
Spatial Partitioning of miRNAs Is Related to Sequence Similarity in Overall Transcriptome

Abstract
:
1. Introduction
2. Results
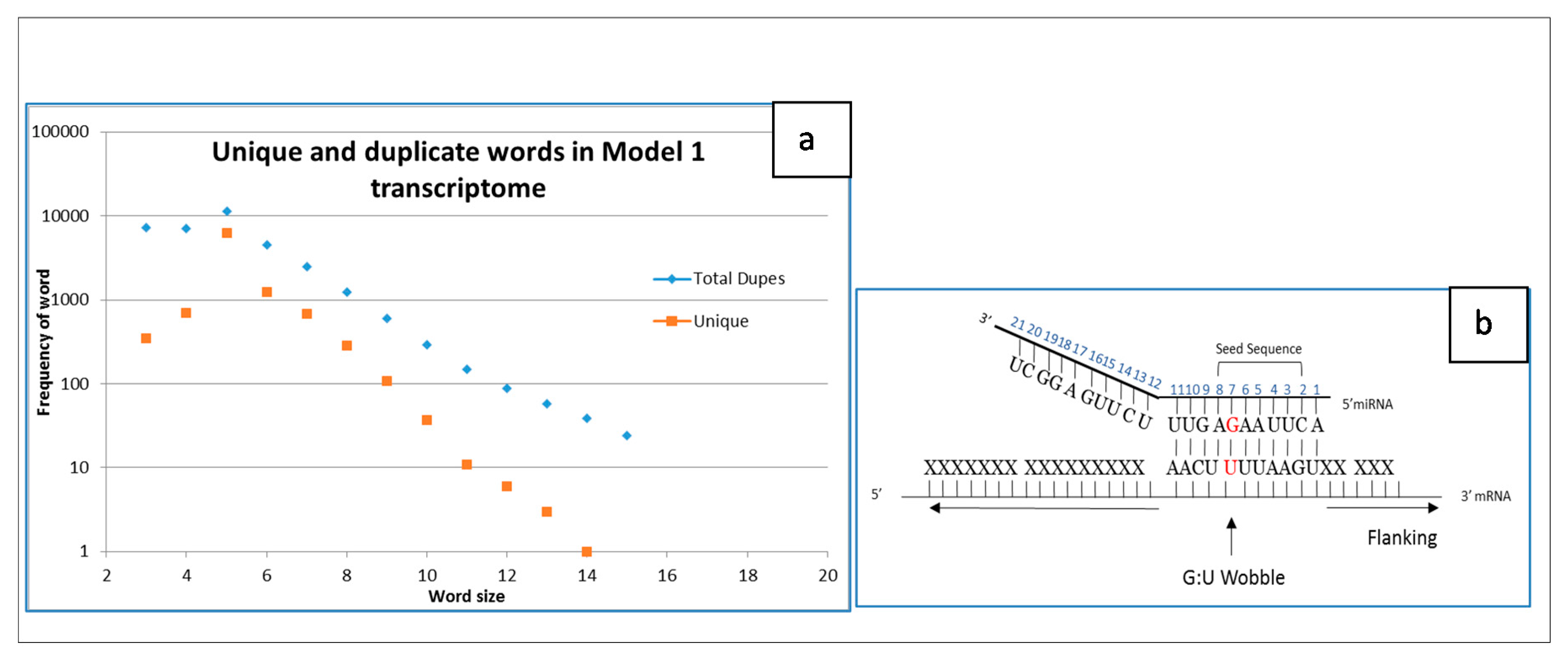
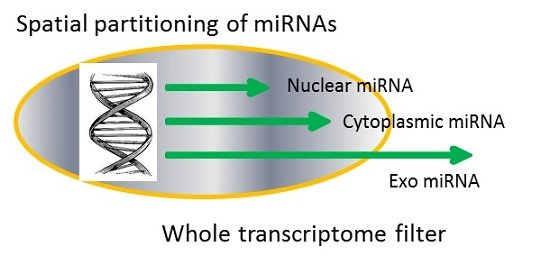
2.1. Whole Transcriptome as an Information Cloud of Sequence Words
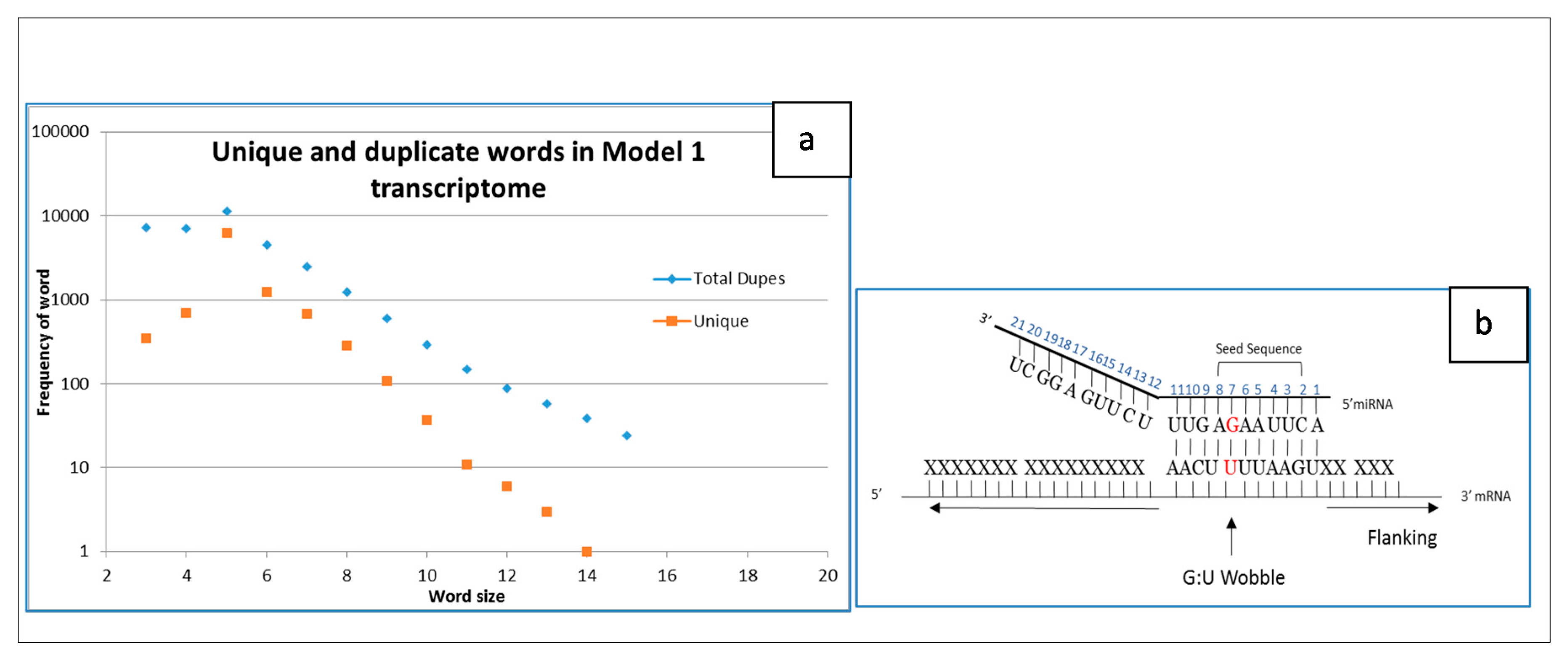
2.2. Simple Transcriptome Model
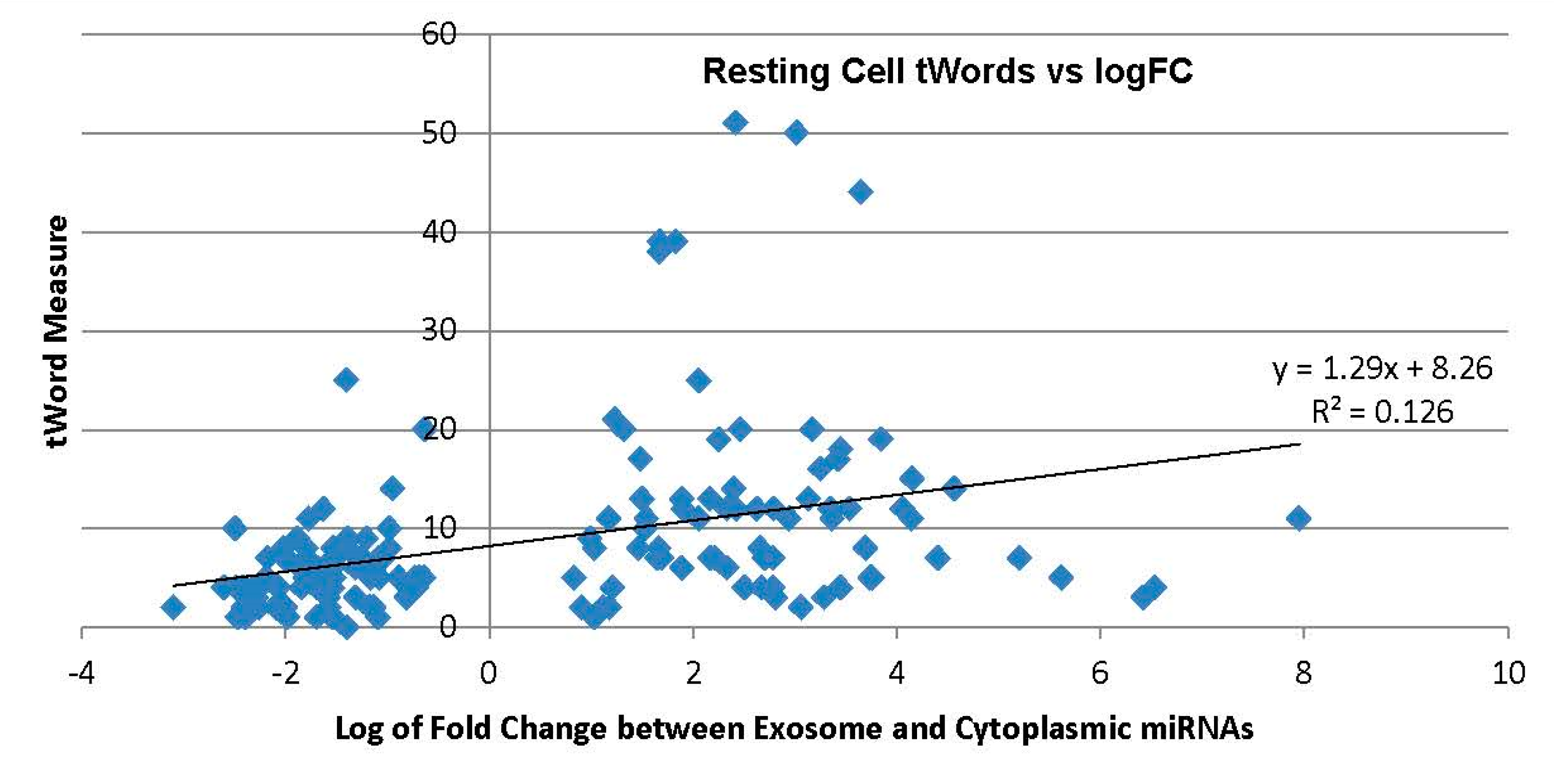
2.3. miRNA Datasets Examined with Simple Model Transcriptome
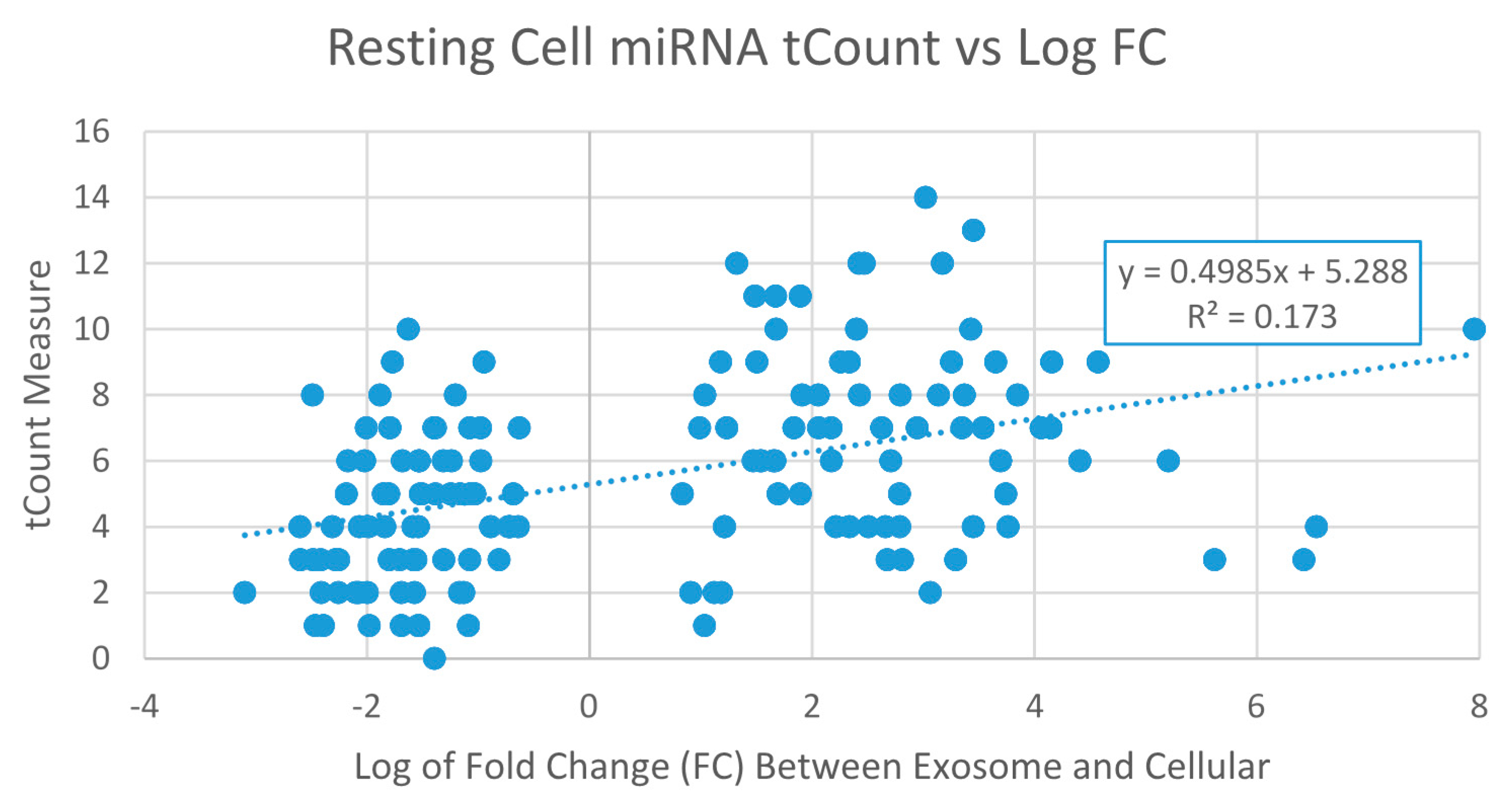
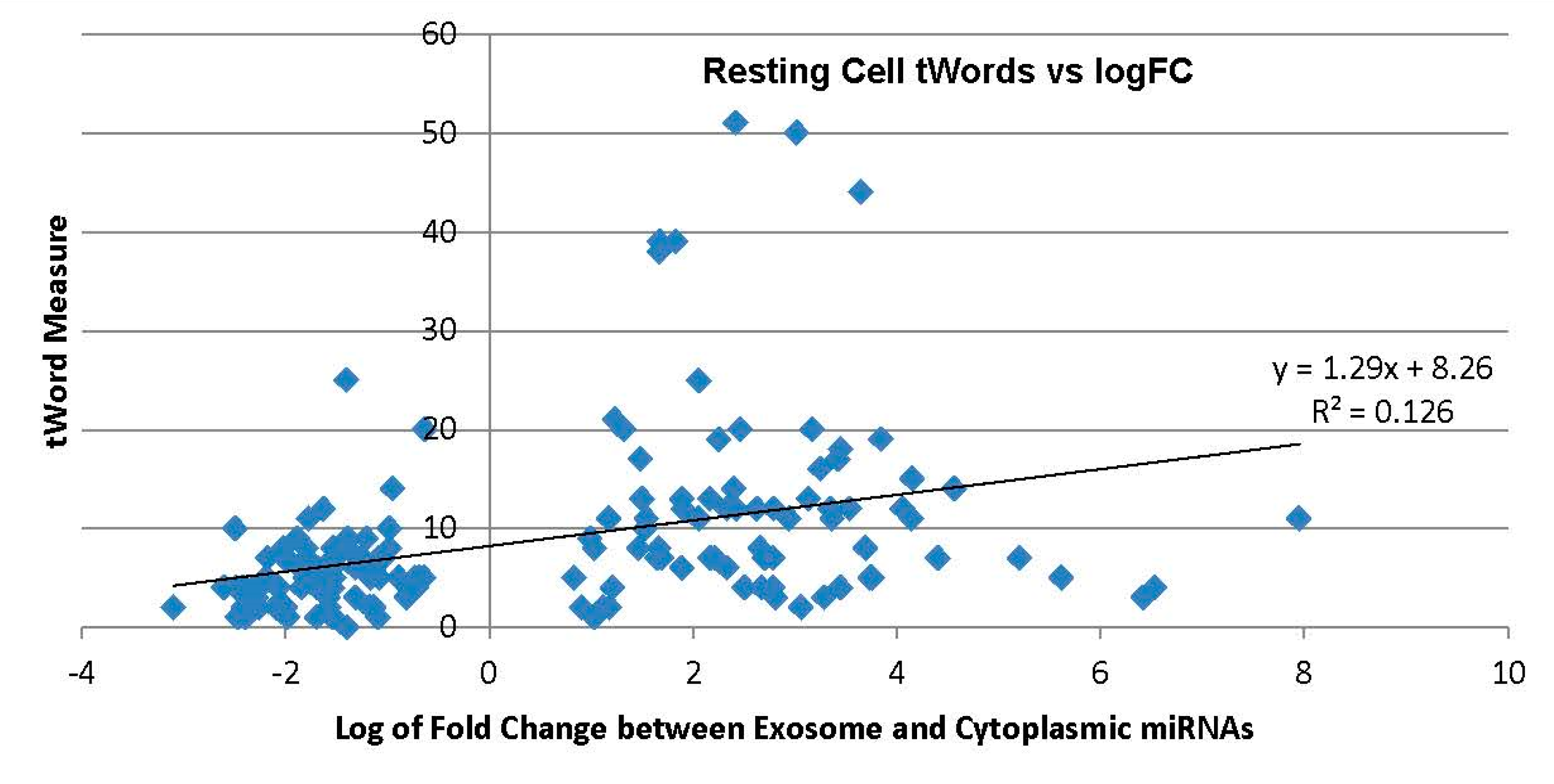
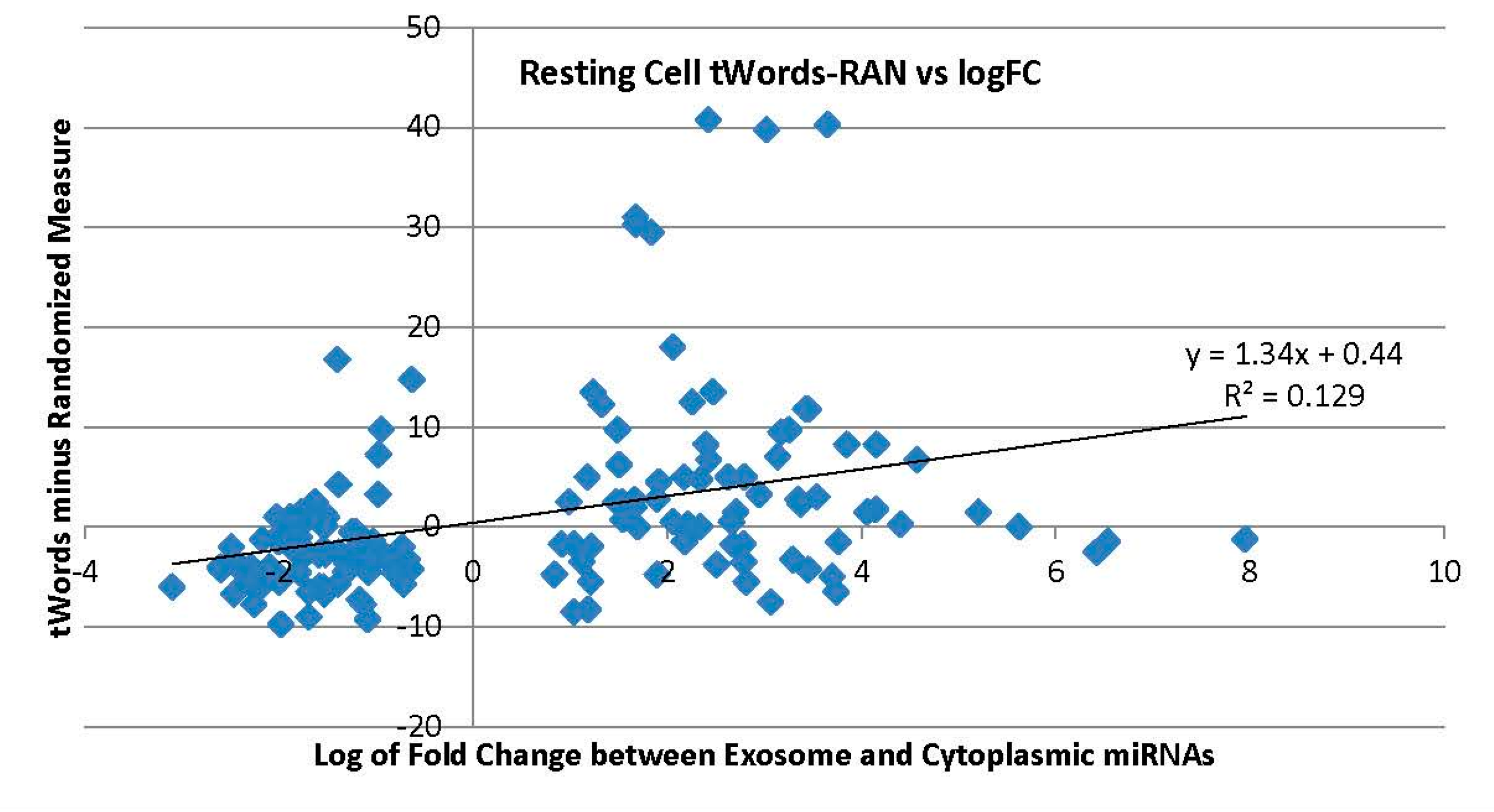
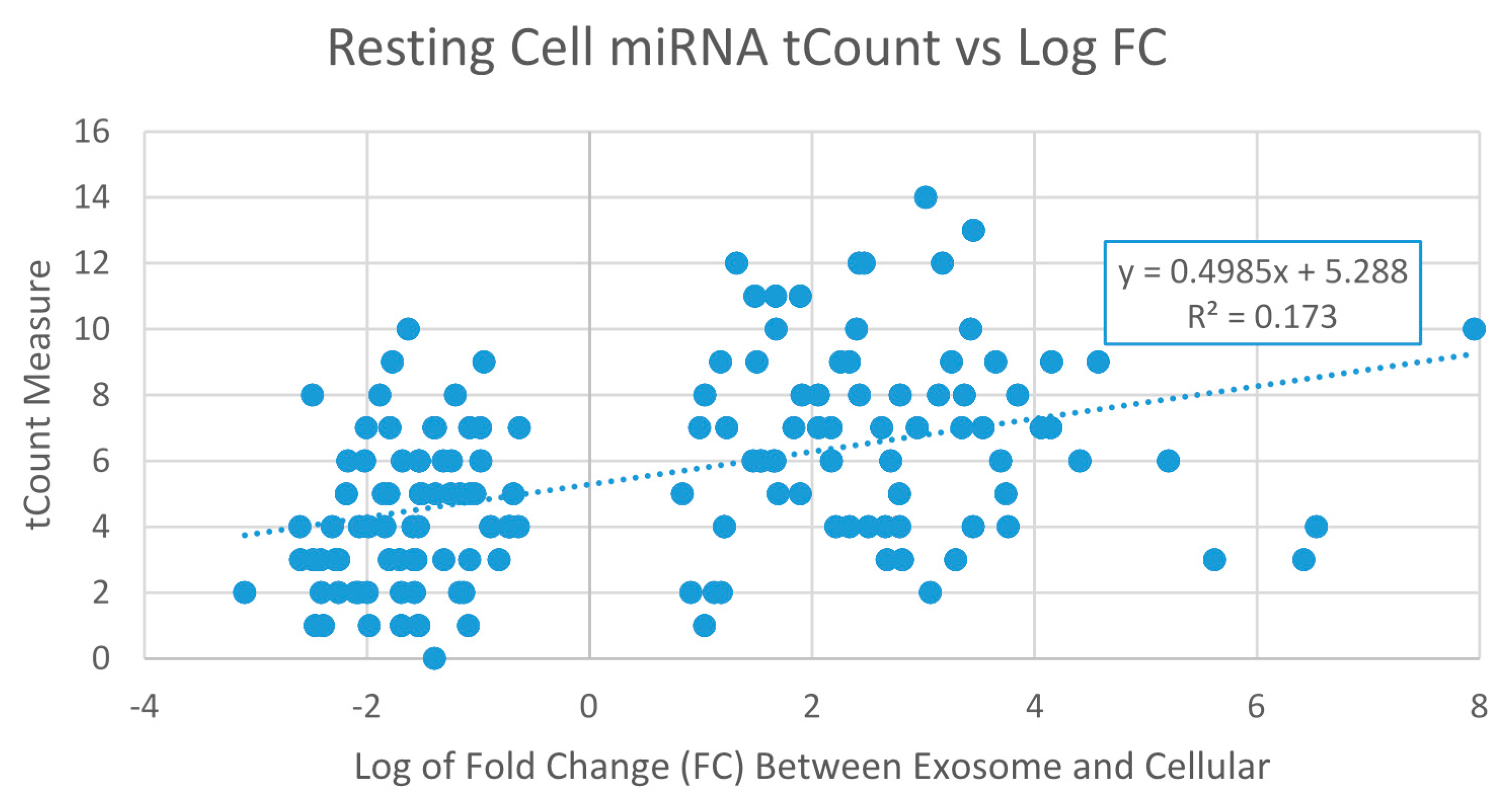
2.3.1. Exosome Enriched miRNAs
2.3.2. Nuclear Enriched miRNAs
2.3.3. Other miRNA Studies
3. Discussion
4. Materials and Methods
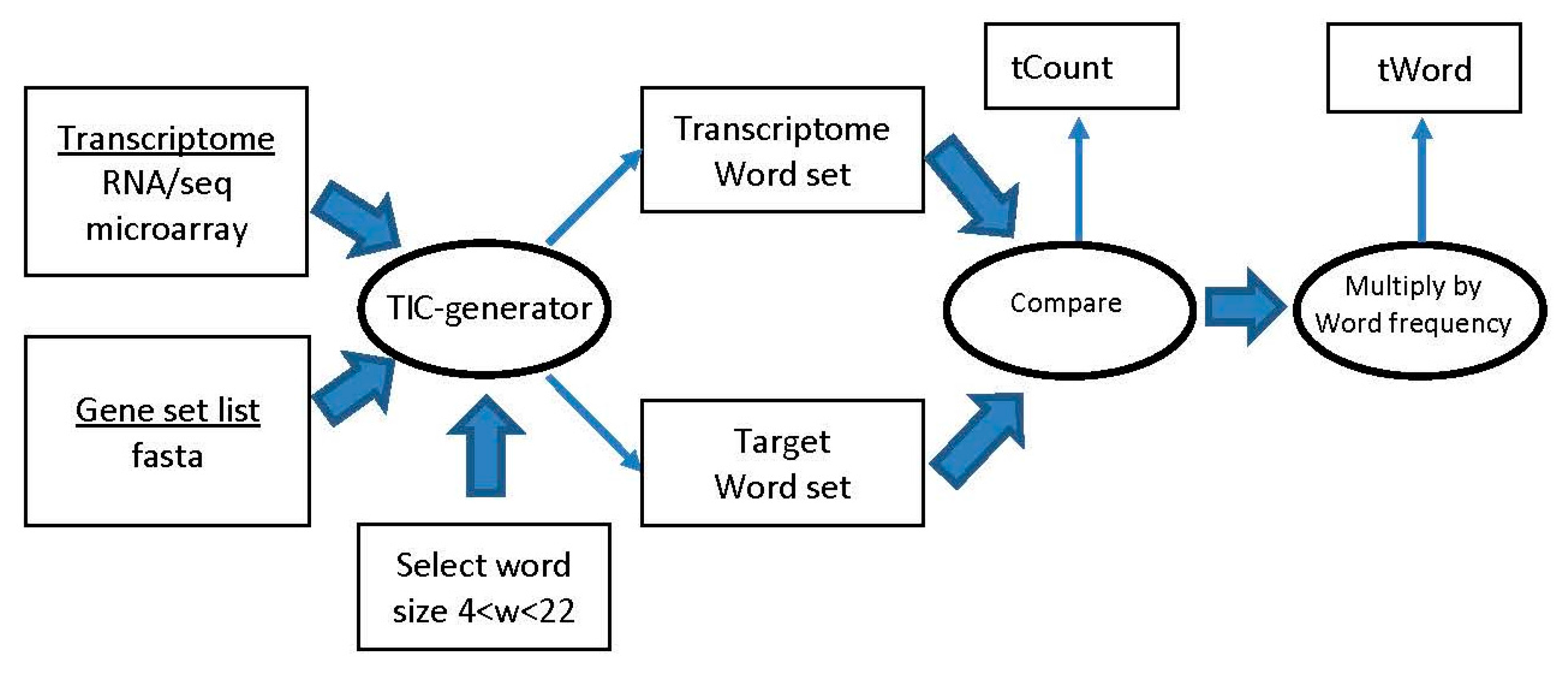
4.1. Sliding Window Word Generator
4.2. Duplicate Words in Cloud
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| miRNA | microRNA |
| nt | nucleotides |
| ceRNA | competitive endogenous RNA |
| ncRNA | non-coding RNA |
References
- Collins, L.J. The RNA infrastructure: An introduction to ncRNA networks. Adv. Exp. Med. Biol. 2011, 722, 1–19. [Google Scholar] [PubMed]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P. A ceRNA hypothesis: The rosetta stone of a hidden RNA language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Trovato, F.; Tozzini, V. Diffusion within the cytoplasm: A mesoscale model of interacting macromolecules. Biophys. J. 2014, 107, 2579–2591. [Google Scholar] [CrossRef] [PubMed]
- Ben-Ari, Y.A.; Brody, Y.; Kinor, N.; Mor, A.; Tsukamoto, T.; Spector, D.L.; Singer, R.H.; Shav-Tal, Y. The life of an mRNA in space and time. J. Cell Sci. 2010, 123, 1761–1774. [Google Scholar] [CrossRef] [PubMed]
- Mayorga, M.; Romero-Salazar, L.; Rubi, J. Stochastic model for the dynamics of interacting Brownian particles. Phys. A 2002, 307, 297–314. [Google Scholar] [CrossRef]
- Savel’ev, S.; Marchesoni, F.; Taloni, A.; Nori, F. Diffusion of interacting Brownian particles: Jamming and anomalous diffusion. Phys. Rev. E 2006, 74, 021119. [Google Scholar] [CrossRef] [PubMed]
- Ekiel-Jezewska, M.; Felderhof, B. Clusters of particles falling in a viscous fluid with periodic boundary conditions. Phys. Fluids 2006, 18, 121502. [Google Scholar] [CrossRef]
- Wang, X.-Q.; Abebe, F.; Seffens, W. Dynamic system modeling the whole transcriptome in a eukaryotic cell. Proc. Dyn. Syst. Appl. 2016, in press. [Google Scholar]
- Regner, B.; Vucinic, D.; Domnisoru, C.; Bartol, T.; Hetzer, M.; Tartakovsky, D.; Sejnowski, T. Anomalous diffusion of single particles in cytoplasm. Biophys. J. 2013, 104, 1652–1660. [Google Scholar] [CrossRef] [PubMed]
- Wang, X. Composition of seed sequence is a major determinant of microRNA targeting patterns. Bioinformatics 2014, 30, 1377–1383. [Google Scholar] [CrossRef] [PubMed]
- Carninci, P.; Kasukawa, T. The FANTOM Consortium. The transcriptional landscape of the mammalian genome. Science 2005, 309, 1559–1563. [Google Scholar] [PubMed]
- Villarroya-Beltri, C.; Gutiérrez-Vázquez, C.; Sánchez-Cabo, F.; Pérez-Hernández, D.; Vázquez, J.; Martin-Cofreces, N.; Jorge Martinez-Herrera, D.; Pascual-Montano, A.; Mittelbrunn, M.; Sánchez-Madrid, F. Sumoylated hnRNPA2B1 controls the sorting of miRNAs into exosomes through binding to specific motifs. Nat. Commun. 2013, 4, 2980. [Google Scholar] [CrossRef] [PubMed]
- Park, C.W.; Zeng, Y.; Zhang, X.; Subramanian, S.; Steer, C. Mature microRNAs identified in highly purified nuclei from HCT116 colon cancer cells. RNA Biol. 2010, 7, 606–614. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Yuan, T.; Tschannen, M.; Sun, Z.; Jacob, H.; Du, M.; Liang, M.; Dittmar, R.L.; Liu, Y.; Liang, M.; et al. Characterization of human plasma-derived exosomal RNAs by deep sequencing. BMC Genom. 2013, 14, 319. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Sharples, R.A.; Scicluna, B.J.; Hill, A.F. Exosomes provide a protective and enriched source of miRNA for biomarker profiling compared to intracellular and cell-free blood. J. Extracell. Vesicles 2014, 3, 23743. [Google Scholar] [CrossRef] [PubMed]
- Guduric-Fuchs, J.; O’Connor, A.; Camp, B.; O’Neill, C.L.; Medina, R.J.; Simpson, D.A. Selective extracellular vesicle-mediated export of an overlapping set of microRNAs from multiple cell types. BMC Genom. 2012, 13, 357. [Google Scholar] [CrossRef] [PubMed]
- Seffens, W.; Digby, D. mRNAs Have greater calculated folding free energies than shuffled or codon choice randomized sequences. Nucleic Acids Res. 1999, 27, 1578–1584. [Google Scholar] [CrossRef] [PubMed]
- Jeggari, A.; Marks, D.S.; Larsson, E. MiRcode: A map of putative microRNA target sites in the long non-coding transcriptome. Bioinformatics 2012, 28, 2062–2063. [Google Scholar] [CrossRef] [PubMed]
- Jalali, S.; Bhartiya, D.; Lawani, M.; Sivasubbu, S.; Scaria, V. Systematic transcriptome-wide analysis of lncRNA-miRNA interactions. PLoS ONE 2013, 8, e53823. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.; Digby, D.; Davis, A.; Seffens, W. Whole transcriptome mRNA secondary structure analysis using distributed computation. In Proceedings of the International IEEE-Granular Computing 2006, Atlanta, GA, USA, 10–12 May 2006; pp. 647–650.
- Lockhart, E.; Yoo, J.; Seffens, W. Comparison of Transcriptomes by mRNA Folding Free Energies. In Proceedings of the 5th International Symposium Bioinformatics Research and Applications (ISBRA 2009), Fort Lauderdale, FL, USA, 13–16 May 2009.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Transcript Molecule | Size (nt) | Abundance (Copies) | Distinct Types | Notes |
|---|---|---|---|---|
| 28S rRNA | 5070 | 3.5 × 106 | 1 | Subunit in 80S ribosome |
| 18S rRNA | 1869 | 3.5 × 106 | 1 | Subunit in 80S ribosome |
| 5.8S rRNA | 156 | 3.5 × 106 | 1 | Subunit in 80S ribosome |
| 5S rRNA | 121 | 3.5 × 106 | 1 | Subunit in 80S ribosome |
| tRNA | ~85 | 3 × 107 | ~100 | 497 genes in 40 families, tissue specific |
| mRNA | 2 kb | 4 × 105 | 4 × 105 | Tissue specific, many isoforms |
| ncRNA | >200 | variable | >35,000 | Complex isoforms [11] |
| miRNA | 22 | variable | 1000 |
| Transcriptome Measures for Published Data Sets of miRNA | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Data Set | tCount | RANtCount | tWord | RANtWord | tW–tC | tC–RAN | tW–RAN | tC Z | tW Z | N |
| has-miR- | 5.3 (2.9) | 5.7 | 9 (10.9) | 7.1 (1.9) | 3.7 (9.0) | −0.4 (3.2) | 1.9 (1.1) | −0.2 (2.4) | 1.1 (7.3) | 2588 |
| V-B All | 5.5 (2.9) | 6.1 (1.3) | 8.9 (8.8) | 7.8 (1.9) | 3.4 (6.8) | −0.6 (3.1) | 1.1 (9.0) | −0.3 (2.0) | 0.6 (4.6) | 151 |
| V-B EXO | 6.8 (2.9) | 6 | 12.5 (10.7) | 7.7 | 5.7 (8.8) | 0.8 (2.9) | 4.8 (10.7) | 0.4 (2.2) | 2.2 (5.8) | 75 |
| V-B CL | 4.3 (2.2) | 6.3 | 5.5 (4.0) | 8 | 1.2 (2.6) | −2.0 (2.7) | −2.4 (4.6) | −1.1 (1.5) | −1.0 (1.9) | 76 |
| Park All | 4.4 | 7.2 | 2.8 | −1.7 | −0.4 | −1.3 | −0.3 | 78 | ||
| Park NU | 4.0 (2.0) | 4.7 (2.6) | 0.7 (1.1) | −2.3 (2.3) | −3.2 (2.9) | −1.6 (1.8) | −1.4 (1.4) | 45 | ||
| Park CL | 5.0 (3.3) | 10.6 (16.4) | 5.6 (13.7) | −1.0 (3.7) | 3.2 (16.8) | −0.8 (2.4) | 1.1 (6.2) | 33 | ||
| G-F All | 5.4 (3.5) | 8.5 (8.6) | 3.1 (6.0) | 27 | ||||||
| G-F EXO | 7.9 (4.1) | 13.5 (11.5) | 5.6 (8.7) | 10 | ||||||
| G-F CL | 4.0 (2.2) | 6.1 (4.9) | 2.1 (3.2) | 10 | ||||||
| w = 7 | Transcript | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | Total | Unique | Duplicates |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Transcript | nt length | 73 | 73 | 71 | 71 | 156 | 121 | 5034 | 1871 | 7470 | ||
| Word size | ||||||||||||
| Total words | w = 7 | 67 | 67 | 65 | 65 | 150 | 115 | 5028 | 1865 | 7422 | ||
| Unique words | w = 7 | 67 | 67 | 65 | 65 | 149 | 114 | 3355 | 1742 | 5625 | 4934 | 691 |
| Duplicates | w = 7 | 0 | 0 | 0 | 0 | 1672 | 123 | 1797 | ||||
| Word size | ||||||||||||
| Total words | w = 8 | 66 | 66 | 64 | 64 | 149 | 114 | 5013 | 1864 | 7400 | ||
| Unique words | w = 8 | 66 | 66 | 64 | 64 | 149 | 114 | 4085 | 1831 | 6439 | 6439 | 288 |
| Duplicates | w = 8 | 0 | 0 | 0 | 0 | 0 | 0 | 928 | 33 | 961 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seffens, W.; Abebe, F.; Evans, C.; Wang, X.-Q. Spatial Partitioning of miRNAs Is Related to Sequence Similarity in Overall Transcriptome. Int. J. Mol. Sci. 2016, 17, 830. https://doi.org/10.3390/ijms17060830
Seffens W, Abebe F, Evans C, Wang X-Q. Spatial Partitioning of miRNAs Is Related to Sequence Similarity in Overall Transcriptome. International Journal of Molecular Sciences. 2016; 17(6):830. https://doi.org/10.3390/ijms17060830
Chicago/Turabian StyleSeffens, William, Fisseha Abebe, Chad Evans, and Xiao-Qian Wang. 2016. "Spatial Partitioning of miRNAs Is Related to Sequence Similarity in Overall Transcriptome" International Journal of Molecular Sciences 17, no. 6: 830. https://doi.org/10.3390/ijms17060830
APA StyleSeffens, W., Abebe, F., Evans, C., & Wang, X.-Q. (2016). Spatial Partitioning of miRNAs Is Related to Sequence Similarity in Overall Transcriptome. International Journal of Molecular Sciences, 17(6), 830. https://doi.org/10.3390/ijms17060830