
Role of Mitochondrial DNA Copy Number Alteration in Human Renal Cell Carcinoma †

Abstract
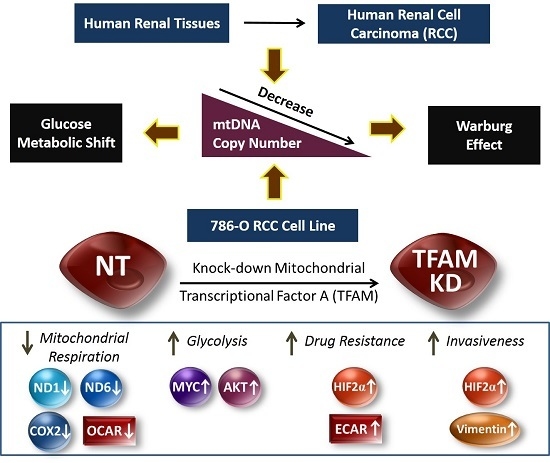
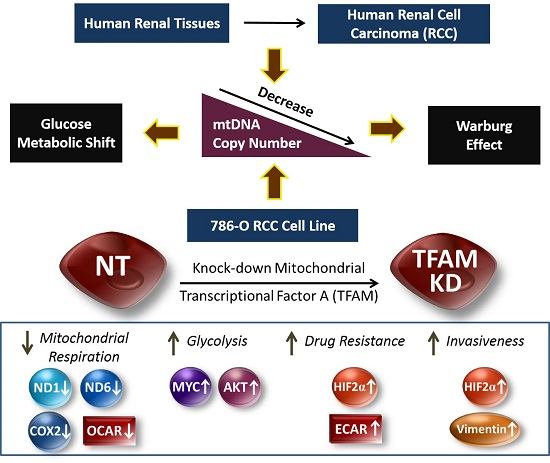
:
1. Introduction
2. Results
2.1. Decrease of mtDNA Copy Number in Human RCC Tissues
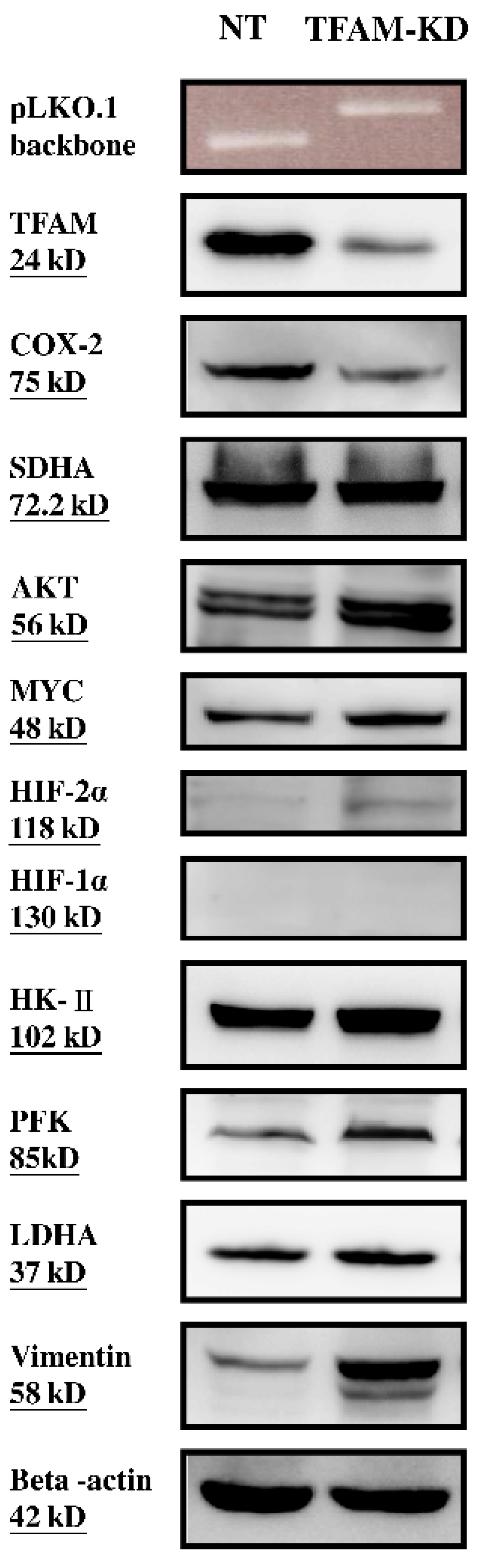
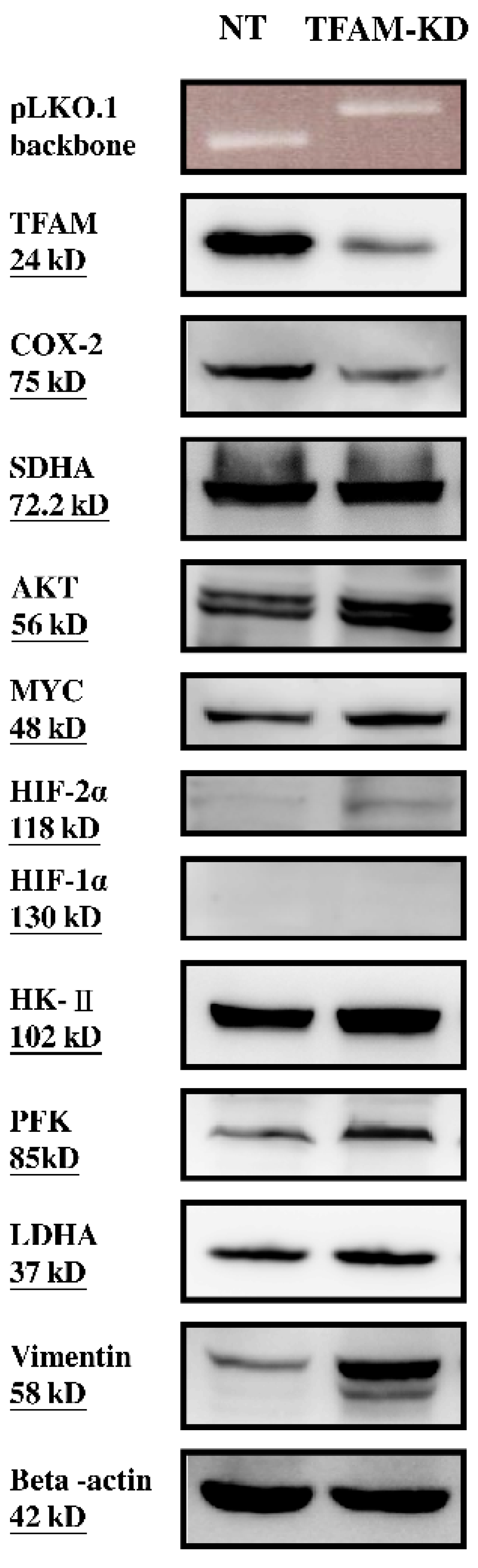
2.2. Lower mtDNA Copy Number and Lower Expression Levels of mtDNA-Encoded Polypeptides of Respiratory Enzymes in the TFAM-KD Clone
2.3. Higher Expressions of Glycolytic Enzymes in the TFAM-KD Clone
2.4. Alterations of Proteins Related to HIF Pathway in the TFAM-KD Clone
2.5. Higher Expression of AKT- and cMYC-Encoded Proteins that Enhance Warburg Effect in the TFAM-KD Clone
2.6. Lower OCR and Higher ECAR of Cellular Metabolism in the TFAM-KD Clone
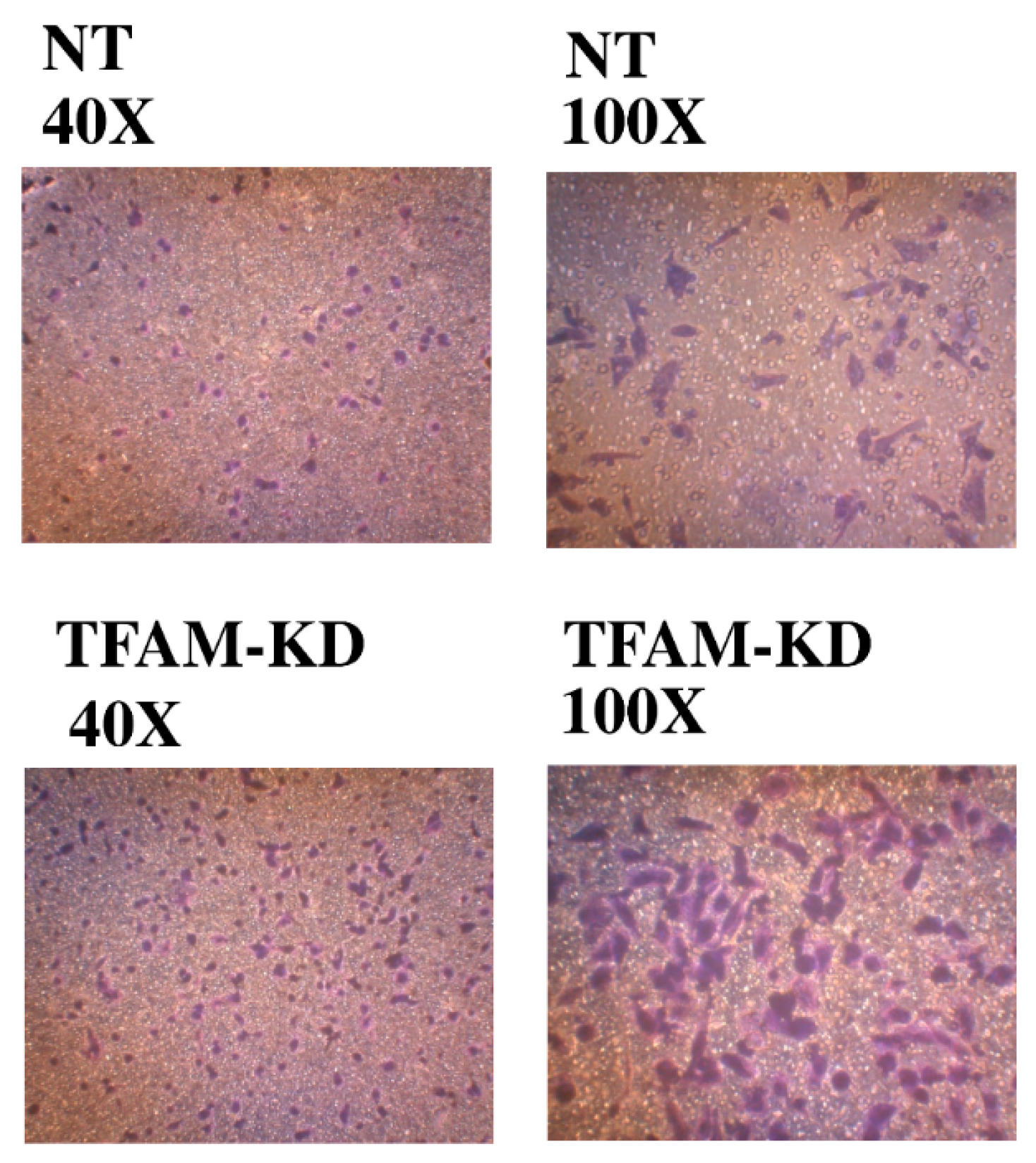
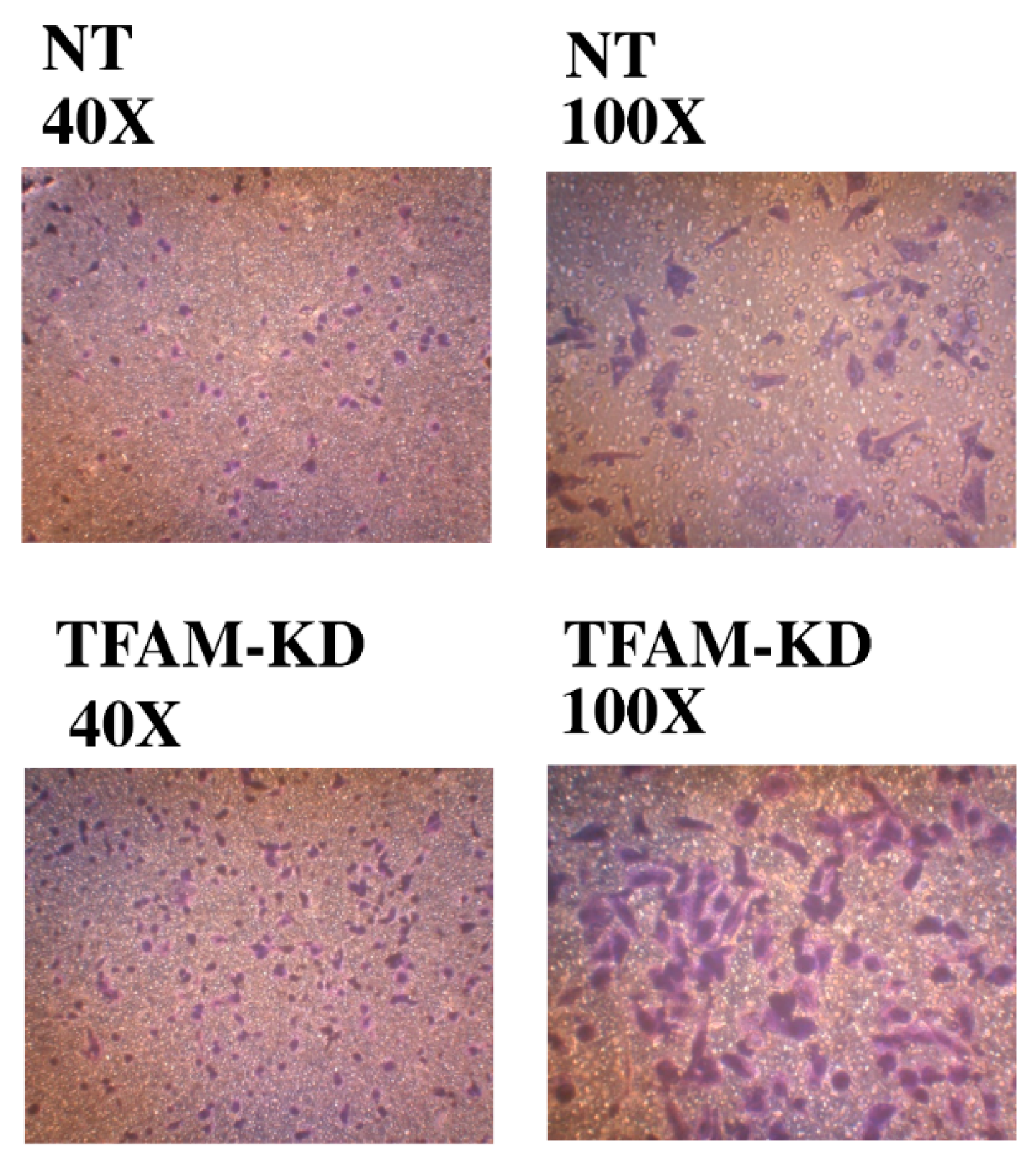
2.7. Higher Trans-Well Migration Activity and Vimentin Expression in the TFAM-KD Clone
2.8. Higher Drug Resistance to Doxorubicin in the TFAM-KD Clone
3. Discussion
4. Materials and Methods
4.1. Collection of Clinical Samples and DNA Extraction
4.2. RCC Cell Line
4.3. Viral Infection to Knockdown TFAM Expression
4.4. DNA, RNA, and Protein Extractions
4.5. Confirmation of pLKO.1-NT Vector in NT Clone and pLKO.1-sh-TFAM Vector in the TFAM-KD Clone
4.6. Analysis of mtDNA Copy Number, mRNA and Protein Expression Levels
4.7. Analysis of Bioenergetic Parameters by the XFe-24 Analyzer
4.8. Trans-Well Migration Activity Assay
4.9. Drug Resistance to Doxorubicin
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bratic, I.; Trifunovic, A. Mitochondrial energy metabolism and ageing. Biochim. Biophys. Acta 2010, 1797, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C.; Wei, Y.H. Mitochondrial role in life and death of the cell. J. Biomed. Sci. 2000, 7, 2–15. [Google Scholar] [CrossRef] [PubMed]
- Rafelski, S.M. Mitochondrial network morphology: Building an integrative, geometrical view. BMC Biol. 2013, 11, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Busch, K.B.; Kowald, A.; Spelbrink, J.N. Quality matters: How does mitochondrial network dynamics and quality control impact on mtDNA integrity? Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C. Mitochondria: Dynamic organelles in disease, aging, and development. Cell 2006, 125, 1241–1252. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C.; Wei, Y.H. Mitochondrial biogenesis and mitochondrial DNA maintenance of mammalian cells under oxidative stress. Int. J. Biochem. Cell Biol. 2005, 37, 822–834. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C.; Wei, Y.H. Oxidative stress, mitochondrial DNA mutation, and apoptosis in aging. Exp. Biol. Med. 2007, 232, 592–606. [Google Scholar]
- Moraes, C.T. What regulates mitochondrial DNA copy number in animal cells? Trends Genet. 2001, 17, 199–205. [Google Scholar] [CrossRef]
- Asin-Cayuela, J.; Gustafsson, C.M. Mitochondrial transcription and its regulation in mammalian cells. Trends Biochem. Sci. 2007, 32, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V.; Semenza, G.L. Oncogenic alterations of metabolism. Trends Biochem. Sci. 1999, 24, 68–72. [Google Scholar] [CrossRef]
- Zeng, W.; Liu, P.; Pan, W.; Singh, S.R.; Wei, Y. Hypoxia and hypoxia inducible factors in tumor metabolism. Cancer Lett. 2015, 356, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Dang, C.V. Cancer’s molecular sweet tooth and the Warburg effect. Cancer Res. 2006, 66, 8927–8930. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C.; Wei, Y.H. Mitochondrial DNA instability and metabolic shift in human cancers. Int. J. Mol. Sci. 2009, 10, 674–701. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-S.; Wang, L.-S. Mitochondrial DNA instability in human cancers. Formos. J. Surg. 2013, 46, 71–75. [Google Scholar] [CrossRef]
- Reznik, E.; Miller, M.L.; Senbabaoglu, Y.; Riaz, N.; Sarungbam, J.; Tickoo, S.K.; Al-Ahmadie, H.A.; Lee, W.; Seshan, V.E.; Hakimi, A.A.; et al. Mitochondrial DNA copy number variation across human cancers. eLife 2016. [Google Scholar] [CrossRef] [PubMed]
- Cheau-Feng Lin, F.; Jeng, Y.C.; Huang, T.Y.; Chi, C.S.; Chou, M.C.; Chin-Shaw Tsai, S. Mitochondrial DNA copy number is associated with diagnosis and prognosis of head and neck cancer. Biomarkers 2014, 19, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.M.; Clinger, J.D.; Masayesva, B.G.; Ha, P.K.; Zahurak, M.L.; Westra, W.H.; Califano, J.A. Mitochondrial DNA quantity increases with histopathologic grade in premalignant and malignant head and neck lesions. Clin. Cancer Res. 2004, 10, 8512–8515. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.S.; Chang, S.C.; Wang, L.S.; Chou, T.Y.; Hsu, W.H.; Wu, Y.C.; Wei, Y.H. The role of mitochondrial DNA alterations in esophageal squamous cell carcinomas. J. Thorac. Cardiovasc. Surg. 2010, 139, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.S.; Wang, L.S.; Chou, T.Y.; Hsu, W.H.; Lin, H.C.; Lee, S.Y.; Lee, M.H.; Chang, S.C.; Wei, Y.H. Cigarette smoking and hOGG1 Ser326Cys polymorphism are associated with 8-OHdG accumulation on mitochondrial DNA in thoracic esophageal squamous cell carcinoma. Ann. Surg. Oncol. 2013, 20 (Suppl. S3), S379–S388. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.S.; Wang, L.S.; Chang, S.C.; Chou, T.Y.; Hsu, W.H.; Liu, C.S.; Lee, M.H.; Chung, M.Y.; Wei, Y.H. Associated microsatellite alterations in mitochondrial DNA and in TP53 in thoracic esophageal squamous cell carcinoma. Oncol. Rep. 2012, 28, 69–76. [Google Scholar] [PubMed]
- Lee, H.C.; Yin, P.H.; Lin, J.C.; Wu, C.C.; Chen, C.Y.; Wu, C.W.; Chi, C.W.; Tam, T.N.; Wei, Y.H. Mitochondrial genome instability and mtDNA depletion in human cancers. Ann. N. Y. Acad. Sci. 2005, 1042, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Tseng, L.M.; Yin, P.H.; Chi, C.W.; Hsu, C.Y.; Wu, C.W.; Lee, L.M.; Wei, Y.H.; Lee, H.C. Mitochondrial DNA mutations and mitochondrial DNA depletion in breast cancer. Genes Chromosomes Cancer 2006, 45, 629–638. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.W.; Yin, P.H.; Hung, W.Y.; Li, A.F.; Li, S.H.; Chi, C.W.; Wei, Y.H.; Lee, H.C. Mitochondrial DNA mutations and mitochondrial DNA depletion in gastric cancer. Genes Chromosomes Cancer 2005, 44, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.S.; Wang, L.S.; Tsai, C.M.; Wei, Y.H. Low copy number and low oxidative damage of mitochondrial DNA are associated with tumor progression in lung cancer tissues after neoadjuvant chemotherapy. Interact. Cardiovasc. Thorac. Surg. 2008, 7, 954–958. [Google Scholar] [CrossRef] [PubMed]
- Yin, P.H.; Lee, H.C.; Chau, G.Y.; Wu, Y.T.; Li, S.H.; Lui, W.Y.; Wei, Y.H.; Liu, T.Y.; Chi, C.W. Alteration of the copy number and deletion of mitochondrial DNA in human hepatocellular carcinoma. Br. J. Cancer 2004, 90, 2390–2396. [Google Scholar] [CrossRef]
- Tanaka, T.; Nangaku, M. Angiogenesis and hypoxia in the kidney. Nat. Rev. Nephrol. 2013, 9, 211–222. [Google Scholar] [CrossRef]
- Simonnet, H.; Alazard, N.; Pfeiffer, K.; Gallou, C.; Beroud, C.; Demont, J.; Bouvier, R.; Schagger, H.; Godinot, C. Low mitochondrial respiratory chain content correlates with tumor aggressiveness in renal cell carcinoma. Carcinogenesis 2002, 23, 759–768. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.A.; Wang, C.Y.; Hsieh, Y.T.; Chen, Y.J.; Wei, Y.H. Metabolic reprogramming orchestrates cancer stem cell properties in nasopharyngeal carcinoma. Cell Cycle 2015, 14, 86–98. [Google Scholar] [CrossRef]
- Mu, W.; Hu, C.; Zhang, H.; Qu, Z.; Cen, J.; Qiu, Z.; Li, C.; Ren, H.; Li, Y.; He, X.; et al. miR-27b synergizes with anticancer drugs via p53 activation and CYP1B1 suppression. Cell Res. 2015, 25, 477–495. [Google Scholar] [CrossRef] [PubMed]
- Inoue, H.; Hwang, S.H.; Wecksler, A.T.; Hammock, B.D.; Weiss, R.H. Sorafenib attenuates p21 in kidney cancer cells and augments cell death in combination with DNA-damaging chemotherapy. Cancer Biol. Ther. 2011, 12, 827–836. [Google Scholar] [CrossRef] [PubMed]
- LaGory, E.L.; Wu, C.; Taniguchi, C.M.; Ding, C.K.; Chi, J.T.; von Eyben, R.; Scott, D.A.; Richardson, A.D.; Giaccia, A.J. Suppression of PGC-1α is critical for reprogramming oxidative metabolism in renal cell carcinoma. Cell Rep. 2015, 12, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.S.; Lee, H.T.; Lee, S.Y.; Shen, Y.A.; Wang, L.S.; Chen, Y.J.; Wei, Y.H. High mitochondrial DNA copy number and bioenergetic function are associated with tumor invasion of esophageal squamous cell carcinoma cell lines. Int. J. Mol. Sci. 2012, 13, 11228–11246. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.S.; Chang, S.C.; Ou, L.H.; Chen, C.M.; Hsieh, S.S.; Chung, Y.P.; King, K.L.; Lin, S.L.; Wei, Y.H. Mitochondrial DNA alterations correlate with the pathological status and the immunological ER, PR, HER-2/neu, p53 and Ki-67 expression in breast invasive ductal carcinoma. Oncol. Rep. 2015, 33, 2924–2934. [Google Scholar] [CrossRef] [PubMed]
- Meierhofer, D.; Mayr, J.A.; Foetschl, U.; Berger, A.; Fink, K.; Schmeller, N.; Hacker, G.W.; Hauser-Kronberger, C.; Kofler, B.; Sperl, W. Decrease of mitochondrial DNA content and energy metabolism in renal cell carcinoma. Carcinogenesis 2004, 25, 1005–1010. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V. Links between metabolism and cancer. Genes Dev. 2012, 26, 877–890. [Google Scholar] [CrossRef] [PubMed]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.K.; Ayyasamy, V.; Owens, K.M.; Koul, M.S.; Vujcic, M. Mutations in mitochondrial DNA polymerase-γ promote breast tumorigenesis. J. Hum. Genet. 2009, 54, 516–524. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Zheng, L.; Liu, W.; Wang, X.; Wang, Z.; French, A.J.; Kang, D.; Chen, L.; Thibodeau, S.N. Frequent truncating mutation of TFAM induces mitochondrial DNA depletion and apoptotic resistance in microsatellite-unstable colorectal cancer. Cancer Res. 2011, 71, 2978–2987. [Google Scholar] [CrossRef] [PubMed]
- Lincet, H.; Icard, P. How do glycolytic enzymes favour cancer cell proliferation by nonmetabolic functions? Oncogene 2015, 34, 3751–3759. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Dang, C.V. Multifaceted roles of glycolytic enzymes. Trends Biochem. Sci. 2005, 30, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Pinthus, J.H.; Whelan, K.F.; Gallino, D.; Lu, J.P.; Rothschild, N. Metabolic features of clear-cell renal cell carcinoma: Mechanisms and clinical implications. Can. Urol. Assoc. J. 2011, 5, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Masson, N.; Ratcliffe, P.J. Hypoxia signaling pathways in cancer metabolism: The importance of co-selecting interconnected physiological pathways. Cancer Metab. 2014, 2, 3. [Google Scholar] [CrossRef] [PubMed]
- Shinojima, T.; Oya, M.; Takayanagi, A.; Mizuno, R.; Shimizu, N.; Murai, M. Renal cancer cells lacking hypoxia inducible factor (HIF)-1α expression maintain vascular endothelial growth factor expression through HIF-2α. Carcinogenesis 2007, 28, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Loboda, A.; Jozkowicz, A.; Dulak, J. HIF-1 and HIF-2 transcription factors-similar but not identical. Mol. Cells 2010, 29, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Wang, V.; Davis, D.A.; Haque, M.; Huang, L.E.; Yarchoan, R. Differential gene up-regulation by hypoxia-inducible factor-1α and hypoxia-inducible factor-2α in HEK293t cells. Cancer Res. 2005, 65, 3299–3306. [Google Scholar] [PubMed]
- Lum, J.J.; Bui, T.; Gruber, M.; Gordan, J.D.; DeBerardinis, R.J.; Covello, K.L.; Simon, M.C.; Thompson, C.B. The transcription factor HIF-1α plays a critical role in the growth factor-dependent regulation of both aerobic and anaerobic glycolysis. Genes Dev. 2007, 21, 1037–1049. [Google Scholar] [CrossRef] [PubMed]
- Franovic, A.; Holterman, C.E.; Payette, J.; Lee, S. Human cancers converge at the HIF-2α oncogenic axis. Proc. Natl. Acad. Sci. USA 2009, 106, 21306–21311. [Google Scholar] [CrossRef] [PubMed]
- Elstrom, R.L.; Bauer, D.E.; Buzzai, M.; Karnauskas, R.; Harris, M.H.; Plas, D.R.; Zhuang, H.; Cinalli, R.M.; Alavi, A.; Rudin, C.M.; et al. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004, 64, 3892–3899. [Google Scholar] [CrossRef] [PubMed]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 2011, 12, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Gogol, M.; Gaudenz, K.; Gerton, J.L. Improved transcription and translation with l-leucine stimulation of mTORC1 in Roberts syndrome. BMC Genom. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Lee, K.K.; Zhang, L.; Gerton, J.L. Stimulation of mtorc1 with l-leucine rescues defects associated with roberts syndrome. PLoS Genet. 2013, 9, e1003857. [Google Scholar] [CrossRef] [PubMed]
- Morita, M.; Gravel, S.P.; Hulea, L.; Larsson, O.; Pollak, M.; St-Pierre, J.; Topisirovic, I. mTOR coordinates protein synthesis, mitochondrial activity and proliferation. Cell Cycle 2015, 14, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Masui, K.; Tanaka, K.; Akhavan, D.; Babic, I.; Gini, B.; Matsutani, T.; Iwanami, A.; Liu, F.; Villa, G.R.; Gu, Y.; et al. mTOR complex 2 controls glycolytic metabolism in glioblastoma through Foxo acetylation and upregulation of c-MYC. Cell Metab. 2013, 18, 726–739. [Google Scholar] [CrossRef] [PubMed]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, P.L. Warburg, me and hexokinase 2: Multiple discoveries of key molecular events underlying one of cancers’ most common phenotypes, the “Warburg effect”, i.e., elevated glycolysis in the presence of oxygen. J. Bioenerg. Biomembr. 2007, 39, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Butler, E.B.; Tan, M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Lu, F. Reactive oxygen species in cancer, too much or too little? Med. Hypoth. 2007, 69, 1293–1298. [Google Scholar] [CrossRef] [PubMed]
- Qinghong, S.; Shen, G.; Lina, S.; Yueming, Z.; Xiaoou, L.; Jianlin, W.; Chengyan, H.; Hongjun, L.; Haifeng, Z. Comparative proteomics analysis of differential proteins in response to doxorubicin resistance in myelogenous leukemia cell lines. Proteome Sci. 2015, 13, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H. The cancer stem cell: Premises, promises and challenges. Nat. Med. 2011, 17, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Pacini, N.; Borziani, F. Cancer stem cell theory and the Warburg effect, two sides of the same coin? Int. J. Mol. Sci. 2014, 15, 8893–8930. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A.; Joven, J.; Cufi, S.; Corominas-Faja, B.; Oliveras-Ferraros, C.; Cuyas, E.; Martin-Castillo, B.; Lopez-Bonet, E.; Alarcon, T.; Vazquez-Martin, A. The Warburg effect version 2.0: Metabolic reprogramming of cancer stem cells. Cell Cycle 2013, 12, 1166–1179. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.C.; Lee, J.T.; Shih, C.P.; Chao, T.T.; Sytwu, H.K.; Li, S.L.; Fang, M.C.; Chen, H.K.; Lin, Y.C.; Kuo, C.Y.; et al. Hypoxia induces a metabolic shift and enhances the stemness and expansion of cochlear spiral ganglion stem/progenitor cells. BioMed Res. Int. 2015. [Google Scholar] [CrossRef] [PubMed]
- Guha, M.; Srinivasan, S.; Ruthel, G.; Kashina, A.K.; Carstens, R.P.; Mendoza, A.; Khanna, C.; van Winkle, T.; Avadhani, N.G. Mitochondrial retrograde signaling induces epithelial-mesenchymal transition and generates breast cancer stem cells. Oncogene 2014, 33, 5238–5250. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.T.; Lin, C.S.; Chen, W.S.; Liao, H.T.; Tsai, C.Y.; Wei, Y.H. Leukocyte mitochondrial DNA alteration in systemic lupus erythematosus and its relevance to the susceptibility to lupus nephritis. Int. J. Mol. Sci. 2012, 13, 8853–8868. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.T.; Lin, C.S.; Lee, C.S.; Tsai, C.Y.; Wei, Y.H. Increased 8-hydroxy-2′-deoxyguanosine in plasma and decreased mrna expression of human 8-oxoguanine DNA glycosylase 1, antioxidant enzymes, mitochondrial biogenesis-related proteins and glycolytic enzymes in leucocytes in patients with systemic lupus erythematosus. Clin. Exp. Immunol. 2014, 176, 66–77. [Google Scholar]
- Dang, C.V. The interplay between myc and HIF in the Warburg effect. Ernst Scher. Found. Symp. Proc. 2007. [Google Scholar] [CrossRef]
- Robey, R.B.; Hay, N. Is Akt the “Warburg kinase”?—Akt-Energy metabolism interactions and oncogenesis. Semin. Cancer Biol. 2009, 19, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Schulz, N.; Kluth, O.; Jastroch, M.; Schurmann, A. Minor role of mitochondrial respiration for fatty-acid induced insulin secretion. Int. J. Mol. Sci. 2013, 14, 18989–18998. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| mtDNA Copy Number * | p-Value ** | ||
|---|---|---|---|
| Non-Cancerous Part | Cancerous Part | ||
| Overall (n = 5) | |||
| M ± S.D. | 0.50 ± 0.27 | 0.17 ± 0.06 | 0.043 |
| Examined subjects | |||
| Patient 1 | 0.22 | 0.12 | - |
| Patient 2 | 0.25 | 0.14 | - |
| Patient 3 | 0.81 | 0.15 | - |
| Patient 4 | 0.49 | 0.28 | - |
| Patient 5 | 0.74 | 0.16 | - |
| Parameters | 786-O RCC (n = 3) | p-Value ** | |
|---|---|---|---|
| NT | TFAM-KD | ||
| mtDNA copy number (M ± S.D.) * | 1.00 ± 0.12 | 0.72 ± 0.09 | 0.034 |
| mRNA expression level (M ± S.D.) * | |||
| TFAM | 1.00 ± 0.20 | 0.29 ± 0.15 | 0.008 |
| ND1 | 1.00 ± 0.07 | 0.55 ± 0.13 | 0.007 |
| ND6 | 1.00 ± 0.16 | 0.51 ± 0.14 | 0.017 |
| PDK1 | 1.00 ± 0.41 | 1.08 ± 0.43 | 0.830 |
| PDHA1 | 1.00 ± 0.38 | 0.98 ± 0.40 | 0.947 |
| HK-II | 1.00 ± 0.16 | 1.51 ± 0.12 | 0.013 |
| GPI | 1.00 ± 0.10 | 0.93 ± 0.25 | 0.664 |
| PFK | 1.00 ± 0.14 | 1.27 ± 0.16 | 0.050 |
| LDHA | 1.00 ± 0.33 | 1.32 ± 0.70 | 0.268 |
| Parameters | 786-O RCC (n = 3) | p-Value * | |
|---|---|---|---|
| NT (M ± S.D.) | TFAM-KD (M ± S.D.) | ||
| OCR of cellular metabolism | |||
| mOCRB (pmole/min/106 cells) | 1986.3 ± 167.4 | 1294.9 ± 187.3 | 0.009 |
| mOCRMax (pmole/min/106 cells) | 2056.8 ± 176.3 | 1335.1 ± 90.5 | 0.003 |
| ECAR of cellular metabolism | |||
| ECARB (mpH/min/106 cells) | 2016.4 ± 15.0 | 2230.2 ± 77.2 | 0.037 |
| Cellular metabolic shift | |||
| mOCRB/ECARB | 0.983 ± 0.075 | 0.580 ± 0.079 | 0.003 |
| ECARB/mOCRB | 1.020 ± 0.078 | 1.747 ± 0.249 | 0.009 |
| Trans-well migration activity (cells/field) | 132.3 ± 27.5 | 380.3 ± 81.2 | 0.007 |
| Relative cell viability (%) ** | |||
| Doxorubicin concentration [31,32,33] | |||
| 0.5 μM | 25.1 ± 2.5 | 35.0 ± 2.4 | 0.008 |
| 1.0 μM | 11.5 ± 3.7 | 12.8 ± 3.0 | 0.513 |
| 2.5 μM | 10.9 ± 4.8 | 14.5 ± 2.5 | 0.275 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, C.-S.; Lee, H.-T.; Lee, M.-H.; Pan, S.-C.; Ke, C.-Y.; Chiu, A.W.-H.; Wei, Y.-H. Role of Mitochondrial DNA Copy Number Alteration in Human Renal Cell Carcinoma. Int. J. Mol. Sci. 2016, 17, 814. https://doi.org/10.3390/ijms17060814
Lin C-S, Lee H-T, Lee M-H, Pan S-C, Ke C-Y, Chiu AW-H, Wei Y-H. Role of Mitochondrial DNA Copy Number Alteration in Human Renal Cell Carcinoma. International Journal of Molecular Sciences. 2016; 17(6):814. https://doi.org/10.3390/ijms17060814
Chicago/Turabian StyleLin, Chen-Sung, Hui-Ting Lee, Ming-Huei Lee, Siao-Cian Pan, Chen-Yeh Ke, Allen Wen-Hsiang Chiu, and Yau-Huei Wei. 2016. "Role of Mitochondrial DNA Copy Number Alteration in Human Renal Cell Carcinoma" International Journal of Molecular Sciences 17, no. 6: 814. https://doi.org/10.3390/ijms17060814
APA StyleLin, C.-S., Lee, H.-T., Lee, M.-H., Pan, S.-C., Ke, C.-Y., Chiu, A. W.-H., & Wei, Y.-H. (2016). Role of Mitochondrial DNA Copy Number Alteration in Human Renal Cell Carcinoma. International Journal of Molecular Sciences, 17(6), 814. https://doi.org/10.3390/ijms17060814