
RNA Interference in the Age of CRISPR: Will CRISPR Interfere with RNAi?

{kind=link}
Abstract
:1. Introduction
2. CRISPR: Prokaryotic Adaptive Immune Response System to Powerful Eukaryotic Gene Editing Tool
2.1. CRISPR: A Prokaryotic Adaptive Immune Response System
2.2. CRISPR: Biochemical Architecture
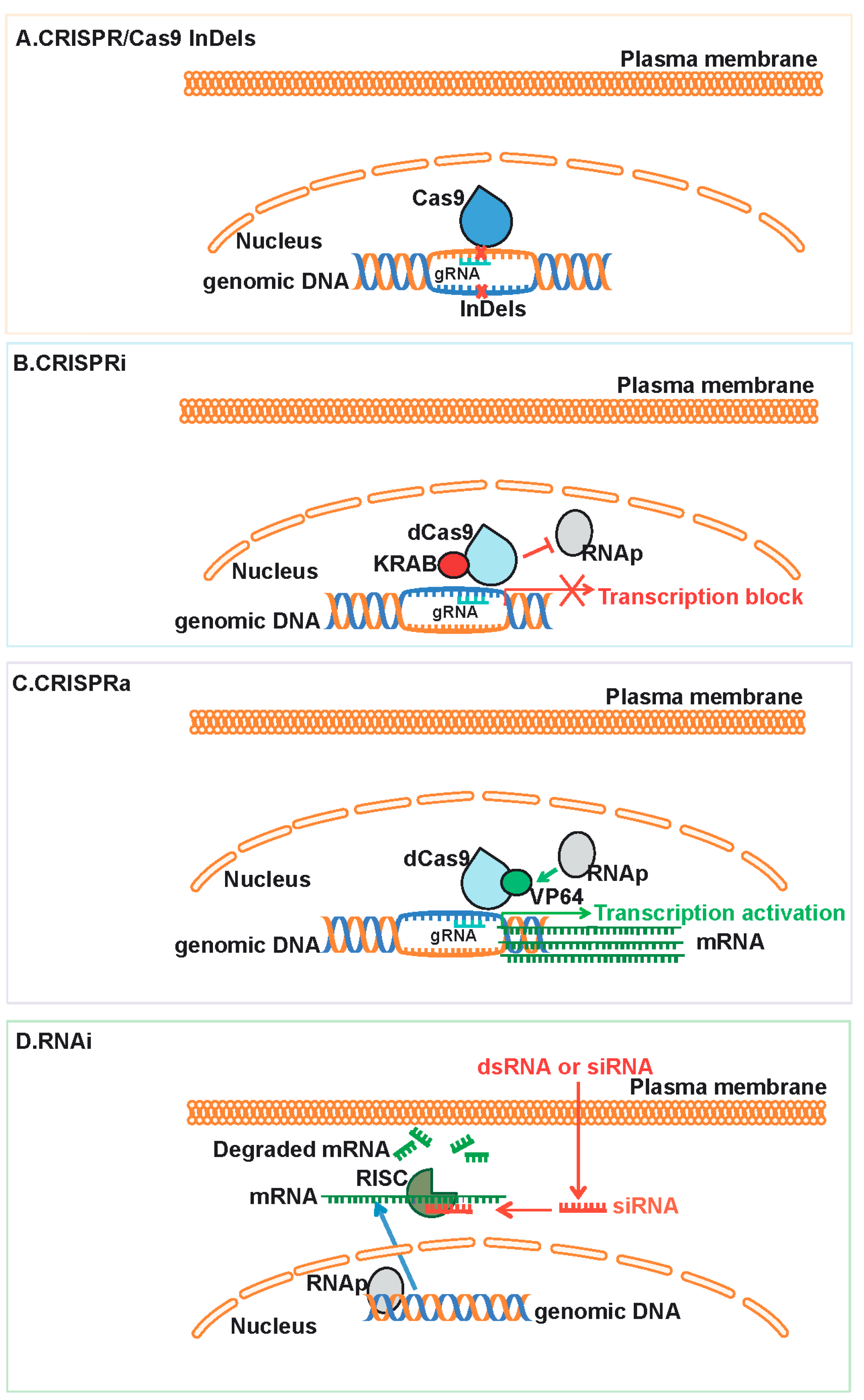
2.3. CRISPR/Cas9, CRISPRi and CRISPRa: Tools for Eukaryotic Gene Editing
3. RNAi: Evolutionarily Conserved Eukaryotic Post-Transcriptional Gene Expression Regulation Mechanism
3.1. RNAi: Eukaryotic Post-Transcriptional Gene Expression Regulation Mechanism
3.2. RNAi: Biochemical Mechinery
4. Comparison of the Applications of CRISPR/Cas9 and RNAi in the Mammalian Biomedical Field: Will CRISPR Delete RNAi or Only Moderately Repress?
4.1. Comparative Analysis of the Applications of CRISPR/Cas9 and RNAi as Research Tools
4.2. Comparative Analysis of the Clinical Applications of CRISPR/Cas9 and RNAi
4.3. Technical Considerations Impacting the Applications of CRISPR/Cas9 and RNAi
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.C.; Doudna, J.A. Molecular mechanisms of RNA interference. Annu. Rev. Biophys. 2013, 42, 217–239. [Google Scholar] [CrossRef] [PubMed]
- Boettcher, M.; McManus, M.T. Choosing the right tool for the job: RNAi, TALEN, or CRISPR. Mol. Cell 2015, 58, 575–585. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D.A.; Horvath, P. CRISPR provides acquired resistance against viruses in prokaryotes. Science 2007, 315, 1709–1712. [Google Scholar] [CrossRef] [PubMed]
- Sorek, R.; Kunin, V.; Hugenholtz, P. CRISPR—A widespread system that provides acquired resistance against phages in bacteria and archaea. Nat. Rev. Microbiol. 2008, 6, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Grissa, I.; Vergnaud, G.; Pourcel, C. The CRISPRdb database and tools to display CRISPRs and to generate dictionaries of spacers and repeats. BMC Bioinform. 2007, 8, 172. [Google Scholar] [CrossRef] [PubMed]
- Ishino, Y.; Shinagawa, H.; Makino, K.; Amemura, M.; Nakata, A. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J. Bacteriol. 1987, 169, 5429–5433. [Google Scholar] [PubMed]
- Jansen, R.; Embden, J.D.; Gaastra, W.; Schouls, L.M. Identification of genes that are associated with DNA repeats in prokaryotes. Mol. Microbiol. 2002, 43, 1565–1575. [Google Scholar] [CrossRef] [PubMed]
- Brouns, S.J.; Jore, M.M.; Lundgren, M.; Westra, E.R.; Slijkhuis, R.J.; Snijders, A.P.; Dickman, M.J.; Makarova, K.S.; Koonin, E.V.; van der Oost, J. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science 2008, 321, 960–964. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Krauss, G. Characterization of the endonuclease SSO2001 from Sulfolobus solfataricus P2. FEBS Lett. 2009, 583, 771–776. [Google Scholar] [CrossRef] [PubMed]
- Makarova, K.S.; Grishin, N.V.; Shabalina, S.A.; Wolf, Y.I.; Koonin, E.V. A putative RNA-interference-based immune system in prokaryotes: Computational analysis of the predicted enzymatic machinery, functional analogies with eukaryotic RNAi, and hypothetical mechanisms of action. Biol. Direct 2006, 1, 7. [Google Scholar] [CrossRef] [PubMed]
- Marraffini, L.A.; Sontheimer, E.J. CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. Science 2008, 322, 1843–1845. [Google Scholar] [CrossRef] [PubMed]
- Haurwitz, R.E.; Jinek, M.; Wiedenheft, B.; Zhou, K.; Doudna, J.A. Sequence- and structure-specific RNA processing by a CRISPR endonuclease. Science 2010, 329, 1355–1358. [Google Scholar] [CrossRef] [PubMed]
- Kunin, V.; Sorek, R.; Hugenholtz, P. Evolutionary conservation of sequence and secondary structures in CRISPR repeats. Genome Biol. 2007, 8, R61. [Google Scholar] [CrossRef] [PubMed]
- Wiedenheft, B.; Sternberg, S.H.; Doudna, J.A. RNA-guided genetic silencing systems in bacteria and archaea. Nature 2012, 482, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Haft, D.H.; Selengut, J.; Mongodin, E.F.; Nelson, K.E. A guild of 45 CRISPR-associated (Cas) protein families and multiple CRISPR/Cas subtypes exist in prokaryotic genomes. PLoS Comput. Biol. 2005, 1, e60. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Marraffini, L.A. CRISPR-Cas: New tools for genetic manipulations from bacterial immunity systems. Annu. Rev. Microbiol. 2015, 69, 209–228. [Google Scholar] [CrossRef] [PubMed]
- Van der Oost, J.; Jore, M.M.; Westra, E.R.; Lundgren, M.; Brouns, S.J. CRISPR-based adaptive and heritable immunity in prokaryotes. Trends Biochem. Sci. 2009, 34, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Garneau, J.E.; Dupuis, M.E.; Villion, M.; Romero, D.A.; Barrangou, R.; Boyaval, P.; Fremaux, C.; Horvath, P.; Magadán, A.H.; Moineau, S. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 2010, 468, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Mojica, F.J.; Diez-Villasenor, C.; Garcia-Martinez, J.; Almendros, C. Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology 2009, 155 Pt 3, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Mojica, F.J.; Diez-Villasenor, C.; Garcia-Martinez, J.; Soria, E. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J. Mol. Evol. 2005, 60, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Carte, J.; Wang, R.; Li, H.; Terns, R.M.; Terns, M.P. Cas6 is an endoribonuclease that generates guide RNAs for invader defense in prokaryotes. Genes Dev. 2008, 22, 3489–3496. [Google Scholar] [CrossRef] [PubMed]
- Deltcheva, E.; Chylinski, K.; Sharma, C.M.; Gonzales, K.; Chao, Y.; Pirzada, Z.A.; Eckert, M.R.; Vogel, J.; Charpentier, E. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 2011, 471, 602–607. [Google Scholar] [CrossRef] [PubMed]
- Gesner, E.M.; Schellenberg, M.J.; Garside, E.L.; George, M.M.; Macmillan, A.M. Recognition and maturation of effector RNAs in a CRISPR interference pathway. Nat. Struct. Mol. Biol. 2011, 18, 688–692. [Google Scholar] [CrossRef] [PubMed]
- Semenova, E.; Jore, M.M.; Datsenko, K.A.; Semenova, A.; Westra, E.R.; Wanner, B.; van der Oost, J.; Brouns, S.J.; Severinov, K. Interference by clustered regularly interspaced short palindromic repeat (CRISPR) RNA is governed by a seed sequence. Proc. Natl. Acad. Sci. USA 2011, 108, 10098–10103. [Google Scholar] [CrossRef] [PubMed]
- Wiedenheft, B.; van Duijn, E.; Bultema, J.B.; Waghmare, S.P.; Zhou, K.; Barendregt, A.; Westphal, W.; Heck, A.J.; Boekema, E.J.; Dickman, M.J.; et al. RNA-guided complex from a bacterial immune system enhances target recognition through seed sequence interactions. Proc. Natl. Acad. Sci. USA 2011, 108, 10092–10097. [Google Scholar] [CrossRef] [PubMed]
- Makarova, K.S.; Haft, D.H.; Barrangou, R.; Brouns, S.J.; Charpentier, E.; Horvath, P.; Moineau, S.; Mojica, F.J.; Wolf, Y.I.; Yakunin, A.F.; et al. Evolution and classification of the CRISPR-Cas systems. Nat. Rev. Microbiol. 2011, 9, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Datsenko, K.A.; Pougach, K.; Tikhonov, A.; Wanner, B.L.; Severinov, K.; Semenova, E. Molecular memory of prior infections activates the CRISPR/Cas adaptive bacterial immunity system. Nat. Commun. 2012, 3, 945. [Google Scholar] [CrossRef] [PubMed]
- Yosef, I.; Goren, M.G.; Qimron, U. Proteins and DNA elements essential for the CRISPR adaptation process in Escherichia coli. Nucleic Acids Res. 2012, 40, 5569–5576. [Google Scholar] [CrossRef] [PubMed]
- Naito, Y.; Ui-Tei, K. Designing functional siRNA with reduced off-target effects. Methods Mol. Biol. 2013, 942, 57–68. [Google Scholar] [PubMed]
- Tsui, T.K.; Li, H. Structure principles of CRISPR-Cas surveillance and effector complexes. Annu. Rev. Biophys. 2015, 44, 229–255. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, L.A.; Horlbeck, M.A.; Adamson, B.; Villalta, J.E.; Chen, Y.; Whitehead, E.H.; Guimaraes, C.; Panning, B.; Ploegh, H.L.; Bassik, M.C.; et al. Genome-scale CRISPR-mediated control of gene repression and activation. Cell 2014, 159, 647–661. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, L.A.; Larson, M.H.; Morsut, L.; Liu, Z.; Brar, G.A.; Torres, S.E.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.H.; Doudna, J.A.; et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 2013, 154, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef] [PubMed]
- Choulika, A.; Perrin, A.; Dujon, B.; Nicolas, J.F. Induction of homologous recombination in mammalian chromosomes by using the I-SceI system of Saccharomyces cerevisiae. Mol. Cell. Biol. 1995, 15, 1968–1973. [Google Scholar] [CrossRef] [PubMed]
- Rouet, P.; Smih, F.; Jasin, M. Introduction of double-strand breaks into the genome of mouse cells by expression of a rare-cutting endonuclease. Mol. Cell. Biol. 1994, 14, 8096–8106. [Google Scholar] [CrossRef] [PubMed]
- Bosher, J.M.; Labouesse, M. RNA interference: Genetic wand and genetic watchdog. Nat. Cell Biol. 2000, 2, E31–E36. [Google Scholar] [CrossRef] [PubMed]
- Dillon, C.P.; Sandy, P.; Nencioni, A.; Kissler, S.; Rubinson, D.A.; van Parijs, L. RNAi as an experimental and therapeutic tool to study and regulate physiological and disease processes. Annu. Rev. Physiol. 2005, 67, 147–173. [Google Scholar] [CrossRef] [PubMed]
- Mohr, S.; Bakal, C.; Perrimon, N. Genomic screening with RNAi: Results and challenges. Annu. Rev. Biochem. 2010, 79, 37–64. [Google Scholar] [CrossRef] [PubMed]
- Napoli, C.; Lemieux, C.; Jorgensen, R. Introduction of a chimeric chalcone synthase gene into petunia results in reversible co-suppression of homologous genes in trans. Plant Cell 1990, 2, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Paddison, P.J.; Hannon, G.J. RNA interference: The new somatic cell genetics? Cancer Cell 2002, 2, 17–23. [Google Scholar] [CrossRef]
- Elbashir, S.M.; Harborth, J.; Lendeckel, W.; Yalcin, A.; Weber, K.; Tuschl, T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 2001, 411, 494–498. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, E.; Caudy, A.A.; Hammond, S.M.; Hannon, G.J. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 2001, 409, 363–366. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Hur, I.; Park, S.Y.; Kim, Y.K.; Suh, M.R.; Kim, V.N. The role of PACT in the RNA silencing pathway. EMBO J. 2006, 25, 522–532. [Google Scholar] [CrossRef] [PubMed]
- Hammond, S.M.; Boettcher, S.; Caudy, A.A.; Kobayashi, R.; Hannon, G.J. Argonaute2, a link between genetic and biochemical analyses of RNAi. Science 2001, 293, 1146–1150. [Google Scholar] [CrossRef] [PubMed]
- Janowski, B.A.; Huffman, K.E.; Schwartz, J.C.; Ram, R.; Nordsell, R.; Shames, D.S.; Minna, J.D.; Corey, D.R. Involvement of AGO1 and AGO2 in mammalian transcriptional silencing. Nat. Struct. Mol. Biol. 2006, 13, 787–792. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Villeneuve, L.M.; Morris, K.V.; Rossi, J.J. Argonaute-1 directs siRNA-mediated transcriptional gene silencing in human cells. Nat. Struct. Mol. Biol. 2006, 13, 793–797. [Google Scholar] [CrossRef] [PubMed]
- Parnas, O.; Jovanovic, M.; Eisenhaure, T.M.; Herbst, R.H.; Dixit, A.; Ye, C.J.; Przybylski, D.; Platt, R.J.; Tirosh, I.; Sanjana, N.E.; et al. A Genome-wide CRISPR screen in primary immune cells to dissect regulatory networks. Cell 2015, 162, 675–686. [Google Scholar] [CrossRef] [PubMed]
- Shalem, O.; Sanjana, N.E.; Zhang, F. High-throughput functional genomics using CRISPR-Cas9. Nat. Rev. Genet. 2015, 16, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Birsoy, K.; Hughes, N.W.; Krupczak, K.M.; Post, Y.; Wei, J.J.; Lander, E.S.; Sabatini, D.M. Identification and characterization of essential genes in the human genome. Science 2015, 350, 1096–1101. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Wei, J.J.; Sabatini, D.M.; Lander, E.S. Genetic screens in human cells using the CRISPR-Cas9 system. Science 2014, 343, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Duda, K.; Lonowski, L.A.; Kofoed-Nielsen, M.; Ibarra, A.; Delay, C.M.; Kang, Q.; Yang, Z.; Pruett-Miller, S.M.; Bennett, E.P.; Wandall, H.H.; et al. High-efficiency genome editing via 2A-coupled co-expression of fluorescent proteins and zinc finger nucleases or CRISPR/Cas9 nickase pairs. Nucleic Acids Res. 2014, 42, e84. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Potter, J.; Kumar, S.; Zou, Y.; Quintanilla, R.; Sridharan, M.; Carte, J.; Chen, W.; Roark, N.; Ranganathan, S.; et al. Rapid and highly efficient mammalian cell engineering via Cas9 protein transfection. J. Biotechnol. 2015, 208, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Swiech, L.; Heidenreich, M.; Banerjee, A.; Habib, N.; Li, Y.; Trombetta, J.; Sur, M.; Zhang, F. In vivo interrogation of gene function in the mammalian brain using CRISPR-Cas9. Nat. Biotechnol. 2015, 33, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Chu, V.T.; Weber, T.; Wefers, B.; Wurst, W.; Sander, S.; Rajewsky, K.; Kühn, R. Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat. Biotechnol. 2015, 33, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Staahl, B.T.; Alla, R.K.; Doudna, J.A. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. eLife 2014, 3, e04766. [Google Scholar] [CrossRef] [PubMed]
- Konermann, S.; Brigham, M.D.; Trevino, A.E.; Joung, J.; Abudayyeh, O.O.; Barcena, C.; Hsu, P.D.; Habib, N.; Gootenberg, J.S.; Nishimasu, H.; et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 2015, 517, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Aach, J.; Stranges, P.B.; Esvelt, K.M.; Moosburner, M.; Kosuri, S.; Yang, L.; Church, G.M. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat. Biotechnol. 2013, 31, 833–838. [Google Scholar] [CrossRef] [PubMed]
- Borna, H.; Imani, S.; Iman, M.; Azimzadeh Jamalkandi, S. Therapeutic face of RNAi: In vivo challenges. Expert Opin. Biol. Ther. 2015, 15, 269–285. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Murawaki, Y. The therapeutic potential of RNA interference: Novel approaches for cancer treatment. Curr. Pharm. Biotechnol. 2012, 13, 2235–2247. [Google Scholar] [CrossRef] [PubMed]
- Yao, S.; He, Z.; Chen, C. CRISPR/Cas9-mediated genome editing of epigenetic factors for cancer therapy. Hum. Gene Ther. 2015, 26, 463–471. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, T.P.; Crystal, R.G. Genetic medicines: Treatment strategies for hereditary disorders. Nat. Rev. Genet. 2006, 7, 261–276. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.W.; Lai, Y.S.; Westin, E.; Khodadadi-Jamayran, A.; Pawlik, K.M.; Lamb, L.S., Jr.; Goldman, F.D.; Townes, T.M. Modeling human severe combined immunodeficiency and correction by CRISPR/Cas9-enhanced gene targeting. Cell Rep. 2015, 12, 1668–1677. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.M.; Flynn, R.; Hollywood, J.A.; Scallan, M.F.; Harrison, P.T. Correction of the DeltaF508 mutation in the cystic fibrosis transmembrane conductance regulator gene by zinc-finger nuclease homology-directed repair. Biores. Open Access 2012, 1, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Graslund, T.; Li, X.; Magnenat, L.; Popkov, M.; Barbas, C.F., 3rd. Exploring strategies for the design of artificial transcription factors: Targeting sites proximal to known regulatory regions for the induction of gamma-globin expression and the treatment of sickle cell disease. J. Biol. Chem. 2005, 280, 3707–3714. [Google Scholar] [CrossRef] [PubMed]
- Thrasher, A.J.; Gaspar, H.B.; Baum, C.; Modlich, U.; Schambach, A.; Candotti, F.; Otsu, M.; Sorrentino, B.; Scobie, L.; Cameron, E.; et al. Gene therapy: X-SCID transgene leukaemogenicity. Nature 2006, 443, E5–E6. [Google Scholar] [CrossRef] [PubMed]
- Borel, F.; Kay, M.A.; Mueller, C. Recombinant AAV as a platform for translating the therapeutic potential of RNA interference. Mol. Ther. 2014, 22, 692–701. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Mou, H.; Li, S.; Li, Y.; Hough, S.; Tran, K.; Li, J.; Yin, H.; Anderson, D.G.; Sontheimer, E.J.; et al. Adenovirus-mediated somatic genome editing of pten by CRISPR/Cas9 in mouse liver in spite of Cas9-Specific immune responses. Hum. Gene Ther. 2015, 26, 432–442. [Google Scholar] [CrossRef] [PubMed]
- Oakland, M.; Sinn, P.L.; McCray, P.B., Jr. Advances in cell and gene-based therapies for cystic fibrosis lung disease. Mol. Ther. 2012, 20, 1108–1115. [Google Scholar] [CrossRef] [PubMed]
- Ousterout, D.G.; Kabadi, A.M.; Thakore, P.I.; Majoros, W.H.; Reddy, T.E.; Gersbach, C.A. Multiplex CRISPR/Cas9-based genome editing for correction of dystrophin mutations that cause Duchenne muscular dystrophy. Nat. Commun. 2015, 6, 6244. [Google Scholar] [CrossRef] [PubMed]
- Schwank, G.; Koo, B.K.; Sasselli, V.; Dekkers, J.F.; Heo, I.; Demircan, T.; Sasaki, N.; Boymans, S.; Cuppen, E.; van der Ent, C.K.; et al. Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell 2013, 13, 653–658. [Google Scholar] [CrossRef] [PubMed]
- Liechtenstein, T.; Perez-Janices, N.; Escors, D. Lentiviral vectors for cancer immunotherapy and clinical applications. Cancers 2013, 5, 815–837. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, A.; Vickery, O.N.; Laugel, B. Beyond the antigen receptor: Editing the genome of T-cells for cancer adoptive cellular therapies. Front. Immunol. 2013, 4, 221. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kim, D.; Cho, S.W.; Kim, J.; Kim, J.S. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 2014, 24, 1012–1019. [Google Scholar] [CrossRef] [PubMed]
- Schumann, K.; Lin, S.; Boyer, E.; Simeonov, D.R.; Subramaniam, M.; Gate, R.E.; Haliburton, G.E.; Ye, C.J.; Bluestone, J.A.; Doudna, J.A.; et al. Generation of knock-in primary human T cells using Cas9 ribonucleoproteins. Proc. Natl. Acad. Sci. USA 2015, 112, 10437–10442. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, E.M.; Kornepati, A.V.; Cullen, B.R. Targeting hepatitis B virus cccDNA using CRISPR/Cas9. Antivir. Res. 2015, 123, 188–192. [Google Scholar] [CrossRef] [PubMed]
- Price, A.A.; Sampson, T.R.; Ratner, H.K.; Grakoui, A.; Weiss, D.S. Cas9-mediated targeting of viral RNA in eukaryotic cells. Proc. Natl. Acad. Sci. USA 2015, 112, 6164–6169. [Google Scholar] [CrossRef] [PubMed]
- Jahan, S.; Khaliq, S.; Samreen, B.; Ijaz, B.; Khan, M.; Ahmad, W.; Ashfaq, U.A.; Hassan, S. Effect of combined siRNA of HCV E2 gene and HCV receptors against HCV. Virol. J. 2011, 8, 295. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Lee, S.K.; Shankar, P.; Manjunath, N. A single siRNA suppresses fatal encephalitis induced by two different flaviviruses. PLoS Med. 2006, 3, e96. [Google Scholar] [CrossRef] [PubMed]
- Ui-Tei, K. Optimal choice of functional and off-target effect-reduced siRNAs for RNAi therapeutics. Front. Genet. 2013, 4, 107. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.W.; Kim, S.; Kim, Y.; Kweon, J.; Kim, H.S.; Bae, S.; Kim, J.S. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 2014, 24, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Frock, R.L.; Hu, J.; Meyers, R.M.; Ho, Y.J.; Kii, E.; Alt, F.W. Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nat. Biotechnol. 2015, 33, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Foden, J.A.; Khayter, C.; Maeder, M.L.; Reyon, D.; Joung, J.K.; Sander, J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013, 31, 822–826. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Cradick, T.J.; Brown, M.T.; Deshmukh, H.; Ranjan, P.; Sarode, N.; Wile, B.M.; Vertino, P.M.; Stewart, F.J.; Bao, G. CRISPR/Cas9 systems have off-target activity with insertions or deletions between target DNA and guide RNA sequences. Nucleic Acids Res. 2014, 42, 7473–7485. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Lin, C.Y.; Gootenberg, J.S.; Konermann, S.; Trevino, A.E.; Scott, D.A.; Inoue, A.; Matoba, S.; Zhang, Y.; et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 2013, 154, 1380–1389. [Google Scholar] [CrossRef] [PubMed]
- Chang, N.; Sun, C.; Gao, L.; Zhu, D.; Xu, X.; Zhu, X.; Xiong, J.W.; Xi, J.J. Genome editing with RNA-guided Cas9 nuclease in zebrafish embryos. Cell Res. 2013, 23, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Kleinstiver, B.P.; Pattanayak, V.; Prew, M.S.; Tsai, S.Q.; Nguyen, N.T.; Zheng, Z.; Joung, J.K. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature 2016, 529, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Kleinstiver, B.P.; Prew, M.S.; Tsai, S.Q.; Nguyen, N.T.; Topkar, V.V.; Zheng, Z.; Joung, J.K. Broadening the targeting range of Staphylococcus aureus CRISPR-Cas9 by modifying PAM recognition. Nat. Biotechnol. 2015, 33, 1293–1298. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Zhang, J.; Chen, L.; Shen, B.; Zhou, J.; Hu, B.; Du, Y.; Tate, P.H.; Huang, X.; Zhang, W. Efficient in vivo deletion of a large imprinted lncRNA by CRISPR/Cas9. RNA Biol. 2014, 11, 829–835. [Google Scholar] [CrossRef] [PubMed]
- Shrivastav, M.; De Haro, L.P.; Nickoloff, J.A. Regulation of DNA double-strand break repair pathway choice. Cell Res. 2008, 18, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Zetsche, B.; Gootenberg, J.S.; Abudayyeh, O.O.; Slaymaker, I.M.; Makarova, K.S.; Essletzbichler, P.; Essletzbichler, P.; Volz, S.E.; Joung, J.; van der Oost, J.; et al. Cpf1 Is a Single RNA-Guided Endonuclease of a Class 2 CRISPR-Cas System. Cell 2015, 163, 759–771. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Unniyampurath, U.; Pilankatta, R.; Krishnan, M.N. RNA Interference in the Age of CRISPR: Will CRISPR Interfere with RNAi? Int. J. Mol. Sci. 2016, 17, 291. https://doi.org/10.3390/ijms17030291
Unniyampurath U, Pilankatta R, Krishnan MN. RNA Interference in the Age of CRISPR: Will CRISPR Interfere with RNAi? International Journal of Molecular Sciences. 2016; 17(3):291. https://doi.org/10.3390/ijms17030291
Chicago/Turabian StyleUnniyampurath, Unnikrishnan, Rajendra Pilankatta, and Manoj N. Krishnan. 2016. "RNA Interference in the Age of CRISPR: Will CRISPR Interfere with RNAi?" International Journal of Molecular Sciences 17, no. 3: 291. https://doi.org/10.3390/ijms17030291
APA StyleUnniyampurath, U., Pilankatta, R., & Krishnan, M. N. (2016). RNA Interference in the Age of CRISPR: Will CRISPR Interfere with RNAi? International Journal of Molecular Sciences, 17(3), 291. https://doi.org/10.3390/ijms17030291