
Discovery of a Potential HER2 Inhibitor from Natural Products for the Treatment of HER2-Positive Breast Cancer

Abstract
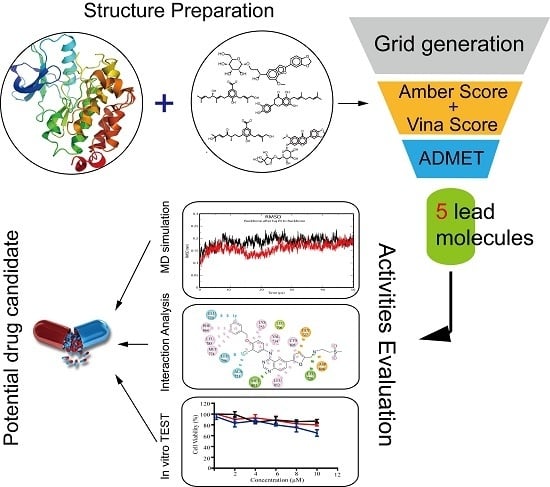
:
1. Introduction
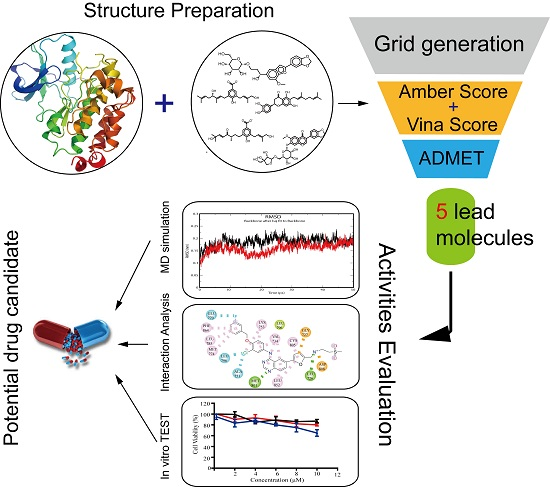
2. Results
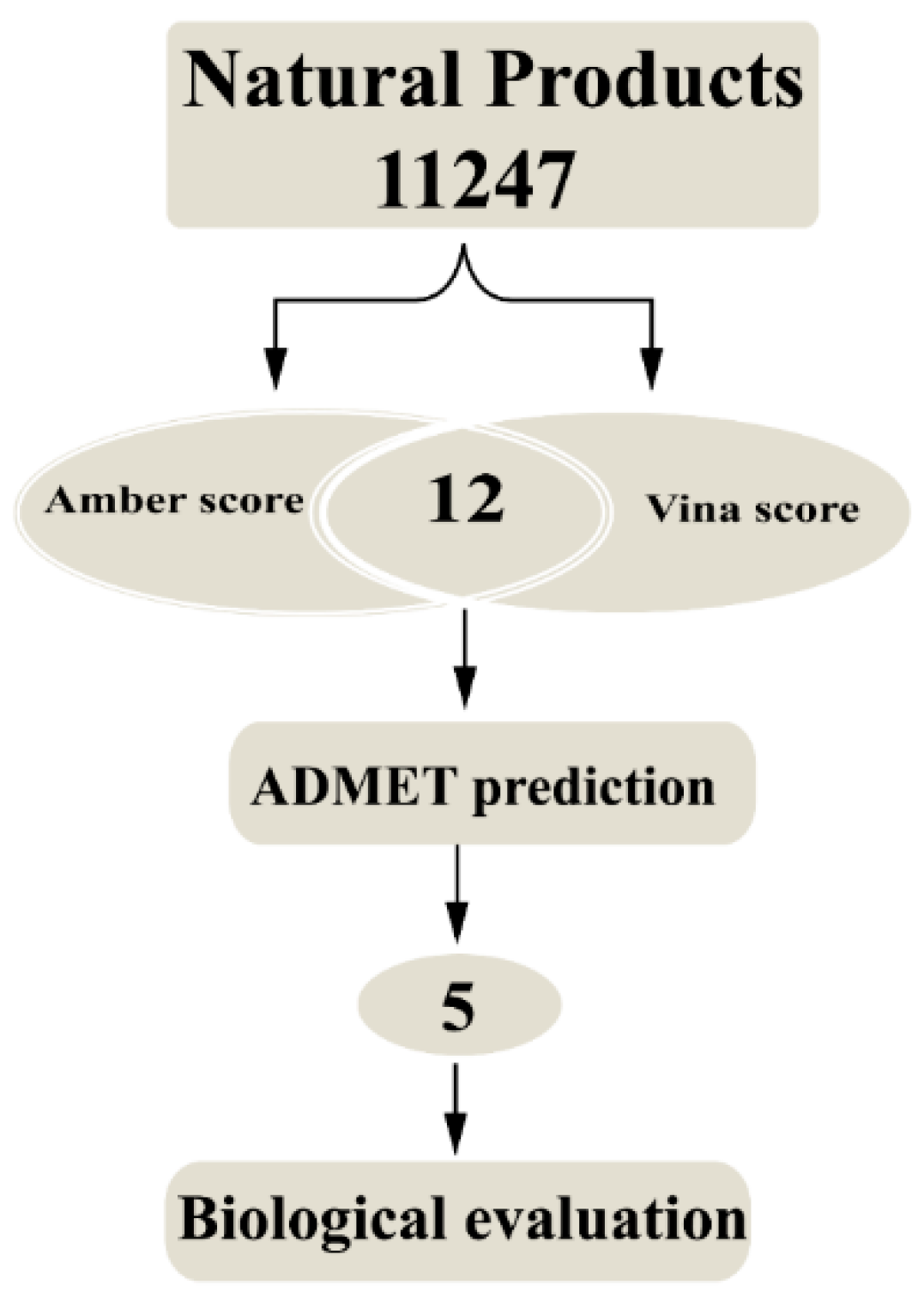
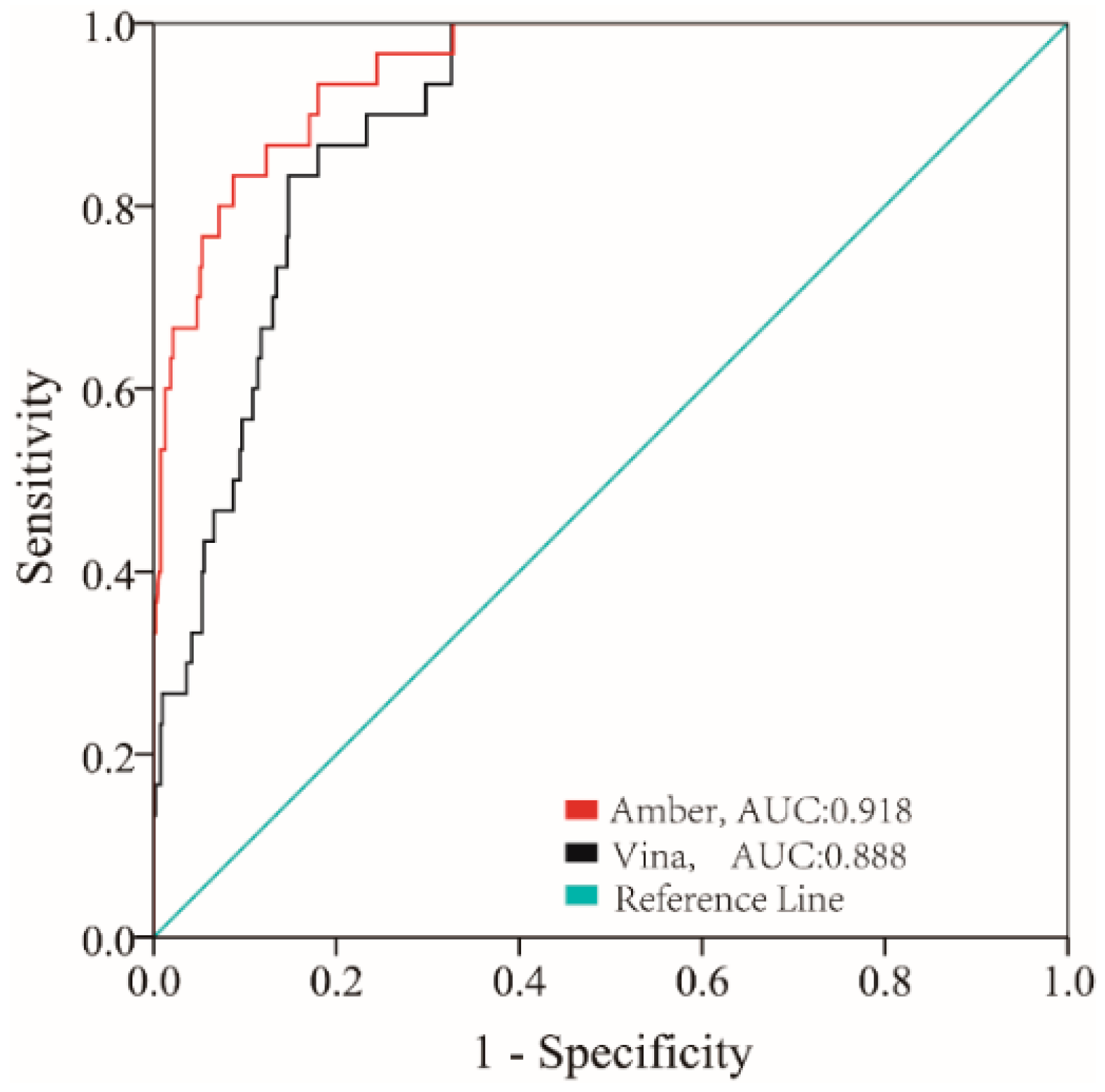
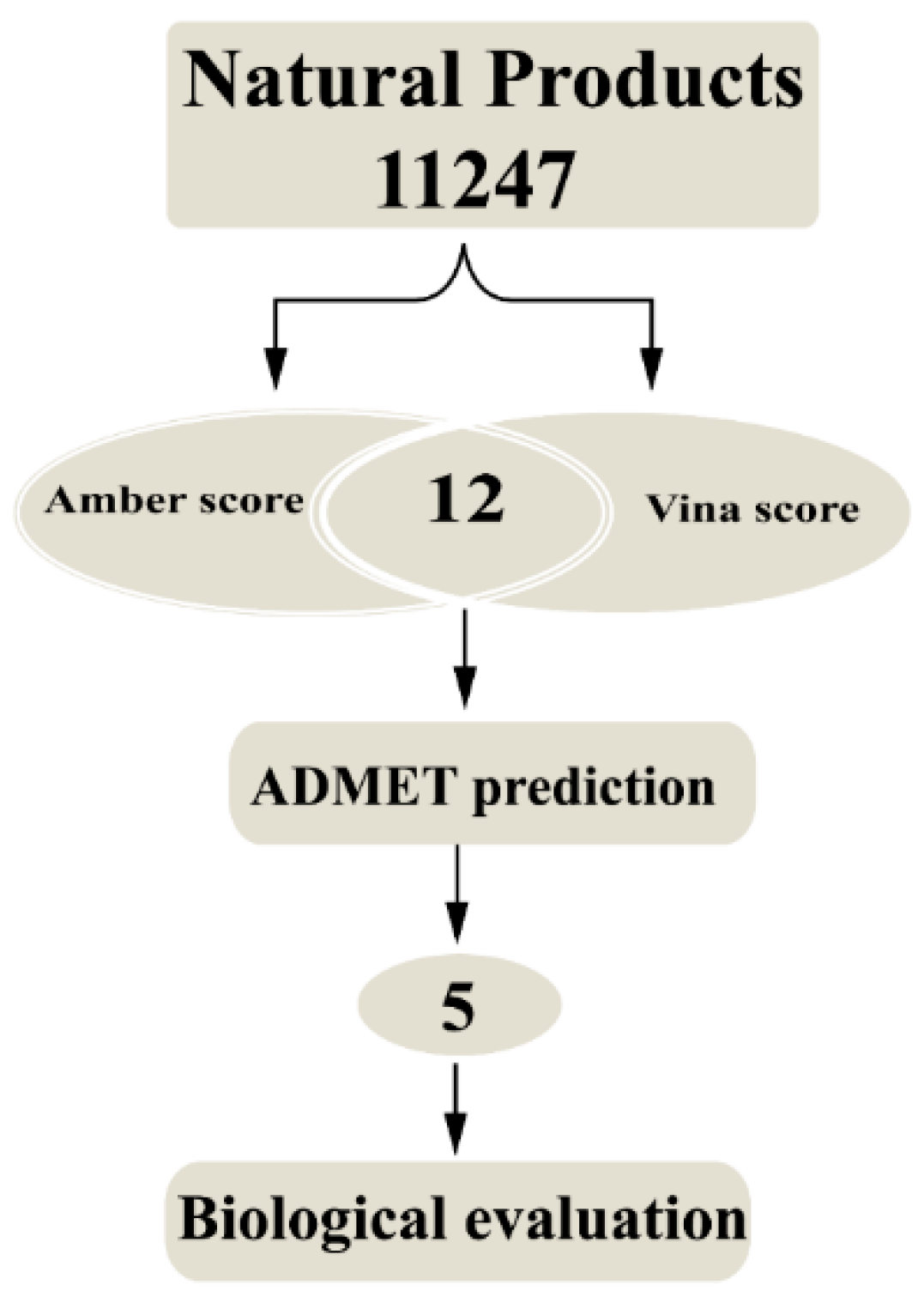
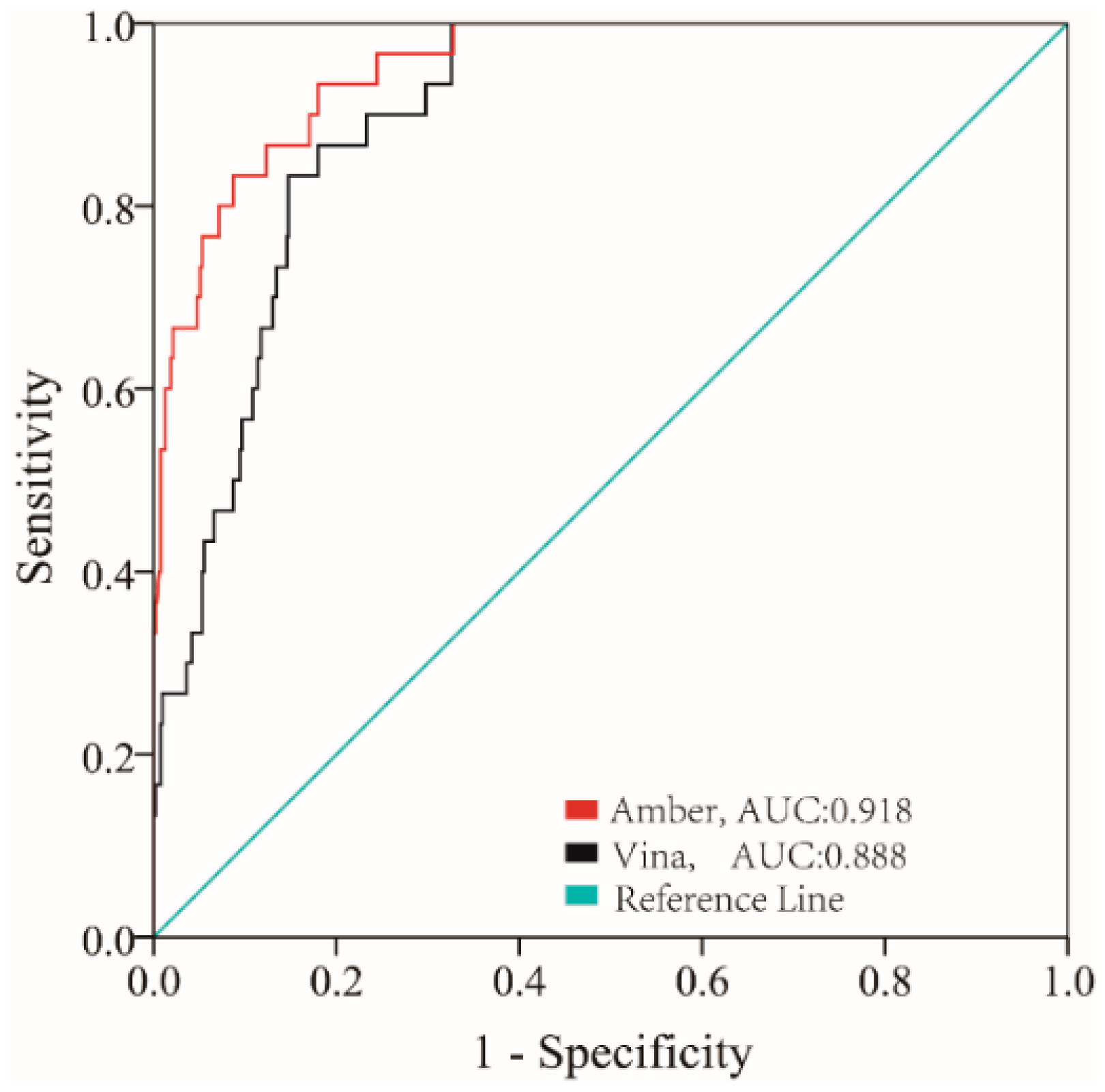
2.1. Molecular Docking Study
2.2. ADMET (Absorption, Distribution, Metabolism, Excretion and Toxicity) Properties Analysis
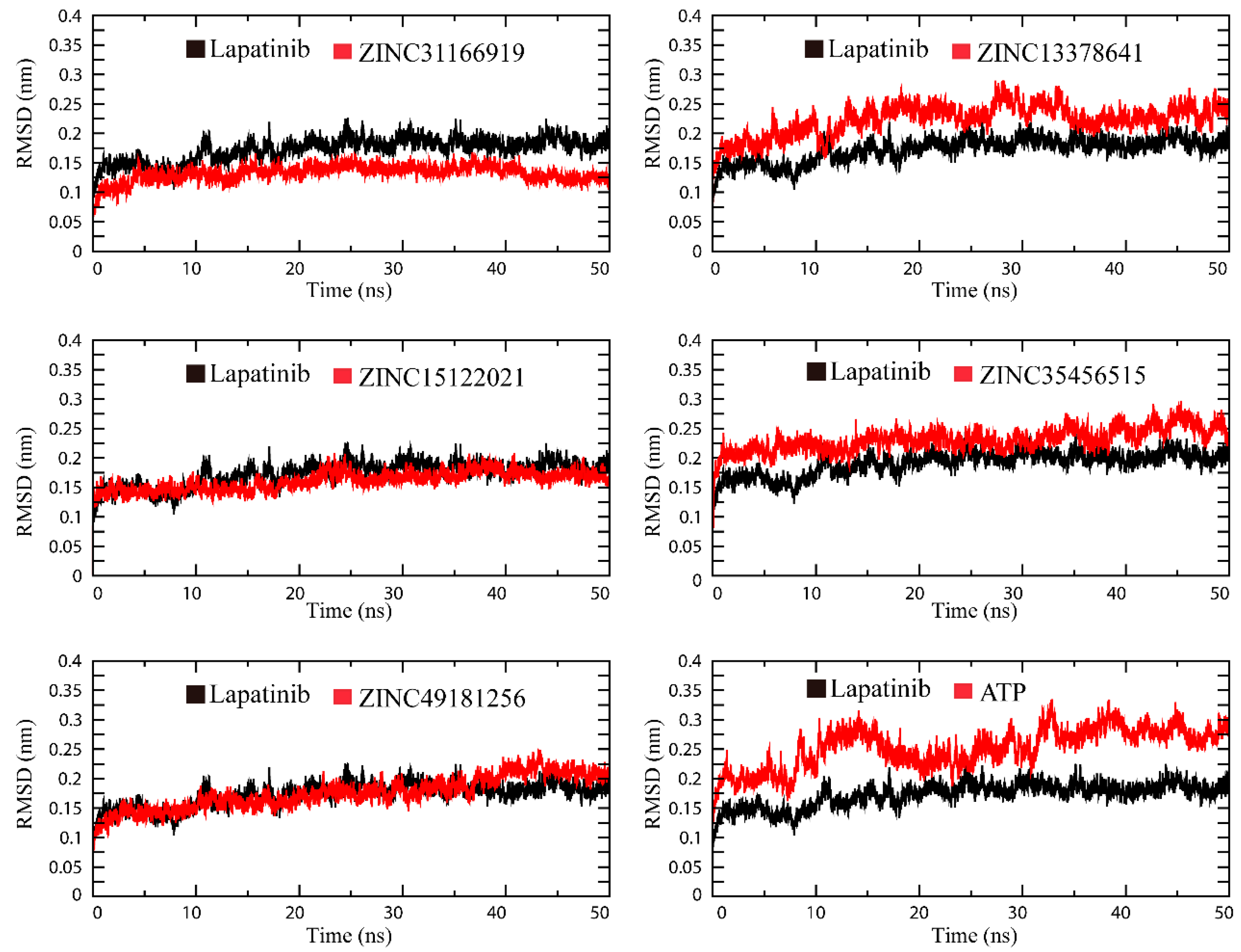
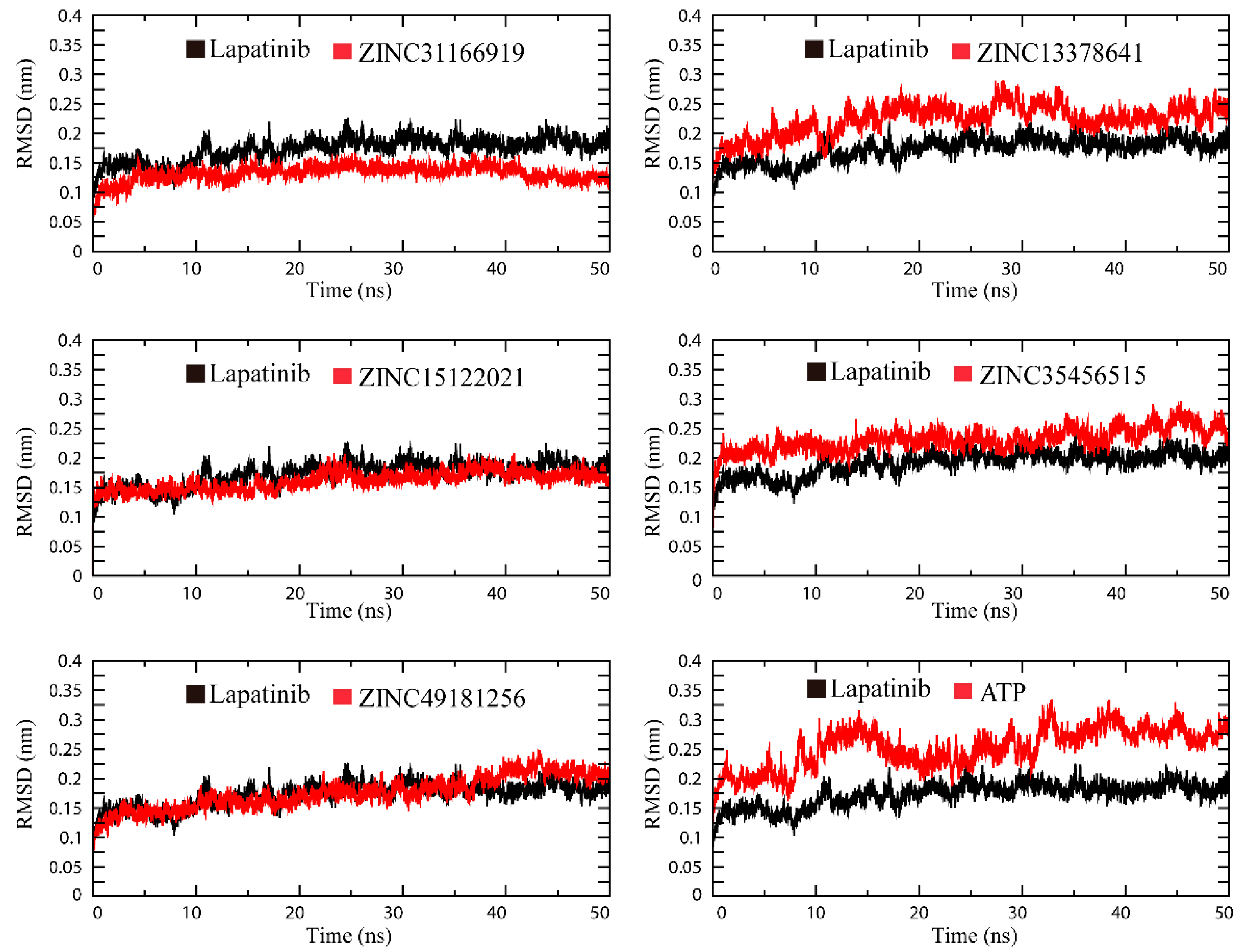
2.3. Molecular Simulation Analysis
2.4. Binding Affinity Prediction
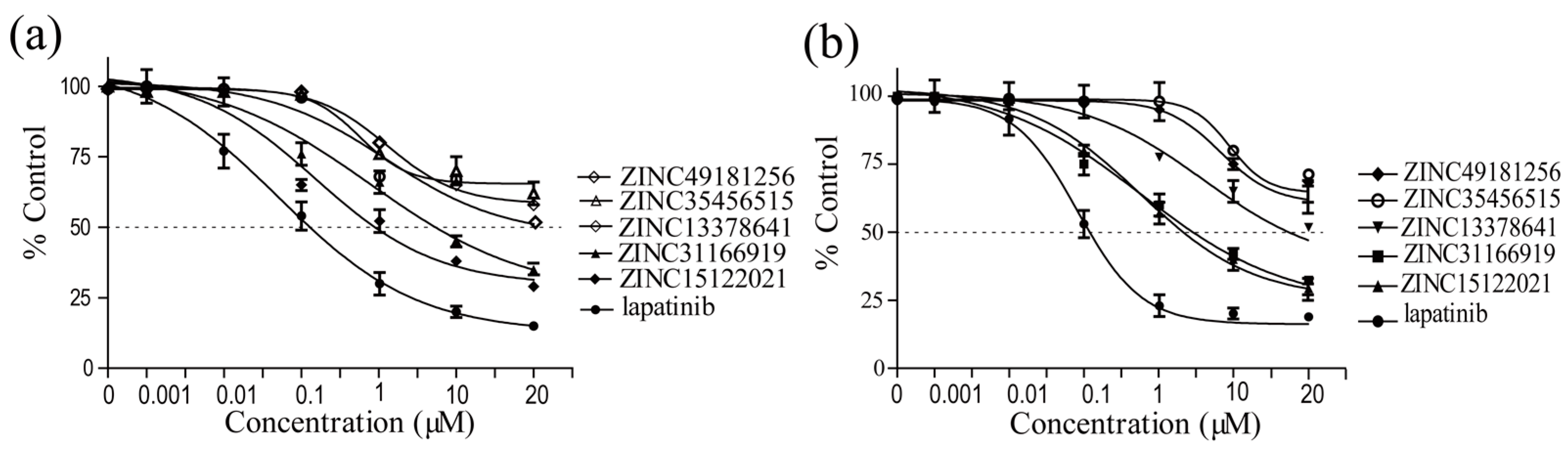
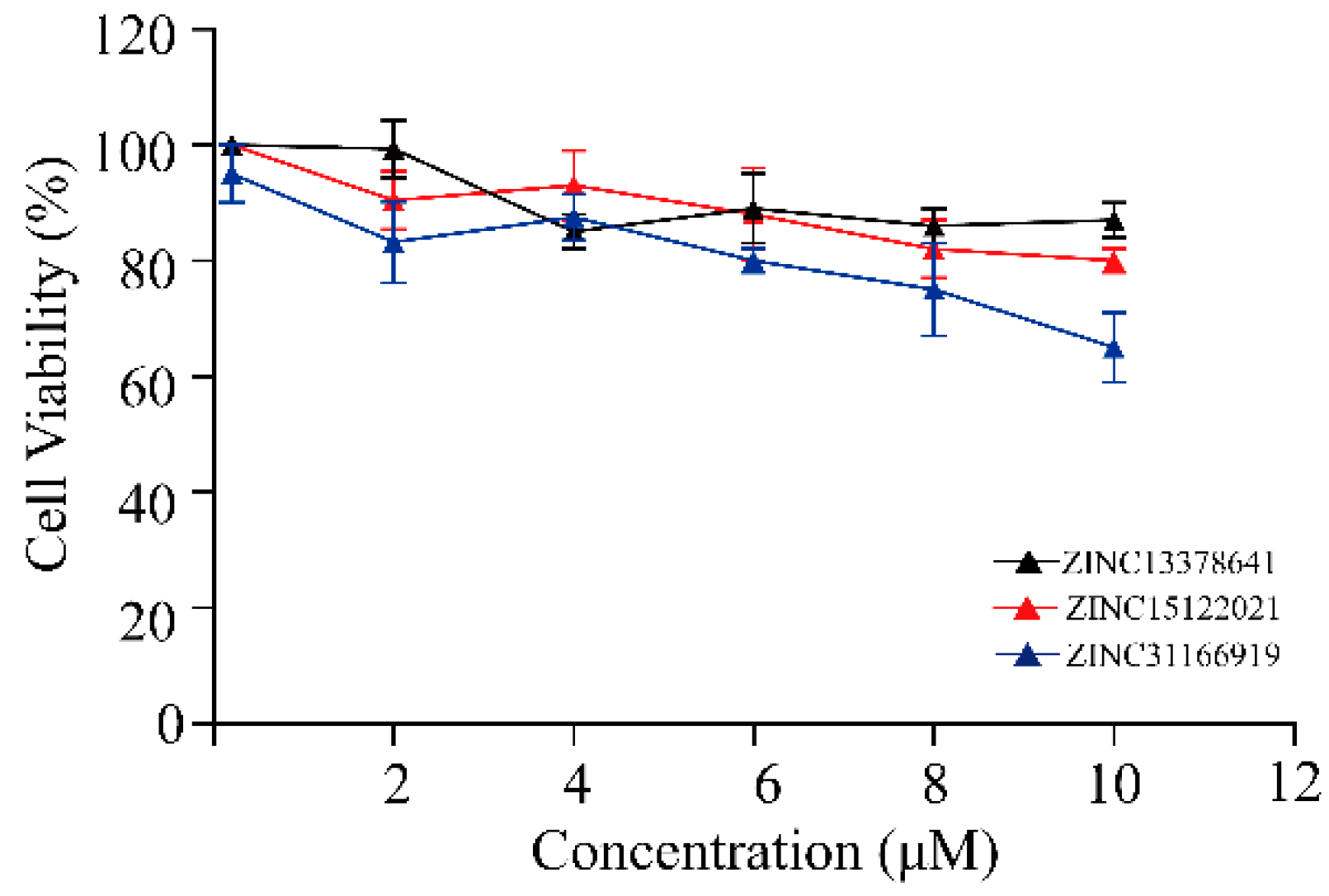
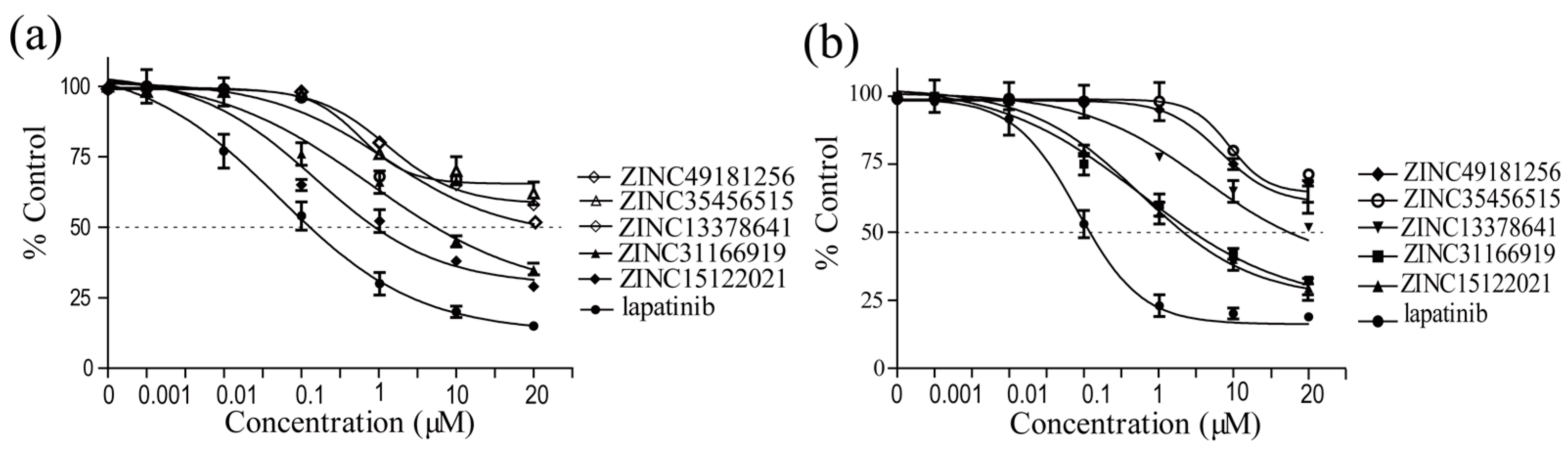
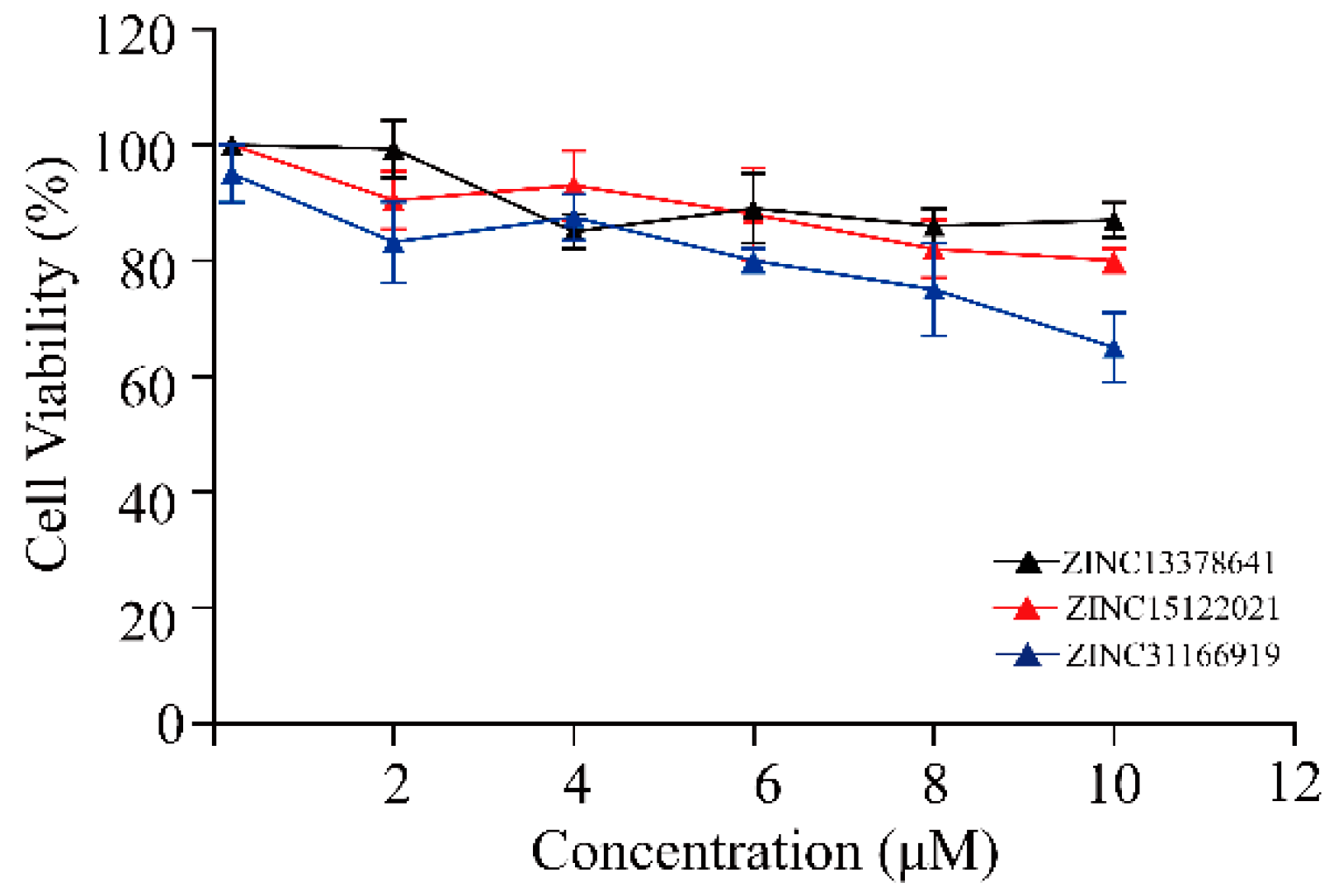
2.5. Biological Evaluation
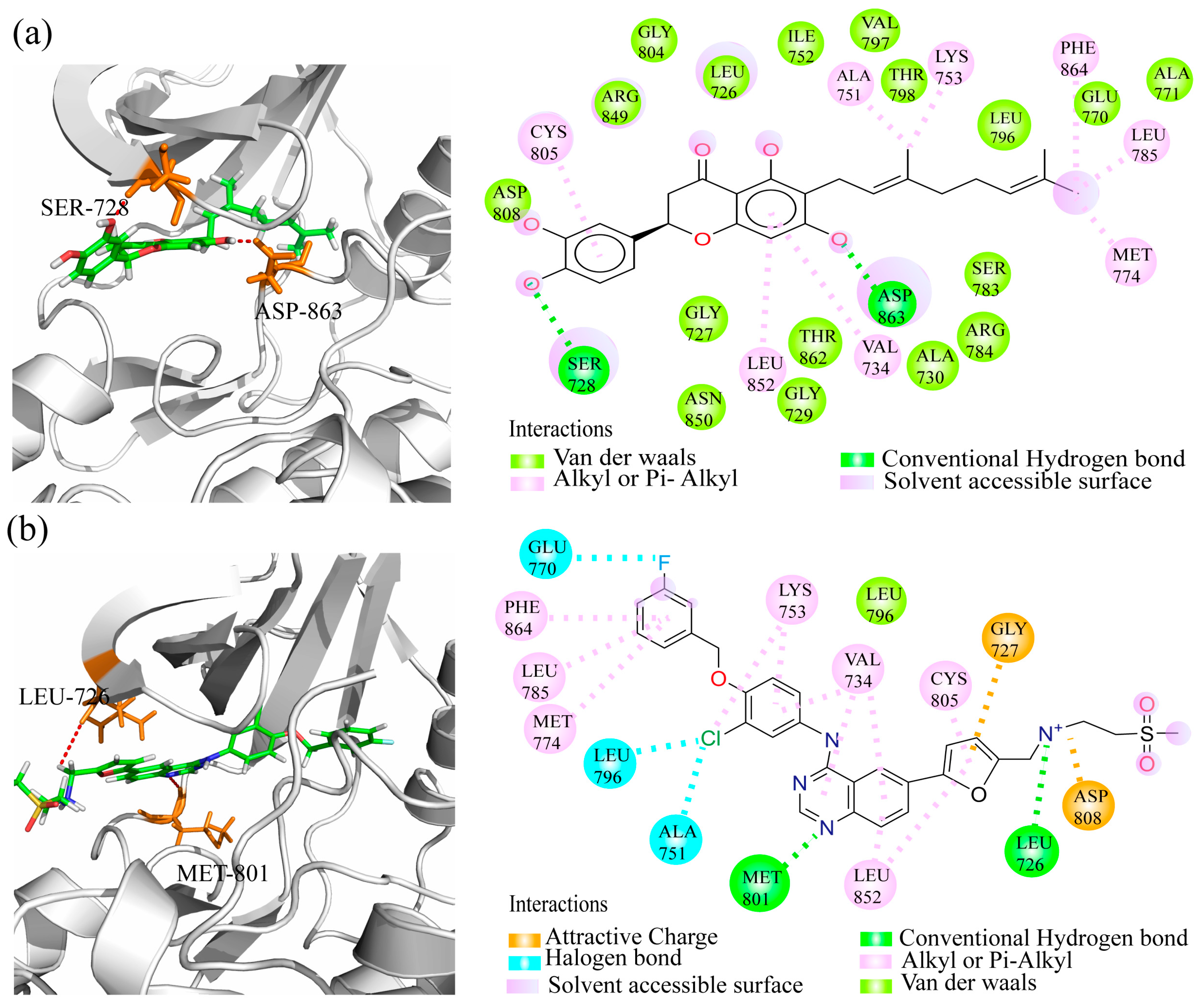
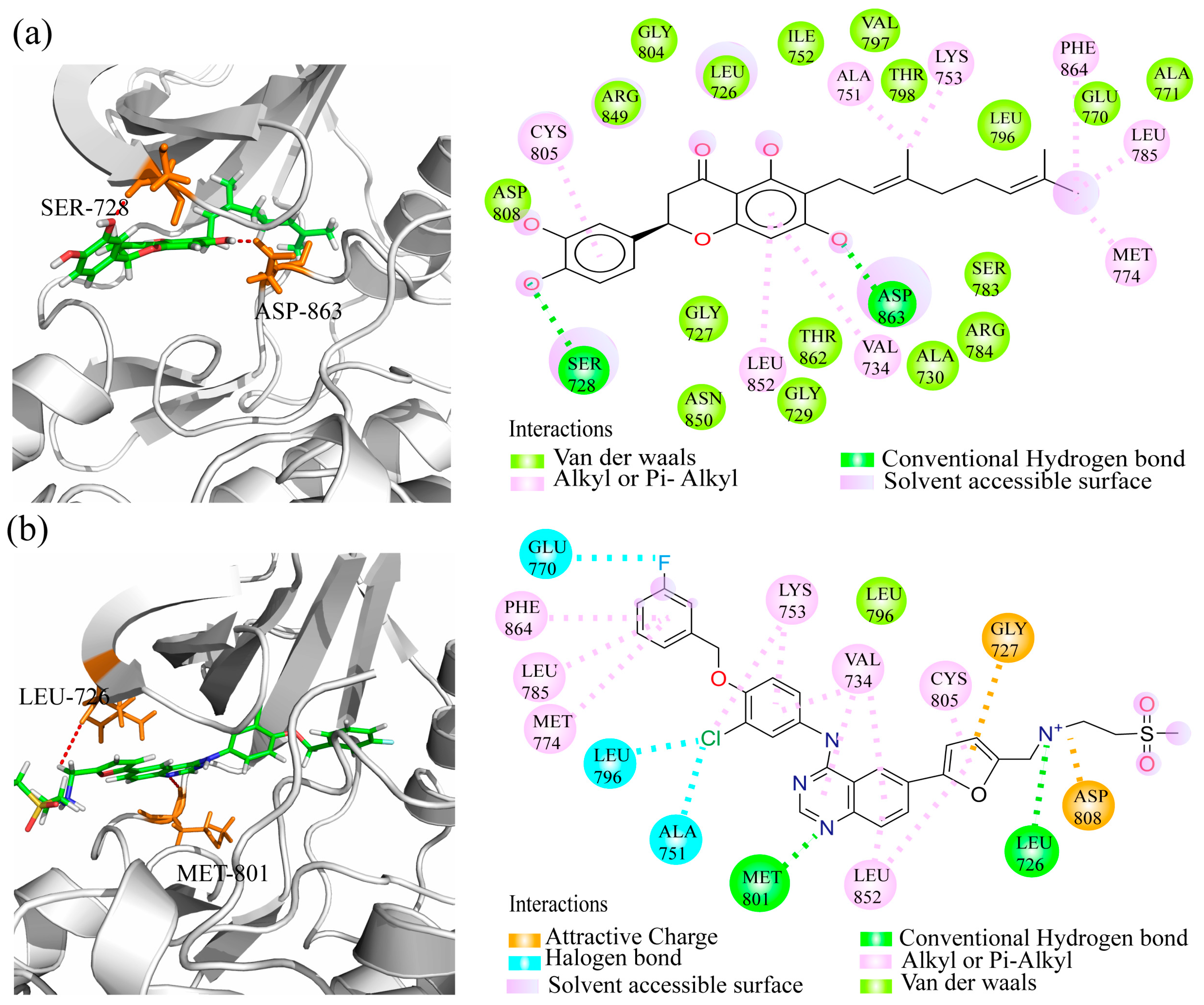
2.6. Binding Model Analysis
3. Discussion
4. Methods
4.1. Receptor and Ligand Preparation
4.2. Molecular Docking Model Validation
4.3. Molecular Docking
4.4. ADMET Prediction
4.5. MM/PBSA Binding Based on Molecular Dynamic Simulation Affinity Prediction
4.6. In Vitro Enzymatic Activity Assay and Cell Proliferation Inhibition
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Stewart, B.; Wild, C.P. World Cancer Report 2014; WHO Press: Geneva, Switzerland, 2014. [Google Scholar]
- Carey, L.A.; Perou, C.M.; Livasy, C.A.; Dressler, L.G.; Cowan, D.; Conway, K.; Karaca, G.; Troester, M.A.; Tse, C.K.; Edmiston, S. Race, breast cancer subtypes, and survival in the carolina breast cancer study. JAMA 2006, 295, 2492–2502. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Bartholomeusz, C.; Gonzalez-Angulo, A.M. Targeting the PI3K signaling pathway in cancer therapy. Expert Opin. Ther. Targets 2012, 16, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Xia, W.; Mullin, R.J.; Keith, B.R.; Liu, L.-H.; Ma, H.; Rusnak, D.W.; Owens, G.; Alligood, K.J.; Spector, N.L. Anti-tumor activity of GW572016: A dual tyrosine kinase inhibitor blocks EGF activation of EGFR/ErbB2 and downstream ERK1/2 and Akt pathways. Oncogene 2002, 21, 6255–6263. [Google Scholar] [CrossRef] [PubMed]
- Graus-Porta, D.; Beerli, R.R.; Daly, J.M.; Hynes, N.E. ErbB-2, the preferred heterodimerization partner of all ErBb receptors, is a mediator of lateral signaling. EMBO J. 1997, 16, 1647–1655. [Google Scholar] [CrossRef] [PubMed]
- Gschwind, A.; Fischer, O.M.; Ullrich, A. The discovery of receptor tyrosine kinases: Targets for cancer therapy. Nat. Rev. Cancer 2004, 4, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Lee-Hoeflich, S.T.; Crocker, L.; Yao, E.; Pham, T.; Munroe, X.; Hoeflich, K.P.; Sliwkowski, M.X.; Stern, H.M. A central role for HER3 in HER2-amplified breast cancer: Implications for targeted therapy. Cancer Res. 2008, 68, 5878–5887. [Google Scholar] [CrossRef] [PubMed]
- Kulendran, M.; Salhab, M.; Mokbel, K. Oestrogen-synthesising enzymes and breast cancer. Anticancer Res. 2009, 29, 1095–1109. [Google Scholar] [PubMed]
- Hynes, N.E.; Lane, H.A. ErbB receptors and cancer: The complexity of targeted inhibitors. Nat. Rev. Cancer 2005, 5, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, R.L.; Stevens, C.L.; Sridhar, J. Small molecule tyrosine kinase inhibitors of ErbB2/HER2/neu in the treatment of aggressive breast cancer. Molecules 2014, 19, 15196–15212. [Google Scholar] [CrossRef] [PubMed]
- Konecny, G.E.; Pegram, M.D.; Venkatesan, N.; Finn, R.; Yang, G.; Rahmeh, M.; Untch, M.; Rusnak, D.W.; Spehar, G.; Mullin, R.J. Activity of the dual kinase inhibitor lapatinib (GW572016) against HER2-overexpressing and trastuzumab-treated breast cancer cells. Cancer Res. 2006, 66, 1630–1639. [Google Scholar] [CrossRef] [PubMed]
- Xia, W.; Liu, L.-H.; Ho, P.; Spector, N.L. Truncated ErbB2 receptor (p95ErbB2) is regulated by heregulin through heterodimer formation with erbb3 yet remains sensitive to the dual EGFR/ErbB2 kinase inhibitor GW572016. Oncogene 2004, 23, 646–653. [Google Scholar] [CrossRef] [PubMed]
- Xia, W.; Gerard, C.M.; Liu, L.; Baudson, N.M.; Ory, T.L.; Spector, N.L. Combining lapatinib (GW572016), a small molecule inhibitor of ErbB1 and ErbB2 tyrosine kinases, with therapeutic anti-ErbB2 antibodies enhances apoptosis of ErbB2-overexpressing breast cancer cells. Oncogene 2005, 24, 6213–6221. [Google Scholar] [CrossRef] [PubMed]
- O’Donovan, N.; Byrne, A.T.; O’Connor, A.E.; McGee, S.; Gallagher, W.M.; Crown, J. Synergistic interaction between trastuzumab and EGFR/HER-2 tyrosine kinase inhibitors in HER-2 positive breast cancer cells. Investig. New Drugs 2011, 29, 752–759. [Google Scholar] [CrossRef] [PubMed]
- Blackwell, K.; Burstein, H.; Sledge, G.; Stein, S.; Ellis, C.; Casey, M.; Baselga, J.; O’Shaughnessy, J. Updated survival analysis of a randomized study of lapatinib alone or in combination with trastuzumab in women with HER2-positive metastatic breast cancer progressing on trastuzumab therapy. Cancer Res. 2009, 69, 61–61. [Google Scholar] [CrossRef]
- Geyer, C.E.; Forster, J.; Lindquist, D.; Chan, S.; Romieu, C.G.; Pienkowski, T.; Jagiello-Gruszfeld, A.; Crown, J.; Chan, A.; Kaufman, B. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N. Engl. J. Med. 2006, 355, 2733–2743. [Google Scholar] [CrossRef] [PubMed]
- Fiszman, G.L.; Jasnis, M.A. Molecular mechanisms of trastuzumab resistance in HER2 overexpressing breast cancer. Int. J. Breast Cancer 2011, 2011. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, A.J.; Hook, K.E.; Althaus, I.W.; Ellis, P.A.; Trachet, E.; Delaney, A.M.; Harvey, P.J.; Ellis, T.A.; Amato, D.M.; Nelson, J.M. Antitumor activity and pharmacokinetic properties of PF-00299804, a second-generation irreversible pan-ErbB receptor tyrosine kinase inhibitor. Mol. Cancer Ther. 2008, 7, 1880–1889. [Google Scholar] [CrossRef] [PubMed]
- Nam, H.-J.; Ching, K.A.; Kan, J.; Kim, H.-P.; Han, S.-W.; Im, S.-A.; Kim, T.-Y.; Christensen, J.G.; Oh, D.-Y.; Bang, Y.-J. Evaluation of the antitumor effects and mechanisms of PF00299804, a pan-HER inhibitor, alone or in combination with chemotherapy or targeted agents in gastric cancer. Mol. Cancer Ther. 2012, 11, 439–451. [Google Scholar] [CrossRef] [PubMed]
- López-Tarruella, S.; Jerez, Y.; Márquez-Rodas, I.; Martín, M. Neratinib (HKI-272) in the treatment of breast cancer. Future Oncol. 2012, 8, 671–681. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A. Lead-and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Zhou, Y.; Li, J.; Li, W.; Liu, G.; Tang, Y. Prediction of chemical–protein interactions: Multitarget-QSAR versus computational chemogenomic methods. Mol. BioSyst. 2012, 8, 2373–2384. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Liu, C.; Jiang, J.; Lu, W.; Li, W.; Liu, G.; Zhou, W.; Huang, J.; Tang, Y. Prediction of drug-target interactions and drug repositioning via network-based inference. PLoS Comput. Biol. 2012, 8, e1002503. [Google Scholar] [CrossRef] [PubMed]
- Mitrasinovic, P.M. Structural elucidation of unique inhibitory activities of two thiazolo[4,5-d]pyrimidines against epidermal growth factor receptor (EGFR): Implications for successful drug design. Med. Chem. 2014, 10, 46–58. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Hong, S.; Kim, J.; Hong, S. Discovery of picomolar ABL kinase inhibitors equipotent for wild type and T315I mutant via structure-based de novo design. J. Am. Chem. Soc. 2013, 135, 8227–8237. [Google Scholar] [CrossRef] [PubMed]
- Mitrasinovic, P.M. Progress in structure-based design of EGFR inhibitors. Curr. Drug Targets 2013, 14, 817–829. [Google Scholar] [CrossRef] [PubMed]
- Cortopassi, W.A.; Feital, R.J.C.; Medeiros, D.D.J.; Guizado, T.R.C.; França, T.C.C.; Pimentel, A.S. Docking and molecular dynamics studies of new potential inhibitors of the human epidermal receptor 2. Mol. Simul. 2012, 38, 1–11. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the last 25 years. J. Nat. Prod. 2007, 70, 461–477. [Google Scholar] [CrossRef] [PubMed]
- Balani, S.K.; Miwa, G.T.; Gan, L.S.; Wu, J.T.; Lee, F.W. Strategy of utilizing in vitro and in vivo ADME tools for lead optimization and drug candidate selection. Curr. Top. Med. Chem. 2005, 5, 1033–1038. [Google Scholar] [CrossRef] [PubMed]
- AnalytiCon Discovery NP. Available online: zinc.docking.org/catalogs/acdiscnp (accessed on 29 June 2016).
- Cereto-Massagué, A.; Guasch, L.; Valls, C.; Mulero, M.; Pujadas, G.; Garcia-Vallvé, S. DecoyFinder: An easy-to-use python GUI application for building target-specific decoy sets. Bioinformatics 2012, 28, 1661–1662. [Google Scholar] [CrossRef] [PubMed]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of useful decoys, enhanced (DUD-E): Better ligands and decoys for better benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef] [PubMed]
- Lang, P.T.; Brozell, S.R.; Mukherjee, S.; Pettersen, E.F.; Meng, E.C.; Thomas, V.; Rizzo, R.C.; Case, D.A.; James, T.L.; Kuntz, I.D. Dock 6: Combining techniques to model RNA–small molecule complexes. RNA 2009, 15, 1219–1230. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Donini, O.; Reyes, C.M.; Kollman, P.A. Biomolecular simulations: Recent developments in force fields, simulations of enzyme catalysis, protein-ligand, protein-protein, and protein-nucleic acid noncovalent interactions. Annu. Rev. Biophys. Biomol. Struct. 2001, 30, 211–243. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.L.; Wang, Z.J.; Liang, X.M.; Li, X.; Shi, Z.; Zhou, N.; Bao, J.K. In silico identification of novel kinase inhibitors targeting wild-type and T315I mutant ABL1 from FDA-approved drugs. Mol. Biosyst. 2014, 10, 1524–1537. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhou, N.; Cai, P.; Bao, J. In silico screening identifies a novel potential PARP1 inhibitor targeting synthetic lethality in cancer treatment. Int. J. Mol. Sci. 2016, 17, 258. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhou, N.; Liu, W.; Li, J.; Feng, Y.; Wang, X.; Wu, C.; Bao, J. Discover natural compounds as potential phosphodiesterase-4b inhibitors via computational approaches. J. Biomol. Struct. Dyn. 2016, 34, 1101–1112. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhou, N.; Luo, K.; Zhang, W.; Li, X.; Wu, C.; Bao, J. In silico discovery of potential VEGFR-2 inhibitors from natural derivatives for anti-angiogenesis therapy. Int. J. Mol. Sci. 2014, 15, 15994–16011. [Google Scholar] [CrossRef] [PubMed]
- Hassan, S.F.; Rashid, U.; Ansari, F.L.; Ul-Haq, Z. Bioisosteric approach in designing new monastrol derivatives: An investigation on their ADMET prediction using in silico derived parameters. J. Mol. Graph. Model. 2013, 45, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.W.; Fraczkiewicz, R.; Woltosz, W.S. Novel ADMET design tool for chemists. J. Cheminform. 2011, 3, 1–1. [Google Scholar] [CrossRef]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; van der Spoel, D. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.; Postma, J.P.; van Gunsteren, W.F.; Hermans, J. Interaction models for water in relation to protein hydration. In Intermolecular Forces; Springer: Dordrecht, The Netherlands, 1981; pp. 331–342. [Google Scholar]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Prot. Struct. Funct. Bioinform. 2006, 65, 712–725. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general Amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Darden, T.; Cheatham, T.E., III; Simmerling, C.; Wang, J.; Duke, R.E.; Luo, R.; Merz, K.M.; Pearlman, D.A.; Crowley, M.; et al. Amber 9; University of California: San Francisco, CA, USA, 2006. [Google Scholar]
- Hess, B.; Bekker, H.; Berendsen, H.J.; Fraaije, J.G. Lincs: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kollman, P.A. Settle: An analytical version of the shake and rattle algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inform. Model. 2010, 51, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Kumari, R.; Kumar, R.; Lynn, A. g_mmpbsa—A GROMACS tool for high-throughput MM–PBSA calculations. J. Chem. Inform. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Tominaga, H.; Ishiyama, M.; Ohseto, F.; Sasamoto, K.; Hamamoto, T.; Suzuki, K.; Watanabe, M. A water-soluble tetrazolium salt useful for colorimetric cell viability assay. Anal. Commun. 1999, 36, 47–50. [Google Scholar] [CrossRef]
- Shi, Z.; Chen, J.; Li, C.-Y.; An, N.; Wang, Z.-J.; Yang, S.-L.; Huang, K.-F.; Bao, J.-K. Antitumor effects of concanavalin A and Sophora flavescens lectin in vitro and in vivo. Acta Pharmacol. Sin. 2014, 35, 248–256. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rank a | ZINC ID | Structure b | Score (kcal/mol) | |
|---|---|---|---|---|
| Amber Score | Vina Score | |||
| 1 | 31166919 | 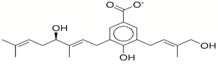 | −44.19 | −10.3 |
| 2 | 15122021 |  | −43.67 | −10.8 |
| 3 | 13378641 | 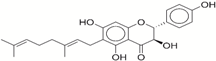 | −43.54 | −10.6 |
| 4 | 72320250 | 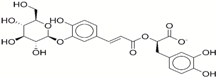 | −35.54 | −10.0 |
| 5 | 49181256 | 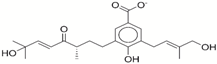 | −35.26 | −9.9 |
| 6 | 35456612 |  | −34.01 | −10.4 |
| 7 | 72320169 | 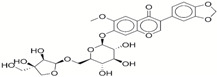 | −33.93 | −9.9 |
| 8 | lapatinib | 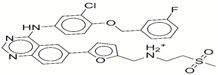 | −32.59 | −10.2 |
| 9 | 35456515 | 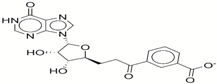 | −33.55 | −10.7 |
| 10 | 35456607 | 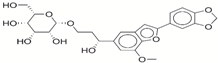 | −32.45 | −10.4 |
| 11 | 72320025 |  | −32.01 | −10.1 |
| 12 | 67912776 | 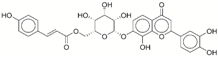 | −31.85 | −10.0 |
| 13 | 44352487 |  | −28.83 | −10.1 |
| 14 | ATP |  | −10.45 | −7.5 |
| ADMET Properties | Molecules | ||||
|---|---|---|---|---|---|
| ZINC13378641 | ZINC15122021 | ZINC35456515 | ZINC31166919 | ZINC49181256 | |
| S + logP | 4.73 | 5.01 | −0.01 | 3.92 | 2.92 |
| S + Sw | 1.10 | 5.97 | 1.08 | 1.00 | 1.11 |
| S + Vd | 0.7 | 0.46 | 0.26 | 0.17 | 0.13 |
| CYP_1A2_Substr | No (66%) | No (59%) | No (96%) | No (96%) | No (96%) |
| MET_UGT1A1 | No (59%) | Yes (58%) | No (92%) | No (88%) | No (82%) |
| TOX_hERG_Filter | No (95%) | No (95%) | No (95%) | No (95%) | No (95%) |
| TOX_BRM_Rat | 289.81 | 522.34 | 9.89 | 200.29 | 206.47 |
| TOX_AlkPhos | Normal (60%) | Normal (74%) | Elevated (65%) | Elevated (97%) | Elevated (97%) |
| TOX_GGT | Normal (78%) | Normal (78%) | Normal (57%) | Elevated (77%) | Elevated (90%) |
| TOX_LDH | Normal (76%) | Normal (76%) | Elevated (75%) | Normal (70%) | Normal (96%) |
| RO5 | 0 | 0 | 0 | 0 | 0 |
| TOX_MUT_Risk | 0 | 0 | 0 | 0 | 0 |
| ADMET Risk | 3.36 | 3.81 | 4.5 | 5 | 5 |
| Components | Molecules | |||||
|---|---|---|---|---|---|---|
| ZINC31166919 | ZINC15122021 | ZINC49181256 | ZINC13378641 | ZINC35456515 | Lapatinib | |
| ∆Evdw | −56.57 ± 1.95 | −63.46 ± 2.58 | −53.88 ± 0.82 | −51.56 ± 2.84 | −57.82 ± 3.09 | −51.02 ± 3.39 |
| ∆Eele | −130.90 ± 7.19 | −109.18 ± 6.70 | −39.73 ± 0.77 | −1.58 ± 5.65 | −17.31 ± 10.65 | −26.03 ± 8.63 |
| ∆Gpb | 61.47 ± 5.13 | 57.30 ± 3.87 | 44.88 ± 1.18 | 24.14 ± 5.35 | 49.84 ± 7.13 | 45.25 ± 5.22 |
| ∆Gsa | −5.36 ± 0.19 | −5.30 ± 0.21 | −5.74 ± 0.06 | −5.37 ± 0.18 | −5.76 ± 0.21 | −5.69 ± 0.21 |
| ∆Emm | −187.47 ± 4.57 | −172.63 ± 4.64 | −93.61 ± 1.59 | −49.99 ± 4.24 | −75.84 ± 6.87 | −77.05 ± 6.01 |
| ∆Gsol | 56.11 ± 5.32 | 52.00 ± 3.66 | 39.04 ± 1.12 | 18.77 ± 5.17 | 44.08 ± 6.92 | 39.56 ± 5.01 |
| ∆Gbind | −131.36 ± 6.63 | −120.63 ± 5.18 | −54.44 ± 0.84 | −31.22 ± 3.89 | −31.05 ± 6.23 | −37.49 ± 5.46 |
| Compounds | Enzymatic IC50 | Cell Inhibition IC50 | |
|---|---|---|---|
| SKBR3 | BT474 | ||
| ZINC31166919 | 2.63 ± 0.03 | 8.61 ± 0.45 | 6.78 ± 0.68 |
| ZINC15122021 | 0.18 ± 0.002 | 1.22 ± 0.05 | 4.11 ± 0.95 |
| ZINC49181256 | 9.18 ± 0.01 | >50 | >50 |
| ZINC13378641 | 3.71 ± 0.03 | 26.48 ± 1.62 | 18.55 ± 2.06 |
| ZINC35456515 | >10 | >50 | >50 |
| lapatinib | 0.06 ± 0.001 | 0.38 ± 0.02 | 0.45 ± 0.03 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Wang, H.; Li, J.; Bao, J.; Wu, C. Discovery of a Potential HER2 Inhibitor from Natural Products for the Treatment of HER2-Positive Breast Cancer. Int. J. Mol. Sci. 2016, 17, 1055. https://doi.org/10.3390/ijms17071055
Li J, Wang H, Li J, Bao J, Wu C. Discovery of a Potential HER2 Inhibitor from Natural Products for the Treatment of HER2-Positive Breast Cancer. International Journal of Molecular Sciences. 2016; 17(7):1055. https://doi.org/10.3390/ijms17071055
Chicago/Turabian StyleLi, Jianzong, Haiyang Wang, Junjie Li, Jinku Bao, and Chuanfang Wu. 2016. "Discovery of a Potential HER2 Inhibitor from Natural Products for the Treatment of HER2-Positive Breast Cancer" International Journal of Molecular Sciences 17, no. 7: 1055. https://doi.org/10.3390/ijms17071055
APA StyleLi, J., Wang, H., Li, J., Bao, J., & Wu, C. (2016). Discovery of a Potential HER2 Inhibitor from Natural Products for the Treatment of HER2-Positive Breast Cancer. International Journal of Molecular Sciences, 17(7), 1055. https://doi.org/10.3390/ijms17071055