
Bidirectional Relationships and Disconnects between NAFLD and Features of the Metabolic Syndrome

Abstract
:
1. Introduction
2. Association between NAFLD and Components of the Metabolic Syndrome
3. NAFLD as a Risk Factor for and Precursor to the Metabolic Syndrome
4. Metabolic Syndrome as an Initiating or Aggravating Factor for Liver Disease
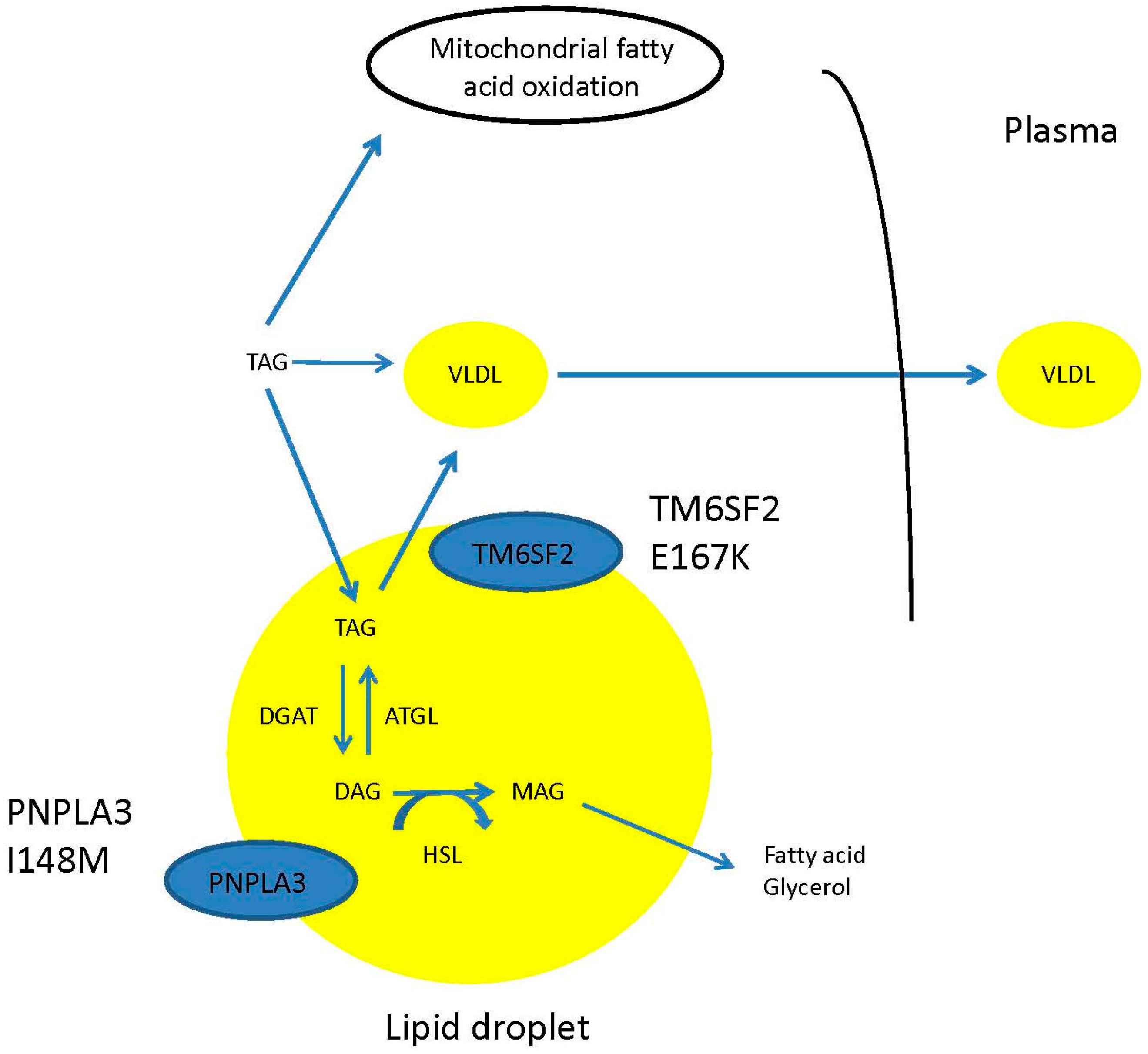
5. Evidence for a Disconnection between Hepatic Steatosis and Metabolic Syndrome
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| NAFLD | Non-alcoholic fatty liver disease |
| PNPLA3 | Patatin-like phospholipase domain-containing protein-3 |
| TM6SF2 | Transmembrane 6 superfamily member 2 protein |
| HDL-C | High density lipoprotein cholesterol |
| NASH | Non-alcoholic steatohepatitis |
| PKC-ε | Protein kinase C-ε |
| FLI | Fatty liver index |
| PAI-1 | Plasminogen activator inhibitor-1 |
| TNF-α | Tumour necrosis factor-α |
| IL | Interleukin |
| DAG | Diacylglycerols |
| DGAT | Diacylglycerol acyltransferase |
| SNP | Single nucleotide polymorphism |
| VLDL | Very low density lipoprotein |
| TAG | Triacylglycerol |
| MAG | Monoacylglycerol |
| ATGL | Adipose triglyceride lipase |
| HSL | Hormone sensitive lipase |
| FHBL | Familial hypobetalipoproteinaemia |
| BMI | Body mass index |
References
- Anstee, Q.M.; McPherson, S.; Day, C.P. How big a problem is nonalcoholic fatty liver disease? BMJ 2011, 343, d3897. [Google Scholar] [CrossRef] [PubMed]
- Targher, G.; Byrne, C.D. Clinical review. Nonalcoholic fatty liver disease: A novel cardiometabolic risk factor for type 2 diabetes and its complications. J. Clin. Endocrinol. Metab. 2013, 98, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Diehl, A.M.; Brunt, E.M.; Cusi, K.; Charlton, M.; Sanyal, A.J. The diagnosis and management of non-alcoholic fatty liver disease: Practice guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Hepatology 2012, 55, 2005–2023. [Google Scholar] [CrossRef] [PubMed]
- Browning, J.D.; Szczepaniak, L.S.; Dobbins, R.; Nuremberg, P.; Horton, J.D.; Cohen, J.C.; Grundy, S.M.; Hobbs, H.H. Prevalence of hepatic steatosis in an urban population in the United States: Impact of ethnicity. Hepatology 2004, 40, 1387–1395. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, M.; Bhuket, T.; Torres, S.; Liu, B.; Wong, R.J. Prevalence of the metabolic syndrome in the United States 2003–2012. JAMA 2015, 313, 1973–1974. [Google Scholar] [CrossRef] [PubMed]
- Cortez-Pinto, H.; Camilo, M.E.; Baptista, A.; de Oliveira, A.G.; de Moura, M.C. Non-alcoholic fatty liver: Another feature of the metabolic syndrome? Clin. Nutr. 1999, 18, 353–358. [Google Scholar] [CrossRef]
- Bedogni, G.; Miglioli, L.; Masutti, F.; Tiribelli, C.; Marchesini, G.; Bellentani, S. Prevalence of and risk factors for nonalcoholic fatty liver disease: The Dionysos nutrition and liver study. Hepatology 2005, 42, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Souza, M.R.; Diniz Mde, F.; Medeiros-Filho, J.E.; Araujo, M.S. Metabolic syndrome and risk factors for non-alcoholic fatty liver disease. Arq. Gastroenterol. 2012, 49, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, T.; Zhang, C.; Tang, F.; Zhong, N.; Li, H.; Song, X.; Lin, H.; Liu, Y.; Xue, F. Identification of reciprocal causality between non-alcoholic fatty liver disease and metabolic syndrome by a simplified Bayesian network in a Chinese population. BMJ Open 2015, 5, e008204. [Google Scholar] [CrossRef] [PubMed]
- Alberti, K.G.; Eckel, R.H.; Grundy, S.M.; Zimmet, P.Z.; Cleeman, J.I.; Donato, K.A.; Fruchart, J.C.; James, W.P.; Loria, C.M.; Smith, S.C.; et al. Harmonizing the metabolic syndrome. Circulation 2009, 120, 1640–1645. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Kim, H.J.; Lee, K.E.; Kim, S.K.; Ahn, C.W.; Lim, S.K.; Kim, K.R.; Lee, H.C.; Huh, K.B.; et al. Metabolic significance of nonalcoholic fatty liver disease in nonobese, nondiabetic adults. Arch. Intern. Med. 2004, 164, 2169–2175. [Google Scholar] [CrossRef] [PubMed]
- Sinn, D.H.; Gwak, G.Y.; Park, H.N.; Kim, J.E.; Min, Y.W.; Kim, K.M.; Kim, Y.J.; Choi, M.S.; Lee, J.H.; Koh, K.C.; et al. Ultrasonographically detected non-alcoholic fatty liver disease is an independent predictor for identifying patients with insulin resistance in non-obese, non-diabetic middle-aged Asian adults. Am. J. Gastroenterol. 2012, 107, 561–567. [Google Scholar] [CrossRef] [PubMed]
- Targher, G.; Bertolini, L.; Padovani, R.; Rodella, S.; Tessari, R.; Zenari, L.; Day, C.; Arcaro, G. Prevalence of nonalcoholic fatty liver disease and its association with cardiovascular disease among type 2 diabetic patients. Diabetes Care 2007, 30, 1212–1218. [Google Scholar] [CrossRef] [PubMed]
- Leite, N.C.; Salles, G.F.; Araujo Antonio, L.E.; Villela-Noqueira, C.A.; Cardosa, C.R. Prevalence and associated factors of non-alcoholic fatty liver disease in patients with type-2 diabetes mellitus. Liver Int. 2009, 29, 113e9. [Google Scholar] [CrossRef] [PubMed]
- Assy, N.; Kaita, K.; Mymin, D.; Levy, C.; Rosser, B.; Minuk, G. Fatty infiltration of liver in hyperlipidemic patients. Dig. Dis. Sci. 2000, 45, 1929–1934. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Suarez, A.; Guerrero, J.M.; Elvira-Gonzalez, J.; Beltran-Robles, M.; Canas-Hormigo, F.; Bascunana-Quirell, A. Nonalcoholic fatty liver disease is associated with blood pressure in hypertensive and nonhypertensive individuals from the general population with normal levels of alanine aminotransferase. Eur. J. Gastroenterol. Hepatol. 2011, 23, 1011–1017. [Google Scholar] [CrossRef] [PubMed]
- Lau, K.; Lorbeer, R.; Haring, R.; Schmidt, C.O.; Wallaschofski, H.; Nauck, M.; John, U.; Baumeister, S.E.; Volzke, H. The association between fatty liver disease and blood pressure in a population-based prospective longitudinal study. J. Hypertens. 2010, 28, 1829–1835. [Google Scholar] [CrossRef] [PubMed]
- Marchesini, G.; Bugianesi, E.; Forlani, G.; Cerelli, F.; Lenzi, M.; Manini, R.; Natale, S.; Vanni, E.; Villanova, N.; Melchionda, N. Nonalcoholic fatty liver, steatohepatitis, and the metabolic syndrome. Hepatology 2003, 37. [Google Scholar] [CrossRef] [PubMed]
- Ryan, M.C.; Wilson, A.M.; Slavin, J.; Best, J.D.; Jenkins, A.J.; Desmond, P.V. Associations between liver histology and severity of the metabolic syndrome in subjects with nonalcoholic fatty liver disease. Diabetes Care 2005, 28, 1222–1224. [Google Scholar] [CrossRef] [PubMed]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two ‘‘hits’’? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef] [PubMed]
- Samuel, V.T.; Liu, Z.-X.; Wang, A.; Beddow, S.A.; Geisler, J.G.; Kahn, M.; Zhang, X.; Monia, B.P.; Bhanot, S.; Shulman, G.I. Inhibition of protein kinase Cε prevents hepatic insulin resistance in nonalcoholic fatty liver disease. J. Clin. Investig. 2007, 117, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Kumashiro, N.; Erion, D.M.; Zhang, D.; Kahn, M.; Beddow, S.A.; Chu, X.; Still, C.D.; Gerhard, G.S.; Han, X.; Dziura, J.; et al. Cellular mechanism of insulin resistance in nonalcoholic fatty liver disease. PNAS 2011, 108, 16381–16385. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.W.; Wang, X.; Joo, J.I.; Kim, D.H.; Oh, T.S.; Choi, D.K.; Yun, J.W. Plasma proteome analysis in diet-induced obesity-prone and obesity resistant rats. Proteomics 2010, 10, 4386–4400. [Google Scholar] [CrossRef] [PubMed]
- Meex, R.C.; Hoy, A.J.; Morris, A.; Brown, R.D.; Lo, J.C.; Burke, M.; Goode, R.J.; Kingwell, B.A.; Kraakman, M.J.; Febbraio, M.A.; et al. Fetuin B is a secreted hepatocyte factor linking steatosis to impaired glucose metabolism. Cell Metab. 2015, 22, 1078–1089. [Google Scholar] [CrossRef] [PubMed]
- Hamaguchi, M.; Takeda, N.; Kojima, T.; Ohbora, A.; Kato, T.; Sarhui, H.; Fukui, M.; Nagata, C.; Takeda, J. Identification of individuals with non-alcoholic fatty liver disease by the diagnostic criteria for the metabolic syndrome. World J. Gastroenterol. 2012, 18, 1508–1516. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Zhang, Y.; Zhang, C.; Tang, F.; Li, H.; Zhang, Q.; Lin, H.; Wu, S.; Liu, Y.; Xue, F. Prediction of metabolic syndrome by non-alcoholic fatty liver disease in northern urban Han Chinese population: A prospective cohort study. PLoS ONE 2014, 9, e96651. [Google Scholar]
- Smits, M.M.; Ioannou, G.N.; Boyko, E.J.; Utzschneider, K.M. Non-alcoholic fatty liver disease as an independent manifestation of the metabolic syndrome: Results of a US national survey in three ethnic groups. J. Gastroenterol. Hepatol. 2013, 28, 664–670. [Google Scholar] [CrossRef] [PubMed]
- Adams, L.A.; Waters, O.R.; Knuiman, M.W.; Elliott, R.R.; Olynyk, J.K. NAFLD as a risk factor for the development of diabetes and the metabolic syndrome: An eleven-year follow-up study. Am. J. Gastroenterol. 2009, 104, 861–867. [Google Scholar] [CrossRef] [PubMed]
- Ryoo, J.H.; Choi, J.M.; Moon, S.Y.; Suh, Y.J.; Shin, J.Y.; Shin, H.C.; Park, S.K. The clinical availability of non alcoholic fatty liver disease as an early predictor of the metabolic syndrome in Korean men: 5-year’s prospective cohort study. Atherosclerosis 2013, 227, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Hamaguchi, M.; Kojima, T.; Itoh, Y.; Harano, Y.; Fujii, K.; Nakajima, T.; Kato, T.; Takeda, N.; Okuda, J.; Ida, K.; et al. The severity of ultrasonographic findings in nonalcoholic fatty liver disease reflects the metabolic syndrome and visceral fat accumulation. Am. J. Gastroenterol. 2007, 102, 2708–2715. [Google Scholar] [CrossRef] [PubMed]
- Mohan, V.; Farooq, S.; Deepa, M.; Ravikumar, R.; Pitchumoni, C.S. Prevalence of non-alcoholic fatty liver disease in urban south Indians in relation to different grades of glucose intolerance and metabolic syndrome. Diabetes Res. Clin. Pract. 2009, 84, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Rhoo, J.H.; Suh, Y.J.; Shin, H.C.; Cho, Y.K.; Choi, J.M.; Park, S.K. Clinical association between non-alcoholic fatty liver disease and the development of hypertension. J. Gastroenterol. Hepatol. 2014, 29, 1926–1931. [Google Scholar]
- Rhoo, J.H.; Ham, W.T.; Choi, J.M.; Kang, M.A.; An, S.H.; Lee, J.K.; Shin, H.C.; Park, S.K. Clinical significance of non-alcoholic fatty liver disease as a risk factor for prehypertension. J. Korean Med. Sci. 2014, 29, 973–979. [Google Scholar]
- Huh, J.H.; Ahn, S.V.; Koh, S.B.; Choi, E.; Kim, J.Y.; Sung, K.C.; Kim, E.J.; Park, J.B. A prospective study of fatty liver index and incident hypertension: The KoGES-ARIRANG study. PLoS ONE 2015, 10, e0143560. [Google Scholar]
- Sung, K.C.; Wild, S.H.; Byrne, C.D. Development of new fatty liver, or resolution of existing fatty liver, over five years of follow-up, and risk of incident hypertension. J. Hepatol. 2014, 60, 1040–1045. [Google Scholar] [CrossRef] [PubMed]
- Sung, K.C.; Wild, S.H.; Byrne, C.D. Resolution of fatty liver and risk of incident diabetes. J. Clin. Endocrinol. Metab. 2013, 93, 3637–3643. [Google Scholar] [CrossRef] [PubMed]
- Vozarova, B.; Stefan, N.; Lindsay, R.S.; Saremi, A.; Pratley, R.E.; Bogardus, C.; Tataranni, P.A. High alanine Aminotransferase is associated with decreased hepatic insulin sensitivity and predicts the development of type 2 diabetes. Diabetes 2002, 51, 1889–1895. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Ha, M.H.; Kim, J.H.; Christiani, D.C.; Gross, M.D.; Steffes, M.; Blomhoff, R.; Jacobs, D.R. Gamma glutamyltransferase and diabetes—A 4 years follow-up study. Diabetologia 2003, 46, 359–364. [Google Scholar] [PubMed]
- Hanley, A.J.; Williams, K.; Festa, A.; Wagenknecht, L.E.; D’Agostino, R.B.; Haffner, S.M. Liver markers and development of the metabolic syndrome: The insulin resistance atherosclerosis study. Diabetes 2005, 54, 3140–3147. [Google Scholar] [CrossRef] [PubMed]
- Ekstedt, M.; Franzén, L.E.; Mathiesen, U.L.; Thorelius, L.; Holmqvist, M.; Bodemar, G.; Kechagias, S. Long-term follow-up of patients with NAFLD and elevated liver enzymes. Hepatology 2006, 44, 865–873. [Google Scholar] [CrossRef] [PubMed]
- Monami, M.; Bardini, G.; Lamanna, C.; Pala, L.; Cresci, B.; Francesconi, P.; Buiatti, E.; Rotella, C.M.; Mannucci, E. Liver enzymes and risk of diabetes and cardiovascular disease: Results of the Firenze Bagno a Ripoli (FIBAR) study. Metabolism 2008, 57, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Goessling, W.; Massaro, J.M.; Vasan, R.S.; D’Agostino, R.B.; Ellison, R.C.; Fox, C.S. Aminotransferase levels and 20-year risk of metabolic syndrome, diabetes, and cardiovascular disease. Gastroenterology 2008, 135, 1935–1944. [Google Scholar] [CrossRef] [PubMed]
- Ryu, S.; Chang, Y.; Woo, H.Y.; Yoo, S.H.; Choi, N.K.; Lee, W.Y.; Kim, I.; Song, J. Longitudinal increase in gamma-glutamyltransferase within the reference interval predicts metabolic syndrome in middle-aged Korean men. Metabolism 2010, 59, 683–689. [Google Scholar] [PubMed]
- Balkau, B.; Lange, C.; Vol, S.; Fumeron, F.; Bonnet, F. Group Study D.E.S.I.R. Nine-year incident diabetes is predicted by fatty liver indices: The French D.E.S.I.R. study. BMC Gastroenterol. 2010, 10, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Sung, K.C.; Kim, S.H. Interrelationship between fatty liver and insulin resistance in the development of type 2 diabetes. J. Clin. Endocrinol. Metab. 2011, 96, 1093–1097. [Google Scholar] [CrossRef] [PubMed]
- Sung, K.C.; Jeong, W.S.; Wild, S.H.; Byrne, C.D. Combined influence of insulin resistance, overweight/obesity, and fatty liver as risk factors for type 2 diabetes. Diabetes Care 2012, 35, 717–722. [Google Scholar] [CrossRef] [PubMed]
- Musso, G.; Gambino, R.; Cassader, M.; Pagano, G. Meta analysis: Natural history of non-alcoholic fatty liver disease (NAFLD) and diagnostic accuracy of non-invasive tests for liver disease severity. Ann. Med. 2011, 43, 617–649. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Vaughan, D.E. PAI-1 in tissue fibrosis. J. Cell. Physiol. 2012, 227, 493–507. [Google Scholar] [CrossRef] [PubMed]
- Cesari, M.; Pahor, M.; Incalzi, R.A. Plasminogen activator inhibitor-1 (PAI-1): A key factor linking fibrinolysis and age-related subclinical and clinical conditions. Cardiovasc. Ther. 2010, 28, e72–e91. [Google Scholar] [CrossRef] [PubMed]
- Oishi, K. Plasminogen activator inhibitor-1 and the circadian clock in metabolic disorders. Clin. Exp. Hypertens. 2009, 31, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Bremer, A.A.; Jialal, I. Adipose tissue dysfunction in nascent metabolic syndrome. J. Obes. 2013, 2013, 393192. [Google Scholar] [CrossRef] [PubMed]
- Takeshita, Y.; Takamura, T.; Hamaguchi, E.; Shimizu, A.; Ota, T.; Sakurai, M.; Kaneko, S. Tumor necrosis factor-alpha-induced production of plasminogen activator inhibitor 1 and its regulation by pioglitazone and cerivastatin in a nonmalignant human hepatocyte cell line. Metabolism 2006, 55, 1464–1472. [Google Scholar] [CrossRef] [PubMed]
- Baranova, A.; Gowder, S.J.; Schlauch, K.; Elariny, H.; Collantes, R.; Afendy, A.; Ong, J.P.; Goodman, Z.; Chandhoke, V.; Younossi, Z.M. Gene expression of leptin, resistin, and adiponectin in the white adipose tissue of obese patients with non-alcoholic fatty liver disease and insulin resistance. Obes. Surg. 2006, 16, 1118–1125. [Google Scholar] [CrossRef] [PubMed]
- Baranova, A.; Schlauch, K.; Elariny, H.; Jarrar, M.; Bennett, C.; Nugent, C.; Gowder, S.J.; Younoszai, Z.; Collantes, R.; Chandhoke, V.; et al. Gene expression patterns in the hepatic tissue and in the visceral adipose tissue of patients with non-alcoholic fatty liver disease. Obes. Surg. 2007, 17, 1111–1118. [Google Scholar] [CrossRef] [PubMed]
- Hubscher, S.G. Histological assessment of non-alcoholic fatty liver disease. J. Clin. Gastroenterol. 2006, 40, S5–S10. [Google Scholar] [CrossRef] [PubMed]
- Weltman, M.D.; Farrell, G.C.; Liddle, C. Increased hepatocyte CYP2E1 expression in a rat nutritional model of hepatic steatosis with inflammation. Gastroenterology 1996, 111, 1645–1653. [Google Scholar] [CrossRef]
- Geerts, A. History, heterogeneity, developmental biology, and functions of quiescent hepatic stellate cells. Semin. Liver Dis. 2001, 21, 3113–3135. [Google Scholar] [CrossRef] [PubMed]
- Fain, J.N. Release of interleukins and other inflammatory cytokines by human adipose tissue is enhanced in obesity and primarily due to the nonfat cells. Vitam. Horm. 2006, 74, 443–477. [Google Scholar] [PubMed]
- You, T.; Nicklas, B.J. Chronic inflammation: Role of adipose tissue and modulation by weight loss. Curr. Diabetes Rev. 2006, 2, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Henao-Mejia, J.; Elinav, E.; Cheng-Cheng, J.; Hao, L.; Mehal, W.Z.; Strowig, T.; Thaiss, C.A.; Kau, A.L.; Eisenbarth, S.C.; Jurczak, M.J.; et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 2012, 482, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Feldstein, A.E.; Canbay, A.; Angulo, P.; Taniai, M.; Burgart, L.J.; Lindor, K.D.; Gores, G.J. Hepatocyte apoptosis and fas expression are prominent features of human non alcoholic steatohepatitis. Gastroenterology 2003, 125, 437–443. [Google Scholar] [CrossRef]
- Bantel, H.; Ruck, P.; Gregor, M.; Shulze-Oshtoff, K. Detection of elevated caspase activation and early apoptosis in lever disease. Eur. J. Cell Biol. 2001, 80, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Civera, M.; Urios, A.; Garcia-Torres, M.L.; Ortega, J.; Martinez-Valls, J.; Cassinello, N.; Del Olmo, J.A.; Ferrandez, A.; Rodrigo, J.M.; Montoliu, C. Relationship between insulin resistance, inflammation and liver cell apoptosis in patients with severe obesity. Diabetes Metab. Res. Rev. 2010, 26, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Adams, L.A.; Sanderson, S.; Lindor, K.D.; Angulo, P. The histological course of non-alcoholic fatty liver disease: A longitudinal study of 103 patients with sequential liver biopsies. J. Hepatol. 2005, 42, 132–138. [Google Scholar] [CrossRef] [PubMed]
- El-Serag, H.B.; Tran, T.; Everhart, J.E. Diabetes increases the risk of chronic liver disease and hepatocellular carcinoma. Gastroenterology 2004, 126, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Hamaguchi, M.; Kojima, T.; Takeda, N.; Nakagawa, T.; Taniguchi, H.; Fujii, K.; Omatsu, T.; Nakajima, T.; Sarui, H.; Shimazaki, M.; et al. The metabolic syndrome as a predictor of nonalcoholic fatty liver disease. Ann. Intern. Med. 2005, 143, 722–728. [Google Scholar] [CrossRef] [PubMed]
- Porepa, L.; Ray, J.G.; Sanchez-Romeu, P.; Booth, G.L. Newly diagnosed diabetes mellitus as a risk factor for serious liver disease. CMAJ 2010, 182, E526–E531. [Google Scholar] [CrossRef] [PubMed]
- Emerging Risk Factors, Collaboration; Seshasai, S.R.; Kaptoge, S.; Thompson, A.; Di Angelantonio, E.; Gao, P.; Sarwar, N.; Whincup, P.H.; Mukamal, K.J.; Gillum, R.F.; et al. Diabetes mellitus, fasting glucose, and risk of cause-specific death. N. Engl. J. Med. 2011, 364, 829–841. [Google Scholar]
- Graham, R.C.; Burke, A.; Stettler, N. Ethnic and sex differences in the association between metabolic syndrome and suspected nonalcoholic fatty liver disease in a nationally representative sample of US adolescents. J. Pediatr. Gastroenterol. Nutr. 2009, 49, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Castro-Martinez, M.G.; Banderas-Lares, D.Z.; Ramirez-Martinez, J.C.; Escobedode, I.P.J. Prevalence of nonalcoholic fatty liver disease in subjects with metabolic syndrome. Cir. Cir. 2012, 80, 128–133. [Google Scholar] [PubMed]
- Tsai, C.H.; Li, T.C.; Lin, C.C. Metabolic syndrome as a risk factor for non-alcoholic fatty liver disease. South Med. J. 2008, 101, 900–905. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Zhang, C.; Zhang, Y.; Tang, F.; Li, H.; Zhang, Q.; Lin, H.; Wu, S.; Liu, Y.; Xue, F. Metabolic syndrome and its components as predictors of non-alcoholic fatty liver disease in a northern urban Han Chinese population: A prospective cohort study. Atherosclerosis 2015, 240, 144–148. [Google Scholar] [CrossRef] [PubMed]
- Monetti, M.; Levin, M.C.; Watt, M.J.; Sajan, M.P.; Marmor, S.; Hubbard, B.K.; Stevens, R.D.; Bain, J.R.; Newgard, C.B.; Farese, R.V.; et al. Dissociation of hepatic steatosis and insulin resistance in mice overexpressing DGAT in the liver. Cell Metab. 2007, 6, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Kantartzis, K.; Machicao, F.; Machann, J.; Schick, F.; Fritsche, A.; Haring, H.U.; Stefan, N. The DGAT2 gene is a candidate for the dissociation between fatty liver and insulin resistance in humans. Clin. Sci. 2009, 116, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Niebergall, L.J.; Jacobs, R.L.; Chaba, T.; Vance, D.E. Phosphatidylcholine protects against steatosis in mice but not non-alcoholic steatohepatitis. Biochim. Biophys. Acta 2011, 1811, 1177–1185. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, R.L.; Zhao, Y.; Koonen, D.P.; Sletten, T.; Su, B.; Lingrell, S.; Cao, G.; Peake, D.A.; Kuo, M.S.; Proctor, S.D.; et al. Impaired de novo choline synthesis explains why phosphatidylethanolamine N-methyltransferase-deficient mice are protected from diet-induced obesity. J. Biol. Chem. 2010, 285, 22403–22413. [Google Scholar] [CrossRef] [PubMed]
- Chamoun, Z.; Vacca, F.; Parton, R.G.; Gruenberg, J. PNPLA3/adiponutrin functions in lipid droplet formation. Biol. Cell 2013, 105, 219–233. [Google Scholar] [CrossRef] [PubMed]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef] [PubMed]
- Romeo, S.; Sentinelli, F.; Dash, S.; Yeo, G.S.; Savage, D.B.; Leonetti, F.; Capoccia, D.; Incani, M.; Maglio, C.; Iacovino, M.; et al. Morbid obesity exposes the association between PNPLA3 I148M (rs738409) and indices of hepatic injury in individuals of European descent. Int. J. Obes. 2010, 34, 190–194. [Google Scholar] [CrossRef] [PubMed]
- Romeo, S.; Sentinelli, F.; Cambuli, V.M.; Incani, M.; Congiu, T.; Matta, V.; Pilia, S.; Huang-Doran, I.; Cossu, E.; Loche, S.; et al. The 148M allele of the PNPLA3 gene is associated with indices of liver damage early in life. J. Hepatol. 2010, 53, 335–338. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Wong, G.L.; Chan, H.L.; Chan, H.Y.; Yeung, D.K.; Chan, R.S.; Chim, A.M.; Chan, A.W.; Choi, P.C.; Woo, J.; et al. PNPLA3 gene polymorphism accounts for fatty liver in community subjects without metabolic syndrome. Aliment. Pharmacol. Ther. 2014, 39, 532–539. [Google Scholar] [CrossRef] [PubMed]
- Kantartzis, K.; Peter, A.; Machicao, F.; Machann, J.; Wagner, S.; Konigsrainer, I.; Konigsrainer, A.; Schick, F.; Fritsche, A.; Haring, H.U.; et al. Dissociation between fatty liver and insulin resistance in humans carrying a variant of the patatin-like phospholipase 3 gene. Diabetes 2009, 58, 2616–2623. [Google Scholar] [CrossRef] [PubMed]
- Mahdessian, H.; Taxiarchis, A.; Popov, S.; Silveira, A.; Franco-Cereceda, A.; Hamsten, A.; Eriksson, P.; van’t Hooft, F. TM6SF2 is a regulator of liver fat metabolism influencing triglyceride secretion and hepatic lipid droplet content. PNAS 2014, 111, 8913–8918. [Google Scholar] [CrossRef] [PubMed]
- Kozlitina, J.; Smagris, E.; Stender, S.; Nordestgaard, B.G.; Zhou, H.H.; Tybjærg-Hansen, A.; Vogt, T.F.; Hobbs, H.H.; Choen, J.C. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2014, 46, 352–356. [Google Scholar] [CrossRef] [PubMed]
- Sookoian, S.; Castano, G.O.; Scian, R.; Mallardi, P.; Fernandez Gianotti, T.; Burqueno, A.L.; San Martino, J.; Pirola, C.J. Genetic variation in transmembrane 6 superfamily member 2 and the risk of nonalcoholic fatty liver disease and histological disease severity. Hepatology 2015, 61, 515–525. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.L.; Reeves, H.L.; Burt, A.D.; Tiniakos, D.; McPherson, S.; Leathart, L.B.; Allison, M.E.; Alexander, G.J.; Piquet, A.C.; Anty, R. TM6SF2 rs58542926 influences hepatic fibrosis progression in patients with non-alcoholic fatty liver disease. Nat. Commun. 2014, 5, 4309. [Google Scholar] [CrossRef] [PubMed]
- Akuta, N.; Kawamura, Y.; Arase, Y.; Suzuki, F.; Sezaki, S.; Hosaka, T.; Kobayashi, M.; Kobayashi, M.; Saitoh, S.; Suzuki, Y. Relationships between genetic variations of PNPLA3, TM6SF2 and histological features of nonalcoholic fatty liver disease in Japan. Gut Liver 2015. [Google Scholar] [CrossRef] [PubMed]
- Wong, V.W.; Wong, G.L.; Tse, C.H.; Chan, H.L. Prevalence of the TM6SF2 variant and non-alcoholic fatty liver disease in Chinese. J. Hepatol. 2014, 61, 708–709. [Google Scholar] [CrossRef] [PubMed]
- Pirola, C.J.; Sookoian, S. The dual and opposite role of the TM6SF2-rs58542926 variant in protecting against cardiovascular disease and conferring risk for nonalcoholic fatty liver: A meta-analysis. Hepatology 2015, 62, 1742–1756. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Llaurado, G.; Oresic, M.; Hyotylainen, T.; Orho-Melander, M.; Yki-Jarvinen, H. Circulating triacylglycerol signatures and insulin sensitivity in NAFLD associated with the E167K variant in TM6SF2. J. Hepatol. 2015, 62, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Amaro, A.; Fabbrini, E.; Kars, M.; Yue, P.; Schechtman, K.; Schonfeld, G.; Klein, S. Dissociation between intrahepatic triglyceride content and insulin resistance in familial hypobetalipoproteinemia. Gastroenterology 2010, 139, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Reiner, Z.; Guardamagna, O.; Nair, D.; Soran, H.; Hovingh, K.; Bertolini, S.; Jones, S.; Coric, M.; Calandra, S.; Hamilton, J. Lysosomal acid lipase deficiency–an under-recognized cause of dyslipidaemias and liver dysfunction. Atherosclerosis 2014, 235, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Stefan, N.; Staiger, H.; Haring, H.U. Dissociation between fatty liver and insulin resistance: The role of adipose triacylglycerol lipase. Diabetologia 2011, 54, 7–9. [Google Scholar] [CrossRef] [PubMed]
- Haemmerle, G.; Lass, A.; Zimmermann, R.; Gorkiewicz, G.; Meyer, C.; Rozman, J.; Heldmaier, G.; Maier, R.; Theussl, C.; Eder, S. Defective lipolysis and altered energy metabolism in mice lacking adipose triglyceride lipase. Science 2006, 312, 734–737. [Google Scholar] [CrossRef] [PubMed]
- Reid, B.N.; Ables, G.P.; Otlivanchik, O.A.; Schoiswohl, G.; Zechner, R.; Blaner, W.S.; Goldberg, I.J.; Schwabe, R.F.; Chua, S.C.; Huang, L.S. Hepatic overexpression of hormone-sensitive lipase and adipose triglyceride lipase promotes fatty acid oxidation, stimulates direct release of free fatty acids, and ameliorates steatosis. J. Biol. Chem. 2008, 283, 13087–13099. [Google Scholar] [CrossRef] [PubMed]
- Turpin, S.N.; Hoy, A.J.; Brown, A.D.; Rudaz, C.G.; Honeyman, J.; Matzaris, M.; Watt, M.J. Adipose triacylglycerol lipase is a major regulator of hepatic lipid metabolism but not insulin sensitivity in mice. Diabetologia 2011, 54, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.J. Pathophysiology of fatty acid oxidation disorders. J. Inherit. Metab. Dis. 2010, 33, 533–537. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Lazar, M.A. Dissociating fatty liver and diabetes. Trends Endocrinol. Metab. 2013, 24, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Kersten, S.; Seydoux, J.; Peters, J.M.; Gonzalez, F.J.; Desvergne, B.; Wahli, W. Peroxisome proliferator-activated receptor alpha mediates the adaptive response to fasting. J. Clin. Invest. 1999, 103, 1489–1498. [Google Scholar] [CrossRef] [PubMed]
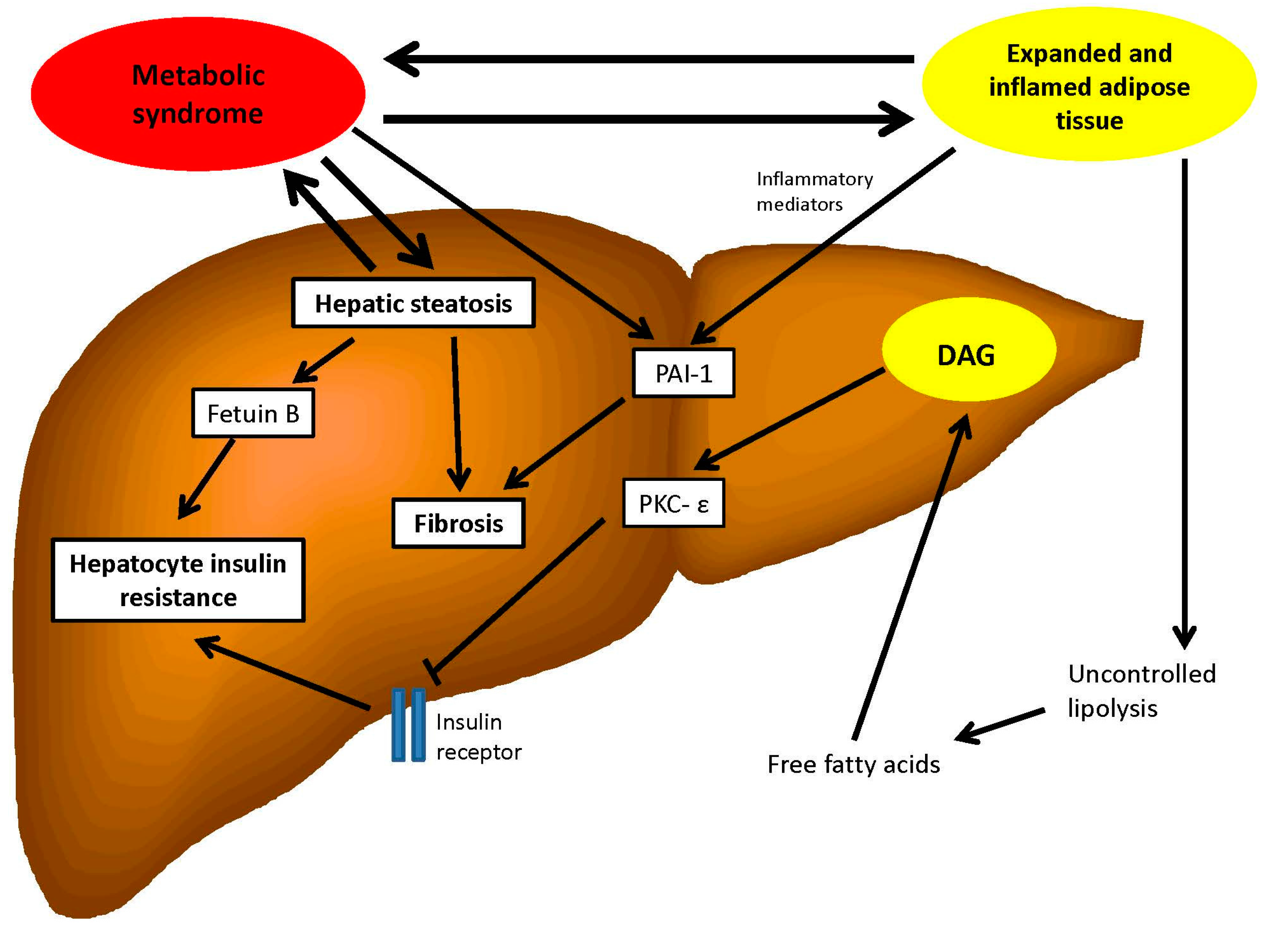

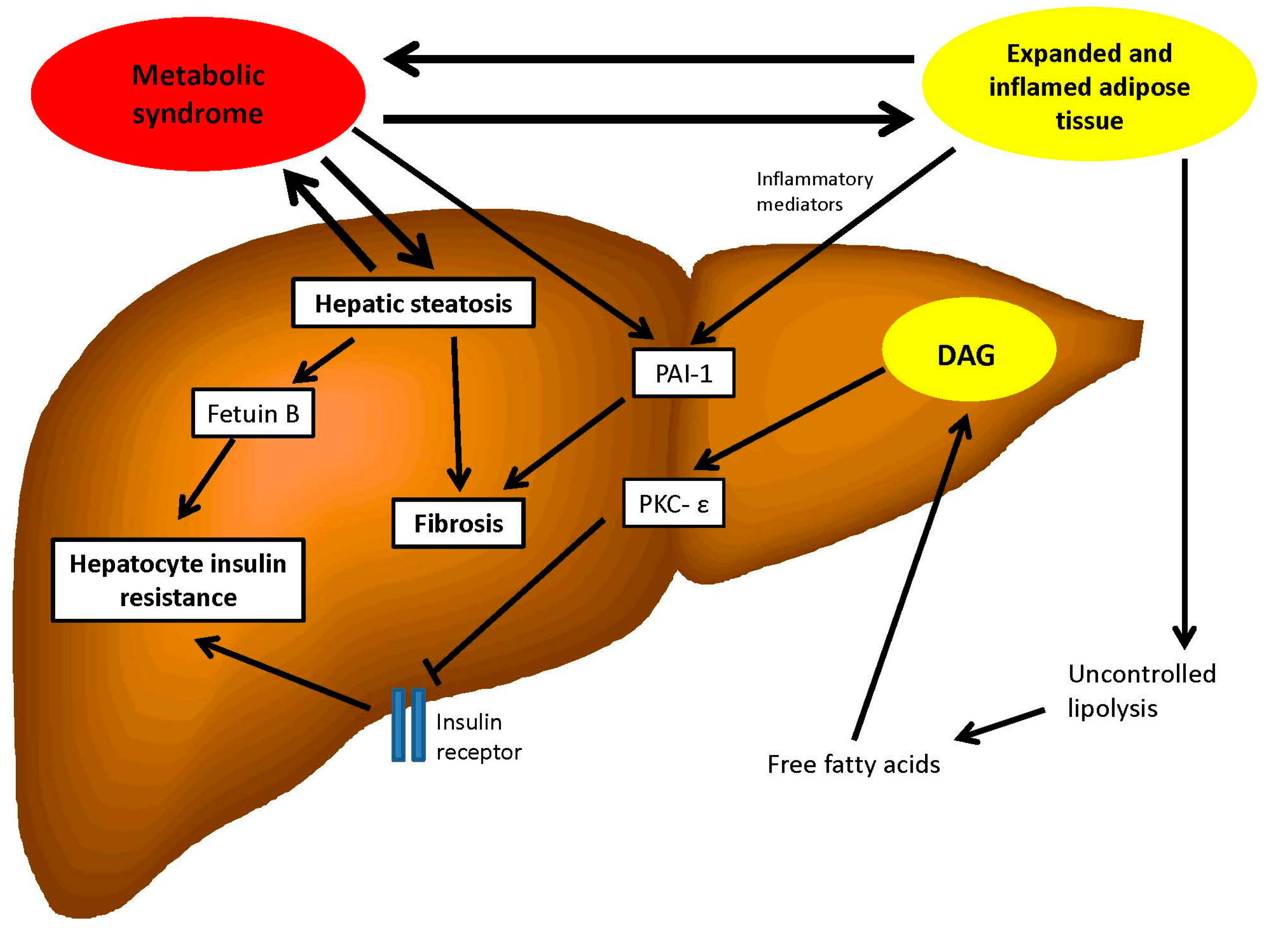
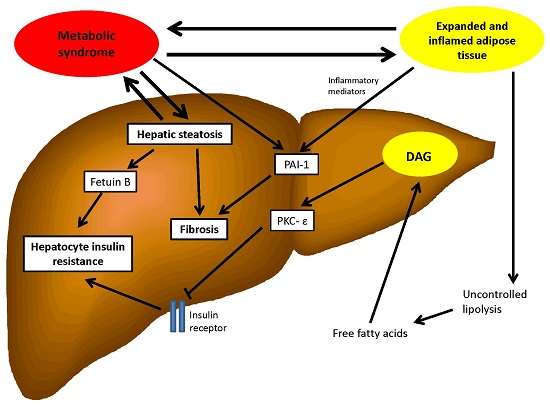{kind=link}
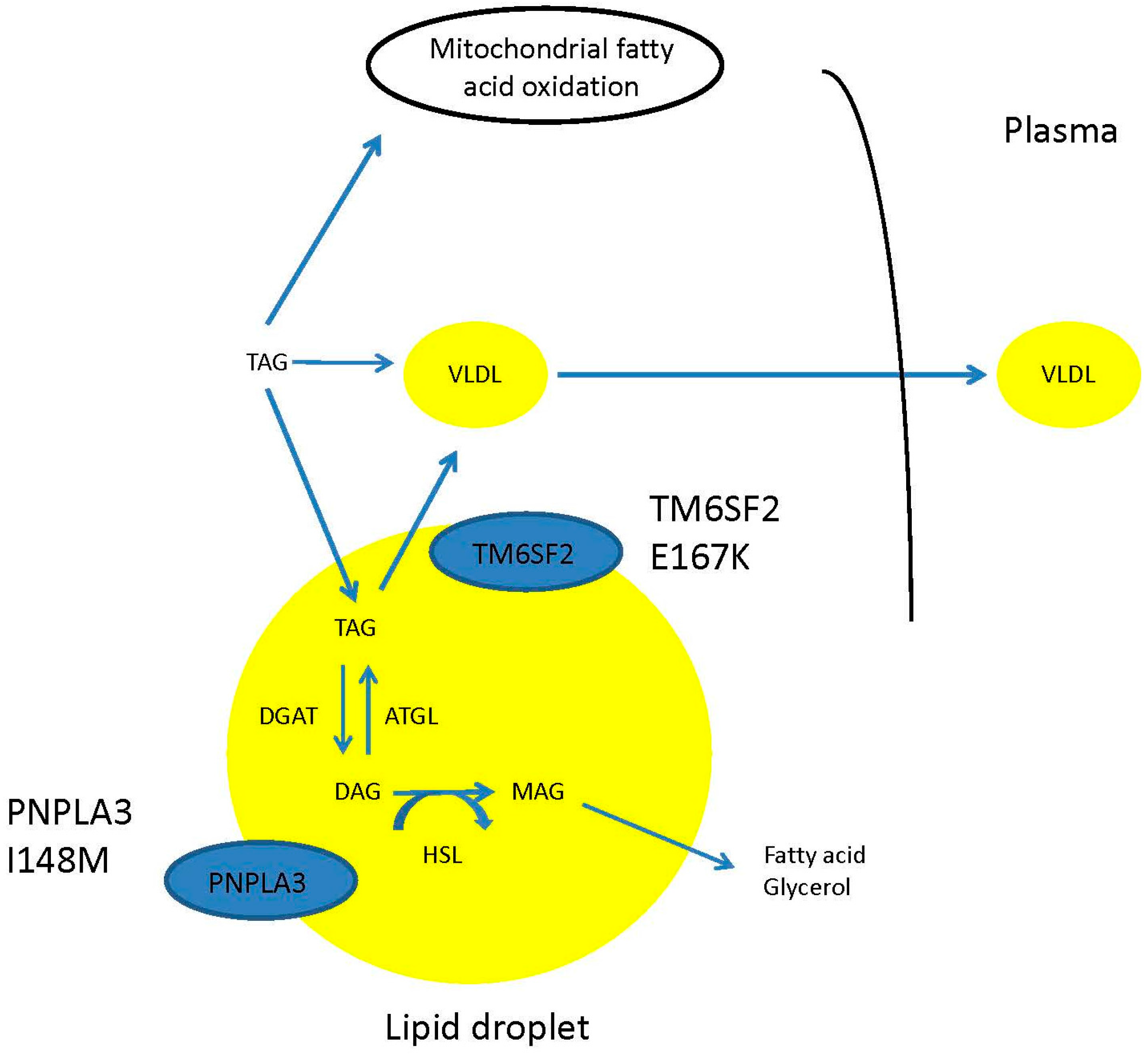
{kind=link}
{kind=link}
| Criteria | WHO (1999) | NCEP (2001) | IDF (2005) | IDF (2009) |
|---|---|---|---|---|
| Required | Insulin resistance | Nil | Waist circumference ≥94 cm in men, ≥80 cm in women | Nil |
| Number of features | ≥2 of: | ≥3 of: | ≥2 of: | ≥3 of: |
| Obesity | Waist/hip ratio of >0.9 in men, >0.85 in women or BMI ≥ 30 | Waist circumference ≥ 102 cm in men, ≥88 cm in women | Waist circumference—population specific definitions | |
| Triglycerides | ≥150 mg/dL (1.7 mmol/L) | ≥150 mg/dL (1.7 mmol/L) | ≥150 mg/dL (1.7 mmol/L) | ≥150 mg/dL (1.7 mmol/L) |
| HDL-cholesterol | <40 mg/dL (1 mmol/L) in men, <50 mg/dL (1.3 mmol/L) in women | <40 mg/dL (1 mmol/L) in men, <50 mg/dL (1.3 mmol/L) in women | <40 mg/dL (1 mmol/L) in men, <50 mg/dL (1.3 mmol/L) in women | <40 mg/dL (1 mmol/L) in men, <50 mg/dL (1.3 mmol/L) in women |
| Hypertension | ≥140/90 mmHg | ≥135/85 mmHg | ≥135/85 mmHg | ≥135/85 mmHg |
| Glucose | 110 mg/dL (6.1 mmol/L) | 100 mg/dL (5.6 mmol/L) | 100 mg/dL (5.6 mmol/L) | |
| Microalbuminuria | Albumin/creatinine ratio > 30 mg/g; albumin excretion rate > 20 mcg/min |
| Study | Country/Population | Sample Size | NAFLD Diagnostic Method/Surrogate Marker Used | Duration of Follow-Up | Key Findings | Limitations of Study |
|---|---|---|---|---|---|---|
| Vozarova 2002 [38] | Pima Indians aged 18–50 | 173 women, 278 men | ALT, AST and GGT concentrations | 6.9 years average | High baseline ALT associated with increased risk of incident DM | Only surrogate markers used, no control for alcohol/hep C |
| Lee 2003 [39] | Korean men aged 25–55 | 4088 men | GGT concentration | 4 years | Strong relationship between baseline GGT and risk of incident DM | Only studied men, only used surrogate marker |
| Hanley 2005 [40] | USA non-Hispanic whites and African American adults | 910 women, 715 men | ALT, AST and ALP concentrations | 5.2 years average | ALT and ALP in upper quartile at baseline significantly increased risk of metabolic syndrome | Only surrogate markers used for NAFLD diagnosis |
| Ekstedt 2006 [41] | Swedish NAFLD patients | 87 men, 42 women | Biopsy-proven NAFLD | 13.7 years average | Marked increase in proportion of patients with DM over period of study | No control group, no baseline glycaemic data to compare |
| Monami 2008 [42] | Florence aged 40–75 | 3124 total | ALT, AST and GGT concentrations | 40 months average | Baseline GGT near upper limit of normal predicts incident DM | Study population participated in screening programme for diabetes, may not be representative |
| Goessling 2008 [43] | New England adults, all white | 1575 women, 1237 men | ALT and AST concentrations | 20 years | Increased ALT associated with higher risk of DM and metabolic syndrome, increased AST associated with incident DM risk | Homogenous study population, only surrogate markers used |
| Adams 2009 [29] | Western Australian adults | 115 women, 243 men | NAFLD diagnosed with ALT after exclusion of other causes | 11 years | NAFLD associated with higher risk of incident diabetes | Not an independent predictor if adjusted for WC, hypertension or insulin resistance |
| Ryu 2010 [44] | Korean men aged 30–65 | 9148 men | GGT concentrations | 4.1 years average | Increase in GGT during study period predicted incident metabolic syndrome | Did not use accepted criteria for diagnosis of metabolic syndrome |
| Balkau 2010 [45] | Western France, aged 30–65 | 1950 women, 1861 men | NAFLD diagnosed using fatty liver index (FLI) score | 9 years | Higher FLI score at baseline predicted incident DM | Used FLI rather than formal diagnostic methods |
| Sung 2011 [46] | Korean adults | 7236 men, 3855 women | NAFLD diagnosed with ultrasound scan | 5 years | Presence of fatty liver on ultrasound strongly predicted incident DM | Ultrasound relatively insensitive for diagnosis |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wainwright, P.; Byrne, C.D. Bidirectional Relationships and Disconnects between NAFLD and Features of the Metabolic Syndrome. Int. J. Mol. Sci. 2016, 17, 367. https://doi.org/10.3390/ijms17030367
Wainwright P, Byrne CD. Bidirectional Relationships and Disconnects between NAFLD and Features of the Metabolic Syndrome. International Journal of Molecular Sciences. 2016; 17(3):367. https://doi.org/10.3390/ijms17030367
Chicago/Turabian StyleWainwright, Patrick, and Christopher D. Byrne. 2016. "Bidirectional Relationships and Disconnects between NAFLD and Features of the Metabolic Syndrome" International Journal of Molecular Sciences 17, no. 3: 367. https://doi.org/10.3390/ijms17030367
APA StyleWainwright, P., & Byrne, C. D. (2016). Bidirectional Relationships and Disconnects between NAFLD and Features of the Metabolic Syndrome. International Journal of Molecular Sciences, 17(3), 367. https://doi.org/10.3390/ijms17030367