
Assessing the Effects of Acute Amyloid β Oligomer Exposure in the Rat

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
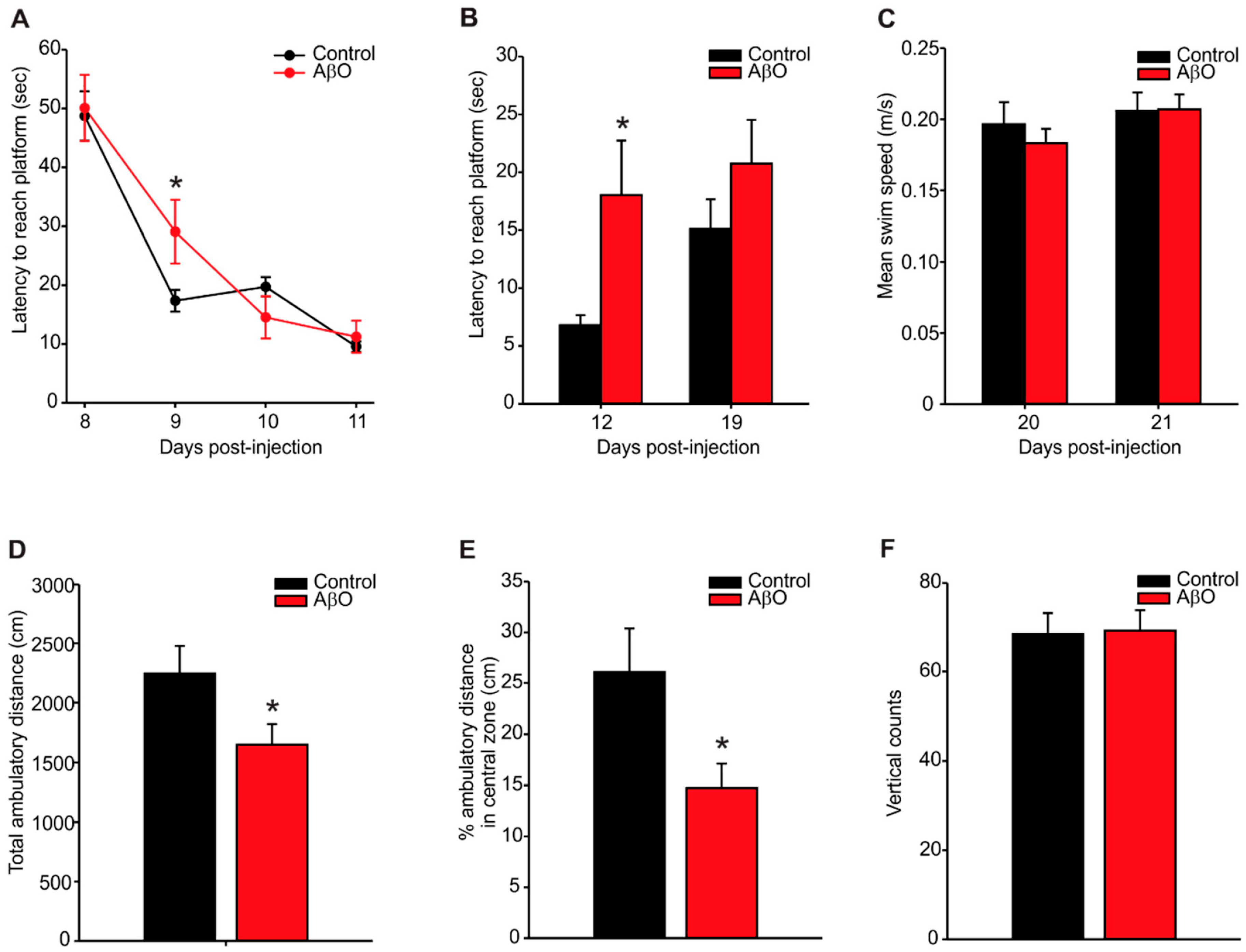
2.1. Transient Deficits in Spatial Memory and Mild Anxiety-Like Behaviour in AβO-Injected Rats
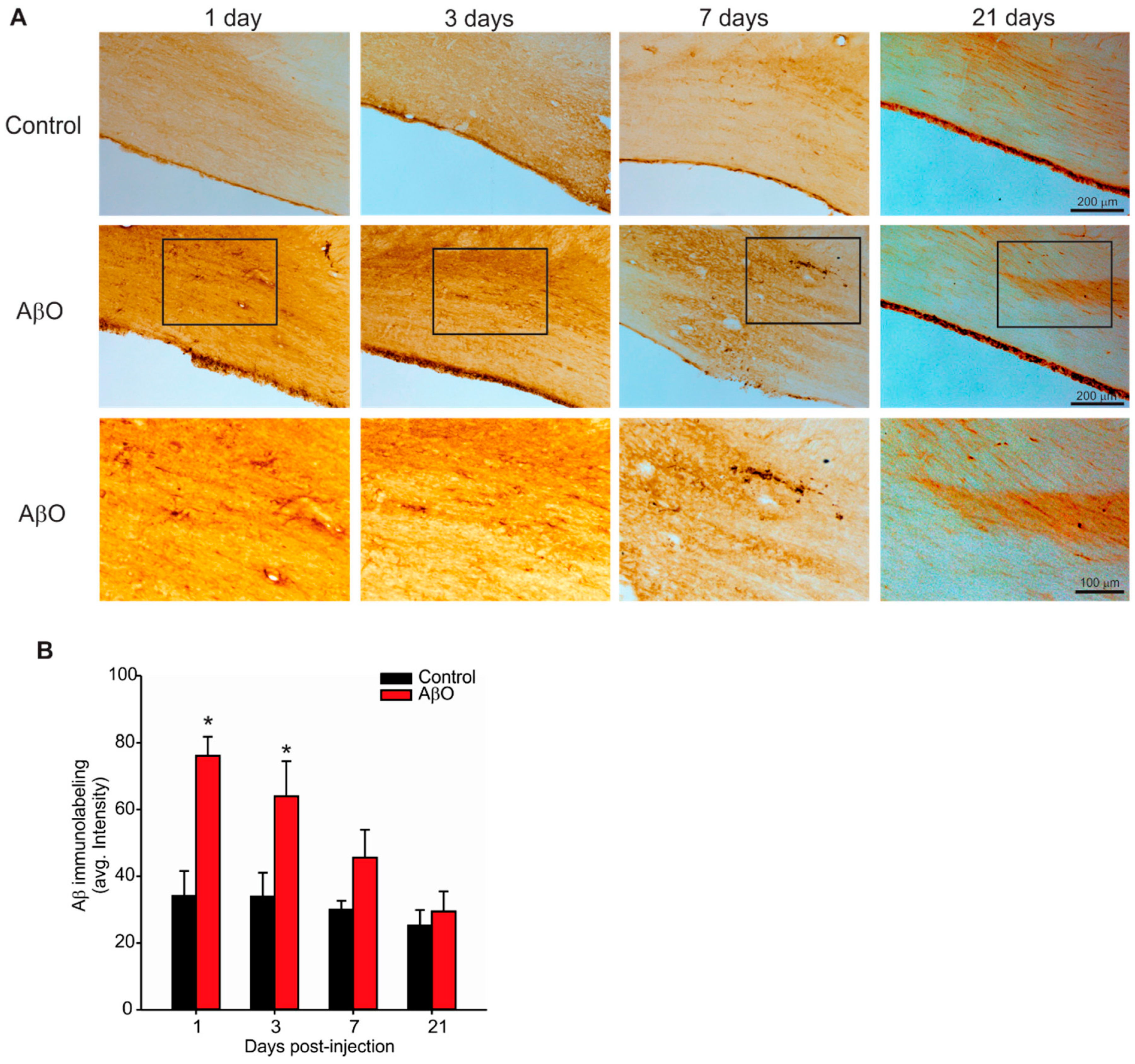
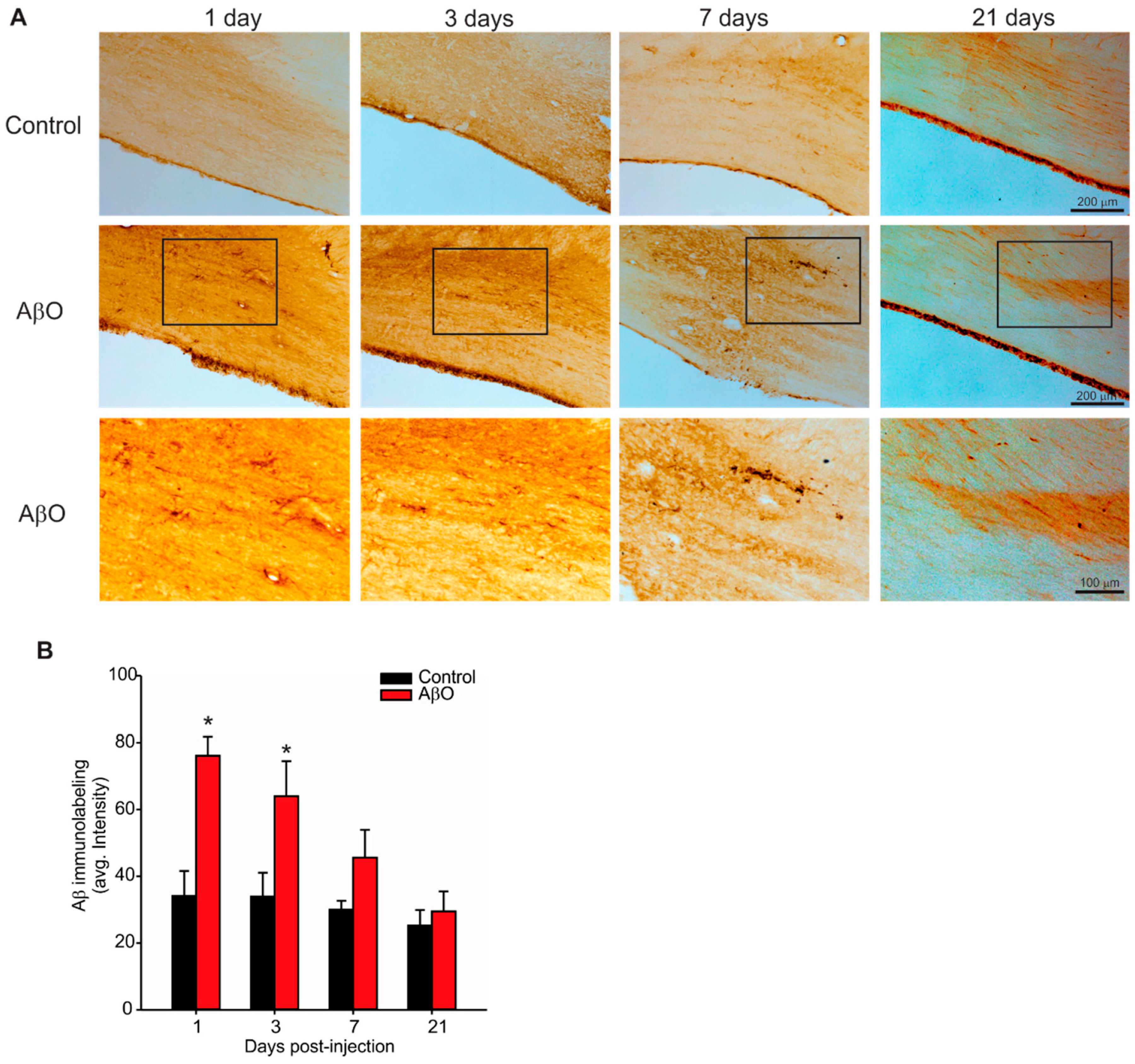
2.2. Transient Deposition of Aβ Following AβO Injections
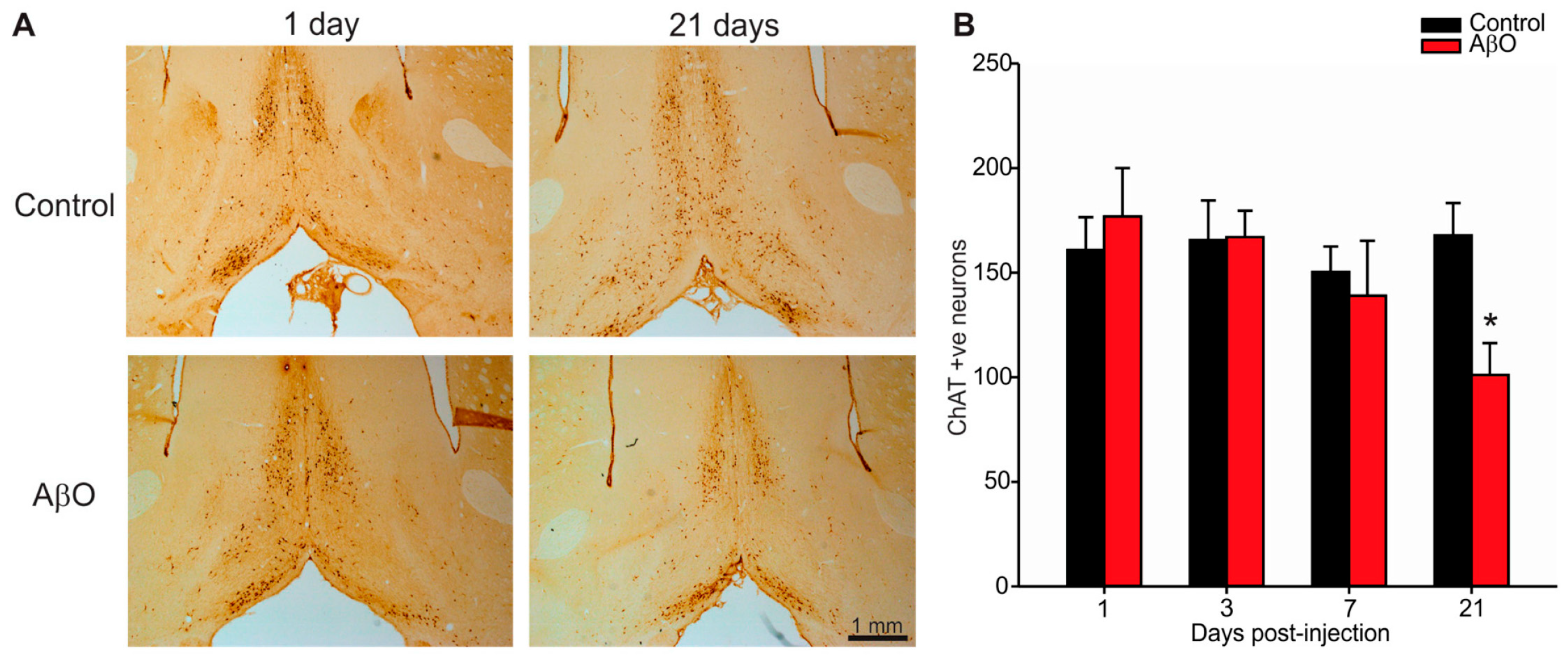
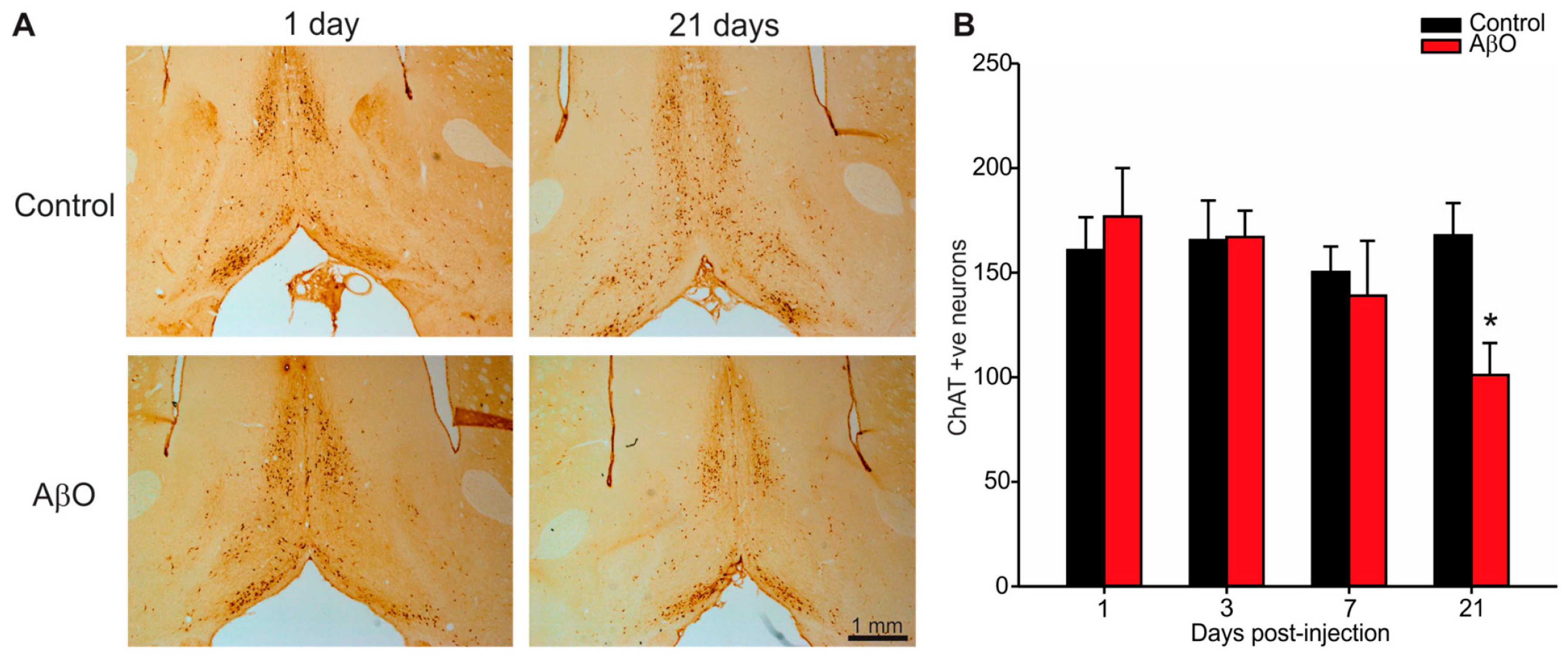
2.3. Cholinergic Neuron Depletion within Basal Forebrain Following AβO Injections
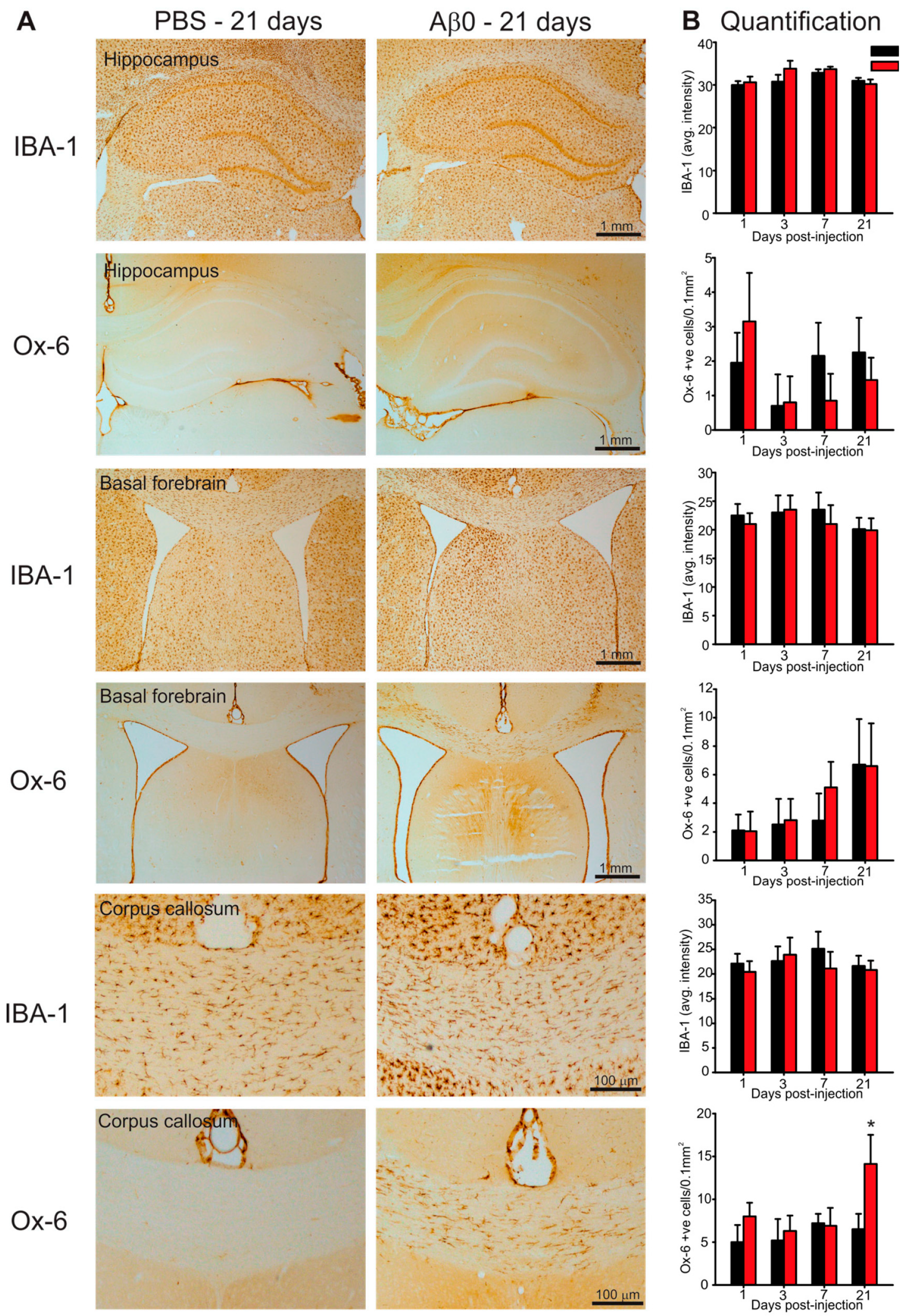
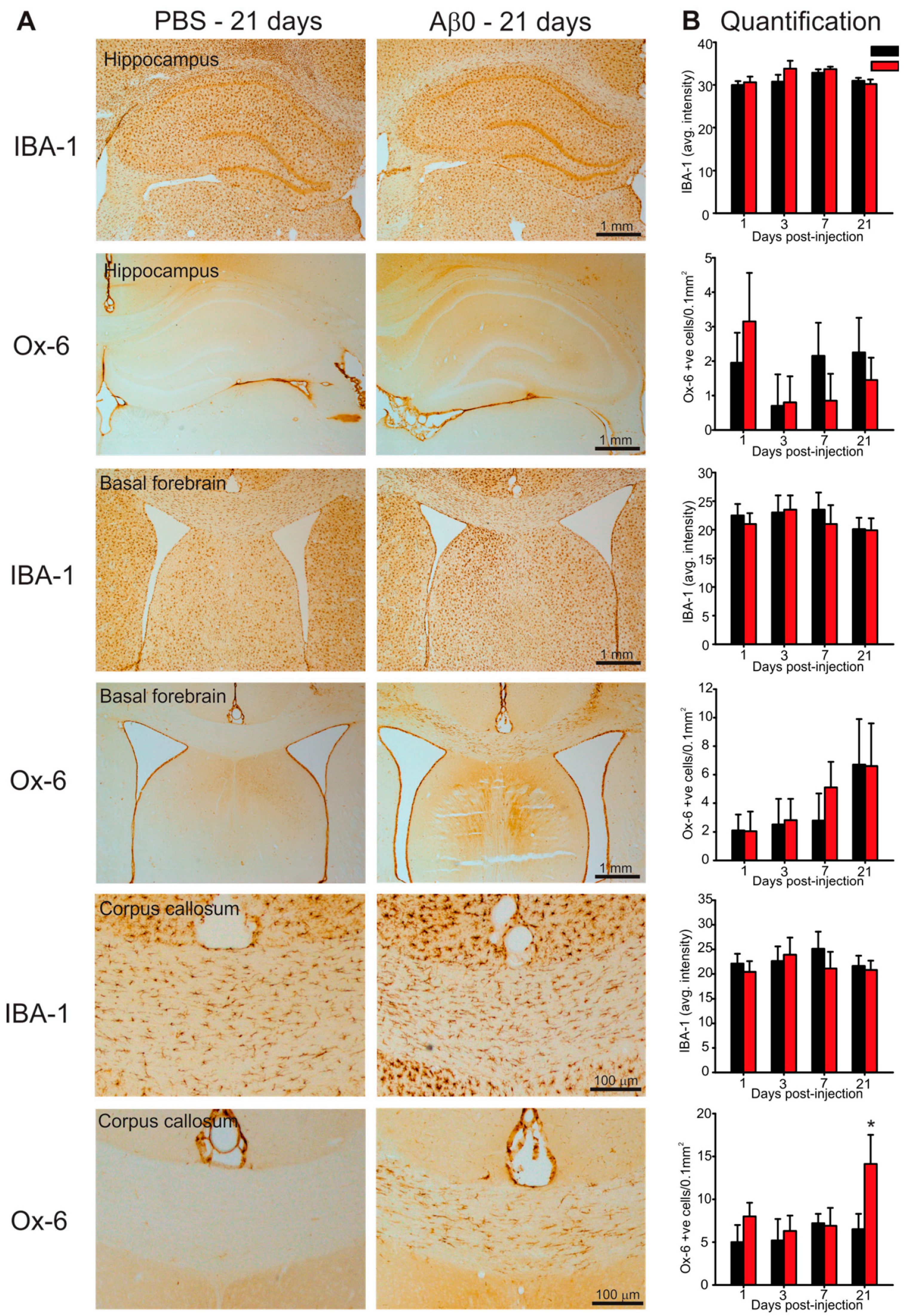
2.4. Microglia Activation in Response to AβO Injections
3. Discussion
4. Materials and Methods
4.1. Synthetic AβO Preparation
4.2. Experimental Groups and Surgical Procedures
4.3. Behavioural Assessments
4.4. Pathological Analysis
4.5. Imaging Investigators Were Blinded to Surgical Identity for All Image Acquisition
4.6. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Haass, C.; Kaether, C.; Thinakaran, G.; Sisodia, S. Trafficking and proteolytic processing of APP. Cold Spring Harb. Perspect. Med. 2012, 2. [Google Scholar] [CrossRef] [PubMed]
- Shoji, M.; Golde, T.; Ghiso, J.; Cheung, T.; Estus, S.; Shaffer, L.; Cai, X.-D.; McKay, D.; Tintner, R.; Frangione, B.; et al. Production of the Alzheimer amyloid β protein by normal proteolytic processing. Science 1992, 258, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Seubert, P.; Vigo-Pelfrey, C.; Esch, F.; Lee, M.; Dovey, H.; Davis, D.; Sinha, S.; Schlossmacher, M.; Whaley, J.; Swindlehurst, C. Isolation and quantification of soluble Alzheimer’s β-peptide from biological fluids. Nature 1992, 359, 325–327. [Google Scholar] [CrossRef] [PubMed]
- Haass, C.; Schlossmacher, M.; Hung, A.; Vigo-Pelfrey, C.; Mellon, A.; Ostaszewski, B.L.; Lieberburg, I.; Koo, E.; Schenk, D.; Teplow, D.; et al. Amyloid β peptide is produced by cultured cells during normal metabolism. Nature 1992, 359, 322–325. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.W.; Quaranta, V.; Glenner, G.G. Neuritic plaques and cerebrovascular amyloid in Alzheimer disease are antigenically related. Proc. Natl. Acad. Sci. USA 1985, 82, 8729–8732. [Google Scholar] [CrossRef] [PubMed]
- Haass, C.; Selkoe, D.J. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 2007, 8, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Lemaire, H.-G.; Unterbeck, A.; Salbaum, J.M.; Masters, C.L.; Grzeschik, K.-H.; Multhaup, G.; Beyreuther, K.; Muller-Hill, B. The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature 1987, 325, 733–736. [Google Scholar] [CrossRef] [PubMed]
- Haass, C.; Selkoe, D.J. Cellular processing of β-amyloid precursor protein and the genesis of amyloid β-peptide. Cell 1993, 75, 1039–1042. [Google Scholar] [CrossRef]
- Gellermann, G.P.; Byrnes, H.; Striebinger, A.; Ullrich, K.; Mueller, R.; Hillen, H.; Barghorn, S. Aβ-globulomers are formed independently of the fibril pathway. Neurobiol. Dis. 2008, 30, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.; Davis, J.; Aucoin, D.; Sato, T.; Ahuja, S.; Aimoto, S.; Elliott, J.I.; van Nostrand, W.E.; Smith, S.O. Structural conversion of neurotoxic amyloid-β1–42 oligomers to fibrils. Nat. Struct. Mol. Biol. 2010, 17, 561–567. [Google Scholar] [CrossRef] [PubMed]
- Rangachari, V.; Moore, B.D.; Reed, D.K.; Sonoda, L.K.; Bridges, A.W.; Conboy, E.; Hartigan, D.; Rosenberry, T.L. Amyloid-B1–42 rapidly forms protofibrils and oligomers by distinct pathways in low concentrations of sodium dodecylsulfate. Biochemistry 2007, 46, 12451–12462. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: Amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 1984, 120, 885–890. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s Disease: Progress and problems on the road to therapeutics. Sci. Compass Rev. 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Stelzmann, R.A.; Schnitzlein, H.N.; Murtagh, F.R. An English translation of Alzheimer’s 1907 paper, “uber eine eigenartige erkankung der hirnrinde”. Clin. Anat. 1995, 8, 429–431. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Chang, L.; Viola, K.L.; Lacor, P.N.; Lambert, M.P.; Finch, C.E.; Krafft, G.A.; Klein, W.L. Alzheimer’s disease-affected brain: Presence of oligomeric A β ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc. Natl. Acad. Sci. USA 2003, 100, 10417–10422. [Google Scholar] [CrossRef] [PubMed]
- Verdile, G.; Fuller, S.; Atwood, C.S.; Laws, S.M.; Gandy, S.E.; Martins, R.N. The role of β amyloid in Alzheimer’s disease: Still a cause of everything or the only one who got caught? Pharmacol. Res. 2004, 50, 397–409. [Google Scholar] [CrossRef] [PubMed]
- McLean, C.A.; Cherny, R.A.; Fraser, F.W.; Fuller, S.J.; Smith, M.J.; Beyreuther, K.; Bush, A.I.; Masters, C.L. Soluble pool of Aβ amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann. Neurol. 1999, 46, 860–866. [Google Scholar] [CrossRef]
- Hibbard, L.S.; McKeel, D.W., Jr. Automated identification and quantitative morphometry of the senile plaques of Alzheimer’s disease. Anal. Quant. Cytol. Histol. Int. Acad. Cytol. 1997, 19, 123–138. [Google Scholar]
- Crystal, H.; Dickson, D.W.; Fuld, P.; Masur, D.; Scott, R.W.; Mehler, M.; Wolfson, L. Clinico-pathologic studies in dementia Nondemented subjects with pathologically confirmed Alzheimer’s disease. Neurology 1988, 38, 1682. [Google Scholar] [CrossRef] [PubMed]
- Westerman, M.A.; Cooper-Blacketer, D.; Mariash, A.; Kotilinek, L.; Kawarabayashi, T.; Younkin, L.H.; Carlson, G.A.; Younkin, S.G.; Ashe, K.H. The relationship between Aβ and memory in the Tg2576 mouse model of Alzheimer’s disease. J. Neurosci. 2002, 22, 1858–1867. [Google Scholar] [PubMed]
- Oda, T.; Wals, P.; Osterburg, H.H.; Johnson, S.A.; Pasinetti, G.M.; Morgan, T.E.; Rozovsky, I.; Stine, W.B.; Snyder, S.W.; Holzman, T.F. Clusterin (apoJ) alters the aggregation of amyloid β-peptide (A β1–42) and forms slowly sedimenting A β complexes that cause oxidative stress. Exp. Neurol. 1995, 136, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Lue, L.F.; Kuo, Y.M.; Roher, A.E.; Brachova, L.; Shen, Y.; Sue, L.; Beach, T.; Kurth, J.H.; Rydel, R.E.; Rogers, J. Soluble amyloid β peptide concentration as a predictor of synaptic change in Alzheimer’s disease. Am. J. Pathol. 1999, 155, 853–862. [Google Scholar] [CrossRef]
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; et al. Amyloid-β protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 2008, 14, 837–842. [Google Scholar] [CrossRef] [PubMed]
- Lambert, M.P.; Barlow, A.K.; Chromy, B.A.; Edwards, C.; Freed, R.; Liosatos, M.; Morgan, T.E.; Rozovsky, I.; Trommer, B.; Viola, K.L.; et al. Diffusible, nonfibrillar ligands derived from Aβ1–42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. USA 1998, 95, 6448–6453. [Google Scholar] [CrossRef] [PubMed]
- Lacor, P.N.; Buniel, M.C.; Chang, L.; Fernandez, S.J.; Gong, Y.; Viola, K.L.; Lambert, M.P.; Velasco, P.T.; Bigio, E.H.; Finch, C.E.; et al. Synaptic targeting by Alzheimer’s-related amyloid β oligomers. J. Neurosci. 2004, 24, 10191–10200. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q. Block of long-term potentiation by naturally secreted and synthetic amyloid-peptide in hippocampal slices is mediated via activation of the kinases c-Jun N-Terminal kinase, cyclin-dependent kinase 5, and p38 mitogen-activated protein kinase as well as M. J. Neurosci. 2004, 24, 3370–3378. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.M.; Townsend, M.; Podlisny, M.B.; Shankar, G.M.; Fadeeva, J.V.; El Agnaf, O.; Hartley, D.M.; Selkoe, D.J. Certain inhibitors of synthetic amyloid β-peptide (Aβ) fibrillogenesis block oligomerization of natural Aβ and thereby rescue long-term potentiation. J. Neurosci. 2005, 25, 2455–2462. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Ojala, J.; Kauppinen, A.; Kaarniranta, K.; Suuronen, T. Inflammation in Alzheimer’s disease: Amyloid-B oligomers trigger innate immunity defence via pattern recognition receptors. Prog. Neurobiol. 2009, 87, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Heppner, F.L.; Ransohoff, R.M.; Becher, B. Immune attack: The role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 2015, 16, 358–372. [Google Scholar] [CrossRef] [PubMed]
- Walter, S.; Letiembre, M.; Liu, Y.; Heine, H.; Hao, W.; Bode, B.; Manietta, N.; Schulz-schäffer, W.; Faßbender, K. Cellular physiology and biochemistr y biochemistry role of the Toll-Like receptor 4 in Neuro- inflammation in Alzheimer’s disease. Cell. Physiol. Biochem. 2007, 20, 947–956. [Google Scholar] [CrossRef] [PubMed]
- Lesné, S.; Koh, M.T.; Kotilinek, L.; Kayed, R.; Glabe, C.G.; Yang, A.; Gallagher, M.; Ashe, K.H. A specific amyloid-β protein assembly in the brain impairs memory. Nature 2006, 440, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Catalano, S.M.; Dodson, E.C.; Henze, D.A.; Joyce, J.G.; Krafft, G.A.; Kinney, G.G. The role of amyloid-β derived diffusible ligands (ADDLs) in Alzheimer’s disease. Curr. Top. Med. Chem. 2006, 6, 597–608. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.M.; Klyubin, I.; Fadeeva, J.V.; Cullen, W.K.; Anwyl, R.; Wolfe, M.S.; Rowan, M.J.; Selkoe, D.J. Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature 2002, 416, 535–539. [Google Scholar] [CrossRef] [PubMed]
- Klyubin, I.; Walsh, D.M.; Lemere, C.A.; Cullen, W.K.; Shankar, G.M.; Betts, V.; Spooner, E.T.; Jiang, L.; Anwyl, R.; Selkoe, D.J.; et al. Amyloid β protein immunotherapy neutralizes Aβ oligomers that disrupt synaptic plasticity in vivo. Nat. Med. 2005, 11, 556–561. [Google Scholar] [CrossRef] [PubMed]
- Kayed, R.; Head, E.; Thompson, J.L.; McIntire, T.M.; Milton, S.C.; Cotman, C.W.; Glabe, C.G. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 2003, 300, 486–489. [Google Scholar] [CrossRef] [PubMed]
- Reed, M.N.; Hofmeister, J.J.; Jungbauer, L.; Welzel, A.T.; Yu, C.; Sherman, M.A.; Lesné, S.; LaDu, M.J.; Walsh, D.M.; Ashe, K.H.; et al. Cognitive effects of cell-derived and synthetically derived Aβ oligomers. Neurobiol. Aging 2011, 32, 1784–1794. [Google Scholar] [CrossRef] [PubMed]
- Francis, P.T.; Palmer, A.M.; Snape, M.; Wilcock, G.K. The cholinergic hypothesis of Alzheimer’s disease: A review of progress. J. Neurol. Neurosurg. Psychiatry 1999, 66, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Golenbock, D.T.; Latz, E.; Anbarasi, M.S. Innate immunity in Alzheimer’s disease. Nat. Immunol. 2015, 16, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Echeverria, V.; Ducatenzeiler, A.; Dowd, E.; Jänne, J.; Grant, S.M.; Szyf, M.; Wandosell, F.; Avila, J.; Grimm, H.; Dunnett, S.B.; et al. Altered mitogen-activated protein kinase signaling, tau hyperphosphorylation and mild spatial learning dysfunction in transgenic rats expressing the β-amyloid peptide intracellularly in hippocampal and cortical neurons. Neuroscience 2004, 129, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Leon, W.C.; Canneva, F.; Partridge, V.; Allard, S.; Ferretti, M.T.; DeWilde, A.; Vercauteren, F.; Atifeh, R.; Ducatenzeiler, A.; Klein, W.; et al. A novel transgenic rat model with a full Alzheimer’s-like amyloid pathology displays pre-plaque intracellular amyloid-β-associated cognitive impairment. J. Alzheimer Dis. 2010, 20, 113–126. [Google Scholar]
- D’Hooge, R.; de Deyn, P.P. Applications of the MORRIS water maze in the study of learning and memory. Brain Res. Rev. 2001, 36, 60–90. [Google Scholar] [CrossRef]
- Nell, H.J.; Whitehead, S.N.; Cechetto, D.F. Age-dependent effect of β-amyloid toxicity on basal forebrain cholinergic neurons and inflammation in the rat brain. Brain Pathol. 2015, 25, 531–542. [Google Scholar] [CrossRef] [PubMed]
- Prut, L.; Belzung, C. The open field as a paradigm to measure the effects of drugs on anxiety-like behaviors: A review. Eur. J. Pharmacol. 2003, 463, 3–33. [Google Scholar] [CrossRef]
- Folkesson, R.; Malkiewicz, K.; Kloskowska, E.; Nilsson, T.; Popova, E.; Bogdanovic, N.; Ganten, U.; Ganten, D.; Bader, M.; Winblad, B.; et al. A transgenic rat expressing human APP with the Swedish Alzheimer’s disease mutation. Biochem. Biophys. Res. Commun. 2007, 358, 777–782. [Google Scholar] [CrossRef] [PubMed]
- Kloskowska, E.; Pham, T.M.; Nilsson, T.; Zhu, S.; Öberg, J.; Codita, A.; Pedersen, L.; Pedersen, J.T.; Malkiewicz, K.; Winblad, B.; et al. Cognitive impairment in the Tg6590 transgenic rat model of Alzheimer’s disease. J. Cell. Mol. Med. 2010, 14, 1816–1823. [Google Scholar] [CrossRef] [PubMed]
- Forny-Germano, L.; Lyra e Silva, N.M.; Batista, A.F.; Brito-Moreira, J.; Gralle, M.; Boehnke, S.E.; Coe, B.C.; Lablans, A.; Marques, S.A.; Martinez, A.M.B.; et al. Alzheimer’s disease-like pathology induced by amyloid-oligomers in nonhuman primates. J. Neurosci. 2014, 34, 13629–13643. [Google Scholar] [CrossRef] [PubMed]
- Thal, D.R.; Rüb, U.; Orantes, M.; Braak, H. Phases of A β-deposition in the human brain and its relevance for the development of AD. Neurology 2002, 58, 1791–1800. [Google Scholar] [CrossRef] [PubMed]
- Kawarabayashi, T.; Younkin, L.H.; Saido, T.C.; Shoji, M.; Ashe, K.H.; Younkin, S.G. Age-dependent changes in brain, CSF, and plasma amyloid β protein in the Tg2576 transgenic mouse model of Alzheimer’s disease. J. Neurosci. 2001, 21, 372–381. [Google Scholar] [PubMed]
- Larson, M.E.; Lesné, S.E. Soluble Aβ oligomer production and toxicity. J. Neurochem. 2012, 120, 125–139. [Google Scholar] [CrossRef] [PubMed]
- Lesné, S.E. Breaking the code of amyloid-β oligomers. Int. J. Cell Biol. 2013, 2013, 950783. [Google Scholar] [CrossRef] [PubMed]
- Sondag, C.M.; Dhawan, G.; Combs, C.K. Beta amyloid oligomers and fibrils stimulate differential activation of primary microglia. J. Neuroinflamm. 2009, 6, 1. [Google Scholar] [CrossRef] [PubMed]
- Doi, Y.; Mizuno, T.; Maki, Y.; Jin, S.; Mizoguchi, H.; Ikeyama, M.; Doi, M.; Michikawa, M.; Takeuchi, H.; Suzumura, A. Microglia activated with the toll-like receptor 9 ligand CpG attenuate oligomeric amyloid β neurotoxicity in in vitro and in vivo models of Alzheimer’s disease. Am. J. Pathol. 2009, 175, 2121–2132. [Google Scholar] [CrossRef] [PubMed]

© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wong, R.S.; Cechetto, D.F.; Whitehead, S.N. Assessing the Effects of Acute Amyloid β Oligomer Exposure in the Rat. Int. J. Mol. Sci. 2016, 17, 1390. https://doi.org/10.3390/ijms17091390
Wong RS, Cechetto DF, Whitehead SN. Assessing the Effects of Acute Amyloid β Oligomer Exposure in the Rat. International Journal of Molecular Sciences. 2016; 17(9):1390. https://doi.org/10.3390/ijms17091390
Chicago/Turabian StyleWong, Ryan S., David F. Cechetto, and Shawn N. Whitehead. 2016. "Assessing the Effects of Acute Amyloid β Oligomer Exposure in the Rat" International Journal of Molecular Sciences 17, no. 9: 1390. https://doi.org/10.3390/ijms17091390
APA StyleWong, R. S., Cechetto, D. F., & Whitehead, S. N. (2016). Assessing the Effects of Acute Amyloid β Oligomer Exposure in the Rat. International Journal of Molecular Sciences, 17(9), 1390. https://doi.org/10.3390/ijms17091390