
The Regulation of Tumor Suppressor p63 by the Ubiquitin-Proteasome System


Abstract
:
{kind=link}
{kind=link}
{kind=link}
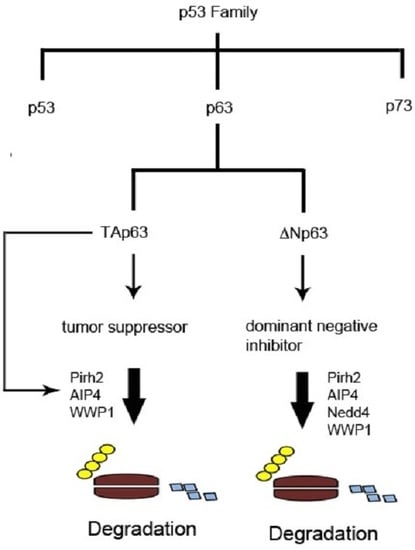
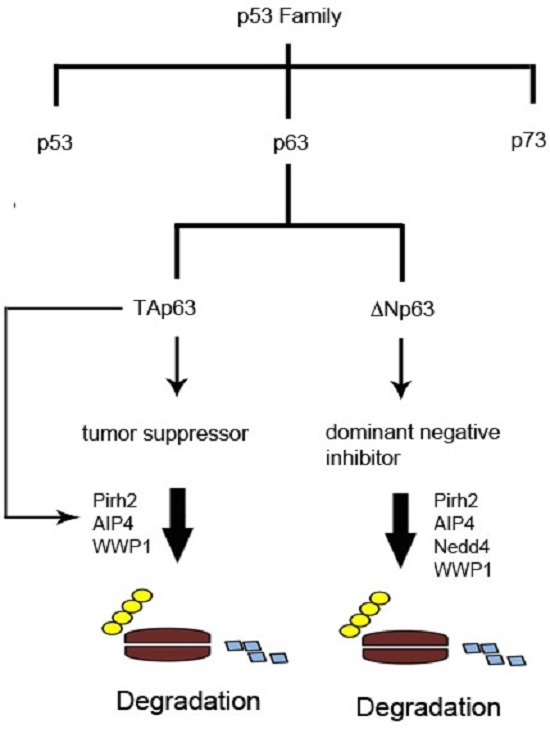
1. The p53 Family of Tumor Suppressors
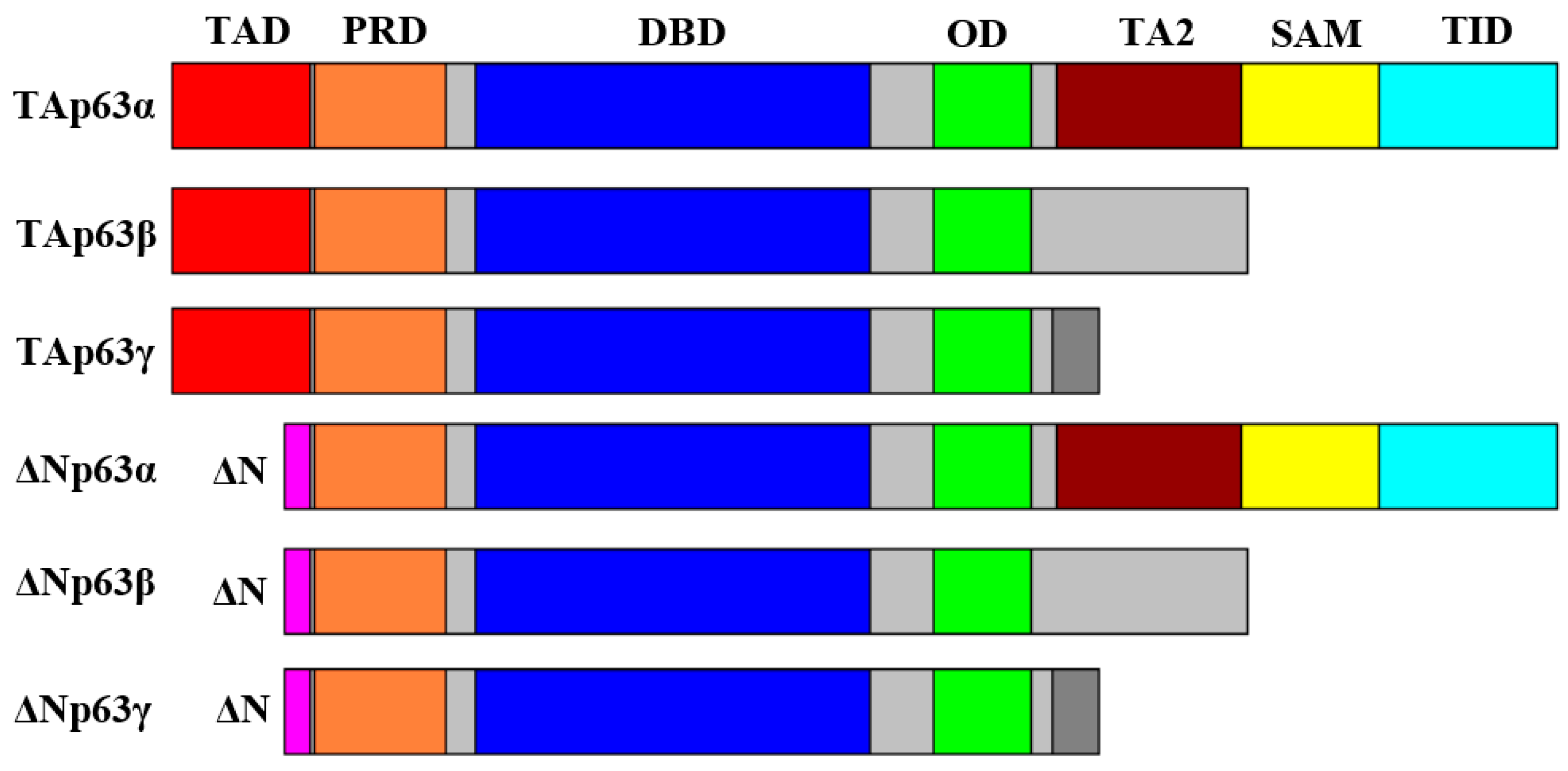
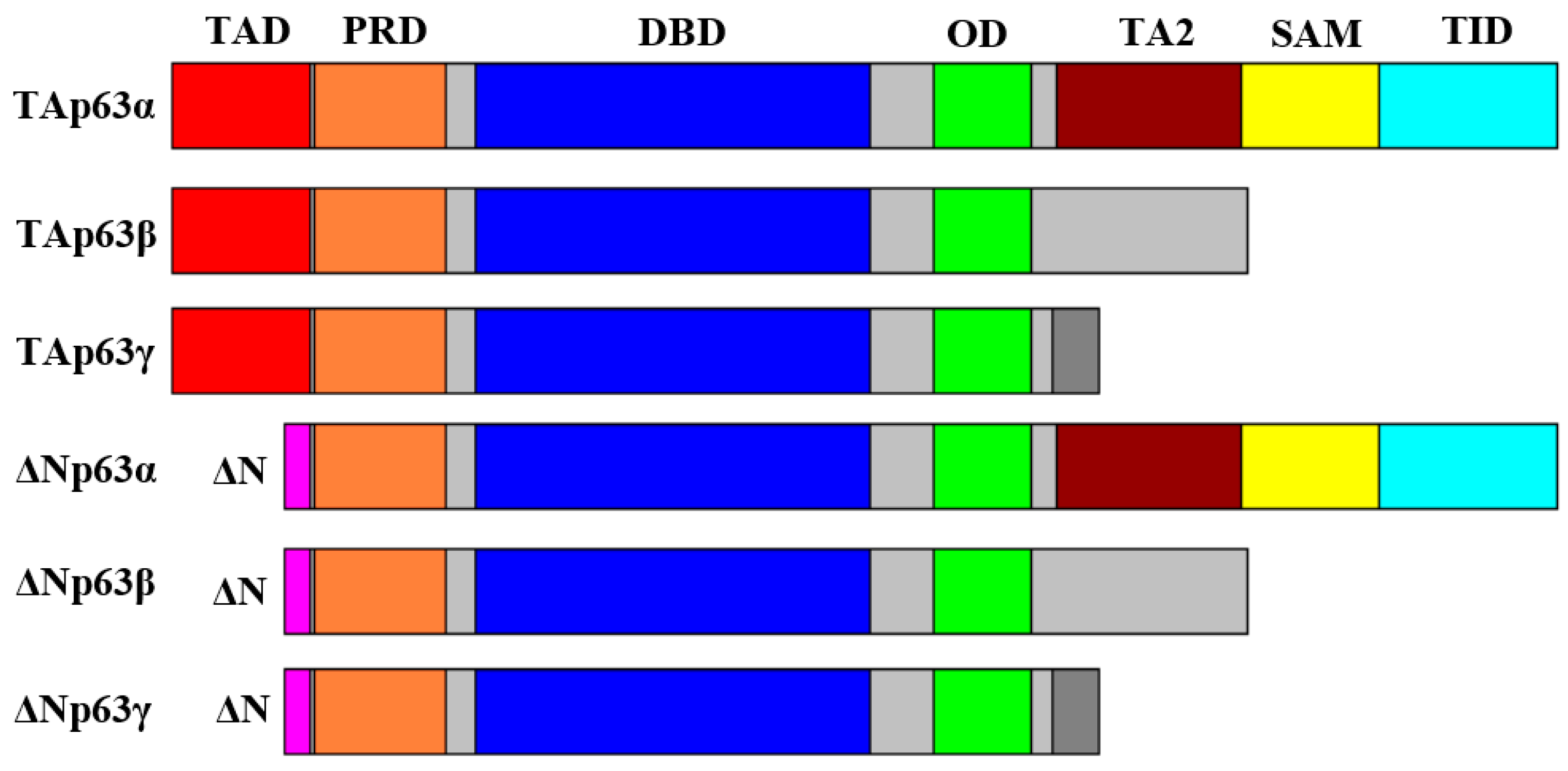
2. Structure and Properties of p53 Family Proteins
3. Expression and Functions of p53 Family Proteins
4. Kinases and p63 Phosphorylation
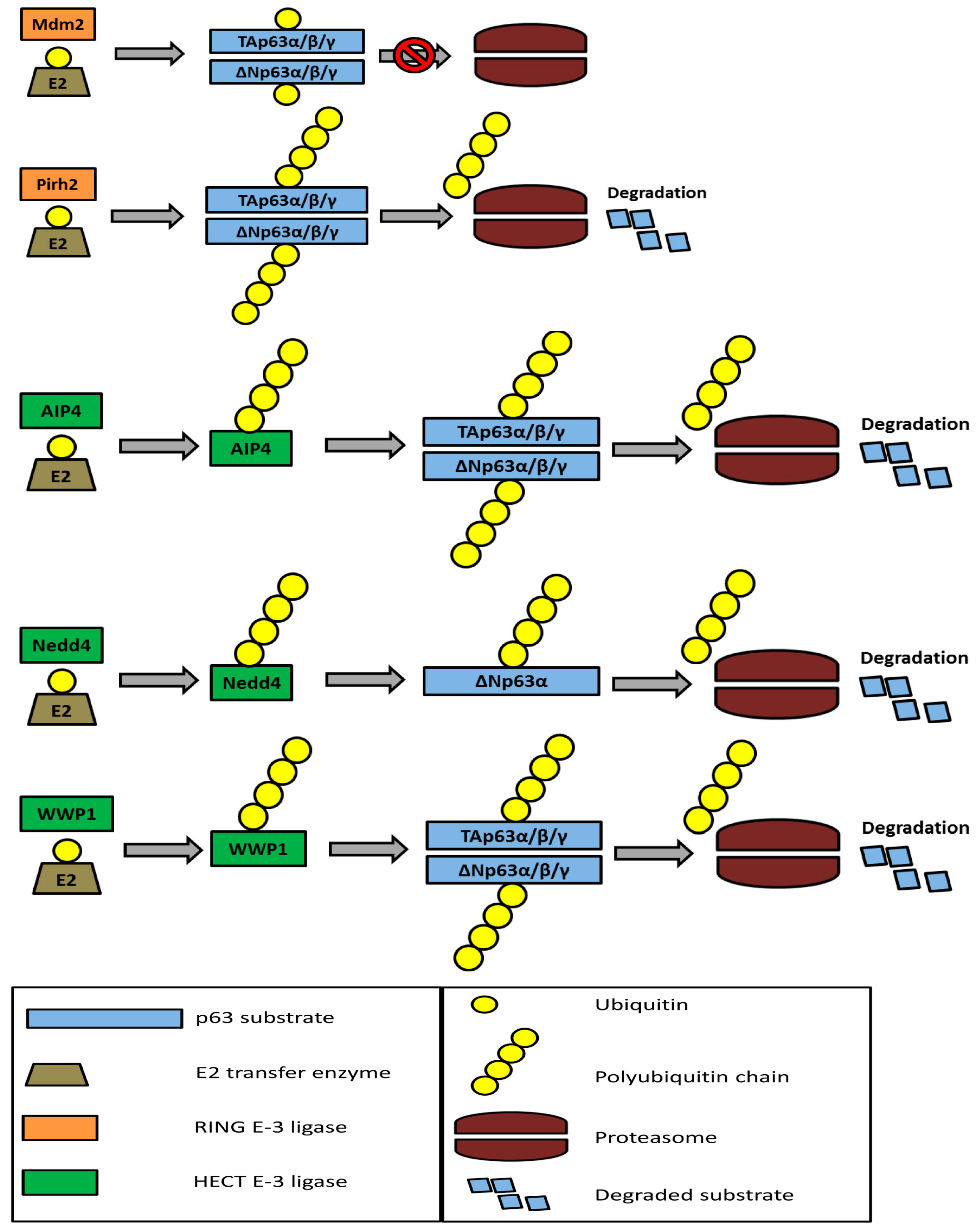
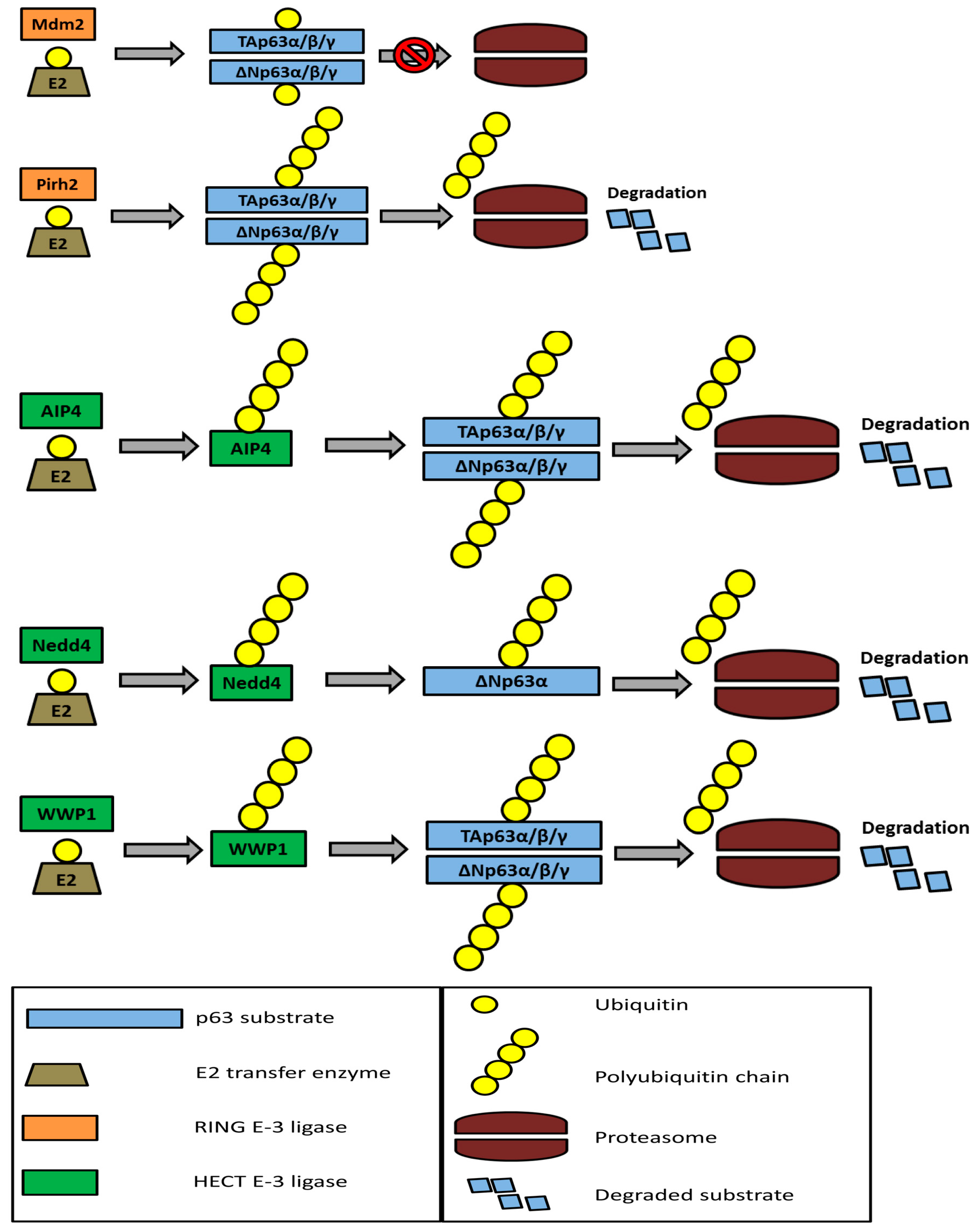
5. The Ubiquitin-Proteasome System and p63 Regulation
6. Summary
Acknowledgments
Conflicts of Interest
References
- Soussi, T. The history of p53. A perfect example of the drawbacks of scientific paradigms. EMBO Rep. 2010, 11, 822–826. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; McKeon, F. p63 and p73: P53 mimics, menaces and more. Nat. Rev. Mol. Cell Biol. 2000, 1, 199–207. [Google Scholar] [CrossRef] [PubMed]
- De Laurenzi, V.; Melino, G. Evolution of functions within the p53/p63/p73 family. Ann. N. Y. Acad. Sci. 2000, 926, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Hollstein, M.; Sidransky, D.; Vogelstein, B.; Harris, C.C. p53 mutations in human cancers. Science 1991, 253, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Khoury, M.P.; Bourdon, J.C. p53 isoforms: An intracellular microprocessor? Genes Cancer 2011, 2, 453–465. [Google Scholar] [CrossRef] [PubMed]
- Melino, G.; Lu, X.; Gasco, M.; Crook, T.; Knight, R.A. Functional regulation of p73 and p63: Development and cancer. Trends Biochem. Sci. 2003, 28, 663–670. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Lu, X. Live or let die: The cell’s response to p53. Nat. Rev. Cancer 2002, 2, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Oren, M.; Rotter, V. Mutant p53 gain-of-function in cancer. Cold Spring Harb. Perspect. Biol. 2010, 2, a001107. [Google Scholar] [CrossRef] [PubMed]
- Kaghad, M.; Bonnet, H.; Yang, A.; Creancier, L.; Biscan, J.C.; Valent, A.; Minty, A.; Chalon, P.; Lelias, J.M.; Dumont, X.; et al. Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other human cancers. Cell 1997, 90, 809–819. [Google Scholar] [CrossRef]
- Trink, B.; Okami, K.; Wu, L.; Sriuranpong, V.; Jen, J.; Sidransky, D. A new human p53 homologue. Nat. Med. 1998, 4, 747–748. [Google Scholar] [CrossRef] [PubMed]
- Schmale, H.; Bamberger, C. A novel protein with strong homology to the tumor suppressor p53. Oncogene 1997, 15, 1363–1367. [Google Scholar] [CrossRef] [PubMed]
- Osada, M.; Ohba, M.; Kawahara, C.; Ishioka, C.; Kanamaru, R.; Katoh, I.; Ikawa, Y.; Nimura, Y.; Nakagawara, A.; Obinata, M.; et al. Cloning and functional analysis of human p51, which structurally and functionally resembles p53. Nat. Med. 1998, 4, 839–843. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Kaghad, M.; Wang, Y.; Gillett, E.; Fleming, M.D.; Dotsch, V.; Andrews, N.C.; Caput, D.; McKeon, F. p63, a p53 homolog at 3q27–29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Mol. Cell 1998, 2, 305–316. [Google Scholar] [CrossRef]
- Yang, A.; Kaghad, M.; Caput, D.; McKeon, F. On the shoulders of giants: p63, p73 and the rise of p53. Trends Genet. 2002, 18, 90–95. [Google Scholar] [CrossRef]
- Moll, U.M.; Slade, N. p63 and p73: Roles in development and tumor formation. Mol. Cancer Res. 2004, 2, 371–386. [Google Scholar] [PubMed]
- Bernard, H.; Garmy-Susini, B.; Ainaoui, N.; van den Berghe, L.; Peurichard, A.; Javerzat, S.; Bikfalvi, A.; Lane, D.P.; Bourdon, J.C.; Prats, A.C. The p53 isoform, Δ133p53α, stimulates angiogenesis and tumour progression. Oncogene 2013, 32, 2150–2160. [Google Scholar] [CrossRef] [PubMed]
- Ishimoto, O.; Kawahara, C.; Enjo, K.; Obinata, M.; Nukiwa, T.; Ikawa, S. Possible oncogenic potential of ΔNp73: A newly identified isoform of human p73. Cancer Res. 2002, 62, 636–641. [Google Scholar] [PubMed]
- Ghioni, P.; Bolognese, F.; Duijf, P.H.; van Bokhoven, H.; Mantovani, R.; Guerrini, L. Complex transcriptional effects of p63 isoforms: Identification of novel activation and repression domains. Mol. Cell. Biol. 2002, 22, 8659–8668. [Google Scholar] [CrossRef] [PubMed]
- Dohn, M.; Zhang, S.; Chen, X. p63α and ΔNp63α can induce cell cycle arrest and apoptosis and differentially regulate p53 target genes. Oncogene 2001, 20, 3193–3205. [Google Scholar] [CrossRef] [PubMed]
- Petitjean, A.; Ruptier, C.; Tribollet, V.; Hautefeuille, A.; Chardon, F.; Cavard, C.; Puisieux, A.; Hainaut, P.; de Fromentel, C.C. Properties of the six isoforms of p63: p53-like regulation in response to genotoxic stress and cross talk with ΔNp73. Carcinogenesis 2008, 29, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Helton, E.S.; Zhang, J.; Chen, X. The proline-rich domain in p63 is necessary for the transcriptional and apoptosis-inducing activities of TAp63. Oncogene 2008, 27, 2843–2850. [Google Scholar] [CrossRef] [PubMed]
- Belyi, V.A.; Ak, P.; Markert, E.; Wang, H.; Hu, W.; Puzio-Kuter, A.; Levine, A.J. The origins and evolution of the p53 family of genes. Cold Spring Harb. Perspect. Biol. 2010, 2, a001198. [Google Scholar] [CrossRef] [PubMed]
- Westfall, M.D.; Pietenpol, J.A. p63: Molecular complexity in development and cancer. Carcinogenesis 2004, 25, 857–864. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Schweitzer, R.; Sun, D.; Kaghad, M.; Walker, N.; Bronson, R.T.; Tabin, C.; Sharpe, A.; Caput, D.; Crum, C.; et al. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature 1999, 398, 714–718. [Google Scholar] [PubMed]
- Mills, A.A.; Zheng, B.; Wang, X.J.; Vogel, H.; Roop, D.R.; Bradley, A. p63 is a p53 homologue required for limb and epidermal morphogenesis. Nature 1999, 398, 708–713. [Google Scholar] [PubMed]
- Guo, X.; Keyes, W.M.; Papazoglu, C.; Zuber, J.; Li, W.; Lower, S.W.; Vogel, H.; Mills, A.A. TAp63 induces senescence and suppresses tumorigenesis in vivo. Nat. Cell Biol. 2009, 11, 1451–1457. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Gi, Y.J.; Chakravarty, D.; Chan, I.L.; Zhang, A.; Xia, X.; Tsai, K.Y.; Flores, E.R. TAp63 is a master transcriptional regulator of lipid and glucose metabolism. Cell Metab. 2012, 16, 511–525. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Walker, N.; Bronson, R.; Kaghad, M.; Oosterwegel, M.; Bonnin, J.; Vagner, C.; Bonnet, H.; Dikkes, P.; Sharpe, A.; et al. p73-deficient mice have neurological, pheromonal and inflammatory defects but lack spontaneous tumours. Nature 2000, 404, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Pozniak, C.D.; Barnabe-Heider, F.; Rymar, W.; Lee, A.F.; Sadikot, A.F.; Miller, F.D. p73 is required for survival and maintenance of CNS neurons. J. Neurosci. 2002, 22, 9800–9809. [Google Scholar] [PubMed]
- Irwin, M.S.; Kaelin, W.G. Role of the newer p53 family proteins in malignancy. Apoptosis 2001, 6, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Hibi, K.; Trink, B.; Patturajan, M.; Westra, W.H.; Caballero, O.L.; Hill, D.E.; Ratovitski, E.A.; Jen, J.; Sidransky, D. AIS is an oncogene amplified in squamous cell carcinoma. Proc. Natl. Acad. Sci. USA 2000, 97, 5462–5467. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, K.; Wu, L.; Caballero, O.L.; Hibi, K.; Trink, B.; Resto, V.; Cairns, P.; Okami, K.; Kock, W.M.; Sidransky, D.; et al. Frequent gain of the p40/p51/p63 gene locus in primary head and neck squamous cell carcinoma. Int. J. Cancer 2000, 86, 684–689. [Google Scholar] [CrossRef]
- Massion, P.P.; Taflan, P.M.; Rahman, S.J.; Yildiz, P.; Shry, Y.; Edgerton, M.E.; Westfall, M.D.; Roberts, J.R.; Pietenpol, J.A.; Carbone, D.P.; et al. Significance of p63 amplification and overexpression in lung cancer development and prognosis. Cancer Res. 2003, 63, 7113–7121. [Google Scholar] [PubMed]
- Rocco, J.W.; Leong, C.O.; Kuperwasser, N.; DeYoung, M.P.; Ellisen, L.W. p63 mediates survival in squamous cell carcinoma by suppression of p73-dependent apoptosis. Cancer Cell 2006, 9, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Donehower, L.A.; Harvey, M.; Slagle, B.L.; McArthur, M.J.; Montgomery, C.A.; Brutel, J.S.; Bradley, A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 1992, 356, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Flores, E.R.; Sengupta, S.; Miller, J.B.; Newman, J.J.; Bronson, R.; Crowley, D.; Yang, A.; McKeon, F.; Jacks, T. Tumor predisposition in mice mutant for p63 and p73: Evidence for broader tumor suppressor functions for the p53 family. Cancer Cell 2005, 7, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Park, B.J.; Lee, S.J.; Kim, J.I.; Lee, S.J.; Lee, C.H.; Chang, S.G.; Park, J.H.; Chi, S.G. Frequent alteration of p63 expression in human primary bladder carcinomas. Cancer Res. 2000, 60, 3370–3374. [Google Scholar] [PubMed]
- Hibi, K.; Nakayama, H.; Taguchi, M.; Kasai, Y.; Ito, K.; Akiyama, S.; Nakao, A. AIS overexpression in advanced esophageal cancer. Clin. Cancer Res. 2001, 7, 469–472. [Google Scholar] [PubMed]
- Pelosi, G.; Pasini, F.; Olsen, S.C.; Pastorino, U.; Maisonneuve, P.; Sonzogni, A.; Maffini, F.; Pruneri, G.; Fraggetta, F.; Cavallon, A.; et al. p63 immunoreactivity in lung cancer: Yet another player in the development of squamous cell carcinomas? J. Pathol. 2002, 198, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.K.; Hsue, S.S.; Lin, L.M. Expression of p63 (TA and ΔN isoforms) in human primary well differentiated buccal carcinomas. Int. J. Oral Maxillofac. Surg. 2004, 33, 493–497. [Google Scholar] [CrossRef] [PubMed]
- Pruneri, G.; Pignataro, L.; Manzotti, M.; Carboni, N.; Ronchetti, D.; Neri, A.; Cesana, B.M.; Viale, G. p63 in laryngeal squamous cell carcinoma: Evidence for a role of TA-p63 down-regulation in tumorigenesis and lack of prognostic implications of p63 immunoreactivity. Lab. Investig. 2002, 82, 1327–1334. [Google Scholar] [CrossRef] [PubMed]
- Park, H.R.; Kin, Y.W.; Park, J.H.; Maeng, Y.H.; Nojima, T.; Hashimoto, H.; Park, Y.K. Low expression of p63 and p73 in osteosarcoma. Tumori 2004, 90, 239–243. [Google Scholar] [PubMed]
- Nylander, K.; Vojtesek, B.; Nenutil, R.; Lindgren, B.; Roos, G.; Zhanxiang, W.; Sjostrom, B.; Dahlgvist, A.; Coates, P.J. Differential expression of p63 isoforms in normal tissues and neoplastic cells. J. Pathol. 2002, 198, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Takahashi, M.; Ozaki, T.; Watanabe, K.K.; Todo, S.; Mizuguchi, H.; Nakagawara, A. Autoinhibitory regulation of p73 by ΔNp73 to modulate cell survival and death through a p73-specific target element within the ΔNp73 promoter. Mol. Cell. Biol. 2002, 22, 2575–2585. [Google Scholar] [CrossRef] [PubMed]
- Stiewe, T.; Theseling, C.C.; Putzer, B.M. Transactivation-deficient ΔTA-p73 inhibits p53 by direct competition for DNA binding: Implications for tumorigenesis. J. Biol. Chem. 2002, 277, 14177–14185. [Google Scholar] [CrossRef] [PubMed]
- Flores, E.R.; Tsai, K.Y.; Crowley, D.; Sengupta, S.; Yang, A.; McKeon, F.; Jacks, T. p63 and p73 are required for p53-dependent apoptosis in response to DNA damage. Nature 2002, 416, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Deyoung, M.P.; Ellisen, L.W. p63 and p73 in human cancer: Defining the network. Oncogene 2007, 26, 5169–5183. [Google Scholar] [CrossRef] [PubMed]
- Beckerman, R.; Prives, C. Transcriptional regulation by p53. Cold Spring Harb. Perspect. Biol. 2010, 2, a000935. [Google Scholar] [CrossRef] [PubMed]
- Perez, C.A.; Ott, J.; Mays, D.J.; Pietenpol, J.A. p63 consensus DNA-binding site: Identification, analysis and application into a p63MH algorithm. Oncogene 2007, 26, 7363–7370. [Google Scholar] [CrossRef] [PubMed]
- Ortt, K.; Sinha, S. Derivation of the consensus DNA-binding sequence for p63 reveals unique requirements that are distinct from p53. FEBS Lett. 2006, 580, 4544–4550. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Chakravarti, D.; Cho, M.S.; Liu, L.; Gi, Y.J.; Lin, Y.L.; Leung, M.L.; El-Naggar, A.; Creighton, C.J.; Suraokar, M.B.; et al. TAp63 suppresses metastasis through coordinate regulation of Dicer and miRNAs. Nature 2010, 467, 986–990. [Google Scholar] [CrossRef] [PubMed]
- Tan, E.H.; Morton, J.P.; Timpson, P.; Tucci, P.; Melino, G.; Flores, E.R.; Sansom, O.J.; Vousden, K.H.; Muller, P.A. Functions of TAp63 and p53 in restraining the development of metastatic cancer. Oncogene 2014, 33, 3325–3333. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.; Anisowicz, A.; Hendrix, M.J.; Thor, A.; Neveu, M.; Sheng, S.; Rafidi, K.; Seftor, E.; Sager, R. Maspin, a serpin with tumor-suppressing activity in human mammary epithelial cells. Science 1994, 263, 526–529. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.; Ageilan, R.I.; Neale, M.; Candi, E.; Salomoni, P.; Knight, R.A.; Croce, C.M.; Melino, G. The E3 ubiquitin ligase Itch controls the protein stability of p63. Proc. Natl. Acad. Sci. USA 2006, 103, 12753–12758. [Google Scholar] [CrossRef] [PubMed]
- Serber, Z.; Lai, H.C.; Yang, A.; Ou, H.D.; Sigal, M.S.; Kelly, A.E.; Darimont, B.D.; Duijf, P.H.; van Bokhoven, H.; McKeon, F.; et al. A C-terminal inhibitory domain controls the activity of p63 by an intramolecular mechanism. Mol. Cell. Biol. 2002, 22, 8601–8611. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, G.B.; Zielonka, E.M.; Coutandin, D.; Weber, T.A.; Schafer, B.; Hannewald, J.; Luh, L.M.; Durst, F.G.; Ibrahim, M.; Hoffmann, J.; et al. DNA damage in oocytes induces a switch of the quality control factor TAp63α from dimer to tetramer. Cell 2011, 144, 566–576. [Google Scholar] [CrossRef] [PubMed]
- Sayan, B.S.; Sayan, A.E.; Yang, A.L.; Ageilan, R.I.; Candi, E.; Cohen, G.M.; Knight, R.A.; Croce, C.M.; Melino, G. Cleavage of the transactivation-inhibitory domain of p63 by caspases enhances apoptosis. Proc. Natl. Acad. Sci. USA 2007, 104, 10871–10876. [Google Scholar] [CrossRef] [PubMed]
- Stindt, M.H.; Muller, P.A.; Ludwig, R.L.; Kehrloesser, S.; Dotsch, V.; Vousden, K.H. Functional interplay between MDM2, p63/p73 and mutant p53. Oncogene 2015, 34, 4300–4310. [Google Scholar] [CrossRef] [PubMed]
- Adorno, M.; Cordenonsi, M.; Montagner, M.; Dupont, S.; Wong, C.; Hann, B.; Solari, A.; Bobisse, S.; Rondina, M.B.; Guzzardo, V.; et al. A Mutant-p53/Smad complex opposes p63 to empower TGFβ-induced metastasis. Cell 2009, 137, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Di Como, C.J.; Gaiddon, C.; Prives, C. p73 function is inhibited by tumor-derived p53 mutants in mammalian cells. Mol. Cell. Biol. 1999, 19, 1438–1449. [Google Scholar] [CrossRef] [PubMed]
- Gonfloni, S.; di Tella, L.; Caldarola, S.; Cannata, S.M.; Klinger, F.G.; di Bartolomeo, C.; Mattei, M.; Candi, E.; de Felici, M.; Melino, G.; et al. Inhibition of the c-Abl-TAp63 pathway protects mouse oocytes from chemotherapy-induced death. Nat. Med. 2009, 15, 1179–1185. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Guo, L.; Rueda, B.R.; Tilly, J.L. Cables1 protects p63 from proteasomal degradation to ensure deletion of cells after genotoxic stress. EMBO Rep. 2010, 11, 633–639. [Google Scholar] [CrossRef] [PubMed]
- MacPartlin, M.; Zeng, S.X.; Lu, H. Phosphorylation and stabilization of TAp63gamma by IκB kinase-β. J. Biol. Chem. 2008, 283, 15754–15761. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Chang, D.L.; Yang, Z.; Qi, J.; Liu, R.; He, H.; Li, D.; Xiao, Z.X. Pin1 modulates p63α protein stability in regulation of cell survival, proliferation and tumor formation. Cell Death Dis. 2013, 4, e943. [Google Scholar] [CrossRef] [PubMed]
- Bernassola, F.; Oberst, A.; Melino, G.; Pandolfi, P.P. The promyelocytic leukaemia protein tumour suppressor functions as a transcriptional regulator of p63. Oncogene 2005, 24, 6982–6986. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Zhang, Y.; Lv, Q.; Liu, B.; Jin, M.; Zhang, W.; He, Q.; Deng, M.; Liu, X.; Li, G.; et al. Toll-like receptor 3 (TLR3) induces apoptosis via death receptors and mitochondria by up-regulating the transactivating p63 isoform α (TAP63α). J. Biol. Chem. 2011, 286, 15918–15928. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, S.; Takenobu, H.; Ozaki, T.; Ando, K.; Koida, N.; Suenaga, Y.; Ichikawa, T.; Hishiki, T.; Chiba, T.; Iwama, A.; et al. Plk1 regulates liver tumor cell death by phosphorylation of TAp63. Oncogene 2009, 28, 3631–3641. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Luong, P.; Hudson, C.; Gudmundsdottir, K.; Basu, S. c-Abl phosphorylation of ΔNp63α is critical for cell viability. Cell Death Dis. 2010, 1, e16. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Sen, T.; Nagpal, J.; Upadhyay, S.; Trink, B.; Ratovitski, E.; Sidransky, D. ATM kinase is a master switch for the ΔNp63α phosphorylation/degradation in human head and neck squamous cell carcinoma cells upon DNA damage. Cell Cycle 2008, 7, 2846–2855. [Google Scholar] [CrossRef] [PubMed]
- Lazzari, C.; Prodosmo, A.; Siepi, F.; Rinaldo, C.; Galli, F.; Gentileschi, M.; Bartolazzi, A.; Costanzo, A.; Sacchi, A.; Guerrini, L.; et al. HIPK2 phosphorylates ΔNp63α and promotes its degradation in response to DNA damage. Oncogene 2011, 30, 4802–4813. [Google Scholar] [CrossRef] [PubMed]
- Hildesheim, J.; Belova, G.I.; Tyner, S.D.; Zhou, X.; Vardanian, L.; Fornance, A.J. Gadd45a regulates matrix metalloproteinases by suppressing ΔNp63α and β-catenin via p38 MAP kinase and APC complex activation. Oncogene 2004, 23, 1829–1837. [Google Scholar] [CrossRef] [PubMed]
- Di Costanzo, A.; Festa, L.; Duverger, O.; Vivo, M.; Guerrini, L.; La Mantia, G.; Morasso, M.I.; Calabro, V. Homeodomain protein Dlx3 induces phosphorylation-dependent p63 degradation. Cell Cycle 2009, 8, 1185–1195. [Google Scholar] [CrossRef] [PubMed]
- Jung, T.; Catalgol, B.; Grune, T. The proteasomal system. Mol. Asp. Med. 2009, 30, 191–296. [Google Scholar] [CrossRef] [PubMed]
- Glickman, M.H.; Ciechanover, A. The ubiquitin-proteasome proteolytic pathway: Destruction for the sake of construction. Physiol. Rev. 2002, 82, 373–428. [Google Scholar] [CrossRef] [PubMed]
- Hochstrasser, M. Lingering mysteries of ubiquitin-chain assembly. Cell 2006, 124, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Wong, B.R.; Parlati, F.; Qu, K.; Demo, S.; Pray, T.; Huang, J.; Payan, D.G.; Bennett, M.K. Drug discovery in the ubiquitin regulatory pathway. Drug Discov. Today 2003, 8, 746–754. [Google Scholar] [CrossRef]
- Metzger, M.B.; Hristova, V.A.; Weissman, A.M. HECT and RING finger families of E3 ubiquitin ligases at a glance. J. Cell Sci. 2012, 125, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Aravind, L.; Koonin, E.V. The U box is a modified RING finger—A common domain in ubiquitination. Curr. Biol. 2000, 10, R132–R134. [Google Scholar] [CrossRef]
- Patterson, C. A new gun in town: The U box is a ubiquitin ligase domain. Sci. STKE. 2002, 116, pe4. [Google Scholar] [CrossRef] [PubMed]
- Sadowski, M.; Suryadinata, R.; Tan, A.R.; Roesley, S.N.; Sarcevic, B. Protein monoubiquitination and polyubiquitination generate structural diversity to control distinct biological processes. IUBMB Life 2012, 64, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, F.; Dikic, I. Atypical ubiquitin chains: New molecular signals. EMBO Rep. 2008, 9, 536–542. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, D.; Riezman, H. Proteasome-independent functions of ubiquitin in endocytosis and signaling. Science 2007, 315, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Thrower, J.S.; Hoffmann, L.; Rechsteiner, M.; Pickart, C.M. Recognition of the polyubiquitin proteolytic signal. EMBO J. 2000, 19, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Duong, D.M.; Seyfried, N.T.; Cheng, D.; Xie, Y.; Robert, J.; Rush, J.; Hochstrasser, M.; Finley, D.; Peng, J. Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation. Cell 2009, 137, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Ohi, M.D.; Vander Kooi, C.W.; Rosenberg, J.A.; Chazin, W.J.; Gould, K.L. Structural insights into the U-box, a domain associated with multi-ubiquitination. Nat. Struct. Biol. 2003, 10, 250–255. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y.; Soda, M.; Hatakeyama, S.; Akagi, T.; Hashikawa, T.; Nakayama, K.I.; Takahashi, R. CHIP is associated with Parkin, a gene responsible for familial Parkinson’s disease, and enhances its ubiquitin ligase activity. Mol. Cell 2002, 10, 55–67. [Google Scholar] [CrossRef]
- Fang, S.; Jensen, J.P.; Ludwig, R.L.; Vousden, K.H.; Weissman, A.M. Mdm2 is a RING finger-dependent ubiquitin protein ligase for itself and p53. J. Biol. Chem. 2000, 275, 8945–8951. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Pomeroy, S.L.; Ferreira, M.; Teider, N.; Mariani, J.; Nakayama, K.I.; Hatakeyama, S.; Tron, V.A.; Saltibus, L.F.; Spyracopoulos, L.; et al. UBE4B promotes Hdm2-mediated degradation of the tumor suppressor p53. Nat. Med. 2011, 17, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Galli, F.; Rossi, M.; D’Alessandra, Y.; de Simone, M.; Lopardo, T.; Haupt, Y.; Alsheich-Bartok, O.; Anzi, S.; Shaulian, E.; Calabro, V.; et al. MDM2 and Fbw7 cooperate to induce p63 protein degradation following DNA damage and cell differentiation. J. Cell Sci. 2010, 123, 2423–2433. [Google Scholar] [CrossRef] [PubMed]
- Kadakia, M.; Slader, C.; Berberich, S.J. Regulation of p63 function by Mdm2 and MdmX. DNA Cell Biol. 2001, 20, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Little, N.A.; Jochemsen, A.G. Hdmx and Mdm2 can repress transcription activation by p53 but not by p63. Oncogene 2001, 20, 4576–4580. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.S.; Ha, J.H.; Lee, D.H.; Ryu, K.S.; Bae, K.H.; Park, B.C.; Park, S.G.; Yi, G.S.; Chi, S.W. Structural convergence of unstructured p53 family transactivation domains in MDM2 recognition. Cell Cycle 2015, 14, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Calabro, V.; Mansueto, G.; Parisi, T.; Vivo, M.; Calogero, R.A.; La Mantia, G. The human MDM2 oncoprotein increases the transcriptional activity and the protein level of the p53 homolog p63. J. Biol. Chem. 2002, 277, 2674–2681. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Arooz, T.; Siu, W.Y.; Chiu, C.H.; Lau, A.; Yamashita, K.; Poon, R.Y. MDM2 and MDMX can interact differently with ARF and members of the p53 family. FEBS Lett. 2001, 490, 202–208. [Google Scholar] [CrossRef]
- Barak, Y.; Juven, T.; Haffner, R.; Oren, M. Mdm2 expression is induced by wild type p53 activity. EMBO J. 1993, 12, 461–468. [Google Scholar] [PubMed]
- Halaby, M.J.; Hakem, R.; Hakem, A. Pirh2: An E3 ligase with central roles in the regulation of cell cycle, DNA damage response, and differentiation. Cell Cycle 2013, 12, 2733–2737. [Google Scholar] [CrossRef] [PubMed]
- Leng, R.P.; Lin, Y.; Ma, W.; Wu, H.; Lemmers, B.; Chung, S.; Parant, J.M.; Lozano, G.; Hakem, R.; Bechimol, S. Pirh2, a p53-induced ubiquitin-protein ligase, promotes p53 degradation. Cell 2003, 112, 779–791. [Google Scholar] [CrossRef]
- Wu, H.; Leng, R.P. MDM2 mediates p73 ubiquitination: A new molecular mechanism for suppression of p73 function. Oncotarget 2015, 6, 21479–21492. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.S.; Qian, Y.; Chen, X. The p73 tumor suppressor is targeted by Pirh2 RING finger E3 ubiquitin ligase for the proteasome-dependent degradation. J. Biol. Chem. 2011, 286, 35388–35395. [Google Scholar] [CrossRef] [PubMed]
- Conforti, F.; Yang, A.L.; Piro, M.C.; Mellone, M.; Terrinoni, A.; Candi, E.; Tucci, P.; Thomas, G.J.; Knight, R.A.; Melino, G.; et al. PIR2/Rnf144B regulates epithelial homeostasis by mediating degradation of p21WAF1 and p63. Oncogene 2013, 32, 4758–4765. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Zeinab, R.A.; Flores, E.R.; Leng, R.P. Pirh2, a ubiquitin E3 ligase, inhibits p73 transcriptional activity by promoting its ubiquitination. Mol. Cancer Res. 2011, 9, 1780–1790. [Google Scholar] [CrossRef] [PubMed]
- Melino, G.; Gallagher, E.; Ageilan, R.I.; Ageilan, R.I.; Knight, R.; Peschiaroli, A.; Rossi, M.; Scialpi, F.; Malatesta, M.; Zoochi, L.; et al. Itch: A HECT-type E3 ligase regulating immunity, skin and cancer. Cell Death Differ. 2008, 15, 1103–1112. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.; de Laurenzi, V.; Munarriz, E.; Green, D.R.; Liu, Y.C.; Vousden, K.H.; Cesareni, G.; Melino, G. The ubiquitin-protein ligase Itch regulates p73 stability. EMBO J. 2005, 24, 836–848. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.; de Simone, M.; Pollice, A.; Santoro, R.; La Mantia, G.; Guerrini, L.; Cababro, V. Itch/AIP4 associates with and promotes p63 protein degradation. Cell Cycle 2006, 5, 1816–1822. [Google Scholar] [CrossRef] [PubMed]
- Melino, G.; Knight, R.A.; Cesareni, G. Degradation of p63 by Itch. Cell Cycle 2006, 5, 1735–1739. [Google Scholar] [PubMed]
- Melino, S.; Bellomaria, A.; Nepravishta, R.; Paci, M.; Melino, G. p63 threonine phosphorylation signals the interaction with the WW domain of the E3 ligase Itch. Cell Cycle 2014, 13, 3207–3217. [Google Scholar] [CrossRef] [PubMed]
- Bakkers, J.; Camacho-Carvajal, M.; Nowak, M.; Kramer, C.; Danger, B.; Hammerschmidt, M. Destabilization of ΔNp63α by Nedd4-mediated ubiquitination and Ubc9-mediated sumoylation, and its implications on dorsoventral patterning of the zebrafish embryo. Cell Cycle 2005, 4, 790–800. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhou, Z.; Chen, C. WW domain-containing E3 ubiquitin protein ligase 1 targets p63 transcription factor for ubiquitin-mediated proteasomal degradation and regulates apoptosis. Cell Death Differ. 2008, 15, 1941–1951. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Armstrong, S.R.; Wu, H.; Wang, B.; Abuetabh, Y.; Sergi, C.; Leng, R.P. The Regulation of Tumor Suppressor p63 by the Ubiquitin-Proteasome System. Int. J. Mol. Sci. 2016, 17, 2041. https://doi.org/10.3390/ijms17122041
Armstrong SR, Wu H, Wang B, Abuetabh Y, Sergi C, Leng RP. The Regulation of Tumor Suppressor p63 by the Ubiquitin-Proteasome System. International Journal of Molecular Sciences. 2016; 17(12):2041. https://doi.org/10.3390/ijms17122041
Chicago/Turabian StyleArmstrong, Stephen R., Hong Wu, Benfan Wang, Yasser Abuetabh, Consolato Sergi, and Roger P. Leng. 2016. "The Regulation of Tumor Suppressor p63 by the Ubiquitin-Proteasome System" International Journal of Molecular Sciences 17, no. 12: 2041. https://doi.org/10.3390/ijms17122041
APA StyleArmstrong, S. R., Wu, H., Wang, B., Abuetabh, Y., Sergi, C., & Leng, R. P. (2016). The Regulation of Tumor Suppressor p63 by the Ubiquitin-Proteasome System. International Journal of Molecular Sciences, 17(12), 2041. https://doi.org/10.3390/ijms17122041