activated protein kinase. Diabetes 2011, 60, 2810–2819. [Google Scholar] [CrossRef] [PubMed]
- Gaster, M.; Petersen, I.; Hojlund, K.; Poulsen, P.; Beck-Nielsen, H. The diabetic phenotype is conserved in myotubes established from diabetic subjects: Evidence for primary defects in glucose transport and glycogen synthase activity. Diabetes 2002, 51, 921–927. [Google Scholar] [CrossRef] [PubMed]
- Scarda, A.; Franzin, C.; Milan, G.; Sanna, M.; Dal Pra, C.; Pagano, C.; Boldrin, L.; Piccoli, M.; Trevellin, E.; Granzotto, M.; et al. Increased adipogenic conversion of muscle satellite cells in obese zucker rats. Int. J. Obes. 2010, 34, 1319–1327. [Google Scholar] [CrossRef] [PubMed]
- Ardite, E.; Barbera, J.A.; Roca, J.; Fernandez-Checa, J.C. Glutathione depletion impairs myogenic differentiation of murine skeletal muscle C2C12 cells through sustained NF-κB activation. Am. J. Pathol. 2004, 165, 719–728. [Google Scholar] [CrossRef]
- Guttridge, D.C.; Albanese, C.; Reuther, J.Y.; Pestell, R.G.; Baldwin, A.S., Jr. NF-κB controls cell growth and differentiation through transcriptional regulation of cyclin d1. Mol. Cell. Biol. 1999, 19, 5785–5799. [Google Scholar] [CrossRef] [PubMed]
- Guttridge, D.C.; Mayo, M.W.; Madrid, L.V.; Wang, C.Y.; Baldwin, A.S., Jr. NF-κB-induced loss of myod messenger RNA: Possible role in muscle decay and cachexia. Science 2000, 289, 2363–2366. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.S.; Befroy, D.E.; Codella, R.; Kim, S.; Reznick, R.M.; Hwang, Y.J.; Liu, Z.X.; Lee, H.Y.; Distefano, A.; Samuel, V.T.; et al. Paradoxical effects of increased expression of PGC-1α on muscle mitochondrial function and insulin-stimulated muscle glucose metabolism. Proc. Natl. Acad. Sci. USA 2008, 105, 19926–19931. [Google Scholar] [CrossRef] [PubMed]
- Sestili, P.; Barbieri, E.; Martinelli, C.; Battistelli, M.; Guescini, M.; Vallorani, L.; Casadei, L.; D’Emilio, A.; Falcieri, E.; Piccoli, G.; et al. Creatine supplementation prevents the inhibition of myogenic differentiation in oxidatively injured C2C12 murine myoblasts. Mol. Nutr. Food Res. 2009, 53, 1187–1204. [Google Scholar] [CrossRef] [PubMed]
- Pawlikowska, P.; Gajkowska, B.; Hocquette, J.F.; Orzechowski, A. Not only insulin stimulates mitochondriogenesis in muscle cells, but mitochondria are also essential for insulin-mediated myogenesis. Cell Prolif. 2006, 39, 127–145. [Google Scholar] [CrossRef] [PubMed]
- Kopan, R.; Ilagan, M.X. The canonical notch signaling pathway: Unfolding the activation mechanism. Cell 2009, 137, 216–233. [Google Scholar] [CrossRef] [PubMed]
- Jarriault, S.; Brou, C.; Logeat, F.; Schroeter, E.H.; Kopan, R.; Israel, A. Signalling downstream of activated mammalian notch. Nature 1995, 377, 355–358. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Sakai, T.; Tamura, K.; Minoguchi, S.; Shirayoshi, Y.; Hamada, Y.; Tsujimoto, Y.; Honjo, T. Functional conservation of mouse notch receptor family members. FEBS Lett. 1996, 395, 221–224. [Google Scholar] [CrossRef]
- Clevers, H.; Nusse, R. Wnt/β-catenin signaling and disease. Cell 2012, 149, 1192–1205. [Google Scholar] [CrossRef] [PubMed]
- Sethi, J.K.; Vidal-Puig, A. Wnt signalling and the control of cellular metabolism. Biochem. J. 2010, 427, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M.; Katoh, M. Wnt signaling pathway and stem cell signaling network. Clin. Cancer Res. 2007, 13, 4042–4045. [Google Scholar] [CrossRef] [PubMed]
- Nusse, R. Wnt signaling. Cold Spring Harb. Perspect. Biol. 2012, 4, a011163. [Google Scholar] [CrossRef] [PubMed]
- Fujimaki, S.; Hidaka, R.; Asashima, M.; Takemasa, T.; Kuwabara, T. Wnt protein-mediated satellite cell conversion in adult and aged mice following voluntary wheel running. J. Biol. Chem. 2014, 289, 7399–7412. [Google Scholar] [CrossRef] [PubMed]
- Fujimaki, S.; Wakabayashi, T.; Takemasa, T.; Asashima, M.; Kuwabara, T. Diabetes and stem cell function. Biomed Res. Int. 2015, 2015, 592915. [Google Scholar] [CrossRef] [PubMed]
- Van Praag, H.; Schinder, A.F.; Christie, B.R.; Toni, N.; Palmer, T.D.; Gage, F.H. Functional neurogenesis in the adult hippocampus. Nature 2002, 415, 1030–1034. [Google Scholar] [CrossRef] [PubMed]
- Carleton, A.; Petreanu, L.T.; Lansford, R.; Alvarez-Buylla, A.; Lledo, P.M. Becoming a new neuron in the adult olfactory bulb. Nat. Neurosci. 2003, 6, 507–518. [Google Scholar] [CrossRef] [PubMed]
- Kee, N.; Teixeira, C.M.; Wang, A.H.; Frankland, P.W. Preferential incorporation of adult-generated granule cells into spatial memory networks in the dentate gyrus. Nat. Neurosci. 2007, 10, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Kempermann, G. Why new neurons? Possible functions for adult hippocampal neurogenesis. J. Neurosci. Off. J. Soc. Neurosci. 2002, 22, 635–638. [Google Scholar]
- Gheusi, G.; Cremer, H.; McLean, H.; Chazal, G.; Vincent, J.D.; Lledo, P.M. Importance of newly generated neurons in the adult olfactory bulb for odor discrimination. Proc. Natl. Acad. Sci. USA 2000, 97, 1823–1828. [Google Scholar] [CrossRef] [PubMed]
- Lie, D.C.; Colamarino, S.A.; Song, H.J.; Desire, L.; Mira, H.; Consiglio, A.; Lein, E.S.; Jessberger, S.; Lansford, H.; Dearie, A.R.; et al. Wnt signalling regulates adult hippocampal neurogenesis. Nature 2005, 437, 1370–1375. [Google Scholar] [CrossRef] [PubMed]
- Wexler, E.M.; Paucer, A.; Kornblum, H.I.; Palmer, T.D.; Geschwind, D.H. Endogenous Wnt signaling maintains neural progenitor cell potency. Stem Cells 2009, 27, 1130–1141. [Google Scholar] [CrossRef] [PubMed]
- Kuwabara, T.; Hsieh, J.; Muotri, A.; Yeo, G.; Warashina, M.; Lie, D.C.; Moore, L.; Nakashima, K.; Asashima, M.; Gage, F.H. Wnt-mediated activation of Neurod1 and retro-elements during adult neurogenesis. Nat. Neurosci. 2009, 12, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Ure, K.; Ables, J.L.; Lagace, D.C.; Nave, K.A.; Goebbels, S.; Eisch, A.J.; Hsieh, J. Neurod1 is essential for the survival and maturation of adult-born neurons. Nat. Neurosci. 2009, 12, 1090–1092. [Google Scholar] [CrossRef] [PubMed]
- Kuwabara, T.; Kagalwala, M.N.; Onuma, Y.; Ito, Y.; Warashina, M.; Terashima, K.; Sanosaka, T.; Nakashima, K.; Gage, F.H.; Asashima, M. Insulin biosynthesis in neuronal progenitors derived from adult hippocampus and the olfactory bulb. EMBO Mol. Med. 2011, 3, 742–754. [Google Scholar] [CrossRef] [PubMed]
- Revsin, Y.; Rekers, N.V.; Louwe, M.C.; Saravia, F.E.; de Nicola, A.F.; de Kloet, E.R.; Oitzl, M.S. Glucocorticoid receptor blockade normalizes hippocampal alterations and cognitive impairment in streptozotocin-induced type 1 diabetes mice. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2009, 34, 747–758. [Google Scholar] [CrossRef] [PubMed]
- Balu, D.T.; Hodes, G.E.; Hill, T.E.; Ho, N.; Rahman, Z.; Bender, C.N.; Ring, R.H.; Dwyer, J.M.; Rosenzweig-Lipson, S.; Hughes, Z.A.; et al. Flow cytometric analysis of BrdU incorporation as a high-throughput method for measuring adult neurogenesis in the mouse. J. Pharmacol. Toxicol. Methods 2009, 59, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Stranahan, A.M.; Arumugam, T.V.; Cutler, R.G.; Lee, K.; Egan, J.M.; Mattson, M.P. Diabetes impairs hippocampal function through glucocorticoid-mediated effects on new and mature neurons. Nat. Neurosci. 2008, 11, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Jackson-Guilford, J.; Leander, J.D.; Nisenbaum, L.K. The effect of streptozotocin-induced diabetes on cell proliferation in the rat dentate gyrus. Neurosci. Lett. 2000, 293, 91–94. [Google Scholar] [CrossRef]
- Piazza, F.V.; Pinto, G.V.; Trott, G.; Marcuzzo, S.; Gomez, R.; Fernandes Mda, C. Enriched environment prevents memory deficits in type 1 diabetic rats. Behav. Brain Res. 2011, 217, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.S.; Bluestone, J.A. The nod mouse: A model of immune dysregulation. Annu. Rev. Immunol. 2005, 23, 447–485. [Google Scholar] [CrossRef] [PubMed]
- Beauquis, J.; Saravia, F.; Coulaud, J.; Roig, P.; Dardenne, M.; Homo-Delarche, F.; de Nicola, A. Prominently decreased hippocampal neurogenesis in a spontaneous model of type 1 diabetes, the nonobese diabetic mouse. Exp. Neurol. 2008, 210, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Yu, C.; Li, H.; Liu, F.; Feng, R.; Wang, H.; Meng, Y.; Li, Z.; Ju, G.; Wang, J. Impaired neural stem/progenitor cell proliferation in streptozotocin-induced and spontaneous diabetic mice. Neurosci. Res. 2010, 68, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Hwang, I.K.; Yi, S.S.; Kim, Y.N.; Kim, I.Y.; Lee, I.S.; Yoon, Y.S.; Seong, J.K. Reduced hippocampal cell differentiation in the subgranular zone of the dentate gyrus in a rat model of type ii diabetes. Neurochem. Res. 2008, 33, 394–400. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, A.; Patterson, S.; Porter, D.; Gault, V.A.; Holscher, C. Novel GLP-1 mimetics developed to treat type 2 diabetes promote progenitor cell proliferation in the brain. J. Neurosci. Res. 2011, 89, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, T.; Hidaka, R.; Fujimaki, S.; Asashima, M.; Kuwabara, T. Diabetes impairs Wnt3 protein-induced neurogenesis in olfactory bulbs via glutamate transporter 1 inhibition. J. Biol. Chem. 2016, 291, 15196–15211. [Google Scholar] [CrossRef] [PubMed]
- Jessberger, S.; Clark, R.E.; Broadbent, N.J.; Clemenson, G.D., Jr.; Consiglio, A.; Lie, D.C.; Squire, L.R.; Gage, F.H. Dentate gyrus-specific knockdown of adult neurogenesis impairs spatial and object recognition memory in adult rats. Learn. Mem. 2009, 16, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Stewart, R.R.; Hoge, G.J.; Zigova, T.; Luskin, M.B. Neural progenitor cells of the neonatal rat anterior subventricular zone express functional GABAA receptors. J. Neurobiol. 2002, 50, 305–322. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.D.; Krueger, D.D.; Bordey, A. GABA depolarizes neuronal progenitors of the postnatal subventricular zone via GABAA receptor activation. J. Physiol. 2003, 550, 785–800. [Google Scholar] [CrossRef] [PubMed]
- Bolteus, A.J.; Bordey, A. GABA release and uptake regulate neuronal precursor migration in the postnatal subventricular zone. J. Neurosci. 2004, 24, 7623–7631. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, Q.; Haydar, T.F.; Bordey, A. Nonsynaptic GABA signaling in postnatal subventricular zone controls proliferation of GFAP-expressing progenitors. Nat. Neurosci. 2005, 8, 1179–1187. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Mendoza, E.H.; Bellver-Landete, V.; Arce, C.; Doeppner, T.R.; Hermann, D.M.; Oset-Gasque, M.J. Vesicular glutamate transporters play a role in neuronal differentiation of cultured SVZ-derived neural precursor cells. PLoS ONE 2017, 12, e0177069. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Kim, J.S.; Yoon, Y.; Santiago, M.C.; Brown, M.D.; Park, J.Y. Inhibition of Drp1-dependent mitochondrial division impairs myogenic differentiation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 305, R927–R938. [Google Scholar] [CrossRef] [PubMed]
- Rharass, T.; Lemcke, H.; Lantow, M.; Kuznetsov, S.A.; Weiss, D.G.; Panakova, D. Ca2+-mediated mitochondrial reactive oxygen species metabolism augments Wnt/β-catenin pathway activation to facilitate cell differentiation. J. Biol. Chem. 2014, 289, 27937–27951. [Google Scholar] [CrossRef] [PubMed]
- Beckervordersandforth, R.; Ebert, B.; Schaffner, I.; Moss, J.; Fiebig, C.; Shin, J.; Moore, D.L.; Ghosh, L.; Trinchero, M.F.; Stockburger, C.; et al. Role of mitochondrial metabolism in the control of early lineage progression and aging phenotypes in adult hippocampal neurogenesis. Neuron 2017, 93, 1518. [Google Scholar] [CrossRef] [PubMed]
- Agnihotri, S.K.; Shen, R.; Li, J.; Gao, X.; Bueler, H. Loss of PINK1 leads to metabolic deficits in adult neural stem cells and impedes differentiation of newborn neurons in the mouse hippocampus. FASEB J. 2017, 31, 2839–2853. [Google Scholar] [CrossRef] [PubMed]
- Pasini, E.; le Douairon Lahaye, S.; Flati, V.; Assanelli, D.; Corsetti, G.; Speca, S.; Bernabei, R.; Calvani, R.; Marzetti, E. Effects of treadmill exercise and training frequency on anabolic signaling pathways in the skeletal muscle of aged rats. Exp. Gerontol. 2012, 47, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Bruce, C.R.; Kriketos, A.D.; Cooney, G.J.; Hawley, J.A. Disassociation of muscle triglyceride content and insulin sensitivity after exercise training in patients with type 2 diabetes. Diabetologia 2004, 47, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Holloszy, J.O. Biochemical adaptations in muscle. Effects of exercise on mitochondrial oxygen uptake and respiratory enzyme activity in skeletal muscle. J. Biol. Chem. 1967, 242, 2278–2282. [Google Scholar] [PubMed]
- Hood, D.A. Invited review: Contractile activity-induced mitochondrial biogenesis in skeletal muscle. J. Appl. Physiol. 2001, 90, 1137–1157. [Google Scholar] [PubMed]
- Perry, C.G.; Lally, J.; Holloway, G.P.; Heigenhauser, G.J.; Bonen, A.; Spriet, L.L. Repeated transient mRNA bursts precede increases in transcriptional and mitochondrial proteins during training in human skeletal muscle. J. Physiol. 2010, 588, 4795–4810. [Google Scholar] [CrossRef] [PubMed]
- Adamovich, Y.; Shlomai, A.; Tsvetkov, P.; Umansky, K.B.; Reuven, N.; Estall, J.L.; Spiegelman, B.M.; Shaul, Y. The protein level of PGC-1α, a key metabolic regulator, is controlled by NADH-NQO1. Mol. Cell. Biol. 2013, 33, 2603–2613. [Google Scholar] [CrossRef] [PubMed]
- Pilegaard, H.; Saltin, B.; Neufer, P.D. Exercise induces transient transcriptional activation of the PGC-1α gene in human skeletal muscle. J. Physiol. 2003, 546, 851–858. [Google Scholar] [CrossRef] [PubMed]
- Baar, K.; Wende, A.R.; Jones, T.E.; Marison, M.; Nolte, L.A.; Chen, M.; Kelly, D.P.; Holloszy, J.O. Adaptations of skeletal muscle to exercise: Rapid increase in the transcriptional coactivator PGC-1. FASEB J. 2002, 16, 1879–1886. [Google Scholar] [CrossRef] [PubMed]
- Leick, L.; Wojtaszewski, J.F.; Johansen, S.T.; Kiilerich, K.; Comes, G.; Hellsten, Y.; Hidalgo, J.; Pilegaard, H. PGC-1α is not mandatory for exercise- and training-induced adaptive gene responses in mouse skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2008, 294, E463–E474. [Google Scholar] [CrossRef] [PubMed]
- Rowe, G.C.; El-Khoury, R.; Patten, I.S.; Rustin, P.; Arany, Z. PGC-1α is dispensable for exercise-induced mitochondrial biogenesis in skeletal muscle. PLoS ONE 2012, 7, e41817. [Google Scholar] [CrossRef] [PubMed]
- Lai, R.Y.; Ljubicic, V.; D’Souza, D.; Hood, D.A. Effect of chronic contractile activity on mRNA stability in skeletal muscle. Am. J. Physiol. Cell Physiol. 2010, 299, C155–C163. [Google Scholar] [CrossRef] [PubMed]
- Pastore, S.; Hood, D.A. Endurance training ameliorates the metabolic and performance characteristics of circadian clock mutant mice. J. Appl. Physiol. 2013, 114, 1076–1084. [Google Scholar] [CrossRef] [PubMed]
- Gordon, J.W.; Rungi, A.A.; Inagaki, H.; Hood, D.A. Effects of contractile activity on mitochondrial transcription factor a expression in skeletal muscle. J. Appl. Physiol. 2001, 90, 389–396. [Google Scholar] [PubMed]
- Bengtsson, J.; Gustafsson, T.; Widegren, U.; Jansson, E.; Sundberg, C.J. Mitochondrial transcription factor a and respiratory complex IV increase in response to exercise training in humans. Pflugers Arch. 2001, 443, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Carter, H.N.; Hood, D.A. Contractile activity-induced mitochondrial biogenesis and mTORC1. Am. J. Physiol. Cell Physiol. 2012, 303, C540–C547. [Google Scholar] [CrossRef] [PubMed]
- Uguccioni, G.; Hood, D.A. The importance of PGC-1α in contractile activity-induced mitochondrial adaptations. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E361–E371. [Google Scholar] [CrossRef] [PubMed]
- Van Tienen, F.H.; Praet, S.F.; de Feyter, H.M.; van den Broek, N.M.; Lindsey, P.J.; Schoonderwoerd, K.G.; de Coo, I.F.; Nicolay, K.; Prompers, J.J.; Smeets, H.J.; et al. Physical activity is the key determinant of skeletal muscle mitochondrial function in type 2 diabetes. J. Clin. Endocrinol. Metab. 2012, 97, 3261–3269. [Google Scholar] [CrossRef] [PubMed]
- Sparks, L.M.; Johannsen, N.M.; Church, T.S.; Earnest, C.P.; Moonen-Kornips, E.; Moro, C.; Hesselink, M.K.; Smith, S.R.; Schrauwen, P. Nine months of combined training improves ex vivo skeletal muscle metabolism in individuals with type 2 diabetes. J. Clin. Endocrinol. Metab. 2013, 98, 1694–1702. [Google Scholar] [CrossRef] [PubMed]
- Phielix, E.; Meex, R.; Moonen-Kornips, E.; Hesselink, M.K.; Schrauwen, P. Exercise training increases mitochondrial content and ex vivo mitochondrial function similarly in patients with type 2 diabetes and in control individuals. Diabetologia 2010, 53, 1714–1721. [Google Scholar] [CrossRef] [PubMed]
- Fujii, N.; Hayashi, T.; Hirshman, M.F.; Smith, J.T.; Habinowski, S.A.; Kaijser, L.; Mu, J.; Ljungqvist, O.; Birnbaum, M.J.; Witters, L.A.; et al. Exercise induces isoform-specific increase in 5’AMP-activated protein kinase activity in human skeletal muscle. Biochem. Biophys. Res. Commun. 2000, 273, 1150–1155. [Google Scholar] [CrossRef] [PubMed]
- Palacios, O.M.; Carmona, J.J.; Michan, S.; Chen, K.Y.; Manabe, Y.; Ward, J.L., 3rd; Goodyear, L.J.; Tong, Q. Diet and exercise signals regulate SIRT3 and activate ampk and PGC-1α in skeletal muscle. Aging 2009, 1, 771–783. [Google Scholar] [CrossRef] [PubMed]
- Jager, S.; Handschin, C.; St-Pierre, J.; Spiegelman, B.M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1α. Proc. Natl. Acad. Sci. USA 2007, 104, 12017–12022. [Google Scholar] [CrossRef] [PubMed]
- McConell, G.K.; Ng, G.P.; Phillips, M.; Ruan, Z.; Macaulay, S.L.; Wadley, G.D. Central role of nitric oxide synthase in aicar and caffeine-induced mitochondrial biogenesis in L6 myocytes. J. Appl. Physiol. 2010, 108, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Kjobsted, R.; Pedersen, A.J.; Hingst, J.R.; Sabaratnam, R.; Birk, J.B.; Kristensen, J.M.; Hojlund, K.; Wojtaszewski, J.F. Intact regulation of the AMPK signaling network in response to exercise and insulin in skeletal muscle of male patients with type 2 diabetes: Illumination of AMPK activation in recovery from exercise. Diabetes 2016, 65, 1219–1230. [Google Scholar] [CrossRef] [PubMed]
- Luquet, S.; Lopez-Soriano, J.; Holst, D.; Fredenrich, A.; Melki, J.; Rassoulzadegan, M.; Grimaldi, P.A. Peroxisome proliferator-activated receptor δ controls muscle development and oxidative capability. FASEB J. 2003, 17, 2299–2301. [Google Scholar] [PubMed]
- Fritz, T.; Kramer, D.K.; Karlsson, H.K.; Galuska, D.; Engfeldt, P.; Zierath, J.R.; Krook, A. Low-intensity exercise increases skeletal muscle protein expression of PPARδ and UCP3 in type 2 diabetic patients. Diabetes Metab. Res. Rev. 2006, 22, 492–498. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.X.; Lee, C.H.; Tiep, S.; Yu, R.T.; Ham, J.; Kang, H.; Evans, R.M. Peroxisome-proliferator-activated receptor δ activates fat metabolism to prevent obesity. Cell 2003, 113, 159–170. [Google Scholar] [CrossRef]
- Tanaka, T.; Yamamoto, J.; Iwasaki, S.; Asaba, H.; Hamura, H.; Ikeda, Y.; Watanabe, M.; Magoori, K.; Ioka, R.X.; Tachibana, K.; et al. Activation of peroxisome proliferator-activated receptor δ induces fatty acid β-oxidation in skeletal muscle and attenuates metabolic syndrome. Proc. Natl. Acad. Sci. USA 2003, 100, 15924–15929. [Google Scholar] [CrossRef] [PubMed]
- Narkar, V.A.; Downes, M.; Yu, R.T.; Embler, E.; Wang, Y.X.; Banayo, E.; Mihaylova, M.M.; Nelson, M.C.; Zou, Y.; Juguilon, H.; et al. Ampk and PPARδ agonists are exercise mimetics. Cell 2008, 134, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Kitaoka, Y.; Ogasawara, R.; Tamura, Y.; Fujita, S.; Hatta, H. Effect of electrical stimulation-induced resistance exercise on mitochondrial fission and fusion proteins in rat skeletal muscle. Appl. Physiol. Nutr. Metab. 2015, 40, 1137–1142. [Google Scholar] [CrossRef] [PubMed]
- Ju, J.S.; Jeon, S.I.; Park, J.Y.; Lee, J.Y.; Lee, S.C.; Cho, K.J.; Jeong, J.M. Autophagy plays a role in skeletal muscle mitochondrial biogenesis in an endurance exercise-trained condition. J. Physiol. Sci. 2016, 66, 417–430. [Google Scholar] [CrossRef] [PubMed]
- Pagano, A.F.; Py, G.; Bernardi, H.; Candau, R.B.; Sanchez, A.M. Autophagy and protein turnover signaling in slow-twitch muscle during exercise. Med. Sci. Sports Exerc. 2014, 46, 1314–1325. [Google Scholar] [CrossRef] [PubMed]
- Scheele, C.; Petrovic, N.; Faghihi, M.A.; Lassmann, T.; Fredriksson, K.; Rooyackers, O.; Wahlestedt, C.; Good, L.; Timmons, J.A. The human PINK1 locus is regulated in vivo by a non-coding natural antisense RNA during modulation of mitochondrial function. BMC Genom. 2007, 8, 74. [Google Scholar] [CrossRef] [PubMed]
- Jamart, C.; Naslain, D.; Gilson, H.; Francaux, M. Higher activation of autophagy in skeletal muscle of mice during endurance exercise in the fasted state. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E964–E974. [Google Scholar] [CrossRef] [PubMed]
- Davies, K.J.; Quintanilha, A.T.; Brooks, G.A.; Packer, L. Free radicals and tissue damage produced by exercise. Biochem. Biophys. Res. Commun. 1982, 107, 1198–1205. [Google Scholar] [CrossRef]
- Powers, S.K.; Jackson, M.J. Exercise-induced oxidative stress: Cellular mechanisms and impact on muscle force production. Physiol. Rev. 2008, 88, 1243–1276. [Google Scholar] [CrossRef] [PubMed]
- Carnio, S.; LoVerso, F.; Baraibar, M.A.; Longa, E.; Khan, M.M.; Maffei, M.; Reischl, M.; Canepari, M.; Loefler, S.; Kern, H.; et al. Autophagy impairment in muscle induces neuromuscular junction degeneration and precocious aging. Cell Rep. 2014, 8, 1509–1521. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G.; Bae, Y.S.; Lee, S.R.; Kwon, J. Hydrogen peroxide: A key messenger that modulates protein phosphorylation through cysteine oxidation. Sci. STKE 2000, 2000, pe1. [Google Scholar] [CrossRef] [PubMed]
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Lo Verso, F.; Carnio, S.; Vainshtein, A.; Sandri, M. Autophagy is not required to sustain exercise and PRKAA1/AMPK activity but is important to prevent mitochondrial damage during physical activity. Autophagy 2014, 10, 1883–1894. [Google Scholar] [CrossRef] [PubMed]
- Hey-Mogensen, M.; Hojlund, K.; Vind, B.F.; Wang, L.; Dela, F.; Beck-Nielsen, H.; Fernstrom, M.; Sahlin, K. Effect of physical training on mitochondrial respiration and reactive oxygen species release in skeletal muscle in patients with obesity and type 2 diabetes. Diabetologia 2010, 53, 1976–1985. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Cabrera, M.C.; Borras, C.; Pallardo, F.V.; Sastre, J.; Ji, L.L.; Vina, J. Decreasing xanthine oxidase-mediated oxidative stress prevents useful cellular adaptations to exercise in rats. J. Physiol. 2005, 567, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Cabrera, M.C.; Domenech, E.; Romagnoli, M.; Arduini, A.; Borras, C.; Pallardo, F.V.; Sastre, J.; Vina, J. Oral administration of vitamin c decreases muscle mitochondrial biogenesis and hampers training-induced adaptations in endurance performance. Am. J. Clin. Nutr. 2008, 87, 142–149. [Google Scholar] [PubMed]
- Ristow, M.; Zarse, K.; Oberbach, A.; Kloting, N.; Birringer, M.; Kiehntopf, M.; Stumvoll, M.; Kahn, C.R.; Bluher, M. Antioxidants prevent health-promoting effects of physical exercise in humans. Proc. Natl. Acad. Sci. USA 2009, 106, 8665–8670. [Google Scholar] [CrossRef] [PubMed]
- Vinetti, G.; Mozzini, C.; Desenzani, P.; Boni, E.; Bulla, L.; Lorenzetti, I.; Romano, C.; Pasini, A.; Cominacini, L.; Assanelli, D. Supervised exercise training reduces oxidative stress and cardiometabolic risk in adults with type 2 diabetes: A randomized controlled trial. Sci. Rep. 2015, 5, 9238. [Google Scholar] [CrossRef] [PubMed]
- Umnova, M.M.; Seene, T.P. The effect of increased functional load on the activation of satellite cells in the skeletal muscle of adult rats. Int. J. Sports Med. 1991, 12, 501–504. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.K.; Maxwell, L.; Rodgers, C.D.; McKee, N.H.; Plyley, M.J. Exercise-enhanced satellite cell proliferation and new myonuclear accretion in rat skeletal muscle. J. Appl. Physiol. 2001, 90, 1407–1414. [Google Scholar] [PubMed]
- Kadi, F.; Eriksson, A.; Holmner, S.; Butler-Browne, G.S.; Thornell, L.E. Cellular adaptation of the trapezius muscle in strength-trained athletes. Histochem. Cell Biol. 1999, 111, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Kadi, F.; Schjerling, P.; Andersen, L.L.; Charifi, N.; Madsen, J.L.; Christensen, L.R.; Andersen, J.L. The effects of heavy resistance training and detraining on satellite cells in human skeletal muscles. J. Physiol. 2004, 558, 1005–1012. [Google Scholar] [CrossRef] [PubMed]
- Kurosaka, M.; Naito, H.; Ogura, Y.; Machida, S.; Katamoto, S. Satellite cell pool enhancement in rat plantaris muscle by endurance training depends on intensity rather than duration. Acta Physiol. 2012, 205, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Martin, N.R.W.; Lewis, M. Satellite cell activation and number following acute and chronic exercise: A mini review. Cell. Mol. Exerc. Physiol. 2012, 1, e3. [Google Scholar] [CrossRef]
- Aschenbach, W.G.; Ho, R.C.; Sakamoto, K.; Fujii, N.; Li, Y.; Kim, Y.B.; Hirshman, M.F.; Goodyear, L.J. Regulation of dishevelled and β-catenin in rat skeletal muscle: An alternative exercise-induced GSK-3β signaling pathway. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E152–E158. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, D.D.; Esser, K.A. Wnt/β-catenin signaling activates growth-control genes during overload-induced skeletal muscle hypertrophy. Am. J. Physiol. Cell Physiol. 2005, 289, C853–C859. [Google Scholar] [CrossRef] [PubMed]
- Fujimaki, S.; Machida, M.; Wakabayashi, T.; Asashima, M.; Takemasa, T.; Kuwabara, T. Functional overload enhances satellite cell properties in skeletal muscle. Stem Cells Int. 2016, 2016, 7619418. [Google Scholar] [CrossRef] [PubMed]
- Van Praag, H.; Kempermann, G.; Gage, F.H. Running increases cell proliferation and neurogenesis in the adult mouse dentate gyrus. Nat. Neurosci. 1999, 2, 266–270. [Google Scholar] [PubMed]
- Creer, D.J.; Romberg, C.; Saksida, L.M.; van Praag, H.; Bussey, T.J. Running enhances spatial pattern separation in mice. Proc. Natl. Acad. Sci. USA 2010, 107, 2367–2372. [Google Scholar] [CrossRef] [PubMed]
- Kempermann, G.; Brandon, E.P.; Gage, F.H. Environmental stimulation of 129/Svj mice causes increased cell proliferation and neurogenesis in the adult dentate gyrus. Curr. Biol. CB 1998, 8, 939–942. [Google Scholar] [CrossRef]
- Kitamura, T.; Sugiyama, H. Running wheel exercises accelerate neuronal turnover in mouse dentate gyrus. Neurosci. Res. 2006, 56, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Kronenberg, G.; Bick-Sander, A.; Bunk, E.; Wolf, C.; Ehninger, D.; Kempermann, G. Physical exercise prevents age-related decline in precursor cell activity in the mouse dentate gyrus. Neurobiol. Aging 2006, 27, 1505–1513. [Google Scholar] [CrossRef] [PubMed]
- Naylor, A.S.; Bull, C.; Nilsson, M.K.; Zhu, C.; Bjork-Eriksson, T.; Eriksson, P.S.; Blomgren, K.; Kuhn, H.G. Voluntary running rescues adult hippocampal neurogenesis after irradiation of the young mouse brain. Proc. Natl. Acad. Sci. USA 2008, 105, 14632–14637. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.W.; Chang, Y.T.; Yu, L.; Chen, H.I.; Jen, C.J.; Wu, S.Y.; Lo, C.P.; Kuo, Y.M. Exercise enhances the proliferation of neural stem cells and neurite growth and survival of neuronal progenitor cells in dentate gyrus of middle-aged mice. J. Appl. Physiol. 2008, 105, 1585–1594. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.B.; Jang, M.H.; Shin, M.C.; Lim, B.V.; Kim, Y.P.; Kim, K.J.; Kim, E.H.; Kim, C.J. Treadmill exercise increases cell proliferation in dentate gyrus of rats with streptozotocin-induced diabetes. J. Diabetes Complicat. 2003, 17, 29–33. [Google Scholar] [CrossRef]
- Yi, S.S.; Hwang, I.K.; Yoo, K.Y.; Park, O.K.; Yu, J.; Yan, B.; Kim, I.Y.; Kim, Y.N.; Pai, T.; Song, W.; et al. Effects of treadmill exercise on cell proliferation and differentiation in the subgranular zone of the dentate gyrus in a rat model of type ii diabetes. Neurochem. Res. 2009, 34, 1039–1046. [Google Scholar] [CrossRef] [PubMed]
- Hwang, I.K.; Yi, S.S.; Song, W.; Won, M.H.; Yoon, Y.S.; Seong, J.K. Effects of age and treadmill exercise in chronic diabetic stages on neuroblast differentiation in a rat model of type 2 diabetes. Brain Res. 2010, 1341, 63–71. [Google Scholar] [CrossRef] [PubMed]
- De Senna, P.N.; Ilha, J.; Baptista, P.P.; do Nascimento, P.S.; Leite, M.C.; Paim, M.F.; Goncalves, C.A.; Achaval, M.; Xavier, L.L. Effects of physical exercise on spatial memory and astroglial alterations in the hippocampus of diabetic rats. Metab. Brain Dis. 2011, 26, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.Y.; Jung, S.Y.; Kim, K.; Kim, C.J. Treadmill exercise ameliorates alzheimer disease-associated memory loss through the Wnt signaling pathway in the streptozotocin-induced diabetic rats. J. Exerc. Rehabil. 2016, 12, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Stranahan, A.M.; Lee, K.; Becker, K.G.; Zhang, Y.; Maudsley, S.; Martin, B.; Cutler, R.G.; Mattson, M.P. Hippocampal gene expression patterns underlying the enhancement of memory by running in aged mice. Neurobiol. Aging 2010, 31, 1937–1949. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, M.; Inoue, K.; Iwamura, H.; Terashima, K.; Soya, H.; Asashima, M.; Kuwabara, T. Reduction in paracrine Wnt3 factors during aging causes impaired adult neurogenesis. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2011, 25, 3570–3582. [Google Scholar] [CrossRef] [PubMed]
- Mir, S.; Cai, W.; Carlson, S.W.; Saatman, K.E.; Andres, D.A. IGF-1 mediated neurogenesis involves a novel RIT1/Akt/Sox2 cascade. Sci. Rep. 2017, 7, 3283. [Google Scholar] [CrossRef] [PubMed]
- Rich, B.; Scadeng, M.; Yamaguchi, M.; Wagner, P.D.; Breen, E.C. Skeletal myofiber vascular endothelial growth factor is required for the exercise training-induced increase in dentate gyrus neuronal precursor cells. J. Physiol. 2017, 595, 5931–5943. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fujimaki, S.; Kuwabara, T. Diabetes-Induced Dysfunction of Mitochondria and Stem Cells in Skeletal Muscle and the Nervous System. Int. J. Mol. Sci. 2017, 18, 2147. https://doi.org/10.3390/ijms18102147
Fujimaki S, Kuwabara T. Diabetes-Induced Dysfunction of Mitochondria and Stem Cells in Skeletal Muscle and the Nervous System. International Journal of Molecular Sciences. 2017; 18(10):2147. https://doi.org/10.3390/ijms18102147
Chicago/Turabian StyleFujimaki, Shin, and Tomoko Kuwabara. 2017. "Diabetes-Induced Dysfunction of Mitochondria and Stem Cells in Skeletal Muscle and the Nervous System" International Journal of Molecular Sciences 18, no. 10: 2147. https://doi.org/10.3390/ijms18102147
APA StyleFujimaki, S., & Kuwabara, T. (2017). Diabetes-Induced Dysfunction of Mitochondria and Stem Cells in Skeletal Muscle and the Nervous System. International Journal of Molecular Sciences, 18(10), 2147. https://doi.org/10.3390/ijms18102147