
Hypertension and Dementia: Epidemiological and Experimental Evidence Revealing a Detrimental Relationship

Abstract
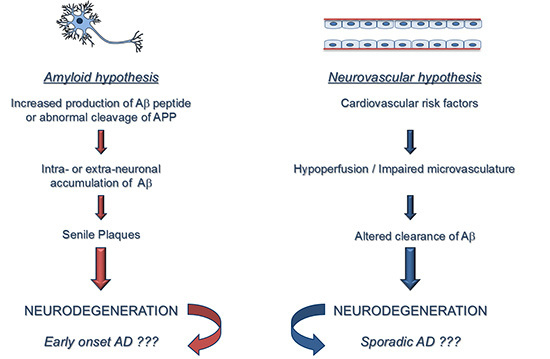
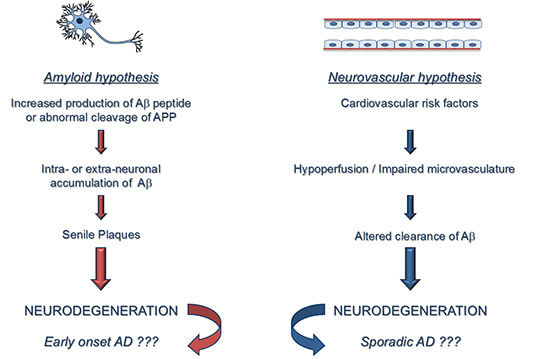
:
{kind=link}
{kind=link}
1. Introduction
2. The Hypertensive Response in the Brain: Structural and Functional Alterations in Cerebral Vasculature
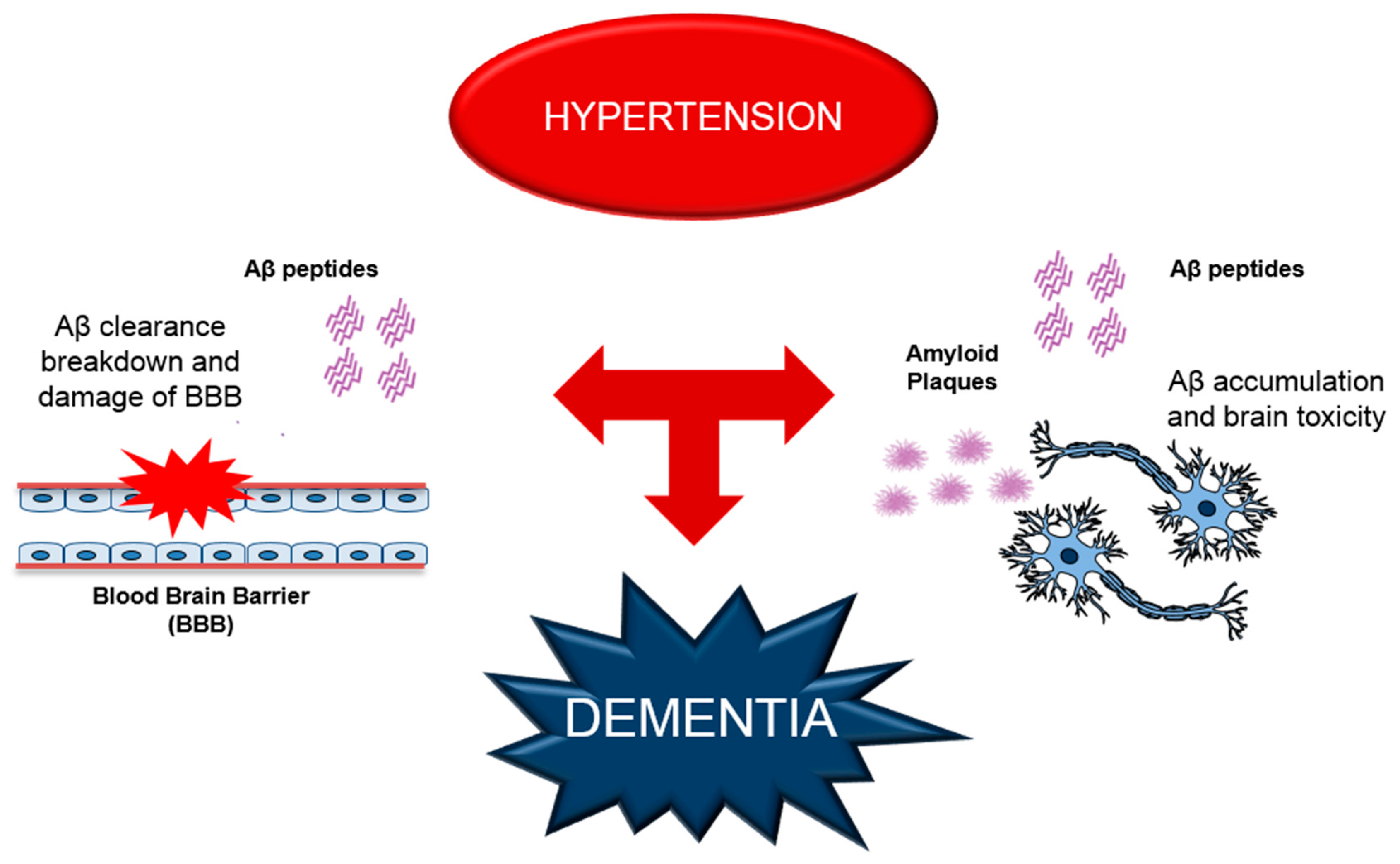
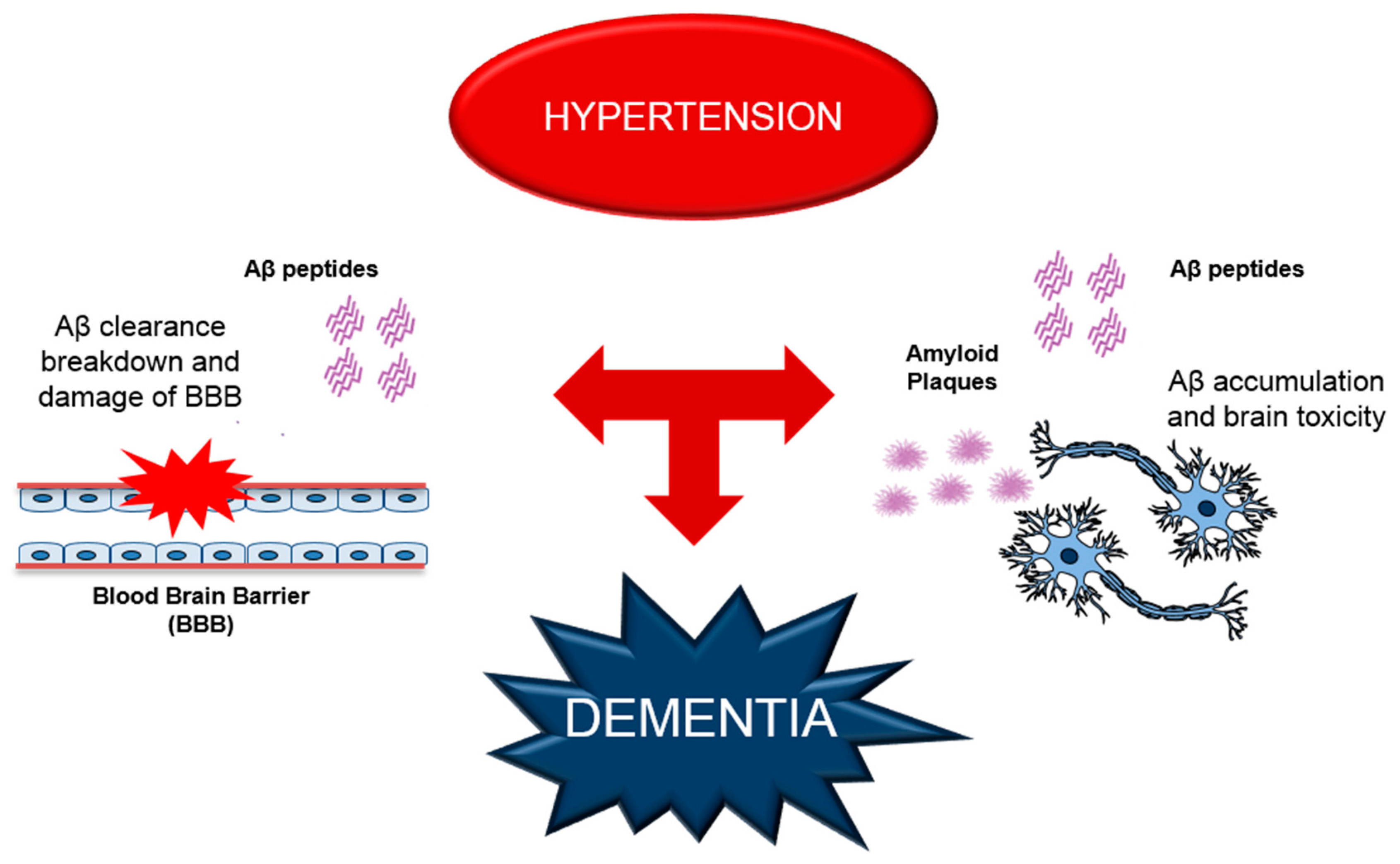
3. Alterations of Aβ Production and BBB Functions Leading to Cerebrovascular Dysfunction in Dementia
4. RAGE as a Possible Molecular Mechanism for Hypertension-Induced Vascular Dysfunction and AD
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Moser, M.; Roccella, E.J. The treatment of hypertension: A remarkable success story. J. Clin. Hypertens. 2013, 15, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Lawes, C.M.; Bennett, D.A.; Feigin, V.L.; Rodgers, A. Blood pressure and stroke: An overview of published reviews. Stroke 2004, 35, 1024. [Google Scholar] [CrossRef] [PubMed]
- Gorelick, P.B.; Scuteri, A.; Black, S.E.; Decarli, C.; Greenberg, S.M.; Iadecola, C.; Launer, L.J.; Laurent, S.; Lopez, O.L.; Nyenhuis, D.; et al. Vascular contributions to cognitive impairment and dementia: A statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2011, 42, 2672–2713. [Google Scholar] [CrossRef] [PubMed]
- Skoog, I.; Gustafson, D. Update on hypertension and Alzheimer’s disease. Neurol. Res. 2006, 28, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, A.; Greenberg, S.M.; Scheltens, P. Role of vascular disease in Alzheimer-like progressive cognitive impairment. Stroke 2016, 47, 577–580. [Google Scholar] [CrossRef] [PubMed]
- Pires, P.W.; Dams Ramos, C.M.; Matin, N.; Dorrance, A.M. The effects of hypertension on the cerebral circulation. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, 1598–1614. [Google Scholar] [CrossRef] [PubMed]
- Kazama, K.; Wang, G.; Frys, K.; Anrather, J.; Iadecola, C. Angiotensin II attenuates functional hyperemia in the mouse somatosensory cortex. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, 1890–1899. [Google Scholar] [CrossRef] [PubMed]
- Iddings, J.A.; Kim, K.J.; Zhou, Y.; Higashimori, H.; Filosa, J.A. Enhanced parenchymal arteriole tone and astrocyte signaling protect neurovascular coupling mediated parenchymal arteriole vasodilation in the spontaneously hypertensive rat. J. Cereb. Blood Flow Metab. 2015, 35, 1127–1136. [Google Scholar] [CrossRef] [PubMed]
- Immink, R.V.; van den Born, B.J.; van Montfrans, G.A.; Koopmans, R.P.; Karemaker, J.M.; van Lieshout, J.J. Impaired cerebral autoregulation in patients with malignant hypertension. Circulation 2004, 110, 2241–2245. [Google Scholar] [CrossRef] [PubMed]
- Carnevale, D.; Mascio, G.; D’Andrea, I.; Fardella, V.; Bell, R.D.; Branchi, I.; Pallante, F.; Zlokovic, B.; Yan, S.S.; Lembo, G. Hypertension induces brain β-amyloid accumulation, cognitive impairment, and memory deterioration through activation of receptor for advanced glycation end products in brain vasculature. Hypertension 2012, 60, 188–197. [Google Scholar] [CrossRef] [PubMed]
- Carnevale, D.; Perrotta, M.; Lembo, G.; Trimarco, B. Pathophysiological links among hypertension and Alzheimer’s disease. High Blood Press Cardiovasc. Prev. 2015. [Google Scholar] [CrossRef] [PubMed]
- Carnevale, D.; Lembo, G. “Alzheimer-like” pathology in a murine model of arterial hypertension. Biochem. Soc. Trans. 2011, 39, 939–944. [Google Scholar] [CrossRef] [PubMed]
- Qiu, C.; Winblad, B.; Fratiglioni, L. The age-dependent relation of blood pressure to cognitive function and dementia. Lancet Neurol. 2005, 4, 487–499. [Google Scholar] [CrossRef]
- Cifuentes, D.; Poittevin, M.; Dere, E.; Broquères-You, D.; Bonnin, P.; Benessiano, J.; Pocard, M.; Mariani, J.; Kubis, N.; Merkulova-Rainon, T.; et al. Hypertension accelerates the progression of Alzheimer-like pathology in a mouse model of the disease. Hypertension 2015, 65, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Wiesmann, M.; Capone, C.; Zerbi, V.; Mellendijk, L.; Heerschap, A.; Claassen, J.A.; Kiliaan, A.J. Hypertension impairs cerebral blood flow in a mouse model for Alzheimer’s disease. Curr. Alzheimer Res. 2015, 12, 914–922. [Google Scholar] [CrossRef] [PubMed]
- Ruitenberg, A.; Skoog, I.; Ott, A.; Aevarsson, O.; Witteman, J.C.; Lernfelt, B.; van Harskamp, F.; Hofman, A.; Breteler, M.M. Blood pressure and risk of dementia: Results from the Rotterdam study and the Gothenburg H-70 Study. Dement. Geriatr. Cogn. Disord. 2001, 12, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Mielke, M.M.; Rosenberg, P.B.; Tschanz, J.; Cook, L.; Corcoran, C.; Hayden, K.M.; Norton, M.; Rabins, P.V.; Green, R.C.; Welsh-Bohmer, K.A.; et al. Vascular factors predict rate of progression in Alzheimer disease. Neurology 2007, 69, 1850–1858. [Google Scholar] [CrossRef] [PubMed]
- Deschaintre, Y.; Richard, F.; Leys, D.; Pasquier, F. Treatment of vascular risk factors is associated with slower decline in Alzheimer disease. Neurology 2009, 73, 674–680. [Google Scholar] [CrossRef] [PubMed]
- Joas, E.; Bäckman, K.; Gustafson, D.; Ostling, S.; Waern, M.; Guo, X.; Skoog, I. Blood pressure trajectories from midlife to late life in relation to dementia in women followed for 37 years. Hypertension 2012, 59, 796–801. [Google Scholar] [CrossRef] [PubMed]
- Messerli, F.H.; Williams, B.; Ritz, E. Essential hypertension. Lancet 2007, 370, 591–603. [Google Scholar] [CrossRef]
- Dahlof, B. Prevention of stroke in patients with hypertension. Am. J. Cardiol. 2007, 100, 17J–24J. [Google Scholar] [CrossRef] [PubMed]
- Kelley, B.J.; Petersen, R.C. Alzheimer’s disease and mild cognitive impairment. Neurol. Clin. 2007, 25, 577–609. [Google Scholar] [CrossRef] [PubMed]
- Dunn, K.M.; Nelson, M.T. Neurovascular signaling in the brain and the pathological consequences of hypertension. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H1–H14. [Google Scholar] [CrossRef] [PubMed]
- Lammie, G.A. Hypertensive cerebral small vessel disease and stroke. Brain Pathol. 2002, 12, 358–370. [Google Scholar] [PubMed]
- Sakamoto, H.; Aikawa, M.; Hill, C.C.; Weiss, D.; Taylor, W.R.; Libby, P.; Lee, R.T. Biomechanical strain induces class a scavenger receptor expression in human monocyte/macrophages and THP-1 cells: A potential mechanism of increased atherosclerosis in hypertension. Circulation 2001, 104, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Faraco, G.; Iadecola, C. Hypertension: A harbinger of stroke and dementia. Hypertension 2013, 62, 810–817. [Google Scholar] [CrossRef] [PubMed]
- Sörös, P.; Whitehead, S.; Spence, J.D.; Hachinski, V. Antihypertensive treatment can prevent stroke and cognitive decline. Nat. Rev. Neurol. 2013, 9, 174–178. [Google Scholar] [CrossRef] [PubMed]
- Jokinen, H.; Gouw, A.A.; Madureira, S.; Ylikoski, R.; van Straaten, E.C.; van der Flier, W.M.; Barkhof, F.; Scheltens, P.; Fazekas, F.; Schmidt, R.; et al. Incident lacunes influence cognitive decline: The LADIS study. Neurology 2011, 76, 1872–1878. [Google Scholar] [CrossRef] [PubMed]
- Westover, M.B.; Bianchi, M.T.; Yang, C.; Schneider, J.A.; Greenberg, S.M. Estimating cerebral microinfarct burden from autopsy samples. Neurology 2013, 80, 1365–1369. [Google Scholar] [CrossRef] [PubMed]
- Feihl, F.; Liaudet, L.; Levy, B.I.; Waeber, B. Hypertension and microvascular remodelling. Cardiovasc. Res. 2008, 78, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Park, G.L.; Heistad, D.D. Cerebral circulation in chronic arterial hypertension. Hypertension 1988, 12, 89–95. [Google Scholar]
- Heistad, D.D.; Baumbach, G.L. Cerebral vascular changes during chronic hypertension: Good guys and bad guys. J. Hypertens. Suppl. 1992, 10, S71–S75. [Google Scholar] [CrossRef] [PubMed]
- Mulvany, M.J. Small artery remodelling in hypertension. Basic Clin. Pharmacol. Toxicol. 2012, 110, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Harvey, A.; Montezano, A.C.; Touyz, R.M. Vascular biology of ageing-Implications in hypertension. J. Mol. Cell. Cardiol. 2015, 83, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Faraci, F.M. Protecting against vascular disease in brain. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H1566–H1582. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, K.; Kuriyama, Y.; Nagatsuka, K.; Nakamura, M.; Sawada, T.; Omae, T. Periventricular white matter lucency and cerebral blood flow autoregulation in hypertensive patients. Hypertension 1994, 23, 565–568. [Google Scholar] [CrossRef] [PubMed]
- Jennings, J.R.; Muldoon, M.F.; Ryan, C.; Price, J.C.; Greer, P.; Sutton-Tyrrell, K.; van der Veen, F.M.; Meltzer, C.C. Reduced cerebral blood flow response and compensation among patients with untreated hypertension. Neurology 2005, 64, 1358–1365. [Google Scholar] [CrossRef] [PubMed]
- Park, L.; Anrather, J.; Girouard, H.; Zhou, P.; Iadecola, C. Nox2-derived reactive oxygen species mediate neurovascular dysregulation in the aging mouse brain. J. Cereb. Blood Flow Metab. 2007, 27, 1908–1918. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C. Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat. Rev. Neurosci. 2004, 5, 347–360. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C. The overlap between neurodegenerative and vascular factors in the pathogenesis of dementia. Acta Neuropathol. 2010, 120, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C.; Park, L.; Capone, C. Threats to the mind: Aging, amyloid, and hypertension. Stroke 2009, 40, S40–S44. [Google Scholar] [CrossRef] [PubMed]
- Capone, C.; Faraco, G.; Park, L.; Cao, X.; Davisson, R.L.; Iadecola, C. The cerebrovascular dysfunction induced by slow pressor doses of angiotensin II precedes the development of hypertension. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Son, S.J.; Kim, J.; Lee, E.; Park, J.Y.; Namkoong, K.; Hong, C.H.; Ku, J.; Kim, E.; Oh, B.H. Effect of hypertension on the resting-state functional connectivity in patients with Alzheimer’s disease (AD). Arch. Gerontol. Geriatr. 2015, 60, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Glenner, G.G. Amyloid deposits and amyloidosis. The ß-fibrilloses (first of two parts). N. Engl. J. Med. 1980, 302, 1283–1292. [Google Scholar] [CrossRef] [PubMed]
- Glenner, G.G. Amyloid deposits and amyloidosis: The ß-fibrilloses (second of two parts). N. Engl. J. Med. 1980, 302, 1333–1343. [Google Scholar] [CrossRef] [PubMed]
- Mandybur, T.I. Cerebral amyloid angiopathy: The vascular pathology and complications. J. Neuropathol. Exp. Neurol. 1986, 45, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Ghiso, J.; Frangione, B. Cerebral amyloidosis, amyloid angiopathy, and their relationship to stroke and dementia. J. Alzheimers. Dis. 2001, 3, 65–73. [Google Scholar] [PubMed]
- Revesz, T.; Holton, J.L.; Lashley, T.; Plant, G.; Rostagno, A.; Ghiso, J.; Frangione, B. Sporadic and familial cerebral amyloid angiopathies. Brain Pathol. 2002, 12, 343–357. [Google Scholar] [CrossRef] [PubMed]
- Chow, N.; Bell, R.D.; Deane, R.; Streb, J.W.; Chen, J.; Brooks, A.; van Nostrand, W.; Miano, J.M.; Zlokovic, B.V. Serum response factor and myocardin mediate cerebral arterial hypercontractility and blood flow dysregulation in Alzheimer’s phenotype. Proc. Natl. Acad. Sci. USA 2007, 104, 823–828. [Google Scholar] [CrossRef] [PubMed]
- Hutton, M. Missense and splice site mutations in tau associated with FTDP-17: Multiple pathogenic mechanisms. Neurology 2001, 56, S21–S25. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, T.; Frangione, B. Molecular biology of Alzheimer’s amyloid—Dutch variant. Mol. Neurobiol. 1992, 6, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Gasparini, L.; Terni, B.; Spillantini, M.G. Frontotemporal dementia with tau pathology. Neurodegener. Dis. 2007, 4, 236–253. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic, B.V. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 2008, 57, 178–201. [Google Scholar] [CrossRef] [PubMed]
- De la Torre, J.C. Alzheimer disease as a vascular disorder: Nosological evidence. Stroke 2003, 33, 1152–1162. [Google Scholar] [CrossRef]
- Sinha, S.; Anderson, J.P.; Barbour, R.; Basi, G.S.; Caccavello, R.; Davis, D.; Doan, M.; Dovey, H.F.; Frigon, N.; Hong, J.; et al. Purification and cloning of amyloid precursor protein β-secretase from human brain. Nature 1999, 402, 537–540. [Google Scholar] [CrossRef] [PubMed]
- Vassar, R. The ß-secretase, BACE: A prime drug target for Alzheimer’s disease. J. Mol. Neurosci. 2001, 17, 157–170. [Google Scholar] [CrossRef]
- Kuznetsova, E.; Schliebs, R. β-Amyloid, cholinergic transmission, and cerebrovascular system—A developmental study in a mouse model of Alzheimer’s disease. Curr. Pharm. Des. 2013, 19, 6749–6765. [Google Scholar] [CrossRef] [PubMed]
- Kruyer, A.; Soplop, N.; Strickland, S.; Norris, E.H. Chronic hypertension leads to neurodegeneration in the TgSwDI mouse model of Alzheimer’s disease. Hypertension 2015, 66, 175–182. [Google Scholar] [CrossRef] [PubMed]
- De la Torre, J.C. How do heart disease and stroke become risk factors for Alzheimer’s disease? Neurol. Res. 2006, 28, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Allt, G.; Lawrenson, J.G. Pericytes: Cell biology and pathology. Cells Tissues Organs 2001, 169, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Von Tell, D.; Armulik, A.; Betsholtz, C. Pericytes and vascular stability. Exp. Cell. Res. 2006, 312, 623–629. [Google Scholar] [CrossRef] [PubMed]
- Armulik, A.; Abramsson, A.; Betsholtz, C. Endothelial/pericyte interactions. Circ. Res. 2005, 97, 512–523. [Google Scholar] [CrossRef] [PubMed]
- Peppiatt, C.M.; Howarth, C.; Mobbs, P.; Attwell, D. Bidirectional control of CNS capillary diameter by pericytes. Nature 2006, 443, 700–704. [Google Scholar] [CrossRef] [PubMed]
- Wyss-Coray, T.; Lin, C.; Sanan, D.A.; Mucke, L.; Masliah, E. Chronic overproduction of transforming growth factor-beta1 by astrocytes promotes Alzheimer’s disease-like microvascular degeneration in transgenic mice. Am. J. Pathol. 2000, 156, 139–150. [Google Scholar] [CrossRef]
- Bell, R.D.; Deane, R.; Chow, N.; Long, X.; Sagare, A.; Singh, I.; Streb, J.W.; Guo, H.; Rubio, A.; van Nostrand, W.; et al. SRF and myocardin regulate LRP-mediated amyloid-beta clearance in brain vascular cells. Nat. Cell Biol. 2009, 11, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C.; Davisson, R.L. Hypertension and cerebrovascular dysfunction. Cell Metab. 2008, 7, 476–484. [Google Scholar] [CrossRef] [PubMed]
- Deane, R.; Du Yan, S.; Submamaryan, R.K.; LaRue, B.; Jovanovic, S.; Hogg, E.; Welch, D.; Manness, L.; Lin, C.; Yu, J.; et al. RAGE mediates amyloid-β peptide transport across the bloodbrain barrier and accumulation in brain. Nat. Med. 2003, 9, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Shibata, M.; Yamada, S.; Kumar, S.R.; Calero, M.; Bading, J.; Frangione, B.; Holtzman, D.M.; Miller, C.A.; Strickland, D.K.; Ghiso, J.; et al. Clearance of Alzheimer’s amyloid-β(1–40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J. Clin. Investig. 2000, 106, 1489–1499. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, N.; Toki, S.; Chowei, H.; Saito, T.; Nakano, N.; Hayashi, Y.; Takeuchi, M.; Makita, Z. Immunohistochemical distribution of the receptor for advanced glycation end products in neurons and astrocytes in Alzheimer’s disease. Brain Res. 2001, 888, 256–262. [Google Scholar] [PubMed]
- Miller, A.A.; Drummond, G.R.; Schmidt, H.H.; Sobey, C.G. NADPH oxidase activity and function are profoundly greater in cerebral versus systemic arteries. Circ. Res. 2005, 97, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Li, X.H.; Tu, Y.; Sun, H.T.; Liang, H.Q.; Cheng, S.X.; Zhang, S. Aβ-AGE aggravates cognitive deficit in rats via RAGE pathway. Neuroscience 2014, 257, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Li, X.H.; Lv, B.L.; Xie, J.Z.; Liu, J.; Zhou, X.W.; Wang, J.Z. AGEs induce Alzheimer-like tau pathology and memory deficit via RAGE-mediated GSK-3 activation. Neurobiol. Aging 2012, 33, 1400–1410. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Ye, R.D. Microglial Aβ receptors in Alzheimer’s disease. Cell. Mol. Neurobiol. 2015, 35, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.; Lue, L.F.; Yan, S.; Xu, H.; Luddy, J.S.; Chen, D.; Walker, D.G.; Stern, D.M.; Yan, S.; Schmidt, A.M.; et al. RAGE-dependent signaling in microglia contributes to neuroinflammation, Aβ accumulation, and impaired learning/memory in a mouse model of Alzheimer’s disease. FASEB J. 2010, 24, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Ihara, Y.; Egashira, K.; Nakano, K.; Ohtani, K.; Kubo, M.; Koga, J.; Iwai, M.; Horiuchi, M.; Gang, Z.; Yamagishi, S.; et al. Upregulation of the ligand-RAGE pathway via the angiotensin II type I receptor is essential in the pathogenesis of diabetic atherosclerosis. J. Mol. Cell. Cardiol. 2007, 43, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Yao, D.; Brownlee, M. Hyperglycemia-induced reactive oxygen species increase expression of RAGE and RAGE ligands. Diabetes 2010, 59, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Poulet, R.; Gentile, M.T.; Vecchione, C.; Distaso, M.; Aretini, A.; Fratta, L.; Russo, G.; Echart, C.; Maffei, A.; de Simoni, M.G.; et al. Acute hypertension induces oxidative stress in brain tissues. J. Cereb. Blood Flow Metab. 2006, 26, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Yamagishi, S.; Nakamura, Y.; Takenaka, K.; Matsui, T.; Jinnouchi, Y.; Imaizumi, T. Telmisartan inhibits expression of a receptor for advanced glycation end products (RAGE) in angiotensin-II-exposed endothelial cells and decreases serum levels of soluble RAGE in patients with essential hypertension. Microvasc. Res. 2005, 70, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.; Ihara, M. New therapeutic approaches for Alzheimer’s disease and cerebral amyloid angiopathy. Front. Aging Neurosci. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- NIH National Institute on Aging Web Site. RI: RAGE inhibitor study. Alzheimer’s disease education and referral center. Available online: http://www.alzheimers.org/clinicaltrials/fullrec.asp?PrimaryKey=287 (accessed on 21 December 2015).
- Deane, R.; Singh, I.; Sagare, A.P.; Bell, R.D.; Ross, N.T.; LaRue, B.; Love, R.; Perry, S.; Paquette, N.; Deane, R.J. A multimodal RAGE-specific inhibitor reduces amyloid β-mediated brain disorder in a mouse model of Alzheimer disease. J. Clin. Investig. 2012, 122, 1377–1392. [Google Scholar] [CrossRef] [PubMed]
- Faraco, G.; Park, L.; Zhou, P.; Luo, W.; Paul, S.M.; Anrather, J.; Iadecola, C. Hypertension enhances Aβ-induced neurovascular dysfunction, promotes β-secretase activity, and leads to amyloidogenic processing of APP. J. Cereb. Blood Flow Metab. 2015. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perrotta, M.; Lembo, G.; Carnevale, D. Hypertension and Dementia: Epidemiological and Experimental Evidence Revealing a Detrimental Relationship. Int. J. Mol. Sci. 2016, 17, 347. https://doi.org/10.3390/ijms17030347
Perrotta M, Lembo G, Carnevale D. Hypertension and Dementia: Epidemiological and Experimental Evidence Revealing a Detrimental Relationship. International Journal of Molecular Sciences. 2016; 17(3):347. https://doi.org/10.3390/ijms17030347
Chicago/Turabian StylePerrotta, Marialuisa, Giuseppe Lembo, and Daniela Carnevale. 2016. "Hypertension and Dementia: Epidemiological and Experimental Evidence Revealing a Detrimental Relationship" International Journal of Molecular Sciences 17, no. 3: 347. https://doi.org/10.3390/ijms17030347
APA StylePerrotta, M., Lembo, G., & Carnevale, D. (2016). Hypertension and Dementia: Epidemiological and Experimental Evidence Revealing a Detrimental Relationship. International Journal of Molecular Sciences, 17(3), 347. https://doi.org/10.3390/ijms17030347