
Type 2 Diabetes in Non-Alcoholic Fatty Liver Disease and Hepatitis C Virus Infection—Liver: The “Musketeer” in the Spotlight


 ,
,  and
and 
Abstract
:1. Introduction
1.1. Definitions
1.2. Epidemiology and Burden of Type 2 Diabetes
1.3. Liver and Type 2 Diabetes: Historical Overview
1.4. Aim of the Review and Evidence Acquisition
2. NAFLD and Type 2 Diabetes
2.1. Epidemiology
2.1.1. NAFLD as a “Manifestation” of Type 2 Diabetes
2.1.2. NAFLD as a Precursor of Type 2 Diabetes
2.2. Pathophysiology
2.2.1. Remodeling of White Adipose Tissue
2.2.2. Role of Skeletal Muscle and Brown Adipose Tissue
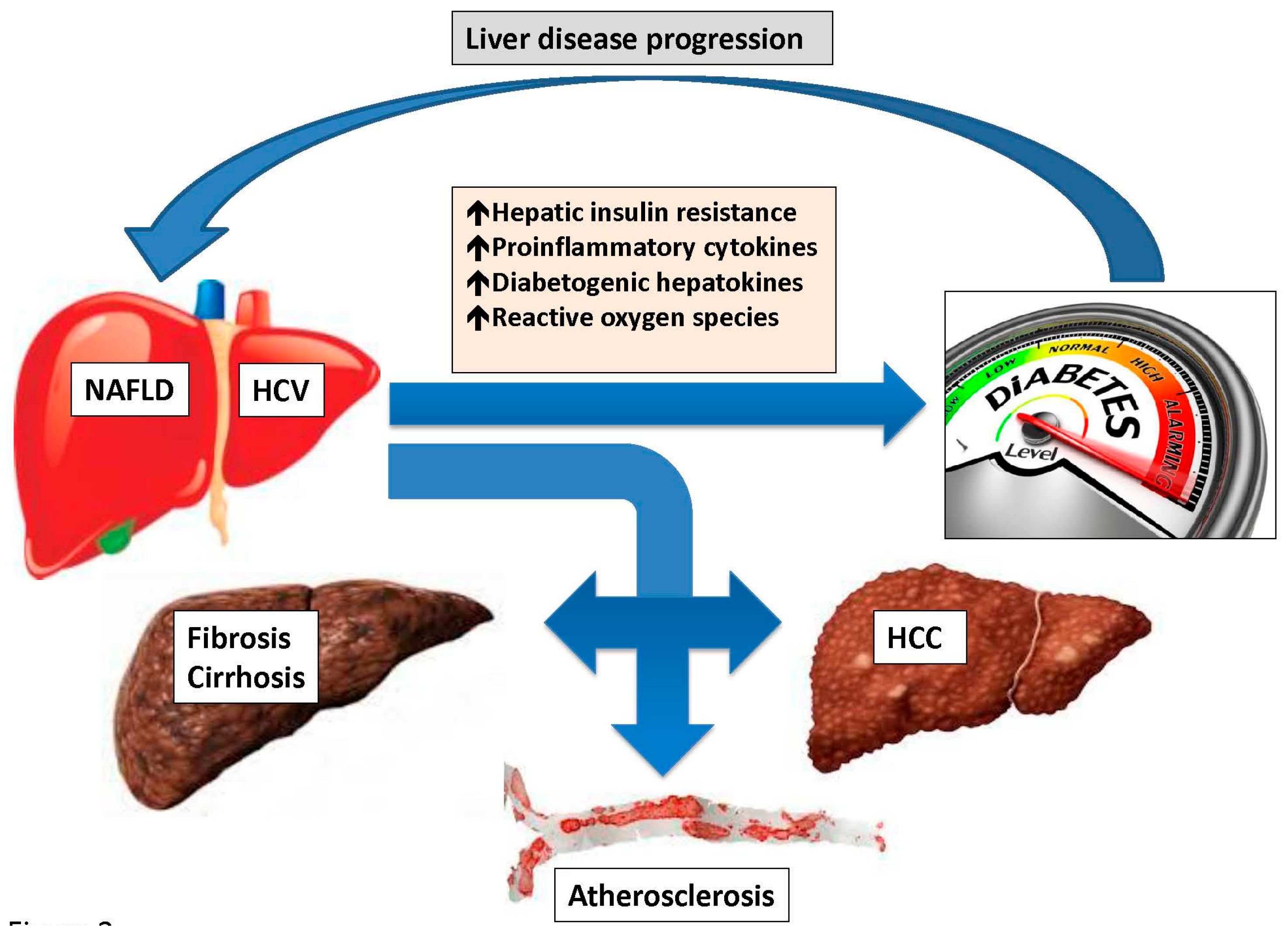
2.2.3. Intrahepatic Fat Accumulation, Hepatic Insulin Resistance and Hepatokines
2.2.4. Gut-Liver Axis
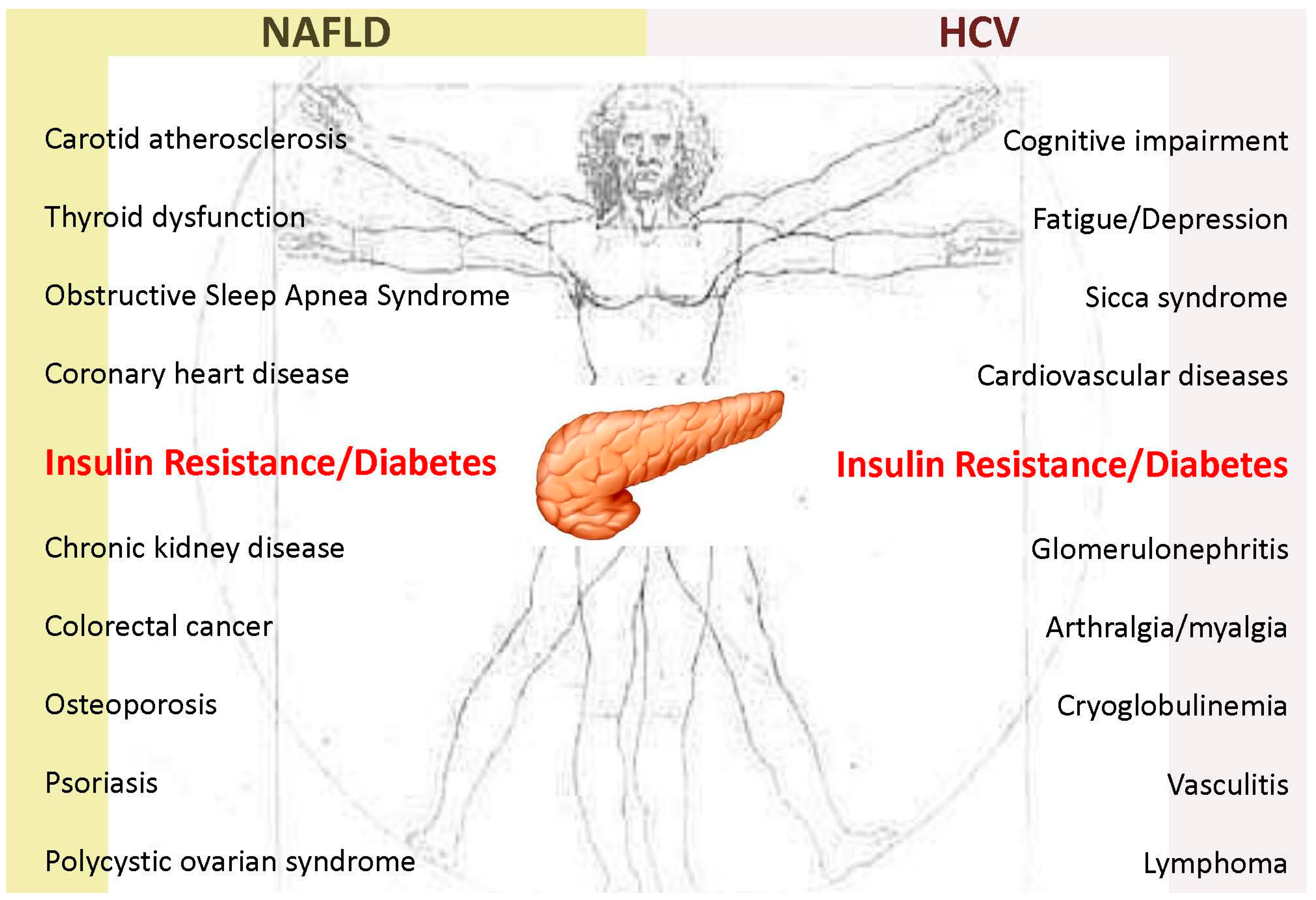
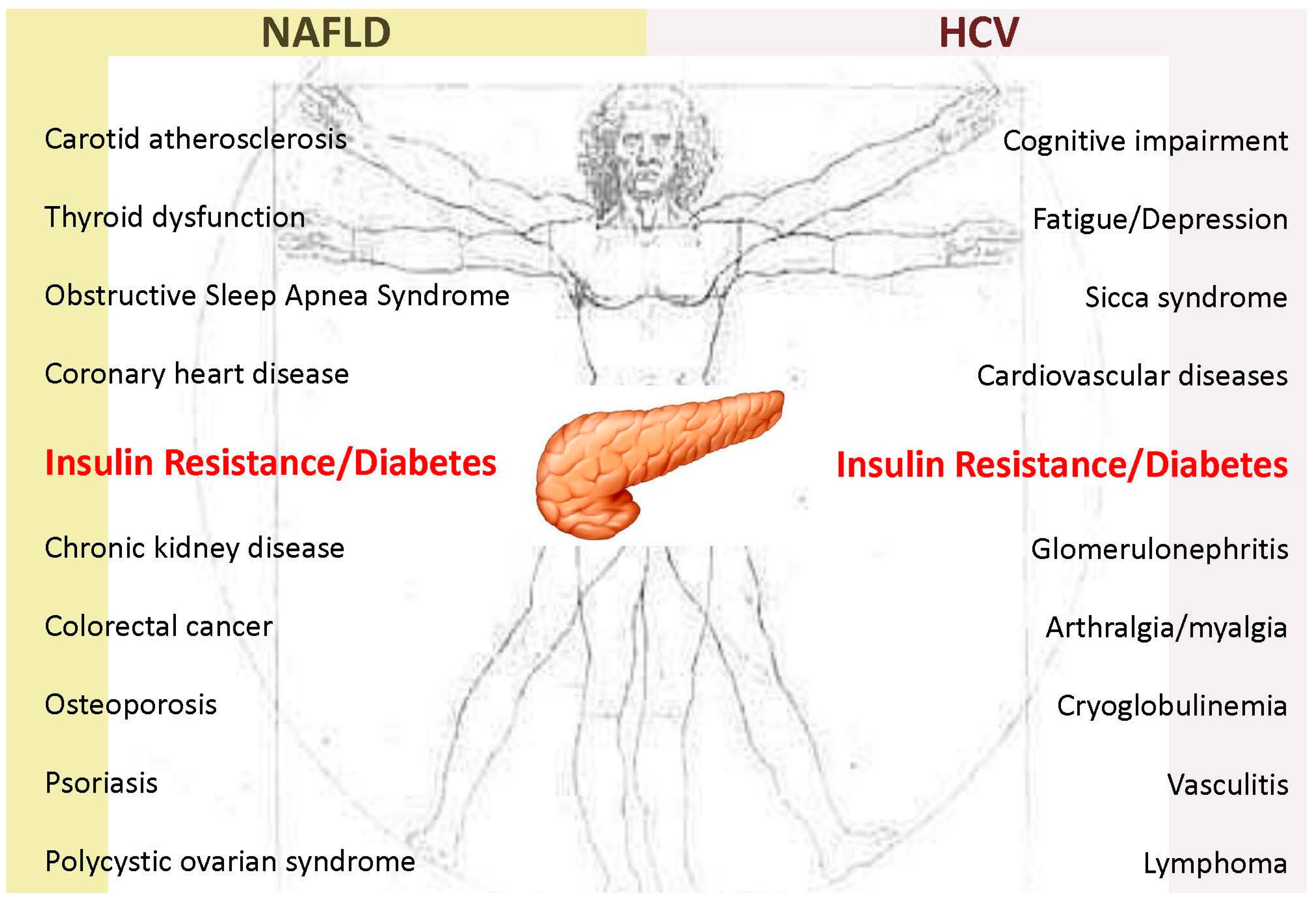
2.3. Clinical Implications
2.3.1. NASH and Fibrosis
2.3.2. Cirrhosis and Hepatocellular Carcinoma
2.3.3. Atherosclerosis
3. HCV and Type 2 Diabetes
3.1 Epidemiology
3.1.1. HCV and Diabetes: A Non-chance Association
3.1.2. The Burden
3.1.3. Extra-Hepatic Manifestations of HCV Infection: Type 2 Diabetes
3.1.4. Heterogeneity in the Distribution of HCV and Type 2 Diabetes and Differential Features of Hepatitis C-Associated Dysmetabolic Syndrome and MetS
3.2. Pathophysiology
3.2.1. HCV Increases T2D Risk via Insulin Resistance
3.2.2. IR Associated with HCV: Antigens, Sites and Determinants
3.2.3. T2D in the Setting of the HCADS
3.3. Clinical Implications
3.3.1. Risk of Fibrosis
3.3.2. Risk of Hepatocellular Carcinoma
3.3.3. Risk of Atherosclerosis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| CCR-2 | C-C chemokine receptor-2 |
| CHD | coronary heart disease |
| chREBP | carbohydrate-responsive element-binding protein |
| CVD | cardiovascular disease |
| DAA | direct acting antivirals |
| DAG | diacylglycerol |
| DGAT2 | diacylglycerolacyltransferase 2 |
| DNL | de-novo lipogenesis |
| FA | fatty acids |
| FGF-21 | fibroblast growth factor 21 |
| FXR | farnesoid X receptor |
| HCC | hepatocellular carcinoma |
| HIF1α | hypoxia inducible factor 1α |
| HCV | hepatitis C virus |
| IR | insulin resistance |
| MCP-1 | monocyte chemoattractant protein-1 |
| MetS | metabolic syndrome |
| PNPLA3 | patatin-like phospholipase domain-containing protein 3 |
| PPAR-γ | peroxisome proliferator–activated receptor γ |
| ROS | reactive oxygen species |
| SREBP1c | sterol regulatory element binding protein 1c |
| T2D | type 2 diabetes |
| TLR-4 | toll-like receptor 4 |
| TNFα | tumor necrosis factor α |
| UCP-1 | uncoupling protein-1 |
| WAT | white adipose tissue |
References
- Inzucchi, S.E. Clinical practice. Diagnosis of diabetes. N. Engl. J. Med. 2012, 367, 542–550. [Google Scholar] [CrossRef] [PubMed]
- Cameron, F.J.; Wherrett, D.K. Care of diabetes in children and adolescents: Controversies, changes, and consensus. Lancet 2015, 385, 2096–2106. [Google Scholar] [CrossRef]
- Lonardo, A.; Ballestri, S.; Marchesini, G.; Angulo, P.; Loria, P. Nonalcoholic fatty liver disease: Aprecursor of the metabolic syndrome. Dig. Liver Dis. 2015, 47, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Nascimbeni, F.; Pais, R.; Bellentani, S.; Day, C.P.; Ratziu, V.; Loria, P.; Lonardo, A. From NAFLD in clinical practice to answers from guidelines. J. Hepatol. 2013, 59, 859–871. [Google Scholar] [CrossRef] [PubMed]
- Byrne, C.D.; Targher, G. NAFLD: A multisystem disease. J. Hepatol. 2015, 62, S47–S64. [Google Scholar] [CrossRef] [PubMed]
- Petta, S.; Valenti, L.; Bugianesi, E.; Targher, G.; Bellentani, S.; Bonino, F.; Special Interest Group on Personalised Hepatology of the Italian Association for the Study of the Liver (AISF). A “systems medicine” approach to the study of non-alcoholic fatty liver disease. Dig. Liver Dis. 2016, 48, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, A.; Loria, P.; Adinolfi, L.E.; Carulli, N.; Ruggiero, G. Hepatitis C and steatosis: A reappraisal. J. Viral Hepat. 2006, 13, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Negro, F.; Forton, D.; Craxi, A.; Sulkowski, M.S.; Feld, J.J.; Manns, M.P. Extrahepatic morbidity and mortality of chronic hepatitis C. Gastroenterology 2015, 149, 1345–1360. [Google Scholar] [CrossRef] [PubMed]
- Shaw, J.E.; Sicree, R.A.; Zimmet, P.Z. Global estimates of the prevalence of diabetes for 2010 and 2030. Diabetes Res. Clin. Pract. 2010, 87, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Kanavos, P.; van den Aardweg, S.; Schurer, W. Diabetes Expenditure, Burden of Disease and Management in 5 EU Countries. Available online: http://www.lse.ac.uk/LSEHealthAndSocialCare/research/LSEHealth/MTRG/LSEDiabetesReport26Jan2012.pdf (accessed on 8 February 2016).
- Centers for Disease Control and Prevention. National Diabetes Fact Sheet: National Estimates and General Information on Diabetes and Prediabetes in the United States; U.S. Department of Health and Human Services, Centers for Disease Control and Prevention: Atlanta, GA, USA, 2011.
- American Diabetes Association. Economic costs of diabetes in the U.S. in 2007. Diabetes Care 2008, 31, 596–615. [Google Scholar]
- Sherwin, R.; Jastreboff, A.M. Year in diabetes 2012: The diabetes tsunami. J. Clin. Endocrinol. Metab. 2012, 97, 4293–4301. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, H.J.; Mac, M.F.; Rappaport, H.; Alpert, L.K. Studies on the liver in diabetes mellitus. II. The significance of fatty metamorphosis and its correlation with insulin sensitivity. J. Lab. Clin. Med. 1950, 36, 922–928. [Google Scholar] [PubMed]
- Ballestri, S.; Nascimbeni, F.; Romagnoli, D.; Lonardo, A. The independent predictors of NASH and its individual histological features. Insulin resistance, serum uric acid, metabolic syndrome, alt and serum total cholesterol are a clue to pathogenesis and candidate targets for treatment. Hepatol. Res. 2016. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, T.; Zhang, C.; Tang, F.; Zhong, N.; Li, H.; Song, X.; Lin, H.; Liu, Y.; Xue, F. Identification of reciprocal causality between non-alcoholic fatty liver disease and metabolic syndrome by a simplified bayesian network in a chinese population. BMJ Open 2015, 5, e008204. [Google Scholar] [CrossRef] [PubMed]
- Musso, G.; Gambino, R.; Cassader, M.; Pagano, G. Meta-analysis: Natural history of non-alcoholic fatty liver disease (NAFLD) and diagnostic accuracy of non-invasive tests for liver disease severity. Ann. Med. 2011, 43, 617–649. [Google Scholar] [CrossRef] [PubMed]
- Ballestri, S.; Zona, S.; Targher, G.; Romagnoli, D.; Baldelli, E.; Nascimbeni, F.; Roverato, A.; Guaraldi, G.; Lonardo, A. Nonalcoholic fatty liver disease is associated with an almost two-fold increased risk of incident type 2 diabetes and metabolic syndrome. Evidence from a systematic review and meta-analysis. J. Gastroenterol. Hepatol. 2015. [Google Scholar] [CrossRef] [PubMed]
- White, D.L.; Ratziu, V.; El-Serag, H.B. Hepatitis Cinfection and risk of diabetes: A systematic review and meta-analysis. J. Hepatol. 2008, 49, 831–844. [Google Scholar] [CrossRef] [PubMed]
- Cacoub, P.; Gragnani, L.; Comarmond, C.; Zignego, A.L. Extrahepatic manifestations of chronic hepatitis Cvirus infection. Dig. Liver Dis. 2014, 46, S165–S173. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, A.; Adinolfi, L.E.; Loria, P.; Carulli, N.; Ruggiero, G.; Day, C.P. Steatosis and hepatitis Cvirus: Mechanisms and significance for hepatic and extrahepatic disease. Gastroenterology 2004, 126, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Asselah, T.; Boyer, N.; Saadoun, D.; Martinot-Peignoux, M.; Marcellin, P. Direct-acting antivirals for the treatment of hepatitis Cvirus infection: Optimizing current IFN-free treatment and future perspectives. Liver Int. 2016, 36, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Ilyas, J.A.; Vierling, J.M. An overview of emerging therapies for the treatment of chronic hepatitis C. Med. Clin. N. Am. 2014, 98, 17–38. [Google Scholar] [CrossRef] [PubMed]
- Welsch, C.; Jesudian, A.; Zeuzem, S.; Jacobson, I. New direct-acting antiviral agents for the treatment of hepatitis Cvirus infection and perspectives. Gut 2012, 61, i36–i46. [Google Scholar] [CrossRef] [PubMed]
- Loria, P.; Lonardo, A.; Anania, F. Liver and diabetes. A vicious circle. Hepatol. Res. 2013, 43, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Ballestri, S.; Nascimbeni, F.; Romagnoli, D.; Baldelli, E.; Lonardo, A. The role of nuclear receptors in the pathophysiology, natural course, and drug treatment of NAFLDin humans. Adv. Ther. 2016. [Google Scholar] [CrossRef] [PubMed]
- Vanni, E.; Bugianesi, E.; Saracco, G. Treatment of type 2 diabetes mellitus by viral eradication in chronic hepatitis C: Myth or reality? Dig. Liver Dis. 2016, 48, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Klip, A.; Vranic, M. Muscle, liver, and pancreas: Three musketeers fighting to control glycemia. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E1141–E1143. [Google Scholar] [CrossRef] [PubMed]
- Reaven, G.M. The fourth musketeer—From Alexandre Dumas to Claude Bernard. Diabetologia 1995, 38, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, M.J.; Adams, L.A.; Canbay, A.; Syn, W.K. Extrahepatic complications of nonalcoholic fatty liver disease. Hepatology 2014, 59, 1174–1197. [Google Scholar] [CrossRef] [PubMed]
- Carulli, L.; Ballestri, S.; Lonardo, A.; Lami, F.; Violi, E.; Losi, L.; Bonilauri, L.; Verrone, A.M.; Odoardi, M.R.; Scaglioni, F.; et al. Is nonalcoholic steatohepatitis associated with a high-though-normal thyroid stimulating hormone level and lower cholesterol levels? Intern. Emerg. Med. 2013, 8, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Nascimbeni, F.; Ballestri, S.; Di Tommaso, L.; Piccoli, M.; Lonardo, A. Inflammatory hepatocellular adenomatosis, metabolic syndrome, polycystic ovary syndrome and non-alcoholic steatohepatitis: Chance tetrad or association by necessity? Dig. Liver Dis. 2014, 46, 288–289. [Google Scholar] [CrossRef] [PubMed]
- Loria, P.; Carulli, L.; Bertolotti, M.; Lonardo, A. Endocrine and liver interaction: The role of endocrine pathways in NASH. Nat.Rev. Gastroenterol. Hepatol. 2009, 6, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Targher, G.; Rossini, M.; Lonardo, A. Evidence that non-alcoholic fatty liver disease and polycystic ovary syndrome are associated by necessity rather than chance: Anovel hepato-ovarian axis? Endocrine 2016, 51, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Targher, G.; Lonardo, A.; Rossini, M. Nonalcoholic fatty liver disease and decreased bone mineral density: Is there a link? J. Endocrinol. Investig. 2015, 38, 817–825. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Gisondi, P.; Lonardo, A.; Targher, G. Relationship between non-alcoholic fatty liver disease and psoriasis: A novel hepato-dermal axis? Int. J. Mol. Sci. 2016, 17, 217. [Google Scholar] [CrossRef] [PubMed]
- Vigano, M.; Colombo, M. Extrahepatic manifestations of hepatitis C virus. Gastroenterol. Clin. N. Am. 2015, 44, 775–791. [Google Scholar] [CrossRef] [PubMed]
- Negro, F. Facts and fictions of HCV and comorbidities: Steatosis, diabetes mellitus, and cardiovascular diseases. J. Hepatol. 2014, 61, S69–S78. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, A.; Bellentani, S.; Argo, C.K.; Ballestri, S.; Byrne, C.D.; Caldwell, S.H.; Cortez-Pinto, H.; Grieco, A.; Machado, M.V.; et al. Epidemiological modifiers of non-alcoholic fatty liver disease: Focus on high-risk groups. Dig Liver Dis. 2015, 47, 997–1006. [Google Scholar]
- Singh, S.; Allen, A.M.; Wang, Z.; Prokop, L.J.; Murad, M.H.; Loomba, R. Fibrosis progression in nonalcoholic fatty liver vs nonalcoholic steatohepatitis: Asystematic review and meta-analysis of paired-biopsy studies. Clin. Gastroenterol. Hepatol. 2015, 13, 643–654. [Google Scholar] [CrossRef] [PubMed]
- Leite, N.C.; Villela-Nogueira, C.A.; Pannain, V.L.; Bottino, A.C.; Rezende, G.F.; Cardoso, C.R.; Salles, G.F. Histopathological stages of nonalcoholic fatty liver disease in type 2 diabetes: Prevalences and correlated factors. Liver Int. 2011, 31, 700–706. [Google Scholar] [CrossRef] [PubMed]
- Portillo Sanchez, P.; Bril, F.; Maximos, M.; Lomonaco, R.; Biernacki, D.; Orsak, B.; Subbarayan, S.; Webb, A.; Hecht, J.; Cusi, K. High prevalence of nonalcoholic fatty liver disease in patients with type 2 diabetes mellitus and normal plasma aminotransferase levels. J. Clin. Endocrinol. Metab. 2015, 100, 2231–2238. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.D.; Stengel, J.; Asike, M.I.; Torres, D.M.; Shaw, J.; Contreras, M.; Landt, C.L.; Harrison, S.A. Prevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle-aged population utilizing ultrasound and liver biopsy: Aprospective study. Gastroenterology 2011, 140, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Kwok, R.; Choi, K.C.; Wong, G.L.; Zhang, Y.; Chan, H.L.; Luk, A.O.; Shu, S.S.; Chan, A.W.; Yeung, M.W.; Chan, J.C.; et al. Screening diabetic patients for non-alcoholic fatty liver disease with controlled attenuation parameter and liver stiffness measurements: Aprospective cohort study. Gut 2015. [Google Scholar] [CrossRef] [PubMed]
- Anstee, Q.M.; Targher, G.; Day, C.P. Progression of NAFLD to diabetes mellitus, cardiovascular disease or cirrhosis. Nat.Rev. Gastroenterol. Hepatol. 2013, 10, 330–344. [Google Scholar] [CrossRef] [PubMed]
- Maximos, M.; Bril, F.; Portillo Sanchez, P.; Lomonaco, R.; Orsak, B.; Biernacki, D.; Suman, A.; Weber, M.; Cusi, K. The role of liver fat and insulin resistance as determinants of plasma aminotransferase elevation in nonalcoholic fatty liver disease. Hepatology 2015, 61, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Sung, K.C.; Wild, S.H.; Byrne, C.D. Resolution of fatty liver and risk of incident diabetes. J. Clin. Endocrinol. Metab. 2013, 98, 3637–3643. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, H.; Tsuboya, T.; Tsuji, K.; Dohke, M.; Maguchi, H. Independent association between improvement of nonalcoholic fatty liver disease and reduced incidence of type 2 diabetes. Diabetes Care 2015, 38, 1673–1679. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, T.; Hamaguchi, M.; Kojima, T.; Hashimoto, Y.; Ohbora, A.; Kato, T.; Nakamura, N.; Fukui, M. The impact of non-alcoholic fatty liver disease on incident type 2 diabetes mellitus in non-overweight individuals. Liver Int. 2016, 36, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.V.; Allison, M.A.; Lima, J.A.; Bluemke, D.A.; Abbasi, S.A.; Ouyang, P.; Jerosch-Herold, M.; Ding, J.; Budoff, M.J.; Murthy, V.L. Liver fat, statin use, and incident diabetes: The multi-ethnic study of atherosclerosis. Atherosclerosis 2015, 242, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Ekstedt, M.; Franzen, L.E.; Mathiesen, U.L.; Thorelius, L.; Holmqvist, M.; Bodemar, G.; Kechagias, S. Long-term follow-up of patients with NAFLD and elevated liver enzymes. Hepatology 2006, 44, 865–873. [Google Scholar] [CrossRef] [PubMed]
- Buechler, C.; Krautbauer, S.; Eisinger, K. Adipose tissue fibrosis. World J. Diabetes 2015, 6, 548–553. [Google Scholar] [PubMed]
- Sun, K.; Tordjman, J.; Clement, K.; Scherer, P.E. Fibrosis and adipose tissue dysfunction. Cell Metab. 2013, 18, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Kusminski, C.M.; Scherer, P.E. Adipose tissue remodeling and obesity. J. Clin. Investig. 2011, 121, 2094–2101. [Google Scholar] [CrossRef] [PubMed]
- Cinti, S.; Mitchell, G.; Barbatelli, G.; Murano, I.; Ceresi, E.; Faloia, E.; Wang, S.; Fortier, M.; Greenberg, A.S.; Obin, M.S. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J. Lipid Res. 2005, 46, 2347–2355. [Google Scholar] [CrossRef] [PubMed]
- Cancello, R.; Henegar, C.; Viguerie, N.; Taleb, S.; Poitou, C.; Rouault, C.; Coupaye, M.; Pelloux, V.; Hugol, D.; Bouillot, J.L.; et al. Reduction of macrophage infiltration and chemoattractant gene expression changes in white adipose tissue of morbidly obese subjects after surgery-induced weight loss. Diabetes 2005, 54, 2277–2286. [Google Scholar] [CrossRef] [PubMed]
- Halberg, N.; Khan, T.; Trujillo, M.E.; Wernstedt-Asterholm, I.; Attie, A.D.; Sherwani, S.; Wang, Z.V.; Landskroner-Eiger, S.; Dineen, S.; Magalang, U.J.; et al. Hypoxia-inducible factor 1alpha induces fibrosis and insulin resistance in white adipose tissue. Mol. Cell. Biol. 2009, 29, 4467–4483. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Qu, A.; Matsubara, T.; Chanturiya, T.; Jou, W.; Gavrilova, O.; Shah, Y.M.; Gonzalez, F.J. Disruption of hypoxia-inducible factor 1 in adipocytes improves insulin sensitivity and decreases adiposity in high-fat diet-fed mice. Diabetes 2011, 60, 2484–2495. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Halberg, N.; Khan, M.; Magalang, U.J.; Scherer, P.E. Selective inhibition of hypoxia-inducible factor 1alpha ameliorates adipose tissue dysfunction. Mol. Cell. Biol. 2013, 33, 904–917. [Google Scholar] [CrossRef] [PubMed]
- Lumeng, C.N.; Bodzin, J.L.; Saltiel, A.R. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Investig. 2007, 117, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Lumeng, C.N.; DelProposto, J.B.; Westcott, D.J.; Saltiel, A.R. Phenotypic switching of adipose tissue macrophages with obesity is generated by spatiotemporal differences in macrophage subtypes. Diabetes 2008, 57, 3239–3246. [Google Scholar] [CrossRef] [PubMed]
- Suganami, T.; Tanaka, M.; Ogawa, Y. Adipose tissue inflammation and ectopic lipid accumulation. Endocr. J. 2012, 59, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Berg, A.H.; Scherer, P.E. Adipose tissue, inflammation, and cardiovascular disease. Circ. Res. 2005, 96, 939–949. [Google Scholar] [CrossRef] [PubMed]
- Rocha, V.Z.; Libby, P. Obesity, inflammation, and atherosclerosis. Nat. Rev. Cardiol. 2009, 6, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S.; Shargill, N.S.; Spiegelman, B.M. Adipose expression of tumor necrosis factor-α: Direct role in obesity-linked insulin resistance. Science 1993, 259, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Uysal, K.T.; Wiesbrock, S.M.; Marino, M.W.; Hotamisligil, G.S. Protection from obesity-induced insulin resistance in mice lacking TNF-alpha function. Nature 1997, 389, 610–614. [Google Scholar] [PubMed]
- Kanda, H.; Tateya, S.; Tamori, Y.; Kotani, K.; Hiasa, K.; Kitazawa, R.; Kitazawa, S.; Miyachi, H.; Maeda, S.; Egashira, K.; et al. Mcp-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J. Clin. Investig. 2006, 116, 1494–1505. [Google Scholar] [CrossRef] [PubMed]
- Kamei, N.; Tobe, K.; Suzuki, R.; Ohsugi, M.; Watanabe, T.; Kubota, N.; Ohtsuka-Kowatari, N.; Kumagai, K.; Sakamoto, K.; Kobayashi, M.; et al. Overexpression of monocyte chemoattractant protein-1 in adipose tissues causes macrophage recruitment and insulin resistance. J. Biol. Chem. 2006, 281, 26602–26614. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, S.P.; Hunter, D.; Huber, R.; Lemieux, J.; Slaymaker, S.; Vaddi, K.; Charo, I.; Leibel, R.L.; Ferrante, A.W., Jr. CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J. Clin. Investig. 2006, 116, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, T.; Kamon, J.; Waki, H.; Terauchi, Y.; Kubota, N.; Hara, K.; Mori, Y.; Ide, T.; Murakami, K.; Tsuboyama-Kasaoka, N.; et al. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat. Med. 2001, 7, 941–946. [Google Scholar] [CrossRef] [PubMed]
- Maeda, N.; Shimomura, I.; Kishida, K.; Nishizawa, H.; Matsuda, M.; Nagaretani, H.; Furuyama, N.; Kondo, H.; Takahashi, M.; Arita, Y.; et al. Diet-induced insulin resistance in mice lacking adiponectin/ACRP 30. Nat. Med. 2002, 8, 731–737. [Google Scholar] [CrossRef] [PubMed]
- Wernstedt Asterholm, I.; Tao, C.; Morley, T.S.; Wang, Q.A.; Delgado-Lopez, F.; Wang, Z.V.; Scherer, P.E. Adipocyte inflammation is essential for healthy adipose tissue expansion and remodeling. Cell Metab. 2014, 20, 103–118. [Google Scholar] [CrossRef] [PubMed]
- Divoux, A.; Tordjman, J.; Lacasa, D.; Veyrie, N.; Hugol, D.; Aissat, A.; Basdevant, A.; Guerre-Millo, M.; Poitou, C.; Zucker, J.D.; et al. Fibrosis in human adipose tissue: Composition, distribution, and link with lipid metabolism and fat mass loss. Diabetes 2010, 59, 2817–2825. [Google Scholar] [CrossRef] [PubMed]
- Samuel, V.T.; Shulman, G.I. The pathogenesis of insulin resistance: Integrating signaling pathways and substrate flux. J. Clin. Investig. 2016, 126, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Chen, Y.; Cline, G.W.; Zhang, D.; Zong, H.; Wang, Y.; Bergeron, R.; Kim, J.K.; Cushman, S.W.; Cooney, G.J.; et al. Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. J. Biol. Chem. 2002, 277, 50230–50236. [Google Scholar] [CrossRef] [PubMed]
- Griffin, M.E.; Marcucci, M.J.; Cline, G.W.; Bell, K.; Barucci, N.; Lee, D.; Goodyear, L.J.; Kraegen, E.W.; White, M.F.; Shulman, G.I. Free fatty acid-induced insulin resistance is associated with activation of protein kinase Ctheta and alterations in the insulin signaling cascade. Diabetes 1999, 48, 1270–1274. [Google Scholar] [CrossRef] [PubMed]
- Szendroedi, J.; Yoshimura, T.; Phielix, E.; Koliaki, C.; Marcucci, M.; Zhang, D.; Jelenik, T.; Muller, J.; Herder, C.; Nowotny, P.; et al. Role of diacylglycerol activation of PKCθ in lipid-induced muscle insulin resistance in humans. Proc. Natl. Acad. Sci. USA 2014, 111, 9597–9602. [Google Scholar] [CrossRef] [PubMed]
- Kitajima, Y.; Hyogo, H.; Sumida, Y.; Eguchi, Y.; Ono, N.; Kuwashiro, T.; Tanaka, K.; Takahashi, H.; Mizuta, T.; Ozaki, I.; et al. Severity of non-alcoholic steatohepatitis is associated with substitution of adipose tissue in skeletal muscle. J. Gastroenterol. Hepatol. 2013, 28, 1507–1514. [Google Scholar] [CrossRef] [PubMed]
- Arias-Loste, M.T.; Ranchal, I.; Romero-Gomez, M.; Crespo, J. Irisin, a link among fatty liver disease, physical inactivity and insulin resistance. Int. J. Mol. Sci. 2014, 15, 23163–23178. [Google Scholar] [CrossRef] [PubMed]
- Bostrom, P.; Wu, J.; Jedrychowski, M.P.; Korde, A.; Ye, L.; Lo, J.C.; Rasbach, K.A.; Bostrom, E.A.; Choi, J.H.; Long, J.Z.; et al. A PGC 1-α-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature 2012, 481, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Schulz, T.J.; Tseng, Y.H. Brown adipose tissue: Development, metabolism and beyond. Biochem. J. 2013, 453, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Seale, P.; Bjork, B.; Yang, W.; Kajimura, S.; Chin, S.; Kuang, S.; Scime, A.; Devarakonda, S.; Conroe, H.M.; Erdjument-Bromage, H.; et al. PRDM16 controls a brown fat/skeletal muscle switch. Nature 2008, 454, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, J.; Seale, P. Medicine. Beige can be slimming. Science 2010, 328, 1113–1114. [Google Scholar] [CrossRef] [PubMed]
- Enerback, S. The origins of brown adipose tissue. N. Engl. J. Med. 2009, 360, 2021–2023. [Google Scholar] [CrossRef] [PubMed]
- Scheja, L.; Heeren, J. Metabolic interplay between white, beige, brown adipocytes and the liver. J. Hepatol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Guerra, C.; Navarro, P.; Valverde, A.M.; Arribas, M.; Bruning, J.; Kozak, L.P.; Kahn, C.R.; Benito, M. Brown adipose tissue-specific insulin receptor knockout shows diabetic phenotype without insulin resistance. J. Clin. Investig. 2001, 108, 1205–1213. [Google Scholar] [CrossRef] [PubMed]
- Bartelt, A.; Bruns, O.T.; Reimer, R.; Hohenberg, H.; Ittrich, H.; Peldschus, K.; Kaul, M.G.; Tromsdorf, U.I.; Weller, H.; Waurisch, C.; et al. Brown adipose tissue activity controls triglyceride clearance. Nat. Med. 2011, 17, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Assyov, Y.; Gateva, A.; Tsakova, A.; Kamenov, Z. Irisin in the glucose continuum. Exp. Clin. Endocrinol. Diabetes 2016, 124, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.J.; Wong, M.D.; Toy, W.C.; Tan, C.S.; Liu, S.; Ng, X.W.; Tavintharan, S.; Sum, C.F.; Lim, S.C. Lower circulating irisin is associated with type 2 diabetes mellitus. J. Diabetes Complicat. 2013, 27, 365–369. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.J.; Zhang, X.F.; Ma, Z.M.; Pan, L.L.; Chen, Z.; Han, H.W.; Han, C.K.; Zhuang, X.J.; Lu, Y.; Li, X.J.; et al. Irisin is inversely associated with intrahepatic triglyceride contents in obese adults. J. Hepatol. 2013, 59, 557–562. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef] [PubMed]
- Haas, J.T.; Francque, S.; Staels, B. Pathophysiology and mechanisms of nonalcoholic fatty liver disease. Ann. Rev. Physiol. 2016, 78, 181–205. [Google Scholar] [CrossRef] [PubMed]
- Seppala-Lindroos, A.; Vehkavaara, S.; Hakkinen, A.M.; Goto, T.; Westerbacka, J.; Sovijarvi, A.; Halavaara, J.; Yki-Jarvinen, H. Fat accumulation in the liver is associated with defects in insulin suppression of glucose production and serum free fatty acids independent of obesity in normal men. J. Clin. Endocrinol. Metab. 2002, 87, 3023–3028. [Google Scholar] [CrossRef] [PubMed]
- Bugianesi, E.; Gastaldelli, A.; Vanni, E.; Gambino, R.; Cassader, M.; Baldi, S.; Ponti, V.; Pagano, G.; Ferrannini, E.; Rizzetto, M. Insulin resistance in non-diabetic patients with non-alcoholic fatty liver disease: Sites and mechanisms. Diabetologia 2005, 48, 634–642. [Google Scholar] [CrossRef] [PubMed]
- Vatner, D.F.; Majumdar, S.K.; Kumashiro, N.; Petersen, M.C.; Rahimi, Y.; Gattu, A.K.; Bears, M.; Camporez, J.P.; Cline, G.W.; Jurczak, M.J.; et al. Insulin-independent regulation of hepatic triglyceride synthesis by fatty acids. Proc. Natl. Acad. Sci. USA 2015, 112, 1143–1148. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaka, T.; Shimano, H.; Yahagi, N.; Amemiya-Kudo, M.; Okazaki, H.; Tamura, Y.; Iizuka, Y.; Ohashi, K.; Tomita, S.; Sekiya, M.; et al. Insulin-independent induction of sterol regulatory element-binding protein-1c expression in the livers of streptozotocin-treated mice. Diabetes 2004, 53, 560–569. [Google Scholar] [CrossRef] [PubMed]
- Stanhope, K.L.; Schwarz, J.M.; Keim, N.L.; Griffen, S.C.; Bremer, A.A.; Graham, J.L.; Hatcher, B.; Cox, C.L.; Dyachenko, A.; Zhang, W.; et al. Consuming fructose-sweetened, not glucose-sweetened, beverages increases visceral adiposity and lipids and decreases insulin sensitivity in overweight/obese humans. J. Clin. Investig. 2009, 119, 1322–1334. [Google Scholar] [CrossRef] [PubMed]
- Uyeda, K.; Repa, J.J. Carbohydrate response element binding protein, chrebp, a transcription factor coupling hepatic glucose utilization and lipid synthesis. Cell Metab. 2006, 4, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Erion, D.M.; Popov, V.; Hsiao, J.J.; Vatner, D.; Mitchell, K.; Yonemitsu, S.; Nagai, Y.; Kahn, M.; Gillum, M.P.; Dong, J.; et al. The role of the carbohydrate response element-binding protein in male fructose-fed rats. Endocrinology 2013, 154, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Nagai, Y.; Yonemitsu, S.; Erion, D.M.; Iwasaki, T.; Stark, R.; Weismann, D.; Dong, J.; Zhang, D.; Jurczak, M.J.; Loffler, M.G.; et al. The role of peroxisome proliferator-activated receptor gamma coactivator-1 β in the pathogenesis of fructose-induced insulin resistance. Cell Metab. 2009, 9, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Bindesboll, C.; Fan, Q.; Norgaard, R.C.; MacPherson, L.; Ruan, H.B.; Wu, J.; Pedersen, T.A.; Steffensen, K.R.; Yang, X.; Matthews, J.; et al. Liver Xreceptor regulates hepatic nuclear O-GlcNAc signaling and carbohydrate responsive element-binding protein activity. J. Lipid Res. 2015, 56, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Samuel, V.T.; Shulman, G.I. Mechanisms for insulin resistance: Common threads and missing links. Cell 2012, 148, 852–871. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, K.; Yang, L.; McCall, S.; Huang, J.; Yu, X.X.; Pandey, S.K.; Bhanot, S.; Monia, B.P.; Li, Y.X.; Diehl, A.M. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology 2007, 45, 1366–1374. [Google Scholar] [CrossRef] [PubMed]
- Samuel, V.T.; Liu, Z.X.; Wang, A.; Beddow, S.A.; Geisler, J.G.; Kahn, M.; Zhang, X.M.; Monia, B.P.; Bhanot, S.; Shulman, G.I. Inhibition of protein kinase cepsilon prevents hepatic insulin resistance in nonalcoholic fatty liver disease. J. Clin. Investig. 2007, 117, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Seale, J.P.; Donnelly, R. Tissue and isoform-selective activation of protein kinase Cin insulin-resistant obese zucker rats—Effects of feeding. J. Endocrinol. 1999, 162, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Kumashiro, N.; Erion, D.M.; Zhang, D.; Kahn, M.; Beddow, S.A.; Chu, X.; Still, C.D.; Gerhard, G.S.; Han, X.; Dziura, J.; et al. Cellular mechanism of insulin resistance in nonalcoholic fatty liver disease. Proc. Natl. Acad. Sci. USA 2011, 108, 16381–16385. [Google Scholar] [CrossRef] [PubMed]
- Magkos, F.; Su, X.; Bradley, D.; Fabbrini, E.; Conte, C.; Eagon, J.C.; Varela, J.E.; Brunt, E.M.; Patterson, B.W.; Klein, S. Intrahepatic diacylglycerol content is associated with hepatic insulin resistance in obese subjects. Gastroenterology 2012, 142, 1444–1446. [Google Scholar] [CrossRef] [PubMed]
- Schmitz-Peiffer, C.; Craig, D.L.; Biden, T.J. Ceramide generation is sufficient to account for the inhibition of the insulin-stimulated PKBpathway in C2C12 skeletal muscle cells pretreated with palmitate. J. Biol. Chem. 1999, 274, 24202–24210. [Google Scholar] [CrossRef] [PubMed]
- Stratford, S.; Hoehn, K.L.; Liu, F.; Summers, S.A. Regulation of insulin action by ceramide: Dual mechanisms linking ceramide accumulation to the inhibition of AKT/protein kinase B. J. Biol. Chem. 2004, 279, 36608–36615. [Google Scholar] [CrossRef] [PubMed]
- Turinsky, J.; O′Sullivan, D.M.; Bayly, B.P. 1,2-Diacylglycerol and ceramide levels in insulin-resistant tissues of the rat in vivo. J. Biol. Chem. 1990, 265, 16880–16885. [Google Scholar] [PubMed]
- Chavez, J.A.; Summers, S.A. A ceramide-centric view of insulin resistance. Cell Metab. 2012, 15, 585–594. [Google Scholar] [CrossRef] [PubMed]
- Hla, T.; Kolesnick, R. C16:0-ceramide signals insulin resistance. Cell Metab. 2014, 20, 703–705. [Google Scholar] [CrossRef] [PubMed]
- Turpin, S.M.; Nicholls, H.T.; Willmes, D.M.; Mourier, A.; Brodesser, S.; Wunderlich, C.M.; Mauer, J.; Xu, E.; Hammerschmidt, P.; Bronneke, H.S.; et al. Obesity-induced CERS6-dependent C16:0 ceramide production promotes weight gain and glucose intolerance. Cell Metab. 2014, 20, 678–686. [Google Scholar] [CrossRef] [PubMed]
- Luukkonen, P.K.; Zhou, Y.; Sadevirta, S.; Leivonen, M.; Arola, J.; Oresic, M.; Hyotylainen, T.; Yki-Jarvinen, H. Ceramides dissociate steatosis and insulin resistance in the human liver in non-alcoholic fatty liver disease. J. Hepatol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, C.M.; Mancuso, D.J.; Yan, W.; Sims, H.F.; Gibson, B.; Gross, R.W. Identification, cloning, expression, and purification of three novel human calcium-independent phospholipase A2 family members possessing triacylglycerol lipase and acylglycerol transacylase activities. J. Biol. Chem. 2004, 279, 48968–48975. [Google Scholar] [CrossRef] [PubMed]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef] [PubMed]
- Kantartzis, K.; Peter, A.; Machicao, F.; Machann, J.; Wagner, S.; Konigsrainer, I.; Konigsrainer, A.; Schick, F.; Fritsche, A.; Haring, H.U.; et al. Dissociation between fatty liver and insulin resistance in humans carrying a variant of the patatin-like phospholipase 3 gene. Diabetes 2009, 58, 2616–2623. [Google Scholar] [CrossRef] [PubMed]
- Kotronen, A.; Johansson, L.E.; Johansson, L.M.; Roos, C.; Westerbacka, J.; Hamsten, A.; Bergholm, R.; Arkkila, P.; Arola, J.; Kiviluoto, T.; et al. A common variant in PNPLA3, which encodes adiponutrin, is associated with liver fat content in humans. Diabetologia 2009, 52, 1056–1060. [Google Scholar] [CrossRef] [PubMed]
- Speliotes, E.K.; Butler, J.L.; Palmer, C.D.; Voight, B.F.; Consortium, G.; Consortium, M.I.; Nash, C.R.N.; Hirschhorn, J.N. PNPLA3 variants specifically confer increased risk for histologic nonalcoholic fatty liver disease but not metabolic disease. Hepatology 2010, 52, 904–912. [Google Scholar] [CrossRef] [PubMed]
- Kumari, M.; Schoiswohl, G.; Chitraju, C.; Paar, M.; Cornaciu, I.; Rangrez, A.Y.; Wongsiriroj, N.; Nagy, H.M.; Ivanova, P.T.; Scott, S.A.; et al. Adiponutrin functions as a nutritionally regulated lysophosphatidic acid acyltransferase. Cell Metab. 2012, 15, 691–702. [Google Scholar] [CrossRef] [PubMed]
- Smagris, E.; BasuRay, S.; Li, J.; Huang, Y.; Lai, K.M.; Gromada, J.; Cohen, J.C.; Hobbs, H.H. PNPLA3I148Mknockin mice accumulate PNPLA3 on lipid droplets and develop hepatic steatosis. Hepatology 2015, 61, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.W.; Yang, H.; Mitchell, G.A. Potential mechanism underlying the PNPLA3I148M-hepatic steatosis connection. Hepatology 2016, 63, 676–677. [Google Scholar] [CrossRef] [PubMed]
- Puri, P.; Mirshahi, F.; Cheung, O.; Natarajan, R.; Maher, J.W.; Kellum, J.M.; Sanyal, A.J. Activation and dysregulation of the unfolded protein response in nonalcoholic fatty liver disease. Gastroenterology 2008, 134, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Malhi, H.; Gores, G.J. Molecular mechanisms of lipotoxicity in nonalcoholic fatty liver disease. Semin. Liver Dis. 2008, 28, 360–369. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Singh, R.; Xiang, Y.; Czaja, M.J. Macroautophagy and chaperone-mediated autophagy are required for hepatocyte resistance to oxidant stress. Hepatology 2010, 52, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Czaja, M.J. Autophagy in health and disease. 2. Regulation of lipid metabolism and storage by autophagy: Pathophysiological implications. Am. J. Physiol. Cell Physiol. 2010, 298, C973–C978. [Google Scholar] [CrossRef] [PubMed]
- Pal, D.; Dasgupta, S.; Kundu, R.; Maitra, S.; Das, G.; Mukhopadhyay, S.; Ray, S.; Majumdar, S.S.; Bhattacharya, S. Fetuin-Aacts as an endogenous ligand of TLR4 to promote lipid-induced insulin resistance. Nat. Med. 2012, 18, 1279–1285. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, P.; Seal, S.; Mukherjee, S.; Kundu, R.; Mukherjee, S.; Ray, S.; Mukhopadhyay, S.; Majumdar, S.S.; Bhattacharya, S. Adipocyte fetuin-Acontributes to macrophage migration into adipose tissue and polarization of macrophages. J. Biol. Chem. 2013, 288, 28324–28330. [Google Scholar] [CrossRef] [PubMed]
- Ix, J.H.; Shlipak, M.G.; Brandenburg, V.M.; Ali, S.; Ketteler, M.; Whooley, M.A. Association between human fetuin-Aand the metabolic syndrome: Data from the heart and soul study. Circulation 2006, 113, 1760–1767. [Google Scholar] [CrossRef] [PubMed]
- Ix, J.H.; Wassel, C.L.; Kanaya, A.M.; Vittinghoff, E.; Johnson, K.C.; Koster, A.; Cauley, J.A.; Harris, T.B.; Cummings, S.R.; Shlipak, M.G.; et al. Fetuin-Aand incident diabetes mellitus in older persons. JAMA 2008, 300, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Stefan, N.; Hennige, A.M.; Staiger, H.; Machann, J.; Schick, F.; Krober, S.M.; Machicao, F.; Fritsche, A.; Haring, H.U. Alpha2-heremans-schmid glycoprotein/fetuin-Ais associated with insulin resistance and fat accumulation in the liver in humans. Diabetes Care 2006, 29, 853–857. [Google Scholar] [CrossRef] [PubMed]
- Meex, R.C.; Hoy, A.J.; Morris, A.; Brown, R.D.; Lo, J.C.; Burke, M.; Goode, R.J.; Kingwell, B.A.; Kraakman, M.J.; Febbraio, M.A.; et al. Fetuin Bis a secreted hepatocyte factor linking steatosis to impaired glucose metabolism. Cell Metab. 2015, 22, 1078–1089. [Google Scholar] [CrossRef] [PubMed]
- Kharitonenkov, A.; Shiyanova, T.L.; Koester, A.; Ford, A.M.; Micanovic, R.; Galbreath, E.J.; Sandusky, G.E.; Hammond, L.J.; Moyers, J.S.; Owens, R.A.; et al. FGF-21 as a novel metabolic regulator. J. Clin. Investig. 2005, 115, 1627–1635. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yeung, D.C.; Karpisek, M.; Stejskal, D.; Zhou, Z.G.; Liu, F.; Wong, R.L.; Chow, W.S.; Tso, A.W.; Lam, K.S.; et al. Serum FGF21 levels are increased in obesity and are independently associated with the metabolic syndrome in humans. Diabetes 2008, 57, 1246–1253. [Google Scholar] [CrossRef] [PubMed]
- Itoh, N. FGF21 as a hepatokine, adipokine, and myokine in metabolism and diseases. Front. Endocrinol. (Lausanne) 2014, 5, 107. [Google Scholar] [CrossRef] [PubMed]
- Chavez, A.O.; Molina-Carrion, M.; Abdul-Ghani, M.A.; Folli, F.; Defronzo, R.A.; Tripathy, D. Circulating fibroblast growth factor-21 is elevated in impaired glucose tolerance and type 2 diabetes and correlates with muscle and hepatic insulin resistance. Diabetes Care 2009, 32, 1542–1546. [Google Scholar] [CrossRef] [PubMed]
- Dushay, J.; Chui, P.C.; Gopalakrishnan, G.S.; Varela-Rey, M.; Crawley, M.; Fisher, F.M.; Badman, M.K.; Martinez-Chantar, M.L.; Maratos-Flier, E. Increased fibroblast growth factor 21 in obesity and nonalcoholic fatty liver disease. Gastroenterology 2010, 139, 456–463. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Dong, K.; Fang, Q.; Hou, X.; Zhou, M.; Bao, Y.; Xiang, K.; Xu, A.; Jia, W. High serum level of fibroblast growth factor 21 is an independent predictor of non-alcoholic fatty liver disease: A3-year prospective study in china. J. Hepatol. 2013, 58, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Fang, Q.; Gao, F.; Fan, J.; Zhou, J.; Wang, X.; Zhang, H.; Pan, X.; Bao, Y.; Xiang, K.; et al. Fibroblast growth factor 21 levels are increased in nonalcoholic fatty liver disease patients and are correlated with hepatic triglyceride. J. Hepatol. 2010, 53, 934–940. [Google Scholar] [CrossRef] [PubMed]
- El Ouaamari, A.; Dirice, E.; Gedeon, N.; Hu, J.; Zhou, J.Y.; Shirakawa, J.; Hou, L.; Goodman, J.; Karampelias, C.; Qiang, G.; et al. SerpinB1 promotes pancreatic beta cell proliferation. Cell Metab. 2016, 23, 194–205. [Google Scholar] [CrossRef] [PubMed]
- Tarasov, A.I.; Rorsman, P. Dramatis personae in beta-cell mass regulation: Enter serpinB1. Cell Metab. 2016, 23, 8–10. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Boursier, J.; Mueller, O.; Barret, M.; Machado, M.; Fizanne, L.; Araujo-Perez, F.; Guy, C.D.; Seed, P.C.; Rawls, J.F.; David, L.A.; et al. The severity of NAFLD is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology 2016, 63, 764–775. [Google Scholar] [CrossRef] [PubMed]
- Le Roy, T.; Llopis, M.; Lepage, P.; Bruneau, A.; Rabot, S.; Bevilacqua, C.; Martin, P.; Philippe, C.; Walker, F.; Bado, A.; et al. Intestinal microbiota determines development of non-alcoholic fatty liver disease in mice. Gut 2013, 62, 1787–1794. [Google Scholar] [CrossRef] [PubMed]
- Musso, G.; Gambino, R.; Cassader, M. Gut microbiota as a regulator of energy homeostasis and ectopic fat deposition: Mechanisms and implications for metabolic disorders. Curr. Opin. Lipidol. 2010, 21, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Nolan, C.J.; Damm, P.; Prentki, M. Type 2 diabetes across generations: From pathophysiology to prevention and management. Lancet 2011, 378, 169–181. [Google Scholar] [CrossRef]
- Shima, T.; Uto, H.; Ueki, K.; Takamura, T.; Kohgo, Y.; Kawata, S.; Yasui, K.; Park, H.; Nakamura, N.; Nakatou, T.; et al. Clinicopathological features of liver injury in patients with type 2 diabetes mellitus and comparative study of histologically proven nonalcoholic fatty liver diseases with or without type 2 diabetes mellitus. J.Gastroenterol. 2013, 48, 515–525. [Google Scholar] [CrossRef] [PubMed]
- Goh, G.B.; Pagadala, M.R.; Dasarathy, J.; Unalp-Arida, A.; Sargent, R.; Hawkins, C.; Sourianarayanane, A.; Khiyami, A.; Yerian, L.; Pai, R.K.; et al. Clinical spectrum of non-alcoholic fatty liver disease in diabetic and non-diabetic patients. BBA Clin. 2015, 3, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Nascimbeni, F.; Aron-Wisniewsky, J.; Pais, R.; Tordjman, J.; Poitou, C.; Charlotte, F.; Bedossa, P.; Poynard, T.; Clement, K.; Ratziu, V. Statins, antidiabetic medications and liver histology in diabetic patients with non-alcoholic fatty liver disease. BMJ Open Gastroenterol. 2016, in press. [Google Scholar]
- Angulo, P.; Keach, J.C.; Batts, K.P.; Lindor, K.D. Independent predictors of liver fibrosis in patients with nonalcoholic steatohepatitis. Hepatology 1999, 30, 1356–1362. [Google Scholar] [CrossRef] [PubMed]
- De Ledinghen, V.; Ratziu, V.; Causse, X.; Le Bail, B.; Capron, D.; Renou, C.; Pilette, C.; Oules, V.; Gelsi, E.; Oberti, F.; et al. Diagnostic and predictive factors of significant liver fibrosis and minimal lesions in patients with persistent unexplained elevated transaminases. A prospective multicenter study. J. Hepatol. 2006, 45, 592–599. [Google Scholar] [CrossRef] [PubMed]
- Hossain, N.; Afendy, A.; Stepanova, M.; Nader, F.; Srishord, M.; Rafiq, N.; Goodman, Z.; Younossi, Z. Independent predictors of fibrosis in patients with nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. 2009, 7, 1224–1229. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, T.; Hyogo, H.; Yoneda, M.; Sumida, Y.; Eguchi, Y.; Fujii, H.; Ono, M.; Kawaguchi, T.; Imajo, K.; Aikata, H.; et al. Type 2 diabetes mellitus is associated with the fibrosis severity in patients with nonalcoholic fatty liver disease in a large retrospective cohort of Japanese patients. J. Gastroenterol. 2014, 49, 1477–1484. [Google Scholar] [CrossRef] [PubMed]
- McPherson, S.; Hardy, T.; Henderson, E.; Burt, A.D.; Day, C.P.; Anstee, Q.M. Evidence of NAFLDprogression from steatosis to fibrosing-steatohepatitis using paired biopsies: Implications for prognosis and clinical management. J. Hepatol. 2015, 62, 1148–1155. [Google Scholar] [CrossRef] [PubMed]
- Pais, R.; Charlotte, F.; Fedchuk, L.; Bedossa, P.; Lebray, P.; Poynard, T.; Ratziu, V.; Group, L.S. A systematic review of follow-up biopsies reveals disease progression in patients with non-alcoholic fatty liver. J. Hepatol. 2013, 59, 550–556. [Google Scholar] [CrossRef] [PubMed]
- Adams, L.A.; Sanderson, S.; Lindor, K.D.; Angulo, P. The histological course of nonalcoholic fatty liver disease: A longitudinal study of 103 patients with sequential liver biopsies. J. Hepatol. 2005, 42, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Hamaguchi, E.; Takamura, T.; Sakurai, M.; Mizukoshi, E.; Zen, Y.; Takeshita, Y.; Kurita, S.; Arai, K.; Yamashita, T.; Sasaki, M.; et al. Histological course of nonalcoholic fatty liver disease in Japanese patients: Tight glycemic control, rather than weight reduction, ameliorates liver fibrosis. Diabetes Care 2010, 33, 284–286. [Google Scholar] [CrossRef] [PubMed]
- Hashiba, M.; Ono, M.; Hyogo, H.; Ikeda, Y.; Masuda, K.; Yoshioka, R.; Ishikawa, Y.; Nagata, Y.; Munekage, K.; Ochi, T.; et al. Glycemic variability is an independent predictive factor for development of hepatic fibrosis in nonalcoholic fatty liver disease. PLoS ONE 2013, 8, e76161. [Google Scholar] [CrossRef] [PubMed]
- Fracanzani, A.L.; Valenti, L.; Bugianesi, E.; Andreoletti, M.; Colli, A.; Vanni, E.; Bertelli, C.; Fatta, E.; Bignamini, D.; Marchesini, G.; et al. Risk of severe liver disease in nonalcoholic fatty liver disease with normal aminotransferase levels: Arole for insulin resistance and diabetes. Hepatology 2008, 48, 792–798. [Google Scholar] [CrossRef] [PubMed]
- Ballestri, S.; Romagnoli, D.; Nascimbeni, F.; Francica, G.; Lonardo, A. Role of ultrasound in the diagnosis and treatment of nonalcoholic fatty liver disease and its complications. Expert Rev. Gastroenterol. Hepatol. 2015, 9, 603–627. [Google Scholar] [CrossRef] [PubMed]
- Nascimbeni, F.; Loria, P.; Ratziu, V. Non-alcoholic fatty liver disease: Diagnosis and investigation. Dig. Dis. 2014, 32, 586–596. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Gramlich, T.; Matteoni, C.A.; Boparai, N.; McCullough, A.J. Nonalcoholic fatty liver disease in patients with type 2 diabetes. Clin. Gastroenterol. Hepatol. 2004, 2, 262–265. [Google Scholar] [CrossRef]
- Hessheimer, A.J.; Forner, A.; Varela, M.; Bruix, J. Metabolic risk factors are a major comorbidity in patients with cirrhosis independent of the presence of hepatocellular carcinoma. Eur. J. Gastroenterol. Hepatol. 2010, 22, 1239–1244. [Google Scholar] [CrossRef] [PubMed]
- Ertle, J.; Dechene, A.; Sowa, J.P.; Penndorf, V.; Herzer, K.; Kaiser, G.; Schlaak, J.F.; Gerken, G.; Syn, W.K.; Canbay, A. Non-alcoholic fatty liver disease progresses to hepatocellular carcinoma in the absence of apparent cirrhosis. Int. J.Cancer 2011, 128, 2436–2443. [Google Scholar] [CrossRef] [PubMed]
- Yasui, K.; Hashimoto, E.; Komorizono, Y.; Koike, K.; Arii, S.; Imai, Y.; Shima, T.; Kanbara, Y.; Saibara, T.; Mori, T.; et al. Characteristics of patients with nonalcoholic steatohepatitis who develop hepatocellular carcinoma. Clin. Gastroenterol. Hepatol. 2011, 9, 428–433. [Google Scholar] [CrossRef] [PubMed]
- Raff, E.J.; Kakati, D.; Bloomer, J.R.; Shoreibah, M.; Rasheed, K.; Singal, A.K. Diabetes mellitus predicts occurrence of cirrhosis and hepatocellular cancer in alcoholic liver and non-alcoholic fatty liver diseases. J. Clin. Transl. Hepatol. 2015, 3, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Giannini, E.G.; Marabotto, E.; Savarino, V.; Trevisani, F.; di Nolfo, M.A.; Del Poggio, P.; Benvegnu, L.; Farinati, F.; Zoli, M.; Borzio, F.; et al. Hepatocellular carcinoma in patients with cryptogenic cirrhosis. Clin. Gastroenterol. Hepatol. 2009, 7, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Piscaglia, F.; Svegliati-Baroni, G.; Barchetti, A.; Pecorelli, A.; Marinelli, S.; Tiribelli, C.; Bellentani, S. HCC-NAFLD Italian Study Group. Clinical patterns of hepatocellular carcinoma (HCC) in nonalcoholic fatty liver disease (NAFLD): Amulticenter prospective study. Hepatology 2016, 63, 827–838. [Google Scholar] [CrossRef] [PubMed]
- Zoppini, G.; Fedeli, U.; Gennaro, N.; Saugo, M.; Targher, G.; Bonora, E. Mortality from chronic liver diseases in diabetes. Am. J. Gastroenterol. 2014, 109, 1020–1025. [Google Scholar] [CrossRef] [PubMed]
- Wild, S.H.; Morling, J.R.; McAllister, D.A.; Kerssens, J.; Fischbacher, C.; Parkes, J.; Roderick, P.J.; Sattar, N.; Byrne, C.D. Scottish and Southampton Diabetes and Liver Disease Group and the Scottish Diabetes Research Network Epidemiology Group. Type 2 diabetes, chronic liver disease and hepatocellular cancer: A national retrospective cohort study using linked routine data. J. Hepatol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Adams, L.A.; Harmsen, S.; St Sauver, J.L.; Charatcharoenwitthaya, P.; Enders, F.B.; Therneau, T.; Angulo, P. Nonalcoholic fatty liver disease increases risk of death among patients with diabetes: A community-based cohort study. Am. J. Gastroenterol. 2010, 105, 1567–1573. [Google Scholar] [CrossRef] [PubMed]
- Ballestri, S.; Lonardo, A.; Bonapace, S.; Byrne, C.D.; Loria, P.; Targher, G. Risk of cardiovascular, cardiac and arrhythmic complications in patients with non-alcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 1724–1745. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, A.; Ballestri, S.; Targher, G.; Loria, P. Diagnosis and management of cardiovascular risk in nonalcoholic fatty liver disease. Expert Rev. Gastroenterol. Hepatol. 2015, 9, 629–650. [Google Scholar] [CrossRef] [PubMed]
- Targher, G.; Bertolini, L.; Rodella, S.; Tessari, R.; Zenari, L.; Lippi, G.; Arcaro, G. Nonalcoholic fatty liver disease is independently associated with an increased incidence of cardiovascular events in type 2 diabetic patients. Diabetes Care 2007, 30, 2119–2121. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Ballestri, S.; Lonardo, A.; Targher, G. Cardiovascular disease and myocardial abnormalities in nonalcoholic fatty liver disease. Dig. Dis. Sci. 2016. [Google Scholar] [CrossRef] [PubMed]
- Naunyn, B. Der Diabetes Melitis; A Holder: Wienna, Austria, 1898. [Google Scholar]
- Lonardo, A.; Adinolfi, L.E.; Petta, S.; Craxi, A.; Loria, P. Hepatitis C and diabetes: The inevitable coincidence? Expert Rev. Anti. Infect. Ther. 2009, 7, 293–308. [Google Scholar] [CrossRef] [PubMed]
- Allison, M.E.; Wreghitt, T.; Palmer, C.R.; Alexander, G.J. Evidence for a link between hepatitis Cvirus infection and diabetes mellitus in a cirrhotic population. J. Hepatol. 1994, 21, 1135–1139. [Google Scholar] [CrossRef]
- Searched on PubMed, Keywords: “HCV and diabetes”. Available online: http://www.ncbi.nlm.nih.gov/pubmed/?term=HCV+and+diabetes (accessed on 24 January 2016).
- Kohli, A.; Shaffer, A.; Sherman, A.; Kottilil, S. Treatment of hepatitis C: Asystematic review. JAMA 2014, 312, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Hajarizadeh, B.; Grebely, J.; Dore, G.J. Epidemiology and natural history of HCVinfection. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Shire, N.J.; Sherman, K.E. Epidemiology of hepatitis Cvirus: Abattle on new frontiers. Gastroenterol. Clin. N. Am. 2015, 44, 699–716. [Google Scholar] [CrossRef] [PubMed]
- Dultz, G.; Zeuzem, S. Hepatitis Cvirus: An European perspective. Gastroenterol. Clin. N. Am. 2015, 44, 807–824. [Google Scholar] [CrossRef] [PubMed]
- Lozano, R.; Naghavi, M.; Foreman, K.; Lim, S.; Shibuya, K.; Aboyans, V.; Abraham, J.; Adair, T.; Aggarwal, R.; Ahn, S.Y.; et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: Asystematic analysis for the global burden of disease study 2010. Lancet 2012, 380, 2095–2128. [Google Scholar] [CrossRef]
- Wong, R.J.; Aguilar, M.; Cheung, R.; Perumpail, R.B.; Harrison, S.A.; Younossi, Z.M.; Ahmed, A. Nonalcoholic steatohepatitis is the second leading etiology of liver disease among adults awaiting liver transplantation in the united states. Gastroenterology 2015, 148, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Mehta, S.H.; Brancati, F.L.; Strathdee, S.A.; Pankow, J.S.; Netski, D.; Coresh, J.; Szklo, M.; Thomas, D.L. Hepatitis Cvirus infection and incident type 2 diabetes. Hepatology 2003, 38, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Rudoni, S.; Petit, J.M.; Bour, J.B.; Aho, L.S.; Castaneda, A.; Vaillant, G.; Verges, B.; Brun, J.M. HCV infection and diabetes mellitus: Influence of the use of finger stick devices on nosocomial transmission. Diabetes Metab. 1999, 25, 502–505. [Google Scholar] [PubMed]
- Lonardo, A.; Carulli, N.; Loria, P. HCV and diabetes. A two-question-based reappraisal. Dig. Liver Dis. 2007, 39, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, A.; Adinolfi, L.E.; Restivo, L.; Ballestri, S.; Romagnoli, D.; Baldelli, E.; Nascimbeni, F.; Loria, P. Pathogenesis and significance of hepatitis C virus steatosis: An update on survival strategy of a successful pathogen. World J. Gastroenterol. 2014, 20, 7089–7103. [Google Scholar] [CrossRef] [PubMed]
- Petta, S.; Macaluso, F.S.; Camma, C.; Marco, V.D.; Cabibi, D.; Craxi, A. Hyperuricaemia: Another metabolic feature affecting the severity of chronic hepatitis because of HCV infection. Liver Int. 2012, 32, 1443–1450. [Google Scholar] [CrossRef] [PubMed]
- Petta, S.; Maida, M.; Macaluso, F.S.; Barbara, M.; Licata, A.; Craxi, A.; Camma, C. Hepatitis C virus infection is associated with increased cardiovascular mortality: Ameta-analysis of observational studies. Gastroenterology 2016, 150, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Singal, A.G.; El-Serag, H.B. Hepatocellular carcinoma from epidemiology to prevention: Translating knowledge into practice. Clin. Gastroenterol. Hepatol. 2015, 13, 2140–2151. [Google Scholar] [CrossRef] [PubMed]
- Dyal, H.K.; Aguilar, M.; Bartos, G.; Holt, E.W.; Bhuket, T.; Liu, B.; Cheung, R.; Wong, R.J. Diabetes mellitus increases risk of hepatocellular carcinoma in chronic hepatitis C virus patients: Asystematic review. Dig. Dis. Sci. 2016, 61, 636–645. [Google Scholar] [CrossRef] [PubMed]
- Lecube, A.; Hernandez, C.; Genesca, J.; Simo, R. Proinflammatory cytokines, insulin resistance, and insulin secretion in chronic hepatitis C patients: A case-control study. Diabetes Care 2006, 29, 1096–1101. [Google Scholar] [CrossRef] [PubMed]
- Knobler, H.; Zhornicky, T.; Sandler, A.; Haran, N.; Ashur, Y.; Schattner, A. Tumor necrosis factor-alpha-induced insulin resistance may mediate the hepatitis C virus-diabetes association. Am. J. Gastroenterol. 2003, 98, 2751–2756. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.Y.; Wang, L.F.; Fan, X.H.; Wu, C.H.; Huo, N.; Lu, H.Y.; Xu, X.Y.; Wei, L. Association of suppressor of cytokine signalling 3 polymorphisms with insulin resistance in patients with chronic hepatitis C. J. Viral Hepat. 2013, 20, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Pazienza, V.; Vinciguerra, M.; Andriulli, A.; Mangia, A. Hepatitis C virus core protein genotype 3a increases SOCS-7 expression through PPAR-{gamma} in Huh-7 cells. J. Gen. Virol. 2010, 91, 1678–1686. [Google Scholar] [CrossRef] [PubMed]
- Vanni, E.; Abate, M.L.; Gentilcore, E.; Hickman, I.; Gambino, R.; Cassader, M.; Smedile, A.; Ferrannini, E.; Rizzetto, M.; Marchesini, G.; et al. Sites and mechanisms of insulin resistance in nonobese, nondiabetic patients with chronic hepatitis C. Hepatology 2009, 50, 697–706. [Google Scholar] [CrossRef] [PubMed]
- Milner, K.L.; van der Poorten, D.; Trenell, M.; Jenkins, A.B.; Xu, A.; Smythe, G.; Dore, G.J.; Zekry, A.; Weltman, M.; Fragomeli, V.; et al. Chronic hepatitis C is associated with peripheral rather than hepatic insulin resistance. Gastroenterology 2010, 138, 932–941. [Google Scholar] [CrossRef] [PubMed]
- Uruha, A.; Noguchi, S.; Hayashi, Y.K.; Tsuburaya, R.S.; Yonekawa, T.; Nonaka, I.; Nishino, I. Hepatitis C virus infection in inclusion body myositis: A case-control study. Neurology 2016, 86, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Adinolfi, L.E.; Gambardella, M.; Andreana, A.; Tripodi, M.F.; Utili, R.; Ruggiero, G. Steatosis accelerates the progression of liver damage of chronic hepatitis C patients and correlates with specific HCV genotype and visceral obesity. Hepatology 2001, 33, 1358–1364. [Google Scholar] [CrossRef] [PubMed]
- Guaraldi, G.; Lonardo, A.; Ballestri, S.; Zona, S.; Stentarelli, C.; Orlando, G.; Carli, F.; Carulli, L.; Roverato, A.; Loria, P. Human immunodeficiency virus is the major determinant of steatosis and hepatitis C virus of insulin resistance in virus-associated fatty liver disease. Arch. Med. Res. 2011, 42, 690–697. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, A.; Loria, P.; Carulli, N. Dysmetabolic changes associated with HCV: Adistinct syndrome? Intern. Emerg. Med. 2008, 3, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Adinolfi, L.E.; Restivo, L.; Zampino, R.; Lonardo, A.; Loria, P. Metabolic alterations and chronic hepatitis C: Treatment strategies. Expert Opin. Pharmacother. 2011, 12, 2215–2234. [Google Scholar] [CrossRef] [PubMed]
- Loria, P.; Marchesini, G.; Nascimbeni, F.; Ballestri, S.; Maurantonio, M.; Carubbi, F.; Ratziu, V.; Lonardo, A. Cardiovascular risk, lipidemic phenotype and steatosis. A comparative analysis of cirrhotic and non-cirrhotic liver disease due to varying etiology. Atherosclerosis 2014, 232, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Mostafa, A.; Mohamed, M.K.; Saeed, M.; Hasan, A.; Fontanet, A.; Godsland, I.; Coady, E.; Esmat, G.; El-Hoseiny, M.; Abdul-Hamid, M.; et al. Hepatitis Cinfection and clearance: Impact on atherosclerosis and cardiometabolic risk factors. Gut 2010, 59, 1135–1140. [Google Scholar] [CrossRef] [PubMed]
- Zampino, R.; Coppola, N.; Cirillo, G.; Boemio, A.; Pisaturo, M.; Marrone, A.; Macera, M.; Sagnelli, E.; Perrone, L.; Adinolfi, L.E.; et al. Abdominal fat interacts with PNPLA3 I148M, but not with the APOC3 variant in the pathogenesis of liver steatosis in chronic hepatitis C. J. Viral Hepat. 2013, 20, 517–523. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.C.; Lamerato, L.E.; Rupp, L.B.; Holmberg, S.D.; Moorman, A.C.; Spradling, P.R.; Teshale, E.; Xu, F.; Boscarino, J.A.; Vijayadeva, V.; et al. Prevalence of cirrhosis in hepatitis Cpatients in the chronic hepatitis cohort study (CHeCS): A retrospective and prospective observational study. Am. J. Gastroenterol. 2015, 110, 1169–1177. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Jinjuvadia, R.; Patel, R.; Liangpunsakul, S. Insulin resistance is associated with significant liver fibrosis in chronic hepatitis C patients: Asystemic review and meta-analysis. J. Clin. Gastroenterol. 2016, 50, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Moucari, R.; Asselah, T.; Cazals-Hatem, D.; Voitot, H.; Boyer, N.; Ripault, M.P.; Sobesky, R.; Martinot-Peignoux, M.; Maylin, S.; Nicolas-Chanoine, M.H.; et al. Insulin resistance in chronic hepatitis C: Association with genotypes 1 and 4, serum HCVRNA level, and liver fibrosis. Gastroenterology 2008, 134, 416–423. [Google Scholar] [CrossRef] [PubMed]
- Serste, T.; Nkuize, M.; Moucari, R.; Van Gossum, M.; Reynders, M.; Scheen, R.; Vertongen, F.; Buset, M.; Mulkay, J.P.; Marcellin, P. Metabolic disorders associated with chronic hepatitis C: Impact of genotype and ethnicity. Liver Int. 2010, 30, 1131–1136. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.S.; Yao, W.J.; Chang, T.T.; Wang, S.T.; Chou, P. The impact of type 2 diabetes on the development of hepatocellular carcinoma in different viral hepatitis statuses. Cancer Epidemiol. Biomarkers Prev. 2009, 18, 2054–2060. [Google Scholar] [CrossRef] [PubMed]
- Arase, Y.; Kobayashi, M.; Suzuki, F.; Suzuki, Y.; Kawamura, Y.; Akuta, N.; Kobayashi, M.; Sezaki, H.; Saito, S.; Hosaka, T.; et al. Effect of type 2 diabetes on risk for malignancies includes hepatocellular carcinoma in chronic hepatitis C. Hepatology 2013, 57, 964–973. [Google Scholar] [CrossRef] [PubMed]
- Bruno, S.; Crosignani, A.; Maisonneuve, P.; Rossi, S.; Silini, E.; Mondelli, M.U. Hepatitis C virus genotype 1b as a major risk factor associated with hepatocellular carcinoma in patients with cirrhosis: Aseventeen-year prospective cohort study. Hepatology 2007, 46, 1350–1356. [Google Scholar] [CrossRef] [PubMed]
- Adinolfi, L.E.; Zampino, R.; Restivo, L.; Lonardo, A.; Guerrera, B.; Marrone, A.; Nascimbeni, F.; Florio, A.; Loria, P. Chronic hepatitis C virus infection and atherosclerosis: Clinical impact and mechanisms. World J. Gastroenterol. 2014, 20, 3410–3417. [Google Scholar] [CrossRef] [PubMed]
- Adinolfi, L.E.; Restivo, L.; Zampino, R.; Guerrera, B.; Lonardo, A.; Ruggiero, L.; Riello, F.; Loria, P.; Florio, A. Chronic HCV infection is a risk of atherosclerosis. Role of HCVand HCV-related steatosis. Atherosclerosis 2012, 221, 496–502. [Google Scholar] [CrossRef] [PubMed]
- Petta, S.; Torres, D.; Fazio, G.; Camma, C.; Cabibi, D.; Di Marco, V.; Licata, A.; Marchesini, G.; Mazzola, A.; Parrinello, G.; et al. Carotid atherosclerosis and chronic hepatitis C: A prospective study of risk associations. Hepatology 2012, 55, 1317–1323. [Google Scholar] [CrossRef] [PubMed]
- Targher, G.; Bertolini, L.; Padovani, R.; Rodella, S.; Arcaro, G.; Day, C. Differences and similarities in early atherosclerosis between patients with non-alcoholic steatohepatitis and chronic hepatitis Band C. J. Hepatol. 2007, 46, 1126–1132. [Google Scholar] [CrossRef] [PubMed]
- Gill, K.; Ghazinian, H.; Manch, R.; Gish, R. Hepatitis C virus as a systemic disease: Reaching beyond the liver. Hepatol. Int. 2015, in press. [Google Scholar] [CrossRef] [PubMed]
- Domont, F.; Cacoub, P. Chronic hepatitis C virus infection, a new cardiovascular risk factor? Liver Int. 2016. [Google Scholar] [CrossRef] [PubMed]
- Backus, L.I.; Belperio, P.S.; Shahoumian, T.A.; Loomis, T.P.; Mole, L.A. Effectiveness of sofosbuvir-based regimens in genotype 1 and 2 hepatitis C virus infection in 4026 U.S. Veterans. Aliment. Pharmacol. Ther. 2015, 42, 559–573. [Google Scholar] [CrossRef] [PubMed]
- Rinella, M.E. Nonalcoholic fatty liver disease: A systematic review. JAMA 2015, 313, 2263–2273. [Google Scholar] [CrossRef] [PubMed]
- Montesi, L.; Caselli, C.; Centis, E.; Nuccitelli, C.; Moscatiello, S.; Suppini, A.; Marchesini, G. Physical activity support or weight loss counseling for nonalcoholic fatty liver disease? World J. Gastroenterol. 2014, 20, 10128–10136. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, C.P.; de Lima Sanches, P.; de Abreu-Silva, E.O.; Marcadenti, A. Nutrition and physical activity in nonalcoholic fatty liver disease. J. Diabetes Res. 2016, 2016, 4597246. [Google Scholar] [CrossRef] [PubMed]
- Hallsworth, K.; Avery, L.; Trenell, M.I. Targeting lifestyle behavior change in adults with NAFLD during a 20-min consultation: Summary of the dietary and exercise literature. Curr. Gastroenterol. Rep. 2016, 18, 11. [Google Scholar] [CrossRef] [PubMed]
- Hickman, I.J.; Clouston, A.D.; Macdonald, G.A.; Purdie, D.M.; Prins, J.B.; Ash, S.; Jonsson, J.R.; Powell, E.E. Effect of weight reduction on liver histology and biochemistry in patients with chronic hepatitis C. Gut 2002, 51, 89–94. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Criteria | Metabolic Syndrome | HCADS | Reference(s) |
|---|---|---|---|
| T2D | Yes | Yes | [3] |
| Hypertension | Yes | Yes | [3] |
| Visceral Obesity | Yes | Preliminary evidence suggests that HCV patients have abdominal fat distribution | [3] |
| Atherogenic dyslipidemia | Yes | Acquired, reversible hypocholesterolemia | [6] |
| Hepatic steatosis | Not included among diagnostic criteria but often found as a concurrent or precursor finding | In chronic HCV patients, steatosis is two- to three-fold more prevalent than in chronic hepatitides of other etiologies. HCV genotype 3 is associated with a higher prevalence and more severe steatosis | [3,6] |
| Hyperuricemia | Not included in diagnostic criteria but often associated on pathophysiological grounds | Strongly associated with severity of steatosis | [3,190] |
| Accelerated atherogenesis | Whether the full-blown MetS adds to the risk of its individual components, particularly T2D, is controversial | Individuals with HCV infection (particularly those with T2D and hypertension) have an excess of cardiovascular morbidity and mortality | [3,191] |
| HCC risk | Both the MetS and T2D increase the risk of HCC. This likely results via NAFLD/NASH even in non-cirrhotic livers | Concurrent T2D and chronic HCV infection lead to increased risk of HCC. Steatosis and overweight/obesity possibly play a role | [168,192,193] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ballestri, S.; Nascimbeni, F.; Romagnoli, D.; Baldelli, E.; Targher, G.; Lonardo, A. Type 2 Diabetes in Non-Alcoholic Fatty Liver Disease and Hepatitis C Virus Infection—Liver: The “Musketeer” in the Spotlight. Int. J. Mol. Sci. 2016, 17, 355. https://doi.org/10.3390/ijms17030355
Ballestri S, Nascimbeni F, Romagnoli D, Baldelli E, Targher G, Lonardo A. Type 2 Diabetes in Non-Alcoholic Fatty Liver Disease and Hepatitis C Virus Infection—Liver: The “Musketeer” in the Spotlight. International Journal of Molecular Sciences. 2016; 17(3):355. https://doi.org/10.3390/ijms17030355
Chicago/Turabian StyleBallestri, Stefano, Fabio Nascimbeni, Dante Romagnoli, Enrica Baldelli, Giovanni Targher, and Amedeo Lonardo. 2016. "Type 2 Diabetes in Non-Alcoholic Fatty Liver Disease and Hepatitis C Virus Infection—Liver: The “Musketeer” in the Spotlight" International Journal of Molecular Sciences 17, no. 3: 355. https://doi.org/10.3390/ijms17030355
APA StyleBallestri, S., Nascimbeni, F., Romagnoli, D., Baldelli, E., Targher, G., & Lonardo, A. (2016). Type 2 Diabetes in Non-Alcoholic Fatty Liver Disease and Hepatitis C Virus Infection—Liver: The “Musketeer” in the Spotlight. International Journal of Molecular Sciences, 17(3), 355. https://doi.org/10.3390/ijms17030355