
Update on Inflammatory Biomarkers and Treatments in Ischemic Stroke

 , ,
, , 
Abstract
:
1. Introduction
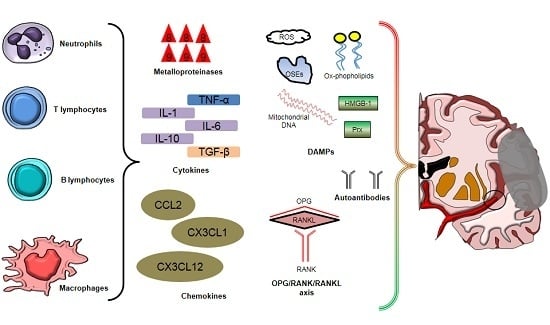
2. Inflammatory Cells Involved in Post-Ischemic Brain Injury and Repair
2.1. Neutrophils
2.2. Microglial Cells and Circulating Monocytes/Macrophages
2.3. T and B Lymphocytes
3. Soluble Mediators of Post-Ischemic Brain Injury
3.1. Cytokines
3.2. Chemokines
3.3. Reactive Oxygen Species
3.4. Damage-Associated Molecular Patterns (DAMPs)
3.5. Autoantibodies
3.6. Miscellaneous: Osteoprotegerin, Adipokines, and Osteopontin
4. Inflammatory Mediators as Potential Diagnostic or Prognostic Biomarkers
4.1. Clinical Evidence
4.2. New Candidate Inflammatory Biomarkers
5. Anti-Inflammatory Treatments in IS: Evidence from Pre-Clinical Studies
5.1. IL-1Ra
5.2. Statins
5.3. Fingolimod (FTY720)
5.4. Donepezil
5.5. Citalopram
5.6. Natalizumab (Anti-CD49d Antibody)
5.7. Cyclosporine A
5.8. Edaravone (MCI-186)
6. Anti-Inflammatory Treatments in IS: Evidence from Clinical Trials
6.1. IL1-Ra
6.2. Statins
6.3. Fingolimod (FTY720)
6.4. Donepezil
6.5. Citalopram
6.6. Natalizumab (Anti-CD49d Antibody)
6.7. Cyclosporine A
6.8. Edaravone (MCI-186)
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Warlow, C.; Sudlow, C.; Dennis, M.; Wardlaw, J.; Sandercock, P. Stroke. Lancet 2003, 362, 1211–1224. [Google Scholar] [CrossRef]
- Writing Group Members; Lloyd-Jones, D.; Adams, R.J.; Brown, T.M.; Carnethon, M.; Dai, S.; de Simone, G.; Ferguson, T.B.; Ford, E.; Furie, K.; et al. Heart disease and stroke statistics—2010 Update: A report from the american heart association. Circulation 2010, 121, e46–e215. [Google Scholar] [PubMed]
- Jin, R.; Yang, G.; Li, G. Inflammatory mechanisms in ischemic stroke: Role of inflammatory cells. J. Leukoc. Biol. 2010, 87, 779–789. [Google Scholar] [CrossRef] [PubMed]
- Petrovic-Djergovic, D.; Goonewardena, S.N.; Pinsky, D.J. Inflammatory disequilibrium in stroke. Circ. Res. 2016, 119, 142–158. [Google Scholar] [CrossRef] [PubMed]
- Yellon, D.M.; Hausenloy, D.J. Myocardial reperfusion injury. N. Engl. J. Med. 2007, 357, 1121–1135. [Google Scholar] [CrossRef] [PubMed]
- Eltzschig, H.K.; Eckle, T. Ischemia and reperfusion—From mechanism to translation. Nat. Med. 2011, 17, 1391–1401. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.Y.; Nunez, G. Sterile inflammation: Sensing and reacting to damage. Nat. Rev. Immunol. 2010, 10, 826–837. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Song, T.J.; Park, J.H.; Lee, H.S.; Nam, C.M.; Nam, H.S.; Kim, Y.D.; Heo, J.H. Different prognostic value of white blood cell subtypes in patients with acute cerebral infarction. Atherosclerosis 2012, 222, 464–467. [Google Scholar] [CrossRef] [PubMed]
- Tsai, N.W.; Chang, W.N.; Shaw, C.F.; Jan, C.R.; Lu, C.H. Leucocyte apoptosis in patients with acute ischaemic stroke. Clin. Exp. Pharmacol. Physiol. 2010, 37, 884–888. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.H.; Liu, K.F.; Bree, M.P. Effects of CD11b/18 monoclonal antibody on rats with permanent middle cerebral artery occlusion. Am. J. Pathol. 1996, 148, 241–248. [Google Scholar] [PubMed]
- Lopes Pinheiro, M.A.; Kooij, G.; Mizee, M.R.; Kamermans, A.; Enzmann, G.; Lyck, R.; Schwaninger, M.; Engelhardt, B.; de Vries, H.E. Immune cell trafficking across the barriers of the central nervous system in multiple sclerosis and stroke. Biochim. Biophys. Acta 2016, 1862, 461–471. [Google Scholar] [CrossRef] [PubMed]
- Perez-de-Puig, I.; Miro-Mur, F.; Ferrer-Ferrer, M.; Gelpi, E.; Pedragosa, J.; Justicia, C.; Urra, X.; Chamorro, A.; Planas, A.M. Neutrophil recruitment to the brain in mouse and human ischemic stroke. Acta Neuropathol. 2015, 129, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Enzmann, G.; Mysiorek, C.; Gorina, R.; Cheng, Y.J.; Ghavampour, S.; Hannocks, M.J.; Prinz, V.; Dirnagl, U.; Endres, M.; Prinz, M.; et al. The neurovascular unit as a selective barrier to polymorphonuclear granulocyte (PMN) infiltration into the brain after ischemic injury. Acta Neuropathol. 2013, 125, 395–412. [Google Scholar] [CrossRef] [PubMed]
- Taichman, N.S.; Young, S.; Cruchley, A.T.; Taylor, P.; Paleolog, E. Human neutrophils secrete vascular endothelial growth factor. J. Leukoc. Biol. 1997, 62, 397–400. [Google Scholar] [PubMed]
- Christoffersson, G.; Vagesjo, E.; Vandooren, J.; Liden, M.; Massena, S.; Reinert, R.B.; Brissova, M.; Powers, A.C.; Opdenakker, G.; Phillipson, M. VEGF-A recruits a proangiogenic MMP-9-delivering neutrophil subset that induces angiogenesis in transplanted hypoxic tissue. Blood 2012, 120, 4653–4662. [Google Scholar] [CrossRef] [PubMed]
- Cauwe, B.; Martens, E.; Proost, P.; Opdenakker, G. Multidimensional degradomics identifies systemic autoantigens and intracellular matrix proteins as novel gelatinase B/MMP-9 substrates. Integr. Biol. 2009, 1, 404–426. [Google Scholar] [CrossRef] [PubMed]
- Segel, G.B.; Halterman, M.W.; Lichtman, M.A. The paradox of the neutrophil’s role in tissue injury. J. Leukoc. Biol. 2011, 89, 359–372. [Google Scholar] [CrossRef] [PubMed]
- Akopov, S.E.; Simonian, N.A.; Grigorian, G.S. Dynamics of polymorphonuclear leukocyte accumulation in acute cerebral infarction and their correlation with brain tissue damage. Stroke 1996, 27, 1739–1743. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, L.A.; Grisham, M.B.; Twohig, B.; Arfors, K.E.; Harlan, J.M.; Granger, D.N. Role of neutrophils in ischemia-reperfusion-induced microvascular injury. Am. J. Physiol. 1987, 253, H699–H703. [Google Scholar] [PubMed]
- Rorvig, S.; Honore, C.; Larsson, L.I.; Ohlsson, S.; Pedersen, C.C.; Jacobsen, L.C.; Cowland, J.B.; Garred, P.; Borregaard, N. Ficolin-1 is present in a highly mobilizable subset of human neutrophil granules and associates with the cell surface after stimulation with fmlp. J. Leukoc. Biol. 2009, 86, 1439–1449. [Google Scholar] [CrossRef] [PubMed]
- Jickling, G.C.; Liu, D.; Ander, B.P.; Stamova, B.; Zhan, X.; Sharp, F.R. Targeting neutrophils in ischemic stroke: Translational insights from experimental studies. J. Cereb. Blood Flow Metab. 2015, 35, 888–901. [Google Scholar] [CrossRef] [PubMed]
- Davalos, D.; Grutzendler, J.; Yang, G.; Kim, J.V.; Zuo, Y.; Jung, S.; Littman, D.R.; Dustin, M.L.; Gan, W.B. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 2005, 8, 752–758. [Google Scholar] [CrossRef] [PubMed]
- Prinz, M.; Priller, J. Microglia and brain macrophages in the molecular age: From origin to neuropsychiatric disease. Nat. Rev. Neurosci. 2014, 15, 300–312. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Fan, Y.; Won, S.J.; Neumann, M.; Hu, D.; Zhou, L.; Weinstein, P.R.; Liu, J. Chronic treatment with minocycline preserves adult new neurons and reduces functional impairment after focal cerebral ischemia. Stroke 2007, 38, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Hoehn, B.D.; Palmer, T.D.; Steinberg, G.K. Neurogenesis in rats after focal cerebral ischemia is enhanced by indomethacin. Stroke 2005, 36, 2718–2724. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.J.; Kim, M.J.; Park, J.M.; Lee, S.H.; Kim, Y.J.; Ryu, S.; Kim, Y.H.; Yoon, B.W. Reduced neurogenesis after suppressed inflammation by minocycline in transient cerebral ischemia in rat. J. Neurol. Sci. 2009, 279, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Faustino, J.V.; Wang, X.; Johnson, C.E.; Klibanov, A.; Derugin, N.; Wendland, M.F.; Vexler, Z.S. Microglial cells contribute to endogenous brain defenses after acute neonatal focal stroke. J. Neurosci. 2011, 31, 12992–13001. [Google Scholar] [CrossRef] [PubMed]
- Breckwoldt, M.O.; Chen, J.W.; Stangenberg, L.; Aikawa, E.; Rodriguez, E.; Qiu, S.; Moskowitz, M.A.; Weissleder, R. Tracking the inflammatory response in stroke in vivo by sensing the enzyme myeloperoxidase. Proc. Natl. Acad. Sci. USA 2008, 105, 18584–18589. [Google Scholar] [CrossRef] [PubMed]
- Kaito, M.; Araya, S.; Gondo, Y.; Fujita, M.; Minato, N.; Nakanishi, M.; Matsui, M. Relevance of distinct monocyte subsets to clinical course of ischemic stroke patients. PLoS ONE 2013, 8, e69409. [Google Scholar] [CrossRef] [PubMed]
- Swirski, F.K.; Nahrendorf, M.; Etzrodt, M.; Wildgruber, M.; Cortez-Retamozo, V.; Panizzi, P.; Figueiredo, J.L.; Kohler, R.H.; Chudnovskiy, A.; Waterman, P.; et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science 2009, 325, 612–616. [Google Scholar] [CrossRef] [PubMed]
- Offner, H.; Subramanian, S.; Parker, S.M.; Wang, C.; Afentoulis, M.E.; Lewis, A.; Vandenbark, A.A.; Hurn, P.D. Splenic atrophy in experimental stroke is accompanied by increased regulatory T cells and circulating macrophages. J. Immunol. 2006, 176, 6523–6531. [Google Scholar] [CrossRef] [PubMed]
- Vendrame, M.; Gemma, C.; Pennypacker, K.R.; Bickford, P.C.; Davis Sanberg, C.; Sanberg, P.R.; Willing, A.E. Cord blood rescues stroke-induced changes in splenocyte phenotype and function. Exp. Neurol. 2006, 199, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Ajmo, C.T., Jr.; Vernon, D.O.; Collier, L.; Hall, A.A.; Garbuzova-Davis, S.; Willing, A.; Pennypacker, K.R. The spleen contributes to stroke-induced neurodegeneration. J. Neurosci. Res 2008, 86, 2227–2234. [Google Scholar] [CrossRef] [PubMed]
- Dotson, A.L.; Wang, J.; Saugstad, J.; Murphy, S.J.; Offner, H. Splenectomy reduces infarct volume and neuroinflammation in male but not female mice in experimental stroke. J. Neuroimmunol. 2015, 278, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Ostrowski, R.P.; Schulte, R.W.; Nie, Y.; Ling, T.; Lee, T.; Manaenko, A.; Gridley, D.S.; Zhang, J.H. Acute splenic irradiation reduces brain injury in the rat focal ischemic stroke model. Transl. Stroke Res. 2012, 3, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Yang, J.; Beltran, C.D.; Cho, S. Role of spleen-derived monocytes/macrophages in acute ischemic brain injury. J. Cereb. Blood Flow Metab. 2014, 34, 1411–1419. [Google Scholar] [CrossRef] [PubMed]
- Perego, C.; Fumagalli, S.; de Simoni, M.G. Temporal pattern of expression and colocalization of microglia/macrophage phenotype markers following brain ischemic injury in mice. J. Neuroinflamm. 2011, 8, 174. [Google Scholar] [CrossRef] [PubMed]
- Gliem, M.; Mausberg, A.K.; Lee, J.I.; Simiantonakis, I.; van Rooijen, N.; Hartung, H.P.; Jander, S. Macrophages prevent hemorrhagic infarct transformation in murine stroke models. Ann. Neurol. 2012, 71, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.X.; Broughton, B.R.; Kim, H.A.; Lee, S.; Drummond, G.R.; Sobey, C.G. Evidence that Ly6Chi monocytes are protective in acute ischemic stroke by promoting M2 macrophage polarization. Stroke 2015, 46, 1929–1937. [Google Scholar] [CrossRef] [PubMed]
- Kleinschnitz, C.; Schwab, N.; Kraft, P.; Hagedorn, I.; Dreykluft, A.; Schwarz, T.; Austinat, M.; Nieswandt, B.; Wiendl, H.; Stoll, G. Early detrimental T-cell effects in experimental cerebral ischemia are neither related to adaptive immunity nor thrombus formation. Blood 2010, 115, 3835–3842. [Google Scholar] [CrossRef] [PubMed]
- Becker, K.; Kindrick, D.; McCarron, R.; Hallenbeck, J.; Winn, R. Adoptive transfer of myelin basic protein-tolerized splenocytes to naive animals reduces infarct size: A role for lymphocytes in ischemic brain injury? Stroke 2003, 34, 1809–1815. [Google Scholar] [CrossRef] [PubMed]
- Schroeter, M.; Jander, S.; Witte, O.W.; Stoll, G. Local immune responses in the rat cerebral cortex after middle cerebral artery occlusion. J. Neuroimmunol. 1994, 55, 195–203. [Google Scholar] [CrossRef]
- Yilmaz, G.; Arumugam, T.V.; Stokes, K.Y.; Granger, D.N. Role of T lymphocytes and interferon-γ in ischemic stroke. Circulation 2006, 113, 2105–2112. [Google Scholar] [CrossRef] [PubMed]
- Shichita, T.; Sugiyama, Y.; Ooboshi, H.; Sugimori, H.; Nakagawa, R.; Takada, I.; Iwaki, T.; Okada, Y.; Iida, M.; Cua, D.J.; et al. Pivotal role of cerebral interleukin-17-producing gammadeltat cells in the delayed phase of ischemic brain injury. Nat. Med. 2009, 15, 946–950. [Google Scholar] [CrossRef] [PubMed]
- Li, G.Z.; Zhong, D.; Yang, L.M.; Sun, B.; Zhong, Z.H.; Yin, Y.H.; Cheng, J.; Yan, B.B.; Li, H.L. Expression of interleukin-17 in ischemic brain tissue. Scand. J. Immunol. 2005, 62, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Liesz, A.; Suri-Payer, E.; Veltkamp, C.; Doerr, H.; Sommer, C.; Rivest, S.; Giese, T.; Veltkamp, R. Regulatory T cells are key cerebroprotective immunomodulators in acute experimental stroke. Nat. Med. 2009, 15, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Akiyoshi, K.; Dziennis, S.; Vandenbark, A.A.; Herson, P.S.; Hurn, P.D.; Offner, H. Regulatory B cells limit cns inflammation and neurologic deficits in murine experimental stroke. J. Neurosci. 2011, 31, 8556–8563. [Google Scholar] [CrossRef] [PubMed]
- Lambertsen, K.L.; Biber, K.; Finsen, B. Inflammatory cytokines in experimental and human stroke. J. Cereb. Blood Flow Metab. 2012, 32, 1677–1698. [Google Scholar] [CrossRef] [PubMed]
- Lambertsen, K.L.; Meldgaard, M.; Ladeby, R.; Finsen, B. A quantitative study of microglial-macrophage synthesis of tumor necrosis factor during acute and late focal cerebral ischemia in mice. J. Cereb. Blood Flow Metab. 2005, 25, 119–135. [Google Scholar] [CrossRef] [PubMed]
- Clausen, B.H.; Lambertsen, K.L.; Meldgaard, M.; Finsen, B. A quantitative in situ hybridization and polymerase chain reaction study of microglial-macrophage expression of interleukin-1β mRNA following permanent middle cerebral artery occlusion in mice. Neuroscience 2005, 132, 879–892. [Google Scholar] [CrossRef] [PubMed]
- Lambertsen, K.L.; Clausen, B.H.; Babcock, A.A.; Gregersen, R.; Fenger, C.; Nielsen, H.H.; Haugaard, L.S.; Wirenfeldt, M.; Nielsen, M.; Dagnaes-Hansen, F.; et al. Microglia protect neurons against ischemia by synthesis of tumor necrosis factor. J. Neurosci. 2009, 29, 1319–1330. [Google Scholar] [CrossRef] [PubMed]
- Murray, K.N.; Parry-Jones, A.R.; Allan, S.M. Interleukin-1 and acute brain injury. Front. Cell. Neurosci. 2015, 9, 18. [Google Scholar] [CrossRef] [PubMed]
- Luheshi, N.M.; Kovacs, K.J.; Lopez-Castejon, G.; Brough, D.; Denes, A. Interleukin-1α expression precedes IL-1β after ischemic brain injury and is localised to areas of focal neuronal loss and penumbral tissues. J. Neuroinflamm. 2011, 8, 186. [Google Scholar] [CrossRef] [PubMed]
- Herx, L.M.; Yong, V.W. Interleukin-1β is required for the early evolution of reactive astrogliosis following CNS lesion. J. Neuropathol. Exp. Neurol. 2001, 60, 961–971. [Google Scholar] [CrossRef] [PubMed]
- Thornton, P.; Pinteaux, E.; Allan, S.M.; Rothwell, N.J. Matrix metalloproteinase-9 and urokinase plasminogen activator mediate interleukin-1-induced neurotoxicity. Mol. Cell. Neurosci. 2008, 37, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.; Thornton, P.; Denes, A.; McColl, B.W.; Pierozynski, A.; Monestier, M.; Pinteaux, E.; Rothwell, N.J.; Allan, S.M. Neutrophil cerebrovascular transmigration triggers rapid neurotoxicity through release of proteases associated with decondensed DNA. J. Immunol. 2012, 189, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Amantea, D.; Micieli, G.; Tassorelli, C.; Cuartero, M.I.; Ballesteros, I.; Certo, M.; Moro, M.A.; Lizasoain, I.; Bagetta, G. Rational modulation of the innate immune system for neuroprotection in ischemic stroke. Front. Neurosci. 2015, 9, 147. [Google Scholar] [CrossRef] [PubMed]
- Grilli, M.; Barbieri, I.; Basudev, H.; Brusa, R.; Casati, C.; Lozza, G.; Ongini, E. Interleukin-10 modulates neuronal threshold of vulnerability to ischaemic damage. Eur. J. Neurosci. 2000, 12, 2265–2272. [Google Scholar] [CrossRef] [PubMed]
- Frenkel, D.; Huang, Z.; Maron, R.; Koldzic, D.N.; Moskowitz, M.A.; Weiner, H.L. Neuroprotection by IL-10-producing MOG CD4+ T cells following ischemic stroke. J. Neurol. Sci. 2005, 233, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Greer, J.M.; McCombe, P.A. Prolonged elevation of cytokine levels after human acute ischaemic stroke with evidence of individual variability. J. Neuroimmunol. 2012, 246, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Doyle, K.P.; Cekanaviciute, E.; Mamer, L.E.; Buckwalter, M.S. TGFβ signaling in the brain increases with aging and signals to astrocytes and innate immune cells in the weeks after stroke. J. Neuroinflamm. 2010, 7, 62. [Google Scholar] [CrossRef] [PubMed]
- Zaremba, J.; Losy, J. Early TNF-α levels correlate with ischaemic stroke severity. Acta Neurol. Scand. 2001, 104, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Beridze, M.; Sanikidze, T.; Shakarishvili, R.; Intskirveli, N.; Bornstein, N.M. Selected acute phase CSF factors in ischemic stroke: Findings and prognostic value. BMC Neurol. 2011, 11, 41. [Google Scholar] [CrossRef] [PubMed]
- Waje-Andreassen, U.; Krakenes, J.; Ulvestad, E.; Thomassen, L.; Myhr, K.M.; Aarseth, J.; Vedeler, C.A. IL-6: An early marker for outcome in acute ischemic stroke. Acta Neurol. Scand. 2005, 111, 360–365. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Li, Y.; Tang, Y.; Tang, G.; Yang, G.Y.; Wang, Y. CXCR4 antagonist AMD3100 protects blood-brain barrier integrity and reduces inflammatory response after focal ischemia in mice. Stroke 2013, 44, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Ruscher, K.; Kuric, E.; Liu, Y.; Walter, H.L.; Issazadeh-Navikas, S.; Englund, E.; Wieloch, T. Inhibition of CXCL12 signaling attenuates the postischemic immune response and improves functional recovery after stroke. J. Cereb. Blood Flow Metab. 2013, 33, 1225–1234. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Huang, J.; Li, Y.; Yang, G.Y. Roles of chemokine CXCL12 and its receptors in ischemic stroke. Curr. Drug Targets 2012, 13, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Shyu, W.C.; Lin, S.Z.; Yen, P.S.; Su, C.Y.; Chen, D.C.; Wang, H.J.; Li, H. Stromal cell-derived factor-1α promotes neuroprotection, angiogenesis, and mobilization/homing of bone marrow-derived cells in stroke rats. J. Pharmacol. Exp. Ther. 2008, 324, 834–849. [Google Scholar] [CrossRef] [PubMed]
- Denes, A.; Ferenczi, S.; Halasz, J.; Kornyei, Z.; Kovacs, K.J. Role of CX3CR1 (fractalkine receptor) in brain damage and inflammation induced by focal cerebral ischemia in mouse. J. Cereb. Blood Flow Metab. 2008, 28, 1707–1721. [Google Scholar] [CrossRef] [PubMed]
- Cipriani, R.; Villa, P.; Chece, G.; Lauro, C.; Paladini, A.; Micotti, E.; Perego, C.; de Simoni, M.G.; Fredholm, B.B.; Eusebi, F.; et al. CX3CL1 is neuroprotective in permanent focal cerebral ischemia in rodents. J. Neurosci. 2011, 31, 16327–16335. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Gan, Y.; Liu, Q.; Yin, J.X.; Liu, Q.; Shi, J.; Shi, F.D. CX3CR1 deficiency suppresses activation and neurotoxicity of microglia/macrophage in experimental ischemic stroke. J. Neuroinflamm. 2014, 11, 26. [Google Scholar] [CrossRef] [PubMed]
- Donohue, M.M.; Cain, K.; Zierath, D.; Shibata, D.; Tanzi, P.M.; Becker, K.J. Higher plasma fractalkine is associated with better 6-month outcome from ischemic stroke. Stroke 2012, 43, 2300–2306. [Google Scholar] [CrossRef] [PubMed]
- Rosito, M.; Lauro, C.; Chece, G.; Porzia, A.; Monaco, L.; Mainiero, F.; Catalano, M.; Limatola, C.; Trettel, F. Trasmembrane chemokines CX3CL1 and CXCL16 drive interplay between neurons, microglia and astrocytes to counteract pMCAO and excitotoxic neuronal death. Front. Cell. Neurosci. 2014, 8, 193. [Google Scholar] [CrossRef] [PubMed]
- Schilling, M.; Strecker, J.K.; Ringelstein, E.B.; Schabitz, W.R.; Kiefer, R. The role of cc chemokine receptor 2 on microglia activation and blood-borne cell recruitment after transient focal cerebral ischemia in mice. Brain Res. 2009, 1289, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Che, X.; Ye, W.; Panga, L.; Wu, D.C.; Yang, G.Y. Monocyte chemoattractant protein-1 expressed in neurons and astrocytes during focal ischemia in mice. Brain Res. 2001, 902, 171–177. [Google Scholar] [CrossRef]
- Strecker, J.K.; Minnerup, J.; Schutte-Nutgen, K.; Gess, B.; Schabitz, W.R.; Schilling, M. Monocyte chemoattractant protein-1-deficiency results in altered blood-brain barrier breakdown after experimental stroke. Stroke 2013, 44, 2536–2544. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.P.; Sailor, K.A.; Lang, B.T.; Park, S.W.; Vemuganti, R.; Dempsey, R.J. Monocyte chemoattractant protein-1 plays a critical role in neuroblast migration after focal cerebral ischemia. J. Cereb. Blood Flow Metab. 2007, 27, 1213–1224. [Google Scholar] [CrossRef] [PubMed]
- Strecker, J.K.; Minnerup, J.; Gess, B.; Ringelstein, E.B.; Schabitz, W.R.; Schilling, M. Monocyte chemoattractant protein-1-deficiency impairs the expression of IL-6, IL-1β and G-CSF after transient focal ischemia in mice. PLoS ONE 2011, 6, e25863. [Google Scholar] [CrossRef] [PubMed]
- Schuette-Nuetgen, K.; Strecker, J.K.; Minnerup, J.; Ringelstein, E.B.; Schilling, M. MCP-1/CCR-2-double-deficiency severely impairs the migration of hematogenous inflammatory cells following transient cerebral ischemia in mice. Exp. Neurol. 2012, 233, 849–858. [Google Scholar] [CrossRef] [PubMed]
- Carbone, F.; Teixeira, P.C.; Braunersreuther, V.; Mach, F.; Vuilleumier, N.; Montecucco, F. Pathophysiology and treatments of oxidative injury in ischemic stroke: Focus on the phagocytic NADPH oxidase 2. Antioxid. Redox Signal. 2015, 23, 460–489. [Google Scholar] [CrossRef] [PubMed]
- Lambeth, J.D. Nox enzymes and the biology of reactive oxygen. Nat. Rev. Immunol. 2004, 4, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Tang, X.N.; Yenari, M.A. The inflammatory response in stroke. J. Neuroimmunol. 2007, 184, 53–68. [Google Scholar] [CrossRef] [PubMed]
- Vallet, P.; Charnay, Y.; Steger, K.; Ogier-Denis, E.; Kovari, E.; Herrmann, F.; Michel, J.P.; Szanto, I. Neuronal expression of the NADPH oxidase NOX4, and its regulation in mouse experimental brain ischemia. Neuroscience 2005, 132, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Drummond, G.R.; Sobey, C.G. Endothelial NADPH oxidases: Which NOX to target in vascular disease? Trends Endocrinol. Metab. 2014, 25, 452–463. [Google Scholar] [CrossRef] [PubMed]
- Douglas, G.; Bendall, J.K.; Crabtree, M.J.; Tatham, A.L.; Carter, E.E.; Hale, A.B.; Channon, K.M. Endothelial-specific NOX2 overexpression increases vascular superoxide and macrophage recruitment in ApoE−/− mice. Cardiovasc. Res. 2012, 94, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Ray, R.; Murdoch, C.E.; Wang, M.; Santos, C.X.; Zhang, M.; Alom-Ruiz, S.; Anilkumar, N.; Ouattara, A.; Cave, A.C.; Walker, S.J.; et al. Endothelial NOX4 NADPH oxidase enhances vasodilatation and reduces blood pressure in vivo. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1368–1376. [Google Scholar] [CrossRef] [PubMed]
- Walder, C.E.; Green, S.P.; Darbonne, W.C.; Mathias, J.; Rae, J.; Dinauer, M.C.; Curnutte, J.T.; Thomas, G.R. Ischemic stroke injury is reduced in mice lacking a functional NADPH oxidase. Stroke 1997, 28, 2252–2258. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.R.; Witting, P.K.; Drummond, G.R. Redox control of endothelial function and dysfunction: Molecular mechanisms and therapeutic opportunities. Antioxid. Redox Signal. 2008, 10, 1713–1765. [Google Scholar] [CrossRef] [PubMed]
- Craige, S.M.; Chen, K.; Pei, Y.; Li, C.; Huang, X.; Chen, C.; Shibata, R.; Sato, K.; Walsh, K.; Keaney, J.F., Jr. NADPH oxidase 4 promotes endothelial angiogenesis through endothelial nitric oxide synthase activation. Circulation 2011, 124, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Kleinschnitz, C.; Grund, H.; Wingler, K.; Armitage, M.E.; Jones, E.; Mittal, M.; Barit, D.; Schwarz, T.; Geis, C.; Kraft, P.; et al. Post-stroke inhibition of induced NADPH oxidase type 4 prevents oxidative stress and neurodegeneration. PLoS Biol. 2010, 8, e1000479. [Google Scholar] [CrossRef] [PubMed]
- Lakhan, S.E.; Kirchgessner, A.; Hofer, M. Inflammatory mechanisms in ischemic stroke: Therapeutic approaches. J. Transl. Med. 2009, 7, 97. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.J.; Xie, Y.; Bosco, G.M.; Chen, C.; Camporesi, E.M. Hyperbaric oxygenation alleviates MCAO-induced brain injury and reduces hydroxyl radical formation and glutamate release. Eur. J. Appl. Physiol. 2010, 108, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Cheret, C.; Gervais, A.; Lelli, A.; Colin, C.; Amar, L.; Ravassard, P.; Mallet, J.; Cumano, A.; Krause, K.H.; Mallat, M. Neurotoxic activation of microglia is promoted by a NOX1-dependent NADPH oxidase. J. Neurosci. 2008, 28, 12039–12051. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Cho, I.H.; Kim, J.E.; Shin, Y.J.; Jeon, J.H.; Kim, Y.; Yang, Y.M.; Lee, K.H.; Lee, J.W.; Lee, W.J.; et al. Ethyl pyruvate has an anti-inflammatory effect by inhibiting ROS-dependent STAT signaling in activated microglia. Free Radic. Biol. Med. 2008, 45, 950–963. [Google Scholar] [CrossRef] [PubMed]
- Kaspar, J.W.; Niture, S.K.; Jaiswal, A.K. Nrf2:INrf2 (Keap1) signaling in oxidative stress. Free Radic. Biol. Med. 2009, 47, 1304–1309. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, M.T.; Leaver, H.A. Brain endothelial cell death: Modes, signaling pathways, and relevance to neural development, homeostasis, and disease. Mol. Neurobiol. 2010, 42, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Liesz, A.; Dalpke, A.; Mracsko, E.; Antoine, D.J.; Roth, S.; Zhou, W.; Yang, H.; Na, S.Y.; Akhisaroglu, M.; Fleming, T.; et al. DAMP signaling is a key pathway inducing immune modulation after brain injury. J. Neurosci. 2015, 35, 583–598. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial damps cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Maeda, A.; Fadeel, B. Mitochondria released by cells undergoing TNF-α-induced necroptosis act as danger signals. Cell Death Dis. 2014, 5, e1312. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Hyakkoku, K.; Hamanaka, J.; Tsuruma, K.; Shimazawa, M.; Tanaka, H.; Uematsu, S.; Akira, S.; Inagaki, N.; Nagai, H.; Hara, H. Toll-like receptor 4 (TLR4), but not TLR3 or TLR9, knock-out mice have neuroprotective effects against focal cerebral ischemia. Neuroscience 2010, 171, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Ceruti, S.; Villa, G.; Genovese, T.; Mazzon, E.; Longhi, R.; Rosa, P.; Bramanti, P.; Cuzzocrea, S.; Abbracchio, M.P. The P2Y-like receptor GPR17 as a sensor of damage and a new potential target in spinal cord injury. Brain 2009, 132, 2206–2218. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Wang, Z.; Wei, X.; Han, H.; Meng, X.; Zhang, Y.; Shi, W.; Li, F.; Xin, T.; Pang, Q.; et al. NLRP3 deficiency ameliorates neurovascular damage in experimental ischemic stroke. J. Cereb. Blood Flow Metab. 2014, 34, 660–667. [Google Scholar] [CrossRef] [PubMed]
- Weismann, D.; Binder, C.J. The innate immune response to products of phospholipid peroxidation. Biochim. Biophys. Acta 2012, 1818, 2465–2475. [Google Scholar] [CrossRef] [PubMed]
- Abe, T.; Shimamura, M.; Jackman, K.; Kurinami, H.; Anrather, J.; Zhou, P.; Iadecola, C. Key role of CD36 in toll-like receptor 2 signaling in cerebral ischemia. Stroke 2010, 41, 898–904. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Zhai, Y.; Liang, S.; Mori, Y.; Han, R.; Sutterwala, F.S.; Qiao, L. TRPM2 links oxidative stress to NLRP3 inflammasome activation. Nat. Commun. 2013, 4, 1611. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.S.; He, Q.; Janczy, J.R.; Elliott, E.I.; Zhong, Z.; Olivier, A.K.; Sadler, J.J.; Knepper-Adrian, V.; Han, R.; Qiao, L.; et al. Mitochondrial cardiolipin is required for NLRP3 inflammasome activation. Immunity 2013, 39, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Lotze, M.T.; Tracey, K.J. High-mobility group box 1 protein (HMGB1): Nuclear weapon in the immune arsenal. Nat. Rev. Immunol. 2005, 5, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Rouhiainen, A.; Kuja-Panula, J.; Wilkman, E.; Pakkanen, J.; Stenfors, J.; Tuominen, R.K.; Lepantalo, M.; Carpen, O.; Parkkinen, J.; Rauvala, H. Regulation of monocyte migration by amphoterin (HMGB1). Blood 2004, 104, 1174–1182. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Liu, B.; Zhao, Q.; Jin, P.; Hua, F.; Zhang, Z.; Liu, Y.; Zan, K.; Cui, G.; Ye, X. Bone marrow stromal cells inhibits HMGB1-mediated inflammation after stroke in type 2 diabetic rats. Neuroscience 2016, 324, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Shichita, T.; Hasegawa, E.; Kimura, A.; Morita, R.; Sakaguchi, R.; Takada, I.; Sekiya, T.; Ooboshi, H.; Kitazono, T.; Yanagawa, T.; et al. Peroxiredoxin family proteins are key initiators of post-ischemic inflammation in the brain. Nat. Med. 2012, 18, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Loser, K.; Vogl, T.; Voskort, M.; Lueken, A.; Kupas, V.; Nacken, W.; Klenner, L.; Kuhn, A.; Foell, D.; Sorokin, L.; et al. The Toll-like receptor 4 ligands Mrp8 and Mrp14 are crucial in the development of autoreactive CD8+ T cells. Nat. Med. 2010, 16, 713–717. [Google Scholar] [CrossRef] [PubMed]
- Qiang, X.; Yang, W.L.; Wu, R.; Zhou, M.; Jacob, A.; Dong, W.; Kuncewitch, M.; Ji, Y.; Yang, H.; Wang, H.; et al. Cold-inducible RNA-binding protein (CIRP) triggers inflammatory responses in hemorrhagic shock and sepsis. Nat. Med. 2013, 19, 1489–1495. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liu, B.; Dai, J.; Srivastava, P.K.; Zammit, D.J.; Lefrancois, L.; Li, Z. Heat shock protein GP96 is a master chaperone for toll-like receptors and is important in the innate function of macrophages. Immunity 2007, 26, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Ehrenstein, M.R.; Notley, C.A. The importance of natural IgM: Scavenger, protector and regulator. Nat. Rev. Immunol. 2010, 10, 778–786. [Google Scholar] [CrossRef] [PubMed]
- Vargas, M.E.; Watanabe, J.; Singh, S.J.; Robinson, W.H.; Barres, B.A. Endogenous antibodies promote rapid myelin clearance and effective axon regeneration after nerve injury. Proc. Natl. Acad. Sci. USA 2010, 107, 11993–11998. [Google Scholar] [CrossRef] [PubMed]
- Bornstein, N.M.; Aronovich, B.; Korczyn, A.D.; Shavit, S.; Michaelson, D.M.; Chapman, J. Antibodies to brain antigens following stroke. Neurology 2001, 56, 529–530. [Google Scholar] [CrossRef] [PubMed]
- Soussan, L.; Tchernakov, K.; Bachar-Lavi, O.; Yuvan, T.; Wertman, E.; Michaelson, D.M. Antibodies to different isoforms of the heavy neurofilament protein (NF-H) in normal aging and Alzheimer’s disease. Mol. Neurobiol. 1994, 9, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Dambinova, S.A.; Khounteev, G.A.; Izykenova, G.A.; Zavolokov, I.G.; Ilyukhina, A.Y.; Skoromets, A.A. Blood test detecting autoantibodies to N-methyl-d-aspartate neuroreceptors for evaluation of patients with transient ischemic attack and stroke. Clin. Chem. 2003, 49, 1752–1762. [Google Scholar] [CrossRef] [PubMed]
- Kimura, A.; Sakurai, T.; Yamada, M.; Koumura, A.; Hayashi, Y.; Tanaka, Y.; Hozumi, I.; Takemura, M.; Seishima, M.; Inuzuka, T. Elevated anti-heat shock protein 60 antibody titer is related to white matter hyperintensities. J. Stroke Cerebrovasc. Dis. 2012, 21, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Becker, K.J.; Kalil, A.J.; Tanzi, P.; Zierath, D.K.; Savos, A.V.; Gee, J.M.; Hadwin, J.; Carter, K.T.; Shibata, D.; Cain, K.C. Autoimmune responses to the brain after stroke are associated with worse outcome. Stroke 2011, 42, 2763–2769. [Google Scholar] [CrossRef] [PubMed]
- Shibata, D.; Cain, K.; Tanzi, P.; Zierath, D.; Becker, K. Myelin basic protein autoantibodies, white matter disease and stroke outcome. J. Neuroimmunol. 2012, 252, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Schoppet, M.; Sattler, A.M.; Schaefer, J.R.; Herzum, M.; Maisch, B.; Hofbauer, L.C. Increased osteoprotegerin serum levels in men with coronary artery disease. J. Clin. Endocrinol. Metab. 2003, 88, 1024–1028. [Google Scholar] [CrossRef] [PubMed]
- Osako, M.K.; Nakagami, H.; Koibuchi, N.; Shimizu, H.; Nakagami, F.; Koriyama, H.; Shimamura, M.; Miyake, T.; Rakugi, H.; Morishita, R. Estrogen inhibits vascular calcification via vascular RANKL system: Common mechanism of osteoporosis and vascular calcification. Circ. Res. 2010, 107, 466–475. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, K.; Takada, Y.; Ray, N.; Kishimoto, Y.; Penninger, J.M.; Yasuda, H.; Matsuo, K. Receptor activator of NF-κB ligand and osteoprotegerin regulate proinflammatory cytokine production in mice. J. Immunol. 2006, 177, 3799–3805. [Google Scholar] [CrossRef] [PubMed]
- Ferrari-Lacraz, S.; Ferrari, S. Do RANKL inhibitors (denosumab) affect inflammation and immunity? Osteoporos. Int. 2011, 22, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Ustundag, M.; Orak, M.; Guloglu, C.; Tamam, Y.; Sayhan, M.B.; Kale, E. The role of serum osteoprotegerin and S-100 protein levels in patients with acute ischaemic stroke: Determination of stroke subtype, severity and mortality. J. Int. Med. Res. 2011, 39, 780–789. [Google Scholar] [CrossRef] [PubMed]
- Jensen, J.K.; Ueland, T.; Atar, D.; Gullestad, L.; Mickley, H.; Aukrust, P.; Januzzi, J.L. Osteoprotegerin concentrations and prognosis in acute ischaemic stroke. J. Intern. Med. 2010, 267, 410–417. [Google Scholar] [CrossRef] [PubMed]
- Guldiken, B.; Guldiken, S.; Turgut, B.; Turgut, N.; Demir, M.; Celik, Y.; Arikan, E.; Tugrul, A. Serum osteoprotegerin levels in patients with acute atherothrombotic stroke and lacunar infarct. Thromb. Res. 2007, 120, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.J.; Lee, S.H.; Ryu, W.S.; Kim, C.K.; Yoon, B.W. Adipocytokines and ischemic stroke: Differential associations between stroke subtypes. J. Neurol. Sci. 2012, 312, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Wang, S.; Signore, A.P.; Chen, J. Neuroprotective effects of leptin against ischemic injury induced by oxygen-glucose deprivation and transient cerebral ischemia. Stroke 2007, 38, 2329–2336. [Google Scholar] [CrossRef] [PubMed]
- Carbone, F.; Burger, F.; Roversi, G.; Tamborino, C.; Casetta, I.; Seraceni, S.; Trentini, A.; Padroni, M.; Bertolotto, M.; Dallegri, F.; et al. Leptin/adiponectin ratio predicts poststroke neurological outcome. Eur. J. Clin. Investig. 2015, 45, 1184–1191. [Google Scholar] [CrossRef] [PubMed]
- Valerio, A.; Dossena, M.; Bertolotti, P.; Boroni, F.; Sarnico, I.; Faraco, G.; Chiarugi, A.; Frontini, A.; Giordano, A.; Liou, H.C.; et al. Leptin is induced in the ischemic cerebral cortex and exerts neuroprotection through NF-κB/c-Rel-dependent transcription. Stroke 2009, 40, 610–617. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Deng, Z.; Liao, J.; Song, C.; Liang, C.; Xue, H.; Wang, L.; Zhang, K.; Yan, G. Leptin attenuates cerebral ischemia injury through the promotion of energy metabolism via the PI3K/Akt pathway. J. Cereb. Blood Flow Metab. 2013, 33, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Avraham, Y.; Davidi, N.; Lassri, V.; Vorobiev, L.; Kabesa, M.; Dayan, M.; Chernoguz, D.; Berry, E.; Leker, R.R. Leptin induces neuroprotection neurogenesis and angiogenesis after stroke. Curr. Neurovasc. Res. 2011, 8, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.Y., Jr.; Si, Y.L.; Liao, J.; Yan, G.T.; Deng, Z.H.; Xue, H.; Wang, L.H.; Zhang, K. Leptin administration alleviates ischemic brain injury in mice by reducing oxidative stress and subsequent neuronal apoptosis. J. Trauma Acute Care Surg. 2012, 72, 982–991. [Google Scholar] [CrossRef] [PubMed]
- Efstathiou, S.P.; Tsioulos, D.I.; Tsiakou, A.G.; Gratsias, Y.E.; Pefanis, A.V.; Mountokalakis, T.D. Plasma adiponectin levels and five-year survival after first-ever ischemic stroke. Stroke 2005, 36, 1915–1919. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Miao, J.; Yuan, F.; Zhao, Y.; Tang, Y.; Wang, Y.; Zhao, Y.; Yang, G.Y. Overexpression of adiponectin promotes focal angiogenesis in the mouse brain following middle cerebral artery occlusion. Gene Ther. 2013, 20, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Miao, J.; Shen, L.H.; Tang, Y.H.; Wang, Y.T.; Tao, M.X.; Jin, K.L.; Zhao, Y.J.; Yang, G.Y. Overexpression of adiponectin improves neurobehavioral outcomes after focal cerebral ischemia in aged mice. CNS Neurosci. Ther. 2013, 19, 969–977. [Google Scholar] [CrossRef] [PubMed]
- Scatena, M.; Liaw, L.; Giachelli, C.M. Osteopontin: A multifunctional molecule regulating chronic inflammation and vascular disease. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2302–2309. [Google Scholar] [CrossRef] [PubMed]
- Mendioroz, M.; Fernandez-Cadenas, I.; Rosell, A.; Delgado, P.; Domingues-Montanari, S.; Ribo, M.; Penalba, A.; Quintana, M.; Alvarez-Sabin, J.; Montaner, J. Osteopontin predicts long-term functional outcome among ischemic stroke patients. J. Neurol. 2011, 258, 486–493. [Google Scholar] [CrossRef] [PubMed]
- Carbone, F.; Vuilleumier, N.; Burger, F.; Roversi, G.; Tamborino, C.; Casetta, I.; Seraceni, S.; Trentini, A.; Padroni, M.; Dallegri, F.; et al. Serum osteopontin levels are upregulated and predict disability after an ischaemic stroke. Eur. J. Clin. Investig. 2015, 45, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Meller, R.; Stevens, S.L.; Minami, M.; Cameron, J.A.; King, S.; Rosenzweig, H.; Doyle, K.; Lessov, N.S.; Simon, R.P.; Stenzel-Poore, M.P. Neuroprotection by osteopontin in stroke. J. Cereb. Blood Flow Metab. 2005, 25, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Doyle, K.P.; Yang, T.; Lessov, N.S.; Ciesielski, T.M.; Stevens, S.L.; Simon, R.P.; King, J.S.; Stenzel-Poore, M.P. Nasal administration of osteopontin peptide mimetics confers neuroprotection in stroke. J. Cereb. Blood Flow Metab. 2008, 28, 1235–1248. [Google Scholar] [CrossRef] [PubMed]
- Gliem, M.; Krammes, K.; Liaw, L.; van Rooijen, N.; Hartung, H.P.; Jander, S. Macrophage-derived osteopontin induces reactive astrocyte polarization and promotes re-establishment of the blood brain barrier after ischemic stroke. Glia 2015, 63, 2198–2207. [Google Scholar] [CrossRef] [PubMed]
- Dassan, P.; Keir, G.; Brown, M.M. Criteria for a clinically informative serum biomarker in acute ischaemic stroke: A review of S100B. Cerebrovasc. Dis. 2009, 27, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Wang, L.; Yang, X.K.; Fan, L.P.; Wang, Y.G.; Guo, L. Serum S100B levels may be associated with cerebral infarction: A meta-analysis. J. Neurol. Sci. 2015, 348, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Kazmierski, R.; Michalak, S.; Wencel-Warot, A.; Nowinski, W.L. Serum tight-junction proteins predict hemorrhagic transformation in ischemic stroke patients. Neurology 2012, 79, 1677–1685. [Google Scholar] [CrossRef] [PubMed]
- Tsai, N.W.; Chang, Y.T.; Huang, C.R.; Lin, Y.J.; Lin, W.C.; Cheng, B.C.; Su, C.M.; Chiang, Y.F.; Chen, S.F.; Huang, C.C.; et al. Association between oxidative stress and outcome in different subtypes of acute ischemic stroke. BioMed Res. Int. 2014, 2014, 256879. [Google Scholar] [CrossRef] [PubMed]
- Lorente, L.; Martin, M.M.; Abreu-Gonzalez, P.; Ramos, L.; Argueso, M.; Sole-Violan, J.; Riano-Ruiz, M.; Jimenez, A. Serum malondialdehyde levels in patients with malignant middle cerebral artery infarction are associated with mortality. PLoS ONE 2015, 10, e0125893. [Google Scholar] [CrossRef] [PubMed]
- Bharosay, A.; Bharosay, V.V.; Varma, M.; Saxena, K.; Sodani, A.; Saxena, R. Correlation of brain biomarker neuron specific enolase (NSE) with degree of disability and neurological worsening in cerebrovascular stroke. Indian J. Clin. Biochem. 2012, 27, 186–190. [Google Scholar] [CrossRef] [PubMed]
- Singh, H.V.; Pandey, A.; Shrivastava, A.K.; Raizada, A.; Singh, S.K.; Singh, N. Prognostic value of neuron specific enolase and IL-10 in ischemic stroke and its correlation with degree of neurological deficit. Clin. Chim. Acta 2013, 419, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Zaheer, S.; Beg, M.; Rizvi, I.; Islam, N.; Ullah, E.; Akhtar, N. Correlation between serum neuron specific enolase and functional neurological outcome in patients of acute ischemic stroke. Ann. Indian Acad. Neurol. 2013, 16, 504–508. [Google Scholar] [PubMed]
- Park, S.Y.; Kim, M.H.; Kim, O.J.; Ahn, H.J.; Song, J.Y.; Jeong, J.Y.; Oh, S.H. Plasma heart-type fatty acid binding protein level in acute ischemic stroke: Comparative analysis with plasma S100B level for diagnosis of stroke and prediction of long-term clinical outcome. Clin. Neurol. Neurosurg. 2013, 115, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.J.; Kim, Y.J.; Ahn, S.H.; Kim, N.Y.; Kang, D.W.; Kim, J.S.; Kwon, S.U. The second elevation of neuron-specific enolase peak after ischemic stroke is associated with hemorrhagic transformation. J. Stroke Cerebrovasc. Dis. 2014, 23, 2437–2443. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.; Xu, X.; Cui, S.; Wang, F.; Zhang, B.; Zhao, Y. Serum neuron specific enolase level as a predictor of prognosis in acute ischemic stroke patients after intravenous thrombolysis. J. Neurol. Sci. 2015, 359, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Haupt, W.F.; Chopan, G.; Sobesky, J.; Liu, W.C.; Dohmen, C. Prognostic value of somatosensory evoked potentials, neuron-specific enolase, and S100 for short-term outcome in ischemic stroke. J. Neurophysiol. 2016, 115, 1273–1278. [Google Scholar] [CrossRef] [PubMed]
- Ozkan, A.K.; Yemisci, O.U.; Saracgil Cosar, S.N.; Oztop, P.; Turhan, N. Can high-sensitivity C-reactive protein and ferritin predict functional outcome in acute ischemic stroke? A prospective study. Top. Stroke Rehabil. 2013, 20, 528–536. [Google Scholar] [CrossRef] [PubMed]
- Taheraghdam, A.; Aminnejad, S.; Pashapour, A.; Rikhtegar, R.; Ghabili, K. Is there a correlation between hs-CRP levels and functional outcome of ischemic stroke? Pak. J. Med. Sci. 2013, 29, 166–169. [Google Scholar] [PubMed]
- VanGilder, R.L.; Davidov, D.M.; Stinehart, K.R.; Huber, J.D.; Turner, R.C.; Wilson, K.S.; Haney, E.; Davis, S.M.; Chantler, P.D.; Theeke, L.; et al. C-reactive protein and long-term ischemic stroke prognosis. J. Clin. Neurosci. 2014, 21, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Karlinski, M.; Bembenek, J.; Grabska, K.; Kobayashi, A.; Baranowska, A.; Litwin, T.; Czlonkowska, A. Routine serum C-reactive protein and stroke outcome after intravenous thrombolysis. Acta Neurol. Scand. 2014, 130, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A.; Shrivastava, A.K.; Saxena, K. Neuron specific enolase and c-reactive protein levels in stroke and its subtypes: Correlation with degree of disability. Neurochem. Res. 2014, 39, 1426–1432. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.M.; Liu, X.Y. Serum levels of procalcitonin and high sensitivity C-reactive protein are associated with long-term mortality in acute ischemic stroke. J. Neurol. Sci. 2015, 352, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Rocco, A.; Ringleb, P.A.; Grittner, U.; Nolte, C.H.; Schneider, A.; Nagel, S. Follow-up C-reactive protein level is more strongly associated with outcome in stroke patients than admission levels. Neurol. Sci. 2015, 36, 2235–2241. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.J.; Shen, R.L.; Li, M.; Teng, J.F. Relationship between procalcitonin serum levels and functional outcome in stroke patients. Cell Mol. Neurobiol. 2015, 35, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Gao, L.; Zhang, Z.G.; Li, Y.Q.; Yang, Y.L.; Chang, T.; Zheng, L.L.; Zhang, X.Y.; Man, M.H.; Li, L.H. Procalcitonin is a stronger predictor of long-term functional outcome and mortality than high-sensitivity C-reactive protein in patients with ischemic stroke. Mol. Neurobiol. 2016, 53, 1509–1517. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, R.; Ago, T.; Hata, J.; Wakisaka, Y.; Kuroda, J.; Kuwashiro, T.; Kitazono, T.; Kamouchi, M. Fukuoka Stroke Registry Investigators. Plasma C-reactive protein and clinical outcomes after acute ischemic stroke: A prospective observational study. PLoS ONE 2016, 11, e0156790. [Google Scholar] [CrossRef] [PubMed]
- Geng, H.H.; Wang, X.W.; Fu, R.L.; Jing, M.J.; Huang, L.L.; Zhang, Q.; Wang, X.X.; Wang, P.X. The relationship between C-reactive protein level and discharge outcome in patients with acute ischemic stroke. Int. J. Environ. Res. Public Health 2016, 13, 636. [Google Scholar] [CrossRef] [PubMed]
- Whiteley, W.; Jackson, C.; Lewis, S.; Lowe, G.; Rumley, A.; Sandercock, P.; Wardlaw, J.; Dennis, M.; Sudlow, C. Inflammatory markers and poor outcome after stroke: A prospective cohort study and systematic review of interleukin-6. PLoS Med. 2009, 6, e1000145. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Kim, J.; Kim, O.J.; Kim, J.K.; Song, J.; Shin, D.A.; Oh, S.H. Predictive value of circulating interleukin-6 and heart-type fatty acid binding protein for three months clinical outcome in acute cerebral infarction: Multiple blood markers profiling study. Crit. Care 2013, 17, R45. [Google Scholar] [CrossRef] [PubMed]
- Bustamante, A.; Sobrino, T.; Giralt, D.; Garcia-Berrocoso, T.; Llombart, V.; Ugarriza, I.; Espadaler, M.; Rodriguez, N.; Sudlow, C.; Castellanos, M.; et al. Prognostic value of blood interleukin-6 in the prediction of functional outcome after stroke: A systematic review and meta-analysis. J. Neuroimmunol. 2014, 274, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Pusch, G.; Debrabant, B.; Molnar, T.; Feher, G.; Papp, V.; Banati, M.; Kovacs, N.; Szapary, L.; Illes, Z. Early dynamics of P-selectin and interleukin 6 predicts outcomes in ischemic stroke. J. Stroke Cerebrovasc. Dis. 2015, 24, 1938–1947. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, M.F.; Kallaur, A.P.; Oliveira, S.R.; Alfieri, D.F.; Delongui, F.; de Sousa Parreira, J.; de Araujo, M.C.; Rossato, C.; de Almeida, J.T.; Pelegrino, L.M.; et al. Inflammatory and metabolic markers and short-time outcome in patients with acute ischemic stroke in relation to toast subtypes. Metab. Brain Dis. 2015, 30, 1417–1428. [Google Scholar] [CrossRef] [PubMed]
- Worthmann, H.; Tryc, A.B.; Dirks, M.; Schuppner, R.; Brand, K.; Klawonn, F.; Lichtinghagen, R.; Weissenborn, K. Lipopolysaccharide binding protein, interleukin-10, interleukin-6 and C-reactive protein blood levels in acute ischemic stroke patients with post-stroke infection. J. Neuroinflamm. 2015, 12, 13. [Google Scholar] [CrossRef] [PubMed]
- Fahmi, R.M.; Elsaid, A.F. Infarction size, interleukin-6, and their interaction are predictors of short-term stroke outcome in young egyptian adults. J. Stroke Cerebrovasc. Dis. 2016, 25, 2475–2481. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Yanez, M.; Castellanos, M.; Sobrino, T.; Brea, D.; Ramos-Cabrer, P.; Pedraza, S.; Castineiras, J.A.; Serena, J.; Davalos, A.; Castillo, J.; et al. Interleukin-10 facilitates the selection of patients for systemic thrombolysis. BMC Neurol. 2013, 13, 62. [Google Scholar] [CrossRef] [PubMed]
- Ashour, W.; Al-Anwar, A.D.; Kamel, A.E.; Aidaros, M.A. Predictors of early infection in cerebral ischemic stroke. J. Med. Life 2016, 9, 163–169. [Google Scholar] [PubMed]
- Carbone, F.; Vuilleumier, N.; Bertolotto, M.; Burger, F.; Galan, K.; Roversi, G.; Tamborino, C.; Casetta, I.; Seraceni, S.; Trentini, A.; et al. Treatment with recombinant tissue plasminogen activator (r-TPA) induces neutrophil degranulation in vitro via defined pathways. Vasc. Pharmacol. 2015, 64, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Inzitari, D.; Giusti, B.; Nencini, P.; Gori, A.M.; Nesi, M.; Palumbo, V.; Piccardi, B.; Armillis, A.; Pracucci, G.; Bono, G.; et al. MMP9 variation after thrombolysis is associated with hemorrhagic transformation of lesion and death. Stroke 2013, 44, 2901–2903. [Google Scholar] [CrossRef] [PubMed]
- Wiseman, S.; Marlborough, F.; Doubal, F.; Webb, D.J.; Wardlaw, J. Blood markers of coagulation, fibrinolysis, endothelial dysfunction and inflammation in lacunar stroke versus non-lacunar stroke and non-stroke: Systematic review and meta-analysis. Cerebrovasc. Dis. 2014, 37, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.B.; Li, M.; Zhuo, W.Y.; Zhang, Y.S.; Xu, A.D. The role of hs-CRP, d-dimer and fibrinogen in differentiating etiological subtypes of ischemic stroke. PLoS ONE 2015, 10, e0118301. [Google Scholar] [CrossRef] [PubMed]
- Yuan, W.; Shi, Z.H. The relationship between plasma D-dimer levels and outcome of Chinese acute ischemic stroke patients in different stroke subtypes. J. Neural Transm. 2014, 121, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.Y.; Gao, S.; Ding, J.; Chen, Y.; Zhou, X.S.; Wang, J.E. Plasma d-dimer predicts short-term poor outcome after acute ischemic stroke. PLoS ONE 2014, 9, e89756. [Google Scholar] [CrossRef] [PubMed]
- Richard, S.; Lagerstedt, L.; Burkhard, P.R.; Debouverie, M.; Turck, N.; Sanchez, J.C. E-selectin and vascular cell adhesion molecule-1 as biomarkers of 3-month outcome in cerebrovascular diseases. J. Inflamm. 2015, 12, 61. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Ning, R.; Wang, Y. Plasma d-dimer level, the promising prognostic biomarker for the acute cerebral infarction patients. J. Stroke Cerebrovasc. Dis. 2016, 25, 2011–2015. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.J.; Chen, C.H.; Yeh, S.J.; Tsai, L.K.; Tang, S.C.; Jeng, J.S. High plasma d-dimer indicates unfavorable outcome of acute ischemic stroke patients receiving intravenous thrombolysis. Cerebrovasc. Dis. 2016, 42, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.; Yang, G.; Li, G. Molecular insights and therapeutic targets for blood-brain barrier disruption in ischemic stroke: Critical role of matrix metalloproteinases and tissue-type plasminogen activator. Neurobiol. Dis. 2010, 38, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Kapural, M.; Krizanac-Bengez, L.; Barnett, G.; Perl, J.; Masaryk, T.; Apollo, D.; Rasmussen, P.; Mayberg, M.R.; Janigro, D. Serum S-100β as a possible marker of blood-brain barrier disruption. Brain Res. 2002, 940, 102–104. [Google Scholar] [CrossRef]
- Steiner, J.; Bernstein, H.G.; Bielau, H.; Berndt, A.; Brisch, R.; Mawrin, C.; Keilhoff, G.; Bogerts, B. Evidence for a wide extra-astrocytic distribution of S100B in human brain. BMC Neurosci. 2007, 8, 2. [Google Scholar] [CrossRef] [PubMed]
- Cherubini, A.; Ruggiero, C.; Polidori, M.C.; Mecocci, P. Potential markers of oxidative stress in stroke. Free Radic. Biol. Med. 2005, 39, 841–852. [Google Scholar] [CrossRef] [PubMed]
- Wunderlich, M.T.; Hanhoff, T.; Goertler, M.; Spener, F.; Glatz, J.F.; Wallesch, C.W.; Pelsers, M.M. Release of brain-type and heart-type fatty acid-binding proteins in serum after acute ischaemic stroke. J. Neurol. 2005, 252, 718–724. [Google Scholar] [CrossRef] [PubMed]
- Wunderlich, M.T.; Lins, H.; Skalej, M.; Wallesch, C.W.; Goertler, M. Neuron-specific enolase and tau protein as neurobiochemical markers of neuronal damage are related to early clinical course and long-term outcome in acute ischemic stroke. Clin. Neurol. Neurosurg. 2006, 108, 558–563. [Google Scholar] [CrossRef] [PubMed]
- Isgro, M.A.; Bottoni, P.; Scatena, R. Neuron-specific enolase as a biomarker: Biochemical and clinical aspects. Adv. Exp. Med. Biol. 2015, 867, 125–143. [Google Scholar] [PubMed]
- Emerging Risk Factors Collaboration; Kaptoge, S.; di Angelantonio, E.; Lowe, G.; Pepys, M.B.; Thompson, S.G.; Collins, R.; Danesh, J. C-reactive protein concentration and risk of coronary heart disease, stroke, and mortality: An individual participant meta-analysis. Lancet 2010, 375, 132–140. [Google Scholar] [PubMed]
- Di Napoli, M.; Schwaninger, M.; Cappelli, R.; Ceccarelli, E.; di Gianfilippo, G.; Donati, C.; Emsley, H.C.; Forconi, S.; Hopkins, S.J.; Masotti, L.; et al. Evaluation of C-reactive protein measurement for assessing the risk and prognosis in ischemic stroke: A statement for health care professionals from the crp pooling project members. Stroke 2005, 36, 1316–1329. [Google Scholar] [CrossRef] [PubMed]
- Bustamante, A.; Simats, A.; Vilar-Bergua, A.; Garcia-Berrocoso, T.; Montaner, J. Blood/brain biomarkers of inflammation after stroke and their association with outcome: From C-reactive protein to damage-associated molecular patterns. Neurotherapeutics 2016, 13, 671–684. [Google Scholar] [CrossRef] [PubMed]
- Doll, D.N.; Barr, T.L.; Simpkins, J.W. Cytokines: Their role in stroke and potential use as biomarkers and therapeutic targets. Aging Dis. 2014, 5, 294–306. [Google Scholar] [PubMed]
- Perini, F.; Morra, M.; Alecci, M.; Galloni, E.; Marchi, M.; Toso, V. Temporal profile of serum anti-inflammatory and pro-inflammatory interleukins in acute ischemic stroke patients. Neurol. Sci. 2001, 22, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Turner, R.J.; Sharp, F.R. Implications of MMP9 for blood brain barrier disruption and hemorrhagic transformation following ischemic stroke. Front. Cell. Neurosci. 2016, 10, 56. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Roth, S.; Veltkamp, R.; Liesz, A. HMGB1 as a key mediator of immune mechanisms in ischemic stroke. Antioxid. Redox Signal. 2016, 24, 635–651. [Google Scholar] [CrossRef] [PubMed]
- Schulze, J.; Zierath, D.; Tanzi, P.; Cain, K.; Shibata, D.; Dressel, A.; Becker, K. Severe stroke induces long-lasting alterations of high-mobility group box 1. Stroke 2013, 44, 246–248. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.M.; Hu, J.; Chen, N.; Hu, M.L. Relationship between plasma high-mobility group box-1 levels and clinical outcomes of ischemic stroke. J. Crit. Care 2013, 28, 792–797. [Google Scholar] [CrossRef] [PubMed]
- Sapojnikova, N.; Kartvelishvili, T.; Asatiani, N.; Zinkevich, V.; Kalandadze, I.; Gugutsidze, D.; Shakarishvili, R.; Tsiskaridze, A. Correlation between MMP-9 and extracellular cytokine HMGB1 in prediction of human ischemic stroke outcome. Biochim. Biophys. Acta 2014, 1842, 1379–1384. [Google Scholar] [CrossRef] [PubMed]
- Marousi, S.G.; Theodorou, G.L.; Karakantza, M.; Zampakis, P.; Papathanasopoulos, P.; Ellul, J. Acute post-stroke adiponectin in relation to stroke severity, progression and 6 month functional outcome. Neurol. Res. 2010, 32, 841–844. [Google Scholar] [CrossRef] [PubMed]
- Kuwashiro, T.; Ago, T.; Kamouchi, M.; Matsuo, R.; Hata, J.; Kuroda, J.; Fukuda, K.; Sugimori, H.; Fukuhara, M.; Awano, H.; et al. Significance of plasma adiponectin for diagnosis, neurological severity and functional outcome in ischemic stroke—Research for biomarkers in ischemic stroke (REBIOS). Metabolism 2014, 63, 1093–1103. [Google Scholar] [CrossRef] [PubMed]
- Vogelgesang, A.; May, V.E.; Grunwald, U.; Bakkeboe, M.; Langner, S.; Wallaschofski, H.; Kessler, C.; Broker, B.M.; Dressel, A. Functional status of peripheral blood T-cells in ischemic stroke patients. PLoS ONE 2010, 5, e8718. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.W.; Lu, F.L.; Zhou, Y.; Wang, L.; Zhong, Q.; Lin, S.; Xiang, J.; Li, J.C.; Fang, C.Q.; Wang, J.Z. HMBG1 mediates ischemia—Reperfusion injury by TRIF-adaptor independent toll-like receptor 4 signaling. J. Cereb. Blood Flow Metab. 2011, 31, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Kanhai, D.A.; Kranendonk, M.E.; Uiterwaal, C.S.; van der Graaf, Y.; Kappelle, L.J.; Visseren, F.L. Adiponectin and incident coronary heart disease and stroke. A systematic review and meta-analysis of prospective studies. Obes. Rev. 2013, 14, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Arregui, M.; Buijsse, B.; Fritsche, A.; di Giuseppe, R.; Schulze, M.B.; Westphal, S.; Isermann, B.; Boeing, H.; Weikert, C. Adiponectin and risk of stroke: Prospective study and meta-analysis. Stroke 2014, 45, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Akasofu, S.; Sawada, K.; Kosasa, T.; Hihara, H.; Ogura, H.; Akaike, A. Donepezil attenuates excitotoxic damage induced by membrane depolarization of cortical neurons exposed to veratridine. Eur. J. Pharmacol. 2008, 588, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Amin-Hanjani, S.; Stagliano, N.E.; Yamada, M.; Huang, P.L.; Liao, J.K.; Moskowitz, M.A. Mevastatin, an HMG-CoA reductase inhibitor, reduces stroke damage and upregulates endothelial nitric oxide synthase in mice. Stroke 2001, 32, 980–986. [Google Scholar] [CrossRef] [PubMed]
- Asahi, M.; Huang, Z.; Thomas, S.; Yoshimura, S.; Sumii, T.; Mori, T.; Qiu, J.; Amin-Hanjani, S.; Huang, P.L.; Liao, J.K.; et al. Protective effects of statins involving both eNOS and tPA in focal cerebral ischemia. J. Cereb. Blood Flow Metab. 2005, 25, 722–729. [Google Scholar] [CrossRef] [PubMed]
- Becker, K.; Kindrick, D.; Relton, J.; Harlan, J.; Winn, R. Antibody to the α4 integrin decreases infarct size in transient focal cerebral ischemia in rats. Stroke 2001, 32, 206–211. [Google Scholar] [CrossRef] [PubMed]
- Campos, F.; Qin, T.; Castillo, J.; Seo, J.H.; Arai, K.; Lo, E.H.; Waeber, C. Fingolimod reduces hemorrhagic transformation associated with delayed tissue plasminogen activator treatment in a mouse thromboembolic model. Stroke 2013, 44, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhang, Z.G.; Li, Y.; Wang, Y.; Wang, L.; Jiang, H.; Zhang, C.; Lu, M.; Katakowski, M.; Feldkamp, C.S.; et al. Statins induce angiogenesis, neurogenesis, and synaptogenesis after stroke. Ann. Neurol. 2003, 53, 743–751. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Guo, Y.; Yang, W.; Zheng, P.; Zeng, J.; Tong, W. Protective effect of ginsenoside Rb1 on integrity of blood-brain barrier following cerebral ischemia. Exp. Brain Res. 2015, 233, 2823–2831. [Google Scholar] [CrossRef] [PubMed]
- Cho, T.H.; Aguettaz, P.; Campuzano, O.; Charriaut-Marlangue, C.; Riou, A.; Berthezene, Y.; Nighoghossian, N.; Ovize, M.; Wiart, M.; Chauveau, F. Pre- and post-treatment with cyclosporine A in a rat model of transient focal cerebral ischaemia with multimodal MRI screening. Int. J. Stroke 2013, 8, 669–674. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Zheng, L.; Lu, S.; Yang, Y. Neuroprotective effects of pretreatment of ginsenoside Rb1 on severe cerebral ischemia-induced injuries in aged mice: Involvement of anti-oxidant signaling. Geriatr. Gerontol. Int. 2015. [Google Scholar] [CrossRef] [PubMed]
- Endres, M.; Laufs, U.; Huang, Z.; Nakamura, T.; Huang, P.; Moskowitz, M.A.; Liao, J.K. Stroke protection by 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase inhibitors mediated by endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. USA 1998, 95, 8880–8885. [Google Scholar] [CrossRef] [PubMed]
- Espinera, A.R.; Ogle, M.E.; Gu, X.; Wei, L. Citalopram enhances neurovascular regeneration and sensorimotor functional recovery after ischemic stroke in mice. Neuroscience 2013, 247, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, N.; Som, A.T.; Pham, L.D.; Lee, B.J.; Mandeville, E.T.; Lo, E.H.; Arai, K. A free radical scavenger edaravone suppresses systemic inflammatory responses in a rat transient focal ischemia model. Neurosci. Lett. 2016, 633, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, S.; Yamashita, T.; Miwa, Y.; Ozaki, M.; Namiki, M.; Hirase, T.; Inoue, N.; Hirata, K.; Yokoyama, M. HMG-CoA reductase inhibitor has protective effects against stroke events in stroke-prone spontaneously hypertensive rats. Stroke 2003, 34, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Kronenberg, G.; Balkaya, M.; Prinz, V.; Gertz, K.; Ji, S.; Kirste, I.; Heuser, I.; Kampmann, B.; Hellmann-Regen, J.; Gass, P.; et al. Exofocal dopaminergic degeneration as antidepressant target in mouse model of poststroke depression. Biol. Psychiatry 2012, 72, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Langhauser, F.; Kraft, P.; Gob, E.; Leinweber, J.; Schuhmann, M.K.; Lorenz, K.; Gelderblom, M.; Bittner, S.; Meuth, S.G.; Wiendl, H.; et al. Blocking of α4 integrin does not protect from acute ischemic stroke in mice. Stroke 2014, 45, 1799–1806. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Park, S.Y.; Shin, Y.W.; Kim, C.D.; Lee, W.S.; Hong, K.W. Concurrent administration of cilostazol with donepezil effectively improves cognitive dysfunction with increased neuroprotection after chronic cerebral hypoperfusion in rats. Brain Res. 2007, 1185, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Liesz, A.; Zhou, W.; Mracsko, E.; Karcher, S.; Bauer, H.; Schwarting, S.; Sun, L.; Bruder, D.; Stegemann, S.; Cerwenka, A.; et al. Inhibition of lymphocyte trafficking shields the brain against deleterious neuroinflammation after stroke. Brain 2011, 134, 704–720. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Sun, W.; Gong, W.; Ding, Y.; Zhuang, Y.; Hou, Q. Ginsenoside Rg1 protects against transient focal cerebral ischemic injury and suppresses its systemic metabolic changes in cerabral injury rats. Acta Pharm. Sin. B 2015, 5, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhu, L.; Zou, Y.; Liu, W.; Zhang, X.; Wei, X.; Hu, B.; Chen, J. Panax notoginseng saponins promotes stroke recovery by influencing expression of Nogo-A, NgR and p75NGF, in vitro and in vivo. Biol. Pharm. Bull. 2014, 37, 560–568. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.Y.; Zhou, X.Y.; Hou, J.C.; Zhu, H.; Wang, Z.; Liu, J.X.; Zheng, Y.Q. Ginsenoside Rd promotes neurogenesis in rat brain after transient focal cerebral ischemia via activation of PI3K/Akt pathway. Acta Pharmacol. Sin. 2015, 36, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Llovera, G.; Hofmann, K.; Roth, S.; Salas-Perdomo, A.; Ferrer-Ferrer, M.; Perego, C.; Zanier, E.R.; Mamrak, U.; Rex, A.; Party, H.; et al. Results of a preclinical randomized controlled multicenter trial (pRCT): Anti-CD49d treatment for acute brain ischemia. Sci. Transl. Med. 2015, 7, 299ra121. [Google Scholar] [CrossRef] [PubMed]
- Min, D.; Mao, X.; Wu, K.; Cao, Y.; Guo, F.; Zhu, S.; Xie, N.; Wang, L.; Chen, T.; Shaw, C.; et al. Donepezil attenuates hippocampal neuronal damage and cognitive deficits after global cerebral ischemia in gerbils. Neurosci. Lett. 2012, 510, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Neumann, J.; Riek-Burchardt, M.; Herz, J.; Doeppner, T.R.; Konig, R.; Hutten, H.; Etemire, E.; Mann, L.; Klingberg, A.; Fischer, T.; et al. Very-late-antigen-4 (VLA-4)-mediated brain invasion by neutrophils leads to interactions with microglia, increased ischemic injury and impaired behavior in experimental stroke. Acta Neuropathol. 2015, 129, 259–277. [Google Scholar] [CrossRef] [PubMed]
- Onetti, Y.; Dantas, A.P.; Perez, B.; Cugota, R.; Chamorro, A.; Planas, A.M.; Vila, E.; Jimenez-Altayo, F. Middle cerebral artery remodeling following transient brain ischemia is linked to early postischemic hyperemia: A target of uric acid treatment. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H862–H874. [Google Scholar] [CrossRef] [PubMed]
- Pradillo, J.M.; Denes, A.; Greenhalgh, A.D.; Boutin, H.; Drake, C.; McColl, B.W.; Barton, E.; Proctor, S.D.; Russell, J.C.; Rothwell, N.J.; et al. Delayed administration of interleukin-1 receptor antagonist reduces ischemic brain damage and inflammation in comorbid rats. J. Cereb. Blood Flow Metab. 2012, 32, 1810–1819. [Google Scholar] [CrossRef] [PubMed]
- Prinz, V.; Laufs, U.; Gertz, K.; Kronenberg, G.; Balkaya, M.; Leithner, C.; Lindauer, U.; Endres, M. Intravenous rosuvastatin for acute stroke treatment: An animal study. Stroke 2008, 39, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Relton, J.K.; Martin, D.; Thompson, R.C.; Russell, D.A. Peripheral administration of interleukin-1 receptor antagonist inhibits brain damage after focal cerebral ischemia in the rat. Exp. Neurol. 1996, 138, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Relton, J.K.; Sloan, K.E.; Frew, E.M.; Whalley, E.T.; Adams, S.P.; Lobb, R.R. Inhibition of α4 integrin protects against transient focal cerebral ischemia in normotensive and hypertensive rats. Stroke 2001, 32, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Reuter, B.; Rodemer, C.; Grudzenski, S.; Meairs, S.; Bugert, P.; Hennerici, M.G.; Fatar, M. Effect of simvastatin on mmps and timps in human brain endothelial cells and experimental stroke. Transl. Stroke Res. 2015, 6, 156–159. [Google Scholar] [CrossRef] [PubMed]
- Rolland, W.B.; Lekic, T.; Krafft, P.R.; Hasegawa, Y.; Altay, O.; Hartman, R.; Ostrowski, R.; Manaenko, A.; Tang, J.; Zhang, J.H. Fingolimod reduces cerebral lymphocyte infiltration in experimental models of rodent intracerebral hemorrhage. Exp. Neurol. 2013, 241, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Romanos, E.; Planas, A.M.; Amaro, S.; Chamorro, A. Uric acid reduces brain damage and improves the benefits of rt-PA in a rat model of thromboembolic stroke. J. Cereb. Blood Flow Metab. 2007, 27, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Yu, W.; Liu, L.; Liu, W.; Zhang, X.; Yang, T.; Chai, L.; Lou, L.; Gao, Y.; Zhu, L. Panax notoginseng saponins administration modulates pro-/anti-inflammatory factor expression and improves neurologic outcome following permanent MCAO in rats. Metab. Brain Dis. 2016. [Google Scholar] [CrossRef] [PubMed]
- Sironi, L.; Cimino, M.; Guerrini, U.; Calvio, A.M.; Lodetti, B.; Asdente, M.; Balduini, W.; Paoletti, R.; Tremoli, E. Treatment with statins after induction of focal ischemia in rats reduces the extent of brain damage. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 322–327. [Google Scholar] [CrossRef] [PubMed]
- Uchino, H.; Elmer, E.; Uchino, K.; Li, P.A.; He, Q.P.; Smith, M.L.; Siesjo, B.K. Amelioration by cyclosporin A of brain damage in transient forebrain ischemia in the rat. Brain Res. 1998, 812, 216–226. [Google Scholar] [CrossRef]
- Wang, T.; Lv, P.; Jin, W.; Zhang, H.; Lang, J.; Fan, M. Protective effect of donepezil hydrochloride on cerebral ischemia/reperfusion injury in mice. Mol. Med. Rep. 2014, 9, 509–514. [Google Scholar] [PubMed]
- Wu, H.Y.; Tang, Y.; Gao, L.Y.; Sun, W.X.; Hua, Y.; Yang, S.B.; Zhang, Z.P.; Liao, G.Y.; Zhou, Q.G.; Luo, C.X.; et al. The synergetic effect of edaravone and borneol in the rat model of ischemic stroke. Eur. J. Pharmacol. 2014, 740, 522–531. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.L.; Li, J.H.; Wang, W.W.; Zheng, G.Q.; Wang, L.X. Neuroprotective effect of ginsenoside-Rg1 on cerebral ischemia/reperfusion injury in rats by downregulating protease-activated receptor-1 expression. Life Sci. 2015, 121, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, Y.; Matsuura, N.; Shozuhara, H.; Onodera, H.; Itoyama, Y.; Kogure, K. Interleukin-1 as a pathogenetic mediator of ischemic brain damage in rats. Stroke 1995, 26, 676–681. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Sato, T.; Sakamoto, K.; Ishii, H.; Yamamoto, J. The free-radical scavenger edaravone accelerates thrombolysis with alteplase in an experimental thrombosis model. Thromb. Res. 2015, 135, 1209–1213. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.X.; Zhang, X.; Zhao, G. Ginsenoside Rd attenuates DNA damage by increasing expression of DNA glycosylase endonuclease VIII-like proteins after focal cerebral ischemia. Chin. Med. J. 2016, 129, 1955–1962. [Google Scholar] [PubMed]
- Yu, G.; Hess, D.C.; Borlongan, C.V. Combined cyclosporine-a and methylprednisolone treatment exerts partial and transient neuroprotection against ischemic stroke. Brain Res. 2004, 1018, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.F.; Bruce-Keller, A.J.; Goodman, Y.; Mattson, M.P. Uric acid protects neurons against excitotoxic and metabolic insults in cell culture, and against focal ischemic brain injury in vivo. J. Neurosci. Res. 1998, 53, 613–625. [Google Scholar] [CrossRef]
- Yuan, H.; Wang, W.P.; Feng, N.; Wang, L.; Wang, X.L. Donepezil attenuated oxygen-glucose deprivation insult by blocking Kv2.1 potassium channels. Eur. J. Pharmacol. 2011, 657, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Yuen, C.M.; Sun, C.K.; Lin, Y.C.; Chang, L.T.; Kao, Y.H.; Yen, C.H.; Chen, Y.L.; Tsai, T.H.; Chua, S.; Shao, P.L.; et al. Combination of cyclosporine and erythropoietin improves brain infarct size and neurological function in rats after ischemic stroke. J. Transl. Med. 2011, 9, 141. [Google Scholar] [CrossRef] [PubMed]
- Sobowale, O.A.; Parry-Jones, A.R.; Smith, C.J.; Tyrrell, P.J.; Rothwell, N.J.; Allan, S.M. Interleukin-1 in stroke: From bench to bedside. Stroke 2016, 47, 2160–2167. [Google Scholar] [CrossRef] [PubMed]
- Relton, J.K.; Rothwell, N.J. Interleukin-1 receptor antagonist inhibits ischaemic and excitotoxic neuronal damage in the rat. Brain Res. Bull. 1992, 29, 243–246. [Google Scholar] [CrossRef]
- Garcia, J.H.; Liu, K.F.; Relton, J.K. Interleukin-1 receptor antagonist decreases the number of necrotic neurons in rats with middle cerebral artery occlusion. Am. J. Pathol. 1995, 147, 1477–1486. [Google Scholar] [PubMed]
- Loddick, S.A.; Rothwell, N.J. Neuroprotective effects of human recombinant interleukin-1 receptor antagonist in focal cerebral ischaemia in the rat. J. Cereb. Blood Flow Metab. 1996, 16, 932–940. [Google Scholar] [CrossRef] [PubMed]
- Boutin, H.; LeFeuvre, R.A.; Horai, R.; Asano, M.; Iwakura, Y.; Rothwell, N.J. Role of IL-1α and IL-1β in ischemic brain damage. J. Neurosci. 2001, 21, 5528–5534. [Google Scholar] [PubMed]
- Banwell, V.; Sena, E.S.; Macleod, M.R. Systematic review and stratified meta-analysis of the efficacy of interleukin-1 receptor antagonist in animal models of stroke. J. Stroke Cerebrovasc. Dis. 2009, 18, 269–276. [Google Scholar] [CrossRef] [PubMed]
- McCann, S.K.; Cramond, F.; Macleod, M.R.; Sena, E.S. Systematic review and meta-analysis of the efficacy of interleukin-1 receptor antagonist in animal models of stroke: An update. Transl. Stroke Res. 2016, 7, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Maysami, S.; Wong, R.; Pradillo, J.M.; Denes, A.; Dhungana, H.; Malm, T.; Koistinaho, J.; Orset, C.; Rahman, M.; Rubio, M.; et al. A cross-laboratory preclinical study on the effectiveness of interleukin-1 receptor antagonist in stroke. J. Cereb. Blood Flow Metab. 2016, 36, 596–605. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Hopkins, S.J.; Hulme, S.; Galea, J.P.; Hoadley, M.; Vail, A.; Hutchinson, P.J.; Grainger, S.; Rothwell, N.J.; King, A.T.; et al. The effect of intravenous interleukin-1 receptor antagonist on inflammatory mediators in cerebrospinal fluid after subarachnoid haemorrhage: A phase II randomised controlled trial. J. Neuroinflamm. 2014, 11, 1. [Google Scholar] [CrossRef] [PubMed]
- Laufs, U.; Gertz, K.; Dirnagl, U.; Bohm, M.; Nickenig, G.; Endres, M. Rosuvastatin, a new HMG-CoA reductase inhibitor, upregulates endothelial nitric oxide synthase and protects from ischemic stroke in mice. Brain Res. 2002, 942, 23–30. [Google Scholar] [CrossRef]
- Baryan, H.K.; Allan, S.M.; Vail, A.; Smith, C.J. Systematic review and meta-analysis of the efficacy of statins in experimental stroke. Int. J. Stroke 2012, 7, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Krauth, D.; Anglemyer, A.; Philipps, R.; Bero, L. Nonindustry-sponsored preclinical studies on statins yield greater efficacy estimates than industry-sponsored studies: A meta-analysis. PLoS Biol. 2014, 12, e1001770. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.A.; Chun, J. Mechanisms of fingolimod’s efficacy and adverse effects in multiple sclerosis. Ann. Neurol. 2011, 69, 759–777. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, C.; Tao, W.; Liu, M. Systematic review and meta-analysis of the efficacy of sphingosine-1-phosphate (S1P) receptor agonist FTY720 (fingolimod) in animal models of stroke. Int. J. Neurosci. 2013, 123, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.J.; Denes, A.; Tyrrell, P.J.; di Napoli, M. Phase II anti-inflammatory and immune-modulating drugs for acute ischaemic stroke. Expert Opin. Investig. Drugs 2015, 24, 623–643. [Google Scholar] [CrossRef] [PubMed]
- Yoshiyama, Y.; Kojima, A.; Ishikawa, C.; Arai, K. Anti-inflammatory action of donepezil ameliorates tau pathology, synaptic loss, and neurodegeneration in a tauopathy mouse model. J. Alzheimers Dis. 2010, 22, 295–306. [Google Scholar] [PubMed]
- Wang, S.S.; Wang, Y.G.; Chen, H.Y.; Wu, Z.P.; Xie, H.G. Expression of genes encoding cytokines and corticotropin releasing factor are altered by citalopram in the hypothalamus of post-stroke depression rats. Neuroendocrinol. Lett. 2013, 34, 773–779. [Google Scholar] [PubMed]
- Dhami, K.S.; Churchward, M.A.; Baker, G.B.; Todd, K.G. Fluoxetine and citalopram decrease microglial release of glutamate and d-serine to promote cortical neuronal viability following ischemic insult. Mol. Cell. Neurosci. 2013, 56, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Osman, M.M.; Lulic, D.; Glover, L.; Stahl, C.E.; Lau, T.; van Loveren, H.; Borlongan, C.V. Cyclosporine-A as a neuroprotective agent against stroke: Its translation from laboratory research to clinical application. Neuropeptides 2011, 45, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, A.; Sharma, U.; Jagannathan, N.R.; Reeta, K.H.; Gupta, Y.K. Rapamycin protects against middle cerebral artery occlusion induced focal cerebral ischemia in rats. Behav. Brain Res. 2011, 225, 603–609. [Google Scholar] [CrossRef] [PubMed]
- Okamura, K.; Tsubokawa, T.; Johshita, H.; Miyazaki, H.; Shiokawa, Y. Edaravone, a free radical scavenger, attenuates cerebral infarction and hemorrhagic infarction in rats with hyperglycemia. Neurol. Res. 2014, 36, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.Y.; Morozov, Y.M.; Yang, D.; Li, Y.; Dunn, R.S.; Rakic, P.; Chan, P.H.; Abe, K.; Lindquist, D.M.; Kuan, C.Y. Synergy of combined tPA-edaravone therapy in experimental thrombotic stroke. PLoS ONE 2014, 9, e98807. [Google Scholar] [CrossRef] [PubMed]
- Dohare, P.; Hyzinski-Garcia, M.C.; Vipani, A.; Bowens, N.H.; Nalwalk, J.W.; Feustel, P.J.; Keller, R.W., Jr.; Jourd’heuil, D.; Mongin, A.A. The neuroprotective properties of the superoxide dismutase mimetic tempol correlate with its ability to reduce pathological glutamate release in a rodent model of stroke. Free Radic. Biol. Med. 2014, 77, 168–182. [Google Scholar] [CrossRef] [PubMed]
- Scandinavian Simvastatin Survival Study Group. Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: The Scandinavian simvastatin survival study (4S). Lancet 1994, 344, 1383–1389. [Google Scholar]
- Amarenco, P.; Bogousslavsky, J.; Callahan, A., III; Goldstein, L.B.; Hennerici, M.; Rudolph, A.E.; Sillesen, H.; Simunovic, L.; Szarek, M.; Welch, K.M.; et al. High-dose atorvastatin after stroke or transient ischemic attack. N. Engl. J. Med. 2006, 355, 549–559. [Google Scholar] [PubMed]
- Amaro, S.; Obach, V.; Cervera, A.; Urra, X.; Gomez-Choco, M.; Planas, A.M.; Chamorro, A. Course of matrix metalloproteinase-9 isoforms after the administration of uric acid in patients with acute stroke: A proof-of-concept study. J. Neurol. 2009, 256, 651–656. [Google Scholar] [CrossRef] [PubMed]
- Barrett, K.M.; Brott, T.G.; Brown, R.D., Jr.; Carter, R.E.; Geske, J.R.; Graff-Radford, N.R.; McNeil, R.B.; Meschia, J.F. Mayo Acute Stroke Trial for Enhancing Recovery Study Group. Enhancing recovery after acute ischemic stroke with donepezil as an adjuvant therapy to standard medical care: Results of a phase IIA clinical trial. J. Stroke Cerebrovasc. Dis. 2011, 20, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Chamorro, A.; Amaro, S.; Castellanos, M.; Segura, T.; Arenillas, J.; Marti-Fabregas, J.; Gallego, J.; Krupinski, J.; Gomis, M.; Canovas, D.; et al. Safety and efficacy of uric acid in patients with acute stroke (URICO-ICTUS): A randomised, double-blind phase 2b/3 trial. Lancet Neurol. 2014, 13, 453–460. [Google Scholar] [CrossRef]
- Edaravone Acute Infarction Study Group. Effect of a novel free radical scavenger, edaravone (MCI-186), on acute brain infarction. Randomized, placebo-controlled, double-blind study at multicenters. Cerebrovasc. Dis. 2003, 15, 222–229. [Google Scholar]
- Emsley, H.C.; Smith, C.J.; Georgiou, R.F.; Vail, A.; Hopkins, S.J.; Rothwell, N.J.; Tyrrell, P.J.; Acute Stroke, I. A randomised phase II study of interleukin-1 receptor antagonist in acute stroke patients. J. Neurol. Neurosurg. Psychiatry 2005, 76, 1366–1372. [Google Scholar] [CrossRef] [PubMed]
- Kaste, M.; Murayama, S.; Ford, G.A.; Dippel, D.W.; Walters, M.R.; Tatlisumak, T. MCI-186 study group. Safety, tolerability and pharmacokinetics of MCI-186 in patients with acute ischemic stroke: New formulation and dosing regimen. Cerebrovasc. Dis. 2013, 36, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, L.; Wen, A.; Yang, J.; Yan, Y.; Song, Y.; Liu, X.; Ren, H.; Wu, Y.; Li, Z.; et al. Ginsenoside-rd improves outcome of acute ischaemic stroke—A randomized, double-blind, placebo-controlled, multicenter trial. Eur. J. Neurol. 2012, 19, 855–863. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Xia, J.; Wang, L.; Song, Y.; Yang, J.; Yan, Y.; Ren, H.; Zhao, G. Efficacy and safety of ginsenoside-Rd for acute ischaemic stroke: A randomized, double-blind, placebo-controlled, phase II multicenter trial. Eur. J. Neurol. 2009, 16, 569–575. [Google Scholar] [CrossRef] [PubMed]
- Llull, L.; Laredo, C.; Renu, A.; Perez, B.; Vila, E.; Obach, V.; Urra, X.; Planas, A.; Amaro, S.; Chamorro, A. Uric acid therapy improves clinical outcome in women with acute ischemic stroke. Stroke 2015, 46, 2162–2167. [Google Scholar] [CrossRef] [PubMed]
- Montaner, J.; Chacon, P.; Krupinski, J.; Rubio, F.; Millan, M.; Molina, C.A.; Hereu, P.; Quintana, M.; Alvarez-Sabin, J. Simvastatin in the acute phase of ischemic stroke: A safety and efficacy pilot trial. Eur. J. Neurol. 2008, 15, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Nighoghossian, N.; Berthezene, Y.; Mechtouff, L.; Derex, L.; Cho, T.H.; Ritzenthaler, T.; Rheims, S.; Chauveau, F.; Bejot, Y.; Jacquin, A.; et al. Cyclosporine in acute ischemic stroke. Neurology 2015, 84, 2216–2223. [Google Scholar] [CrossRef] [PubMed]
- Plehn, J.F.; Davis, B.R.; Sacks, F.M.; Rouleau, J.L.; Pfeffer, M.A.; Bernstein, V.; Cuddy, T.E.; Moye, L.A.; Piller, L.B.; Rutherford, J.; et al. Reduction of stroke incidence after myocardial infarction with pravastatin: The Cholesterol and Recurrent Events (CARE) study. The care investigators. Circulation 1999, 99, 216–223. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Danielson, E.; Fonseca, F.A.; Genest, J.; Gotto, A.M., Jr.; Kastelein, J.J.; Koenig, W.; Libby, P.; Lorenzatti, A.J.; MacFadyen, J.G.; et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N. Engl. J. Med. 2008, 359, 2195–2207. [Google Scholar] [CrossRef] [PubMed]
- Sever, P.S.; Dahlof, B.; Poulter, N.R.; Wedel, H.; Beevers, G.; Caulfield, M.; Collins, R.; Kjeldsen, S.E.; Kristinsson, A.; McInnes, G.T.; et al. Prevention of coronary and stroke events with atorvastatin in hypertensive patients who have average or lower-than-average cholesterol concentrations, in the Anglo-Scandinavian Cardiac Outcomes Trial—Lipid Lowering Arm (ASCOT-LLA): A multicentre randomised controlled trial. Lancet 2003, 361, 1149–1158. [Google Scholar] [PubMed]
- Shepherd, J.; Blauw, G.J.; Murphy, M.B.; Bollen, E.L.; Buckley, B.M.; Cobbe, S.M.; Ford, I.; Gaw, A.; Hyland, M.; Jukema, J.W.; et al. Pravastatin in elderly individuals at risk of vascular disease (PROSPER): A randomised controlled trial. Lancet 2002, 360, 1623–1630. [Google Scholar] [CrossRef]
- Smith, C.J.; Emsley, H.C.; Udeh, C.T.; Vail, A.; Hoadley, M.E.; Rothwell, N.J.; Tyrrell, P.J.; Hopkins, S.J. Interleukin-1 receptor antagonist reverses stroke-associated peripheral immune suppression. Cytokine 2012, 58, 384–389. [Google Scholar] [CrossRef] [PubMed]
- Takenaka, K.; Kato, M.; Yamauti, K.; Hayashi, K. Simultaneous administration of recombinant tissue plasminogen activator and edaravone in acute cerebral ischemic stroke patients. J. Stroke Cerebrovasc. Dis. 2014, 23, 2748–2752. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Thuren, T.; Zalewski, A.; Libby, P. Interleukin-1β inhibition and the prevention of recurrent cardiovascular events: Rationale and design of the canakinumab anti-inflammatory thrombosis outcomes study (CANTOS). Am. Heart J. 2011, 162, 597–605. [Google Scholar] [CrossRef] [PubMed]
- Montecucco, F.; Mach, F. Update on statin-mediated anti-inflammatory activities in atherosclerosis. Semin. Immunopathol. 2009, 31, 127–142. [Google Scholar] [CrossRef] [PubMed]
- Collins, R.; Armitage, J.; Parish, S.; Sleight, P.; Peto, R. Heart Protection Study Collaborative Group. Effects of cholesterol-lowering with simvastatin on stroke and other major vascular events in 20,536 people with cerebrovascular disease or other high-risk conditions. Lancet 2004, 363, 757–767. [Google Scholar] [PubMed]
- Montaner, J.; Bustamante, A.; Garcia-Matas, S.; Martinez-Zabaleta, M.; Jimenez, C.; de la Torre, J.; Rubio, F.R.; Segura, T.; Masjuan, J.; Canovas, D.; et al. Combination of thrombolysis and statins in acute stroke is safe: Results of the STARS randomized trial (stroke treatment with acute reperfusion and simvastatin). Stroke 2016, 47, 2870–2873. [Google Scholar] [CrossRef] [PubMed]
- White, H.D.; Simes, R.J.; Anderson, N.E.; Hankey, G.J.; Watson, J.D.; Hunt, D.; Colquhoun, D.M.; Glasziou, P.; MacMahon, S.; Kirby, A.C.; et al. Pravastatin therapy and the risk of stroke. N. Engl. J. Med. 2000, 343, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Waters, D.D.; Schwartz, G.G.; Olsson, A.G.; Zeiher, A.; Oliver, M.F.; Ganz, P.; Ezekowitz, M.; Chaitman, B.R.; Leslie, S.J.; Stern, T.; et al. Effects of atorvastatin on stroke in patients with unstable angina or non-q-wave myocardial infarction: A myocardial ischemia reduction with aggressive cholesterol lowering (MIRACL) substudy. Circulation 2002, 106, 1690–1695. [Google Scholar] [CrossRef] [PubMed]
- Athyros, V.G.; Tziomalos, K.; Gossios, T.D.; Griva, T.; Anagnostis, P.; Kargiotis, K.; Pagourelias, E.D.; Theocharidou, E.; Karagiannis, A.; Mikhailidis, D.P.; et al. Safety and efficacy of long-term statin treatment for cardiovascular events in patients with coronary heart disease and abnormal liver tests in the Greek Atorvastatin and Coronary Heart Disease Evaluation (GREACE) study: A post-hoc analysis. Lancet 2010, 376, 1916–1922. [Google Scholar] [CrossRef]
- Byington, R.P.; Jukema, J.W.; Salonen, J.T.; Pitt, B.; Bruschke, A.V.; Hoen, H.; Furberg, C.D.; Mancini, G.B. Reduction in cardiovascular events during pravastatin therapy. Pooled analysis of clinical events of the pravastatin atherosclerosis intervention program. Circulation 1995, 92, 2419–2425. [Google Scholar] [CrossRef] [PubMed]
- Heo, J.H.; Song, D.; Nam, H.S.; Kim, E.Y.; Kim, Y.D.; Lee, K.Y.; Lee, K.J.; Yoo, J.; Kim, Y.N.; Lee, B.C.; et al. Effect and safety of rosuvastatin in acute ischemic stroke. J. Stroke 2016, 18, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.W.; Hwang, J.; Lee, M.J.; Cha, J.; Bang, O.Y. Previous statin use and high-resolution magnetic resonance imaging characteristics of intracranial atherosclerotic plaque: The intensive statin treatment in acute ischemic stroke patients with intracranial atherosclerosis study. Stroke 2016, 47, 1789–1796. [Google Scholar] [CrossRef] [PubMed]
- Moonis, M.; Kane, K.; Schwiderski, U.; Sandage, B.W.; Fisher, M. HMG-CoA reductase inhibitors improve acute ischemic stroke outcome. Stroke 2005, 36, 1298–1300. [Google Scholar] [CrossRef] [PubMed]
- Stead, L.G.; Vaidyanathan, L.; Kumar, G.; Bellolio, M.F.; Brown, R.D., Jr.; Suravaram, S.; Enduri, S.; Gilmore, R.M.; Decker, W.W. Statins in ischemic stroke: Just low-density lipoprotein lowering or more? J. Stroke Cerebrovasc. Dis. 2009, 18, 124–127. [Google Scholar] [CrossRef] [PubMed]
- Ni Chroinin, D.; Asplund, K.; Asberg, S.; Callaly, E.; Cuadrado-Godia, E.; Diez-Tejedor, E.; di Napoli, M.; Engelter, S.T.; Furie, K.L.; Giannopoulos, S.; et al. Statin therapy and outcome after ischemic stroke: Systematic review and meta-analysis of observational studies and randomized trials. Stroke 2013, 44, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Blanco, M.; Nombela, F.; Castellanos, M.; Rodriguez-Yanez, M.; Garcia-Gil, M.; Leira, R.; Lizasoain, I.; Serena, J.; Vivancos, J.; Moro, M.A.; et al. Statin treatment withdrawal in ischemic stroke: A controlled randomized study. Neurology 2007, 69, 904–910. [Google Scholar] [CrossRef] [PubMed]
- Squizzato, A.; Romualdi, E.; Dentali, F.; Ageno, W. Statins for acute ischemic stroke. Cochrane Database Syst. Rev. 2011, 8, CD007551. [Google Scholar]
- Hong, K.S.; Lee, J.S. Statins in acute ischemic stroke: A systematic review. J. Stroke 2015, 17, 282–301. [Google Scholar] [CrossRef] [PubMed]
- Bonaventura, A.; Montecucco, F.; Dallegri, F. Update on the effects of treatment with recombinant tissue-type plasminogen activator (rt-PA) in acute ischemic stroke. Expert Opin. Biol. Ther. 2016, 16, 1323–1340. [Google Scholar] [CrossRef] [PubMed]
- Meseguer, E.; Mazighi, M.; Lapergue, B.; Labreuche, J.; Sirimarco, G.; Gonzalez-Valcarcel, J.; Lavallee, P.C.; Cabrejo, L.; Guidoux, C.; Klein, I.F.; et al. Outcomes after thrombolysis in AIS according to prior statin use: A registry and review. Neurology 2012, 79, 1817–1823. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Ramirez, S.; Delgado-Mederos, R.; Marin, R.; Suarez-Calvet, M.; Sainz, M.P.; Alejaldre, A.; Vidal-Jordana, A.; Marti-Vilalta, J.L.; Marti-Fabregas, J. Statin pretreatment may increase the risk of symptomatic intracranial haemorrhage in thrombolysis for ischemic stroke: Results from a case-control study and a meta-analysis. J. Neurol. 2012, 259, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Ward, M.D.; Jones, D.E.; Goldman, M.D. Overview and safety of fingolimod hydrochloride use in patients with multiple sclerosis. Expert Opin. Drug Saf. 2014, 13, 989–998. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.Y.; Kim, J.K.; An, Y.S.; Kim, Y.W. Effect of donepezil on wernicke aphasia after bilateral middle cerebral artery infarction: Subtraction analysis of brain F-18 fluorodeoxyglucose positron emission tomographic images. Clin. Neuropharmacol. 2015, 38, 147–150. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.H.; Park, Y.H.; Ohn, S.H.; Park, C.H.; Lee, P.K.; Kim, Y.H. Neural correlates of donepezil-induced cognitive improvement in patients with right hemisphere stroke: A pilot study. Neuropsychol. Rehabil. 2011, 21, 502–514. [Google Scholar] [CrossRef] [PubMed]
- Kraglund, K.L.; Mortensen, J.K.; Grove, E.L.; Johnsen, S.P.; Andersen, G. Talos: A multicenter, randomized, double-blind, placebo-controlled trial to test the effects of citalopram in patients with acute stroke. Int. J. Stroke 2015, 10, 985–987. [Google Scholar] [CrossRef] [PubMed]
- Simats, A.; Garcia-Berrocoso, T.; Montaner, J. Natalizumab: A new therapy for acute ischemic stroke? Expert Rev. Neurother. 2016, 16, 1013–1021. [Google Scholar] [CrossRef] [PubMed]
- Kern, R.; Nagayama, M.; Toyoda, K.; Steiner, T.; Hennerici, M.G.; Shinohara, Y. Comparison of the European and Japanese guidelines for the management of ischemic stroke. Cerebrovasc. Dis. 2013, 35, 402–418. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, Y.; Saito, I.; Kobayashi, S.; Uchiyama, S. Edaravone (radical scavenger) versus sodium ozagrel (antiplatelet agent) in acute noncardioembolic ischemic stroke (EDO trial). Cerebrovasc. Dis. 2009, 27, 485–492. [Google Scholar] [CrossRef] [PubMed]
- Isahaya, K.; Yamada, K.; Yamatoku, M.; Sakurai, K.; Takaishi, S.; Kato, B.; Hirayama, T.; Hasegawa, Y. Effects of edaravone, a free radical scavenger, on serum levels of inflammatory biomarkers in acute brain infarction. J. Stroke Cerebrovasc. Dis. 2012, 21, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Tsuruoka, A.; Atsumi, C.; Mizukami, H.; Imai, T.; Hagiwara, Y.; Hasegawa, Y. Effects of edaravone, a free radical scavenger, on circulating levels of MMP-9 and hemorrhagic transformation in patients with intravenous thrombolysis using low-dose alteplase. J. Stroke Cerebrovasc. Dis. 2014, 23, 2894–2899. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Chen, X. Edaravone offers neuroprotection for acute diabetic stroke patients. Ir. J. Med. Sci. 2016, 185, 819–824. [Google Scholar] [CrossRef] [PubMed]
- Satani, N.; Savitz, S.I. Is immunomodulation a principal mechanism underlying how cell-based therapies enhance stroke recovery? Neurotherapeutics 2016, 13, 775–782. [Google Scholar] [CrossRef] [PubMed]
- Azodi, S.; Jacobson, S. Cytokine therapies in neurological disease. Neurotherapeutics 2016, 13, 555–561. [Google Scholar] [CrossRef] [PubMed]
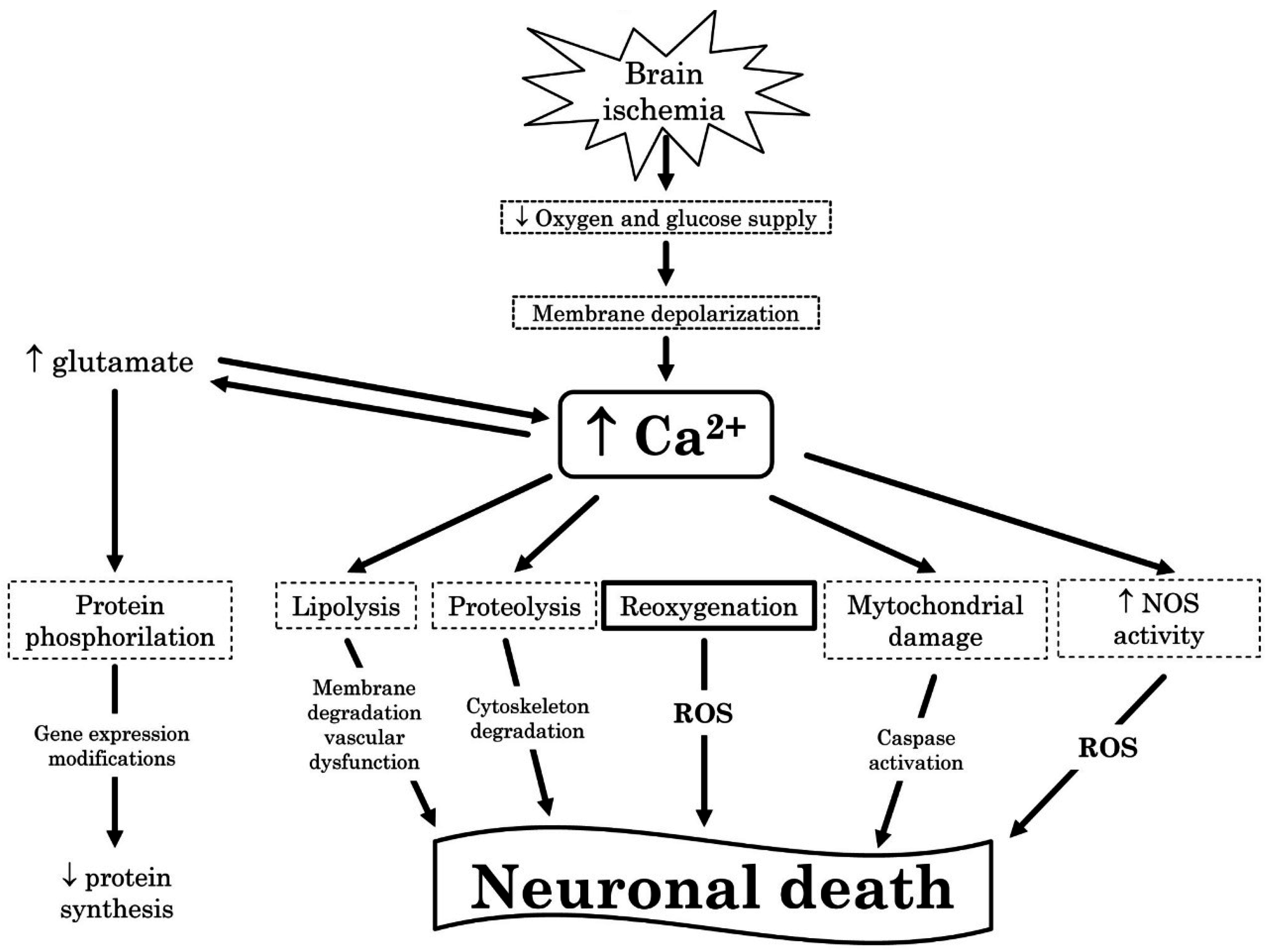
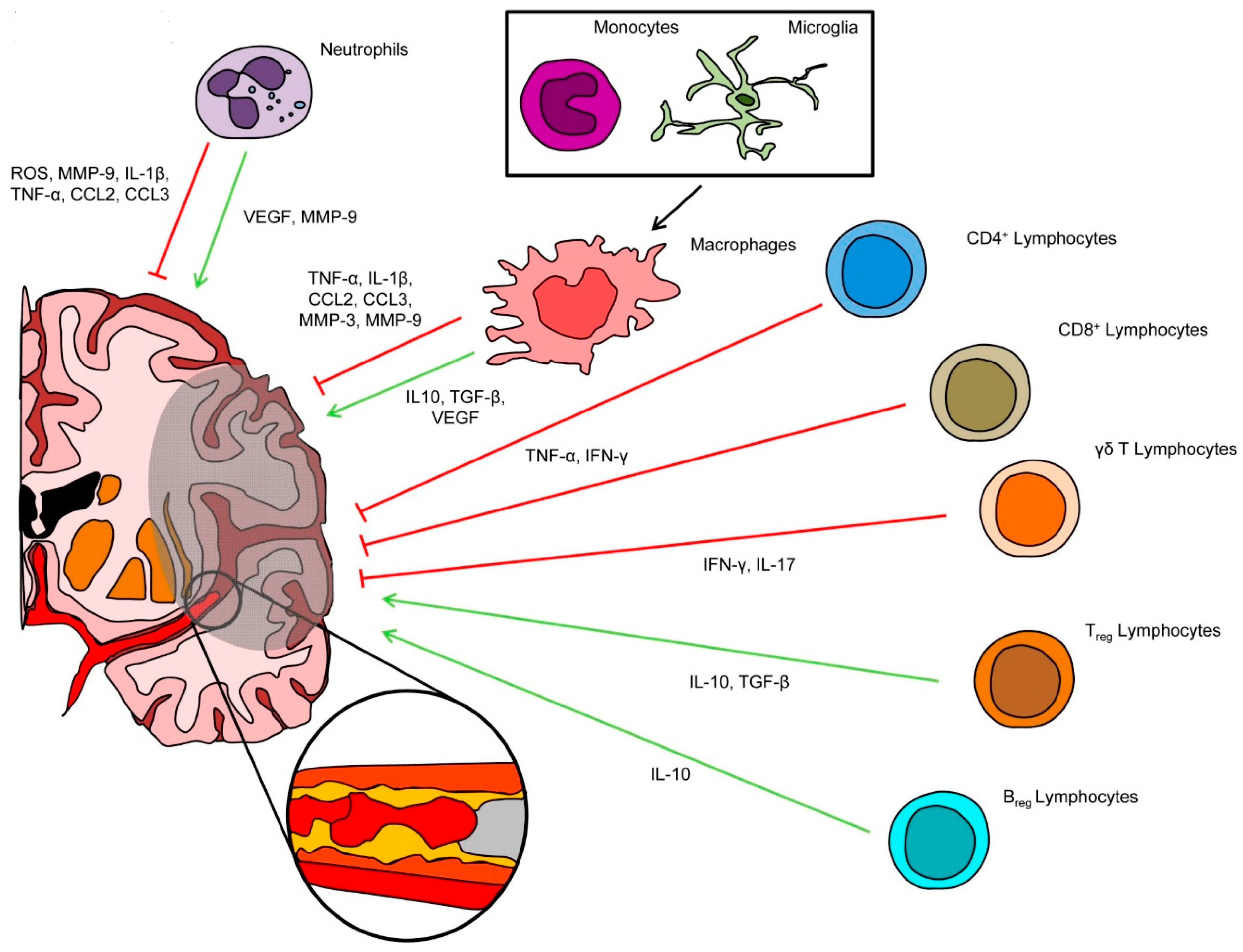
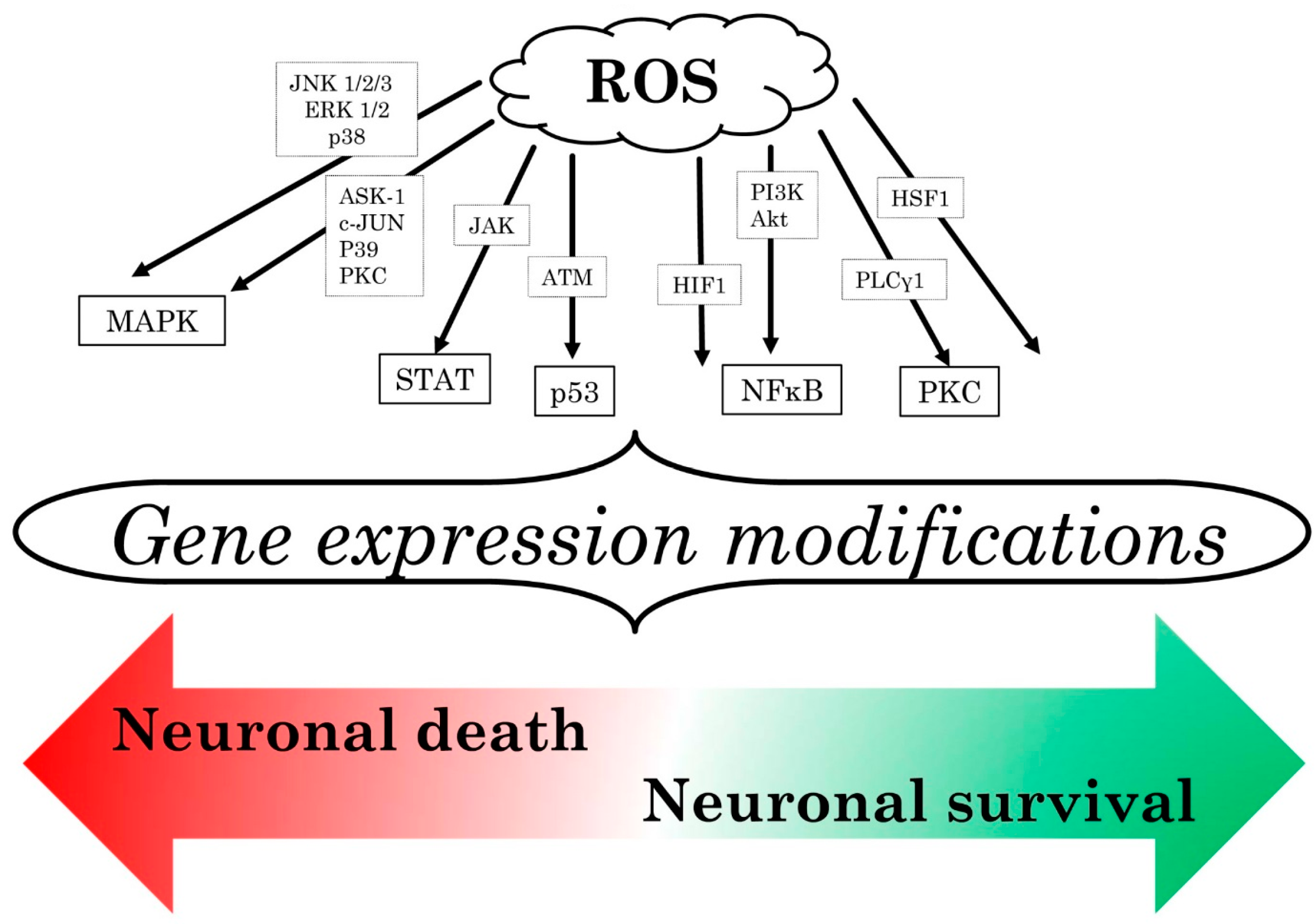
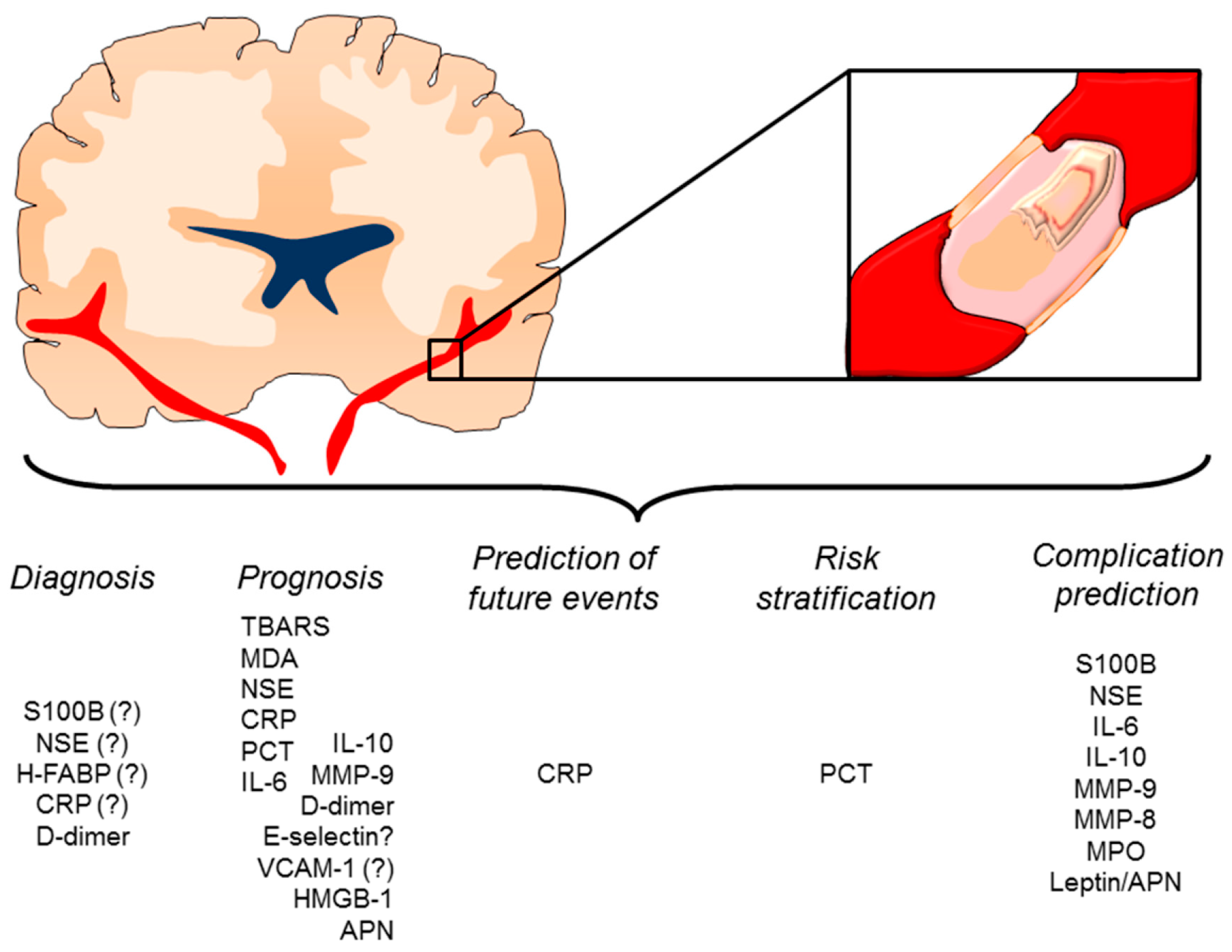
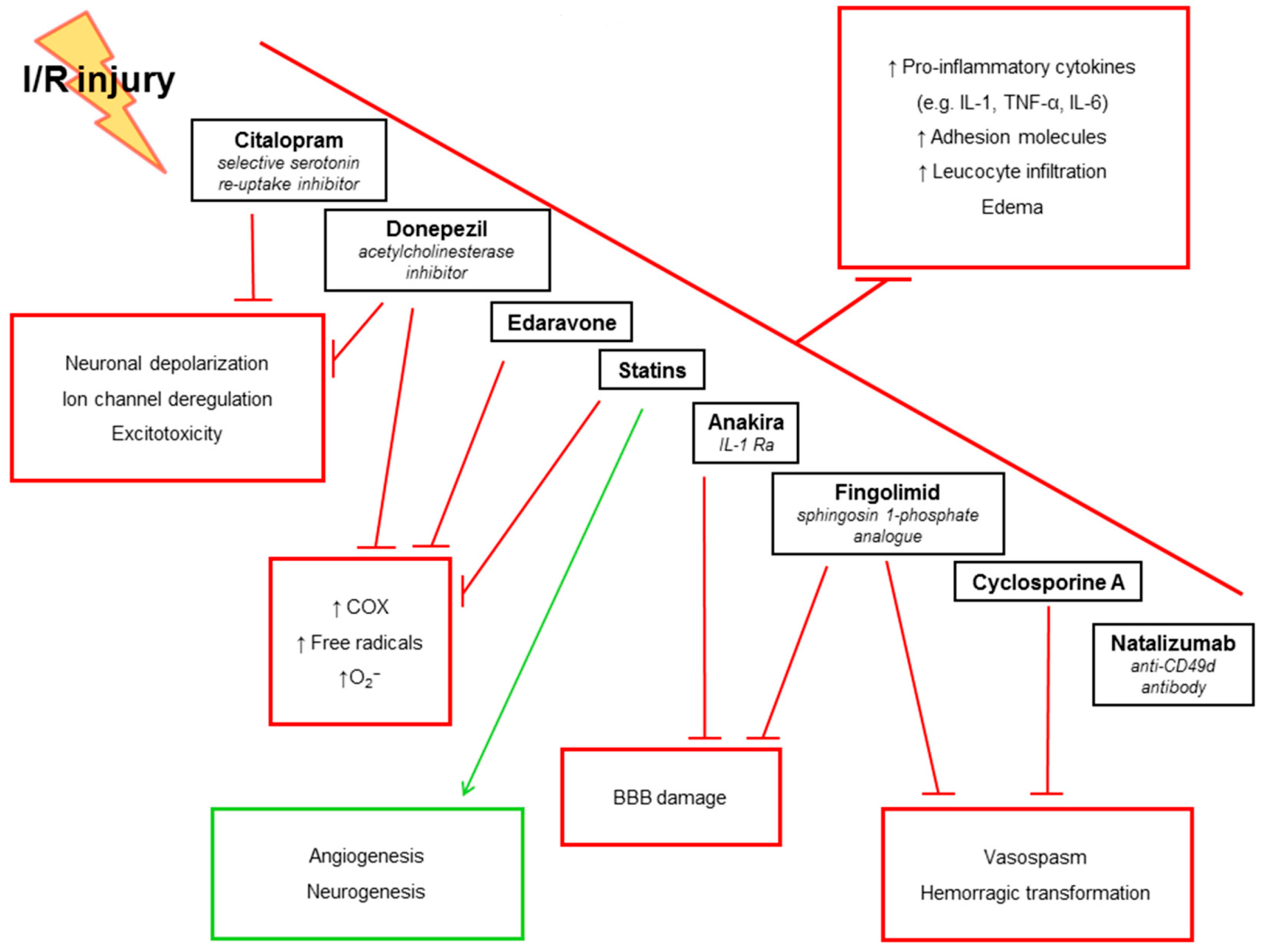

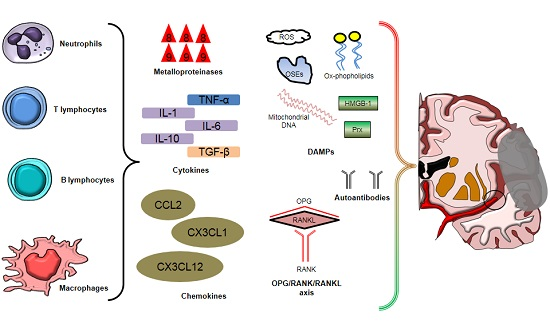{kind=link}
{kind=link}
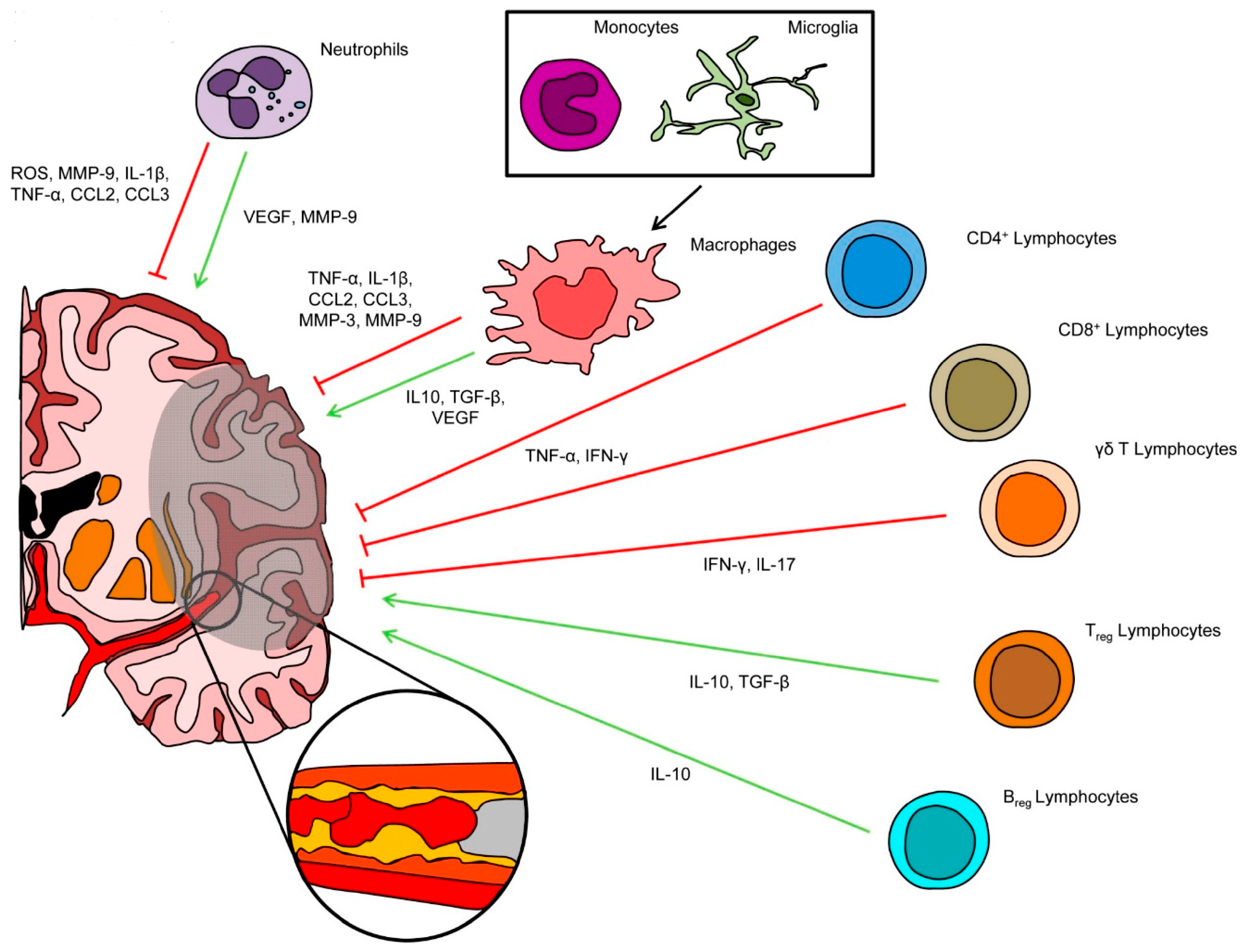{kind=link}
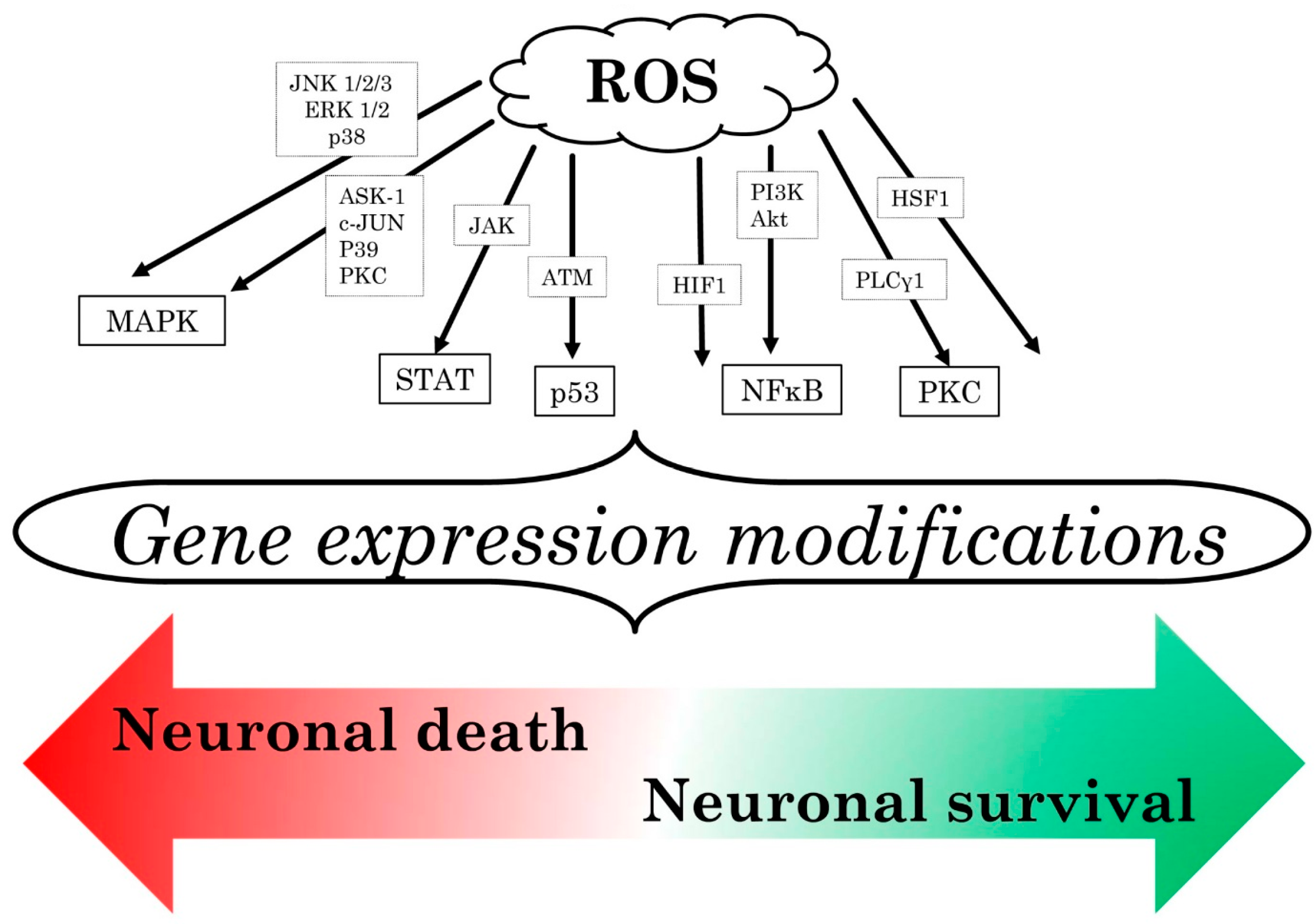{kind=link}
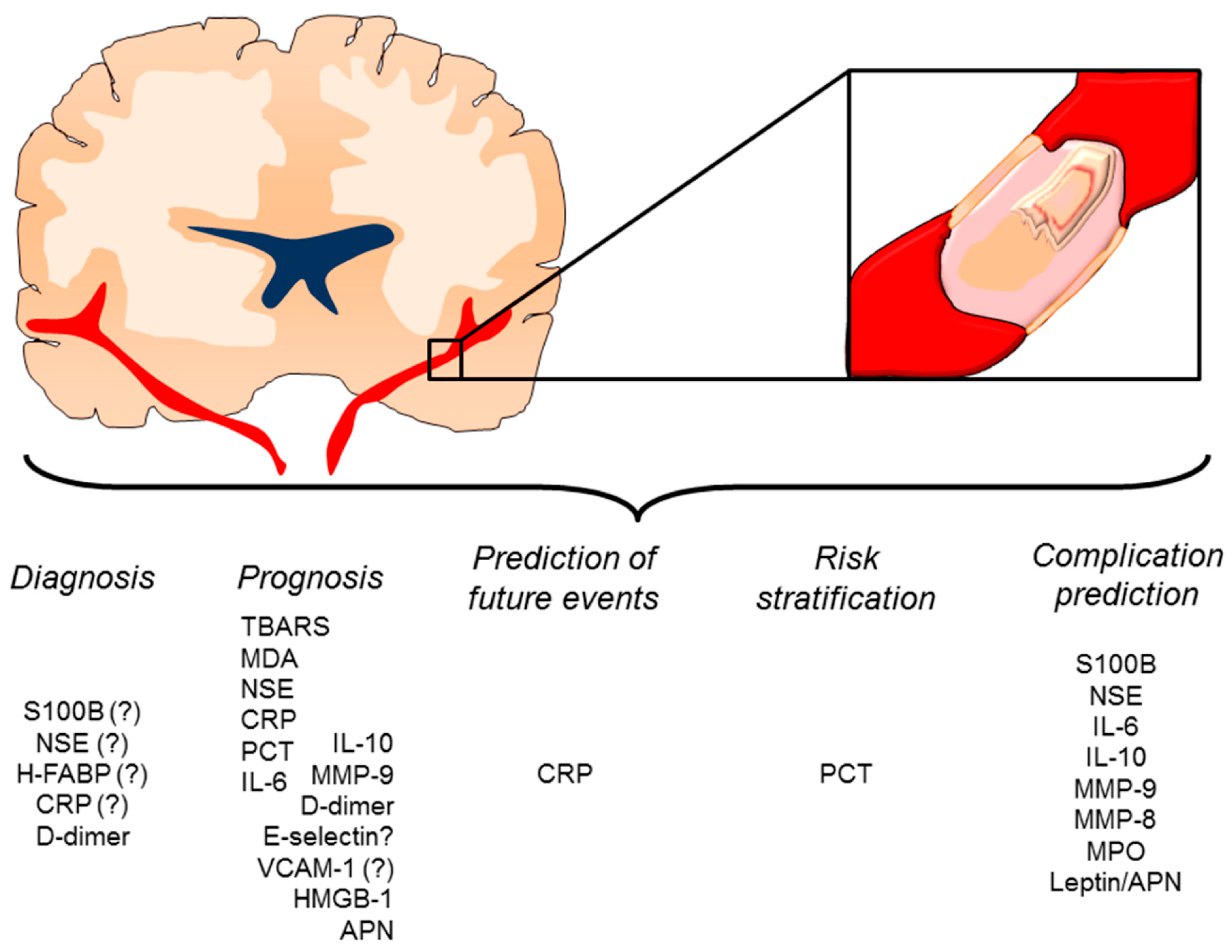{kind=link}
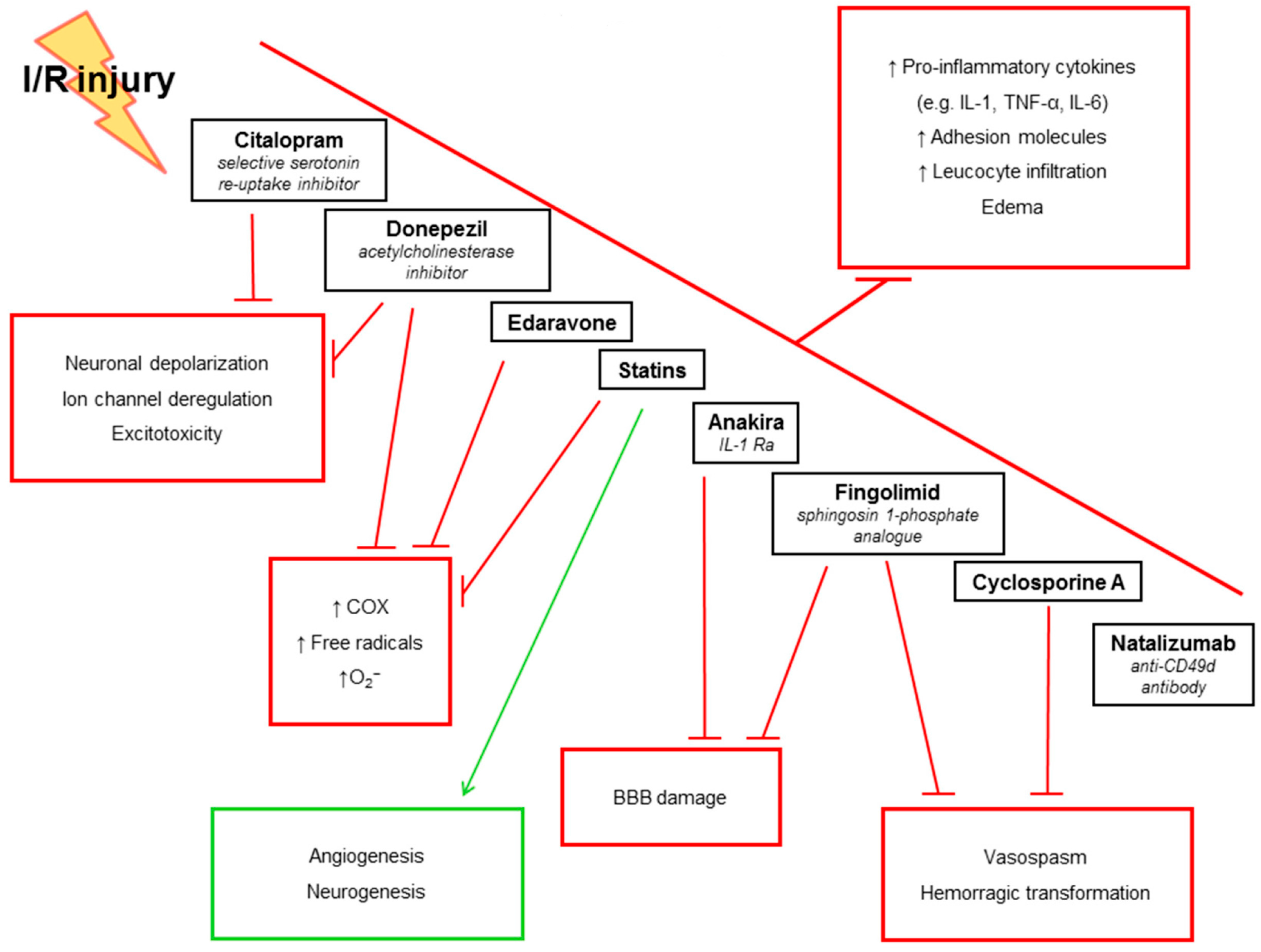{kind=link}
| Author | Year | Study Design | Biomarker | Outcome | Results |
|---|---|---|---|---|---|
| Astroglial activation | |||||
| Dassan et al. [146] | 2009 | Systematic review (13 longitudinal studies) | S100B | IS diagnosis HT mRS | S100B may be useful in predicting clot lysis (p = 0.001) and HT after thrombolysis (p = 0.017) with sensitivity and specificity of 46% and 82%, respectively. S100B also predict final infarct volume and eventually functional outcome (sensitivity 87%, specificity 78%). |
| Ye et al. [147] | 2015 | Meta-analysis (10 pooled case-control studies enrolling 773 patients with IS and 438 healthy controls) | S100B | IS diagnosis | Serum levels of S100B were higher in IS patients as compared to controls (SMD = 1.71 [95% CI 0.62–2.79]; p = 0.002). Subgroup analysis based on ethnicity revealed that S100B predict IS progression in Asians but not in Caucasians. However, no statistical significance was observed in large samples. |
| Kazmierski et al. [148] | 2015 | Prospective observational (458 IS patients) | S100B | HT | HT was associated with higher serum concentrations of S100B (AUC = 0.746; sensitivity 92.9%, specificity 48.1%). |
| Tsai et al. [149] | 2014 | Case-control (100 IS patients and 80 healthy subject) | TBARS thiol | 3-month NIHSS | As compared to controls, IS patients had higher TBARS and low free thiol. Furthermore, serum levels of thiol were lower in large- than small-vessel disease. TBARS at day 7 was identified as independent predictor of poor neurological outcome (OR 1.37 [95% CI 1.14–1.65]; p = 0.001). |
| Lorente et al. [150] | 2015 | Case-control (50 IS patients and 100 healthy controls) | MDA | 30-day mortality | MDA levels were significantly higher in IS patients as compared to healthy controls, as well as in non-surviving IS patients than in survivors (p < 0.001 for both). Furthermore, MDA predicted 30-day mortality (OR 7.23 [95% CI 1.84–28.73]; p = 0.005) with a sensitivity of 65% and a specificity of 75% (AUC of 0.77). |
| Neuronal cell injury | |||||
| Bharosay et al. [151] | 2012 | Case-control (150 IS patients and 101 controls) | NSE | NIHSS at days 1–7 | NSE was higher in IS patients (p < 0.001), also correlating with stroke severity at admission (r = 0.919; p < 0.001) and after 7 days (r = 0.706; p < 0.001). |
| Singh et al. [152] | 2013 | Case-control (100 IS patients and 101 controls) | NSE | NIHSS at admission | Serum NSE was higher in IS group, also correlating with IS severity (r = 0.800; p < 0.001). |
| Zaheer et al. [153] | 2013 | Prospective observational (75 IS patients) | NSE | 30-day mRS | A positive correlation was found between NSE infarct size (r = 0.955, p < 0.001), whereas a negative relationship with GCS was demonstrated (r = −0.806, p < 0.001). Finally, there was a positive correlation between NSE and neurological outcome (r = 0.744, p < 0.001). |
| Kim et al. [155] | 2014 | Prospective observational (83 IS patients) | NSE | HT | In patients with HT, NSE time course was characterized by two peak levels. This specific pattern was significantly associated with the occurrence of HT (OR 6.84 [95% CI 1.12–41.70]; p = 0.04). |
| Lu et al. [156] | 2015 | Prospective observational (74 IS patients) | NSE | 3-month mRS | NSE sowed predictive accuracy toward poor neurological outcome (77.1% sensitivity and 59.4% specificity). However, the adjusted RR for NSE was not effective in predicting poor neurological outcome. |
| Haupt et al. [157] | 2016 | Prospective observational (31 IS patients) | NSE | mRS days 7 and 10 | NSE peak at day 4 in the good outcome patients, whereas a continuous increase was observed in those with poor outcome. Sensitivity of NSE analysis showing an increase over time to >90% at day 4. |
| Park et al. [154] | 2013 | Case-control (111 IS patients and 127 controls) | H-FABP | Stroke diagnosis | H-FABP was significantly higher in the IS group (OR 1.08 [95% CI 1.02–1.13]; p < 0.001). However, H-FABP was not sensitive enough to discriminate stroke from control group or IS subtype. |
| Author | Year | Study Design | Biomarker | Outcome | Results |
|---|---|---|---|---|---|
| Ozkan et al. [158] | 2013 | Prospective observational (62 IS patients) | CRP | Stroke subtype 3 months NIHSS | CRP was unable to predict IS subtype and functional disability at 3 months after IS. |
| Taheraghdam et al. [159] | 2013 | Prospective observational (102 IS patients) | CRP | 3 months mRS | Early CRP measurement failed to predict IS outcome. |
| VanGilder et al. [160] | 2014 | Systematic review (5 longitudinal/case-control studies) | CRP | 3 months mRS | In all studies, acutely elevated CRP was positively associated with long-term (30 days to 3 months) unfavorable outcome (OR ranging from 2.3 to 3.5; p < 0.05). |
| Karlinski et al. [161] | 2014 | Prospective observational (301 IS patients undergoing thrombolysis) | CRP | HT 3 months mRS | CRP measurement failed to independently predict the outcome of IS patients treated with thrombolysis. |
| Pandey et al. [162] | 2014 | Case control (880 IS patients, 32 HS and 50 healthy controls) | CRP | Day 7 NIHSS | CRP was significantly higher in stroke patients as compared to controls (p < 0.001 for both). When categorized on the basis of NIHSS, high serum levels of CRP were found in severe stroke group (p < 0.001 for both). |
| Li et al. [163] | 2015 | Prospective observational (374 IS patients) | PCT CRP | 1-year mortality | Serum PCT levels were higher in non-survival patients (p < 0.001). Long-term mortality was independently predicted by both PCT (OR 3.64 [95% CI 1.54–5.88]) and CRP (OR 12.33 [95% CI 2.44–37.66]). As compared to CRP, PCT was a better predictor of mortality with a sensitivity of 81.5% and a specificity of 84.7% (AUC 0.887). |
| Rocco et al. [164] | 2015 | Prospective observational (1242 IS patients) | CRP | 3 months mRS | Follow-up CRP, assessed during the first 7 days showed significant predictive value toward worse mRS (OR 2.67 [95% CI 1.76–4.06]) and mortality (OR 2.53 [95% CI 1.50–4.25]), with a c-statistic of 0.71 and 0.70, respectively. |
| Deng et al. [165] | 2015 | Case control (378 IS patients and 200 controls) | PCT | 3-month mRS | Serum PCT was higher in IS group and correlated with lesion size and NIHSS (p < 0.001 for all). PCT predict worse functional outcome (OR 3.45 [95% CI 2.29–4.77]; p < 0.001) with a sensitivity of 75.4% and a specificity of 80.7% (AUC 0.845). |
| Wang et al. [166] | 2016 | Case-control (376 IS patients and 200 controls) | PCT CRP | 1-year mRS 1-year mortality | Serum PCT was higher in patients with IS, and correlated with lesion size (p < 0.001). Both PCT and CRP correlated with and NIHSS (p < 0.001 for both). PCT predict worse functional outcome (OR 4.31 [95% CI 1.58–9.12]; p < 0.001). Mortality was independently predicted by both PCT (OR 1.10 [95% CI 1.05–1.15]) and CRP (OR 1.31 [95% CI 1.03–1.74]). |
| Matsuo et al. [167] | 2016 | Prospective observational (3653 IS patients) | CRP | 3 months mRS | At multivariate analysis, CRP was associated with a poor outcome (OR 2.03 [95% CI 1.55–2.67]). |
| Geng et al. [168] | 2016 | Prospective observational (301 IS patients) | CRP | Discharge mRS Recurrent IS | At multivariate analysis, poor outcome at discharge was independently predicted by CRP (OR 4.89 [95% CI 3.06–7.81]). |
| Whiteley et al. [169] | 2009 | Systematic review (4 longitudinal studies) | IL-6 | 3-month mRS | Il-6 was identified as independent predictor of poor neurological outcome after IS (OR 1.05 [1.01–1.09]). |
| Park et al. [170] | 2013 | Prospective observational (175 IS patients) | IL-6 | 3-month mRS | In multivariate analysis IL-6 was independently associated with poor outcome (OR 1.75 [1.25–2.25]; p = 0.001). |
| Bustamante et al. [171] | 2014 | Meta-analysis (24 pooled longitudinal studies enrolling 4523 patients) | IL-6 | 1 to 6 months mRS | The highest quartile of IL-6 was an independent predictor of poor outcome (OR 2.35 [1.81–3.03], p < 0.001), but its additional predictive value was modest in terms of AUC (0.840 to 0.847). |
| Pusch et al. [172] | 2015 | Case-control study (76 patients with IS, 44 with carotid stenosis and 66 with Parkinson disease) | IL-6 | Post-IS infection | High concentration of IL-6, MCP-1, and S100B at 6 h, and increase of P-selectin during the first 72 h were associated with post-stroke infections. Specifically, IL-6 predict the occurrence of post-stroke infection with an AUC of 0.920. |
| Lehmann et al. [173] | 2015 | Case-control study (95 patients with IS and 96 controls) | IL-6 CRP MMP-9 | Stroke subtype | As compared to controls, LAAS, LAC and CEI had higher serum levels of IL-6, CRP, and MMP-9 (p < 0.05 for all). |
| Worthmann et al. [174] | 2015 | Prospective observational (56 IS patients) | IL-6 IL-10 CRP | Post-IS infection | IL-10, IL-6 and CRP show a different time course in patients with and without post-stroke infection. Furthermore, post-stroke infection is independently predicted by serum IL-10 (AUC 0.76) and CRP (AUC 0.74). |
| Fahmi et al. [175] | 2016 | Case-control (50 IS patients and 20 healthy controls) | IL-6 | 15-day NIHSS | At multivariate regression analysis, IL-6 was identified as independent predictor of short-tern neurological outcome (β = 0.451; p < 0.001). |
| Rodríguez-Yáñez et al. [176] | 2013 | Prospective observational (184 thrombolysed IS patients) | IL-10 | 3-month mRS | High levels of IL-10 predicted good functional outcome with a specificity of 88% and a sensibility of 86% (OR 2.86 [1.06–7.82]). |
| Ashour et al. [177] | 2016 | Case-control (60 IS patients and 30 healthy control) | IL-10 | Post-IS infection | The occurrence of infectious was independently predicted by increased levels of IL-10 (OR 6.01 [1.53–23.51]; p = 0.01). |
| Inzitari et al. [179] | 2013 | Prospective observational (327 thrombolysed IS patients) | MMP-9 | HT 3-months death 3-month mRS | Overtime MMP-9 variations (during 24 h across thrombolysis) significantly predicted HT (OR 1.40 [1.02–1.92]) and death (OR 1.58 [1.11–2.26]). |
| Carbone et al. [178] | 2015 | Case-control (60 thrombolysed IS patients and 30 not) | MMP-9 | HT | Peak of MMP-9 (and also MMP-8 and MPO) at day 1 in thrombolysed patients was associated with increased rate of early HT (p = 0.023). |
| Author | Year | Study Design | Biomarker | Outcome | Results |
|---|---|---|---|---|---|
| Wiseman et al. [180] | 2014 | Meta-analysis (42 pooled case-control studies enrolling 2196 lacunar IS and 2500 healthy controls) | t-PA, PAI-1, vWF d-dimer E-, P-selectins ICAM-1 VCAM-1 | Stroke subtype | As compared to other subtypes, lacunar IS was characterized by higher markers of coagulation/fibrinolysis (t-PA, PAI-1, and d-dimer) and lower marker of endothelial dysfunction (vWF, E- and P-selectins, ICAM-1 and VCAM-1). |
| Liu et al. [181] | 2016 | Case-control (317 IS of different subtypes) | d-dimer | Stroke subtype | d-dimer was different in each group with the highest levels in the CE group (<0.001). d-dimer also independently predicted CE (OR 6.83 [95% CI 2.96–15.77]). |
| Yuan et al. [182] | 2014 | Prospective observational (300 IS patients) | d-dimer | Stroke subtype Day 14 NIHSS | Serum levels of d-dimer were higher in the CE group; Furthermore they correlated with neurological improvement (r = −0.410; p = 0.013). |
| Yang et al. [183] | 2014 | Prospective observational (220 IS patients) | d-dimer | 3-month mRS 3-month mortality | Admission d-dimer was higher in patients with poor prognosis also in adjusted analysis (OR 2.18 [95% CI 1.55–2.83]) with high prognostic accuracy (AUC: 0.830). d-dimer also predicted mortality analysis (OR 3.22 [95% CI 2.05–6.43]). |
| Richard et al. [184] | 2015 | Prospective observational (100 IS patients) | E-, P-selectin ICAM-1 VCAM-1 | 3-month mRS | Early after SI, E-selectin was found to be an independent predictor of poor outcome (OR = 24.95 [95% CI 2–354]; p = 0.022 and AUC = 0.780), as was VCAM-1 during the third week after onset (OR = 8 [95% CI 2–37]; p = 0.01 and AUC 0.730). |
| Wang et al. [185] | 2016 | Case-control (1173 IS patients) | d-dimer | 30-day mRS | d-dimer was effective in predicting poor neurological score (OR = 1.60 [95% CI 1.36–1.89]; p < 0.001). |
| Hsu et al. [186] | 2016 | Retrospective observational (307 thrombolysed IS patients) | d-dimer | 3-month mRS HT | At adjusted analysis, higher levels of d-dimer at admission predicted poor outcome (OR = 1.90 [95% CI 1.27–2.86]; p = 0.002) and HT (OR = 2.97 [95% CI 1.15–7.70]; p = 0.025). |
| Author | Year | Study Design | Biomarker | Outcome | Results |
|---|---|---|---|---|---|
| Schulze et al. [201] | 2013 | Prospective observational (114 IS patients) | HMGB1 | 3-month mRS | Plasma HMGB1 weakly correlated with infarct volume and stroke severity at day 3 after IS. However, HMGB1 failed to independently predict long-term outcome. |
| Huang et al. [202] | 2013 | Prospective observational (338 IS patients) | HMGB1 | 1-year mRS | HMGB1 was independently associate with worse clinical outcome (OR 2.21 [95% CI 1.13–4.20]; p = 0.002) with 71.4% sensitivity and 83.7% specificity (AUC, 0.83). Furthermore, in a combined model, HMGB1 significantly improved the AUC of NIHSS score to 0.929 (p < 0.001). |
| Sapojnikova et al. [203] | 2014 | Case-control (42 IS patients and 32 healthy controls) | HMGB1 | GOS | The increased HMGB1 levels and plasma MMP-9 are associated with a poor functional outcome and significantly correlated with each other (p < 0.05). |
| Marousi et al. [204] | 2010 | Prospective observational (82 IS patients) | Adiponectin | mRS at 1 and 6 months | Higher Adiponectin was indicative worse outcome on month 1 (OR = 1.14 [95% CI 1.01–1.29]; p = 0.031). However, adiponectin failed to predict IS severity, infarct size, recurrent IS, mortality, state, disability or functional outcome at 6 months. |
| Kuwashiro et al. [205] | 2013 | Case-control (171 IS patients and 171 healthy controls) | Adiponectin | 3-month mRS | As compared to controls, average adiponectin values at onset were significantly lower and higher in patients with ATBI (p = 0.047) and CE (p = 0.008) IS, respectively. At onset adiponectin correlated with NIHSS (r = 0.420, p = 0.003) and was higher in patients with worse long-term outcome (p = 0.007). |
| Carbone et al. [132] | 2015 | Prospective observational (35 non-obese ATBI patients) | Adiponectin Leptin | 3-month mRS | Serum leptin and leptin/adiponectin ratio at day 1 inversely correlated with both radiological and clinical parameters. Leptin/adiponectin ratio also independently predicted worse neurological outcome (OR = 0.15 [95% CI 0.03–0.83]; p = 0.030) and the occurrence of HT (OR = 0.08 [95% CI 0.01–0.81]; p = 0.028). |
| Study | Year | Treatment | Sample Size | Outcome |
|---|---|---|---|---|
| IL-1Ra | ||||
| Garcia et al. [256] | 1995 | 13 with pMCAO and treated with IL-1Ra; 13 with pMCAO and treated with CSE buffer or placebo group (n = 13); 2 sham-operated animals treated with IL-1Ra or CSE (n = 2) | 30 outbred male Wistar rats and fed Agway rat chow during the 4–6 quarantine days | IL-1Ra in rats with pMCAO significantly decreased the number of necrotic neurons both at 24 h and 7 days after the arterial occlusion (p < 0.0001). Neurological scores were also significantly improved with and a non-significant decrease in the number of PMN leukocytes in the ischemic hemisphere was observed. |
| Yamasaki et al. [247] | 1995 | 60 min of tMCAO followed by reperfusion; first IL-1β, then anti-IL1β was injected | 120 adult male Wistar rats | tMCAO induced an increase in brain water content, necrosis, and neutrophilic infiltration in the cortex perfused by the MCA and the DCP and VCP. rIL-1β into the left lateral ventricle immediately after reperfusion markedly enhanced ischemic brain edema formation and infarction size in MCA zone, DCP, and VCP in a dose-dependent manner (p < 0.01). Anti-IL-1β attenuated the post-ischemic increase of brain water content and decreased the infarction size (p < 0.01). The number of neutrophils infiltrating the ischemic area decreased with anti-IL-1β. |
| Relton et al. [236] | 1996 | MCAO or sham surgery. Animals were injected subcutaneously with either vehicle or rIL-1Ra at 0, 4, 8, 12, and 18 h after ischemia. In separate experiments, initial treatment was delayed until 30 min, 1 h, or 4 h after ischemia and treatments were repeated until 18 h | Male Sprague-Dawley rats | rIL-1Ra significantly inhibited infarct size by 46% at 24 h (p < 0.05), cerebral edema formation by 49% at 24 h (p < 0.05). Infarction inhibition by rIL-1Ra was dependent on dose and time of administration. |
| Pradillo et al. [234] | 2012 | Lean and Cp rats received placebo or IL-1Ra (25 and 12.5 mg/kg) subcutaneously at reperfusion and 6 h later and allocated to different groups: lean + tMCAO + placebo; lean + tMCAO + IL-1Ra; Cp + tMCAO + placebo; and Cp + tMCAO + IL-1Ra. For the delayed administration study, animals were injected subcutaneously with placebo or IL-1Ra at 3 h of reperfusion and again 3 h later | Male, lean and Cp rats | IL-1Ra at reperfusion resulted in a 50% reduction of infarct volume as measured by MRI both in lean and Cp compared with placebo-treated animals (p < 0.05). IL-1Ra decreased the number of MMP-9-positive neutrophils when compared with placebo (p < 0.05). In both lean and Cp rats, IL-1Ra largely reduced the microglial activation compared with the placebo-treated groups (p < 0.05). In 16-month-old lean rats, delayed IL-1Ra significantly reduced the number of MMP-9-positive blood vessels and the number of MMP-9-positive neutrophils when compared with the placebo group (p < 0.05). |
| Statins | ||||
| Endres et al. [219] | 1998 | After tMCAO followed by reperfusion, mice were injected subcutaneously with 0.1 mL of activated simvastatin or lovastatin (0.2–20 mg/kg) or a corresponding volume of PBS once daily for 3 or 14 days | Not declared | In a concentration-dependent manner, simvastatin for 14 days reduced cerebral infarct size by 18, 27 and 46% (p < 0.05) and increased NOS activity (p < 0.05). Simvastatin 20 mg/kg increased basal hemispheric CBF by 31% (p < 0.05). Lovastatin 20 mg/kg daily for 14 days also decreased cerebral infarct size and neurological deficits, even if to a lesser extent than simvastatin. |
| Kawashima et al. [222] | 2003 | Two groups, one statin-treated (cerivastatin 2 mg/kg by gavage once daily) and another vehicle-treated | Stroke-prone spontaneously hypertensive rats (4 weeks of age) | The incidence of stroke and stroke size decreased (p < 0.01). High-dose statin treatment delayed early death and reduced the occurrence of stroke-associated symptoms (p < 0.01) and decreased stroke-associated infiltration of inflammatory cells (p < 0.05). Statin treatment increased eNOS protein levels and eNOS activity (p < 0.05). Superoxide production was reduced in statin-treated rats (p < 0.01). |
| Amin-Hanjani et al. [211] | 2001 | Two groups: mevastatin at a dose of 2 or 20 mg/kg daily and a corresponding concentration of vehicle for 7, 14, or 28 days before tMCAO | Wild-type male mice and eNOS-deficient male mice | Mevastatin increased levels of eNOS mRNA and protein, reduced infarct size, and improved neurological deficits in a dose- and time-dependent manner especially with 14- and 28-day high-dose treatment (26% and 37% infarct reduction, respectively, p < 0.05). Cholesterol levels were reduced only after 28 days of treatment (p < 0.05), but did not correlate with infarct reduction. Baseline absolute cerebral blood flow was 30% higher after 14-day high-dose treatment (p < 0.05). |
| Prinz et al. [235] | 2008 | After tMCAO followed by reperfusion, mice were treated with intravenously or intraperitoneally rosuvastatin given up to 6 h after MCAO (0.02–20 mg/kg) | Wild-type mice aged 6 to 8 weeks | Intravenous rosuvastatin significantly reduced lesion size up to 4 h after MCAO in doses as low as 0.2 mg/kg (p < 0.05). Intraperitoneal administration provided protection only on reperfusion at a dose of 20 mg/kg (p < 0.05). Lesion protection was evident 5 days after brain ischemia and was associated with functional improvements at 2.0 mg/kg dose (p < 0.05). Neuroprotection with intravenous rosuvastatin was achieved with peak plasma concentrations <0.5 ng/mL and was associated with increased levels of phosphorylated Akt kinase and eNOS in the vasculature (p < 0.05). |
| Asahi et al. [212] | 2005 | Heterologous blood clots were used to induce MCAO after long-term simvastatin (20 mg/kg), atorvastatin (20 mg/kg) or vehicle treatment subcutaneously | Male SV-129 mice and male C57Bl/6 mice | In wild-type mice, both simvastatin and atorvastatin reduced ischemic lesions and residual clot after 14 days (p < 0.05). In eNOS knockout mice, atorvastatin reduced the volume of ischemic tissue and improved neurologic outcomes after arterial occlusion (p < 0.05). Both statins did not have protective effects in t-PA knockout mice after embolic focal ischemia, but only in a filament model where focal ischemia was achieved via mechanical occlusion (p < 0.05). |
| Chen et al. [215] | 2003 | 24 h after MCAO, rats were fed atorvastatin (1, 3 or 8 mg/kg) daily for 7 days. Rats were also treated with simvastatin 1 mg/kg with the same protocol | 48 Adult male Wistar rats | Rats treated with 1 and 3 mg/kg atorvastatin and 1 mg/kg simvastatin improved functional recovery (p < 0.05). VEGF production within the ischemic boundary area at 14 days after stroke increased in the 1 mg/kg atorvastatin group (p < 0.05) as well as cyclic guanosine monophosphate, angiogenesis, neurogenesis, and synaptophysin levels (p < 0.05). |
| Sironi et al. [242] | 2003 | Two groups of rats were treated with vehicle alone or simvastatin for 3 days before MCAO, while other two groups underwent MCAO and were treated with vehicle or simvastatin at 3 and 25 h after the induction of the injury. The brain infarct size was evaluated using MRI | Male Sprague-Dawley rats | Treatment with simvastatin (20 mg/kg) after MCAO prevented the increase in brain infarct volume occurring at 24 h and induced a 46.6% reduction after 48 h (p < 0.01). The neuroprotective effects of simvastatin were paralleled by an increase in eNOS immunoreactivity, detectable in the brain of simvastatin-treated rats. |
| Reuter et al. [238] | 2015 | Cultured hBMECs pretreated with simvastatin and subjected to OGD | hBMECs | Simvastatin significantly blocked the expression of MMP-2 under OGD (p < 0.004). MMP-9 synthesis rate was low and unaffected by simvastatin treatment, while the gene expression and protein secretion of TIMP-1 and TIMP-2 were both strongly induced (p < 0.001). |
| Fingolimod (FTY720) | ||||
| Rolland et al. [239] | 2013 | Fingolimod was given intraperitoneally at a dose of 1 mg/kg as single dose 1 h after ICH induction or daily administration 1, 24, and 48 h after ICH induction | 103 male CD-1 mice and 28 male Sprague-Dawley rats | Fingolimod enhanced neurological functions and reduced brain edema at 24 and 72 h following experimental ICH in CD-1 mice (p < 0.05). Fewer lymphocytes were found in blood and brain samples of treated animals (p < 0.05). Fingolimod decreased ICAM-1, IFN-γ, IL-17 levels 72 h after ICH (p < 0.05). Treated Sprague-Dawley rats showed less spatial and motor learning deficits along with significantly reduced brain atrophy and neuronal cell loss within the basal ganglia (p < 0.05). |
| Campos et al. [214] | 2013 | 3 cohorts: pMCAO not treated with t-PA; tMCAO followed by early (30 min after thrombin) t-PA administration; and tMCAO followed by delayed (3 h after thrombin) t-PA administration. Each of these cohort received fingolimod at different time points | C57BL/6 male mice | Fingolimod reduced neurological deficits and infarct volume after in situ thromboembolic MCAO (p < 0.05). Combination of fingolimod and t-PA improved neurological outcomes of the thrombolytic therapy and the risk of hemorrhagic transformation associated with delayed administration of t-PA (p < 0.05). |
| Donepezil | ||||
| Wang et al. [244] | 2014 | 3 groups: the sham operation group (SO), the model group (MG) and the treatment group (TG). Pathological appearance of the hippocampal CA1 region and calpain I and CDK5/p25 expression were observed on the 4th, 6th and 8th week from I/R surgery | 250 3-month old male mice | At each postoperative time point, the normal neuron count of the hippocampal CA1 region in the treatment group increased significantly (p < 0.05), whereas calpain I and CDK5/p25 expression, SOD activity and MDA content were significantly lower than those in the model group (p < 0.05). |
| Min et al. [231] | 2012 | After transient global ischemia, donepezil (5 mg/kg once a day) was administered intragastrically for 21 days | Male Mongolian gerbils | Donepezil significantly inhibited delayed neuronal death in the hippocampal CA1 region (p < 0.01). Memory impairment was significantly improved by donepezil treatment (p < 0.05–0.01). Western blot analysis showed that donepezil treatment prevented reductions in p-CaMKII and p-CREB protein levels in the hippocampus (p < 0.01). |
| Yuan et al. [252] | 2011 | Cultured cells were exposed to both OGD and electrophysiological experiment | HEK293 cells from a human embryonic kidney cell line | Donepezil showed to attenuate OGD-induced apoptosis in Kv2.1/HEK293 cells and to inhibit Kv2.1 currents in a dose-dependent manner under normoxic condition (p < 0.01). Donepezil further inhibited Kv2.1 currents after OGD treatment (p < 0.05). |
| Akasofu et al. [210] | 2008 | Prolonged opening of sodium channels with veratridine led to depolarization-induced neuronal cell injury, which was prevented by 0.1 µM tetrodotoxin | Cortical cell cultures from fetal rats of the Wistar strain | Pre-treatment with donepezil (0.1–10 µM) for 1 day significantly decreased cell death and increased cell viability in a concentration-dependent manner (p < 0.05). At 0.1–10 µM, donepezil concentration-dependently decreased the veratridine-induced increase of calcium concentration, whilst at 10 µM it reduced the veratridine-induced increase of sodium concentration (p < 0.05 for both). |
| Lee et al. [225] | 2007 | After permanent ligation of bilateral common carotid arteries, rats were administered cilostazol (30 mg/kg/day orally) and donepezil (0.3 mg/kg/day intraperitoneally) | Rats | Concurrent treatment with cilostazol and donepezil prevented neuropathological alterations in the white matter by activation of phosphorylated CREB and Bcl-2, resulting in improvement of spatial learning memory (p < 0.05). |
| Citalopram | ||||
| Espinera et al. [220] | 2013 | After focal ischemic stroke, citalopram 10 mg/kg was injected intraperitoneally 24 h after stroke and then daily for 7, 14, 21, or 28 days | Adult male C57 mice | Citalopram had no significant effect on infarct formation or edema 3 days after stroke, but enhanced sensorimotor functional recovery after 14 days (p < 0.05). Citalopram improved neuroblast proliferation and migration (p < 0.01) as well as neurogenesis (p < 0.05) and peri-infarction vessel density (p < 0.05) in the post-ischemic brain. |
| Kronenberg et al. [223] | 2012 | Mice were subjected to 30-min MCAO/reperfusion and serial MRI scans; a subset of animals received citalopram from day 7 after MCAO | Male 129/SV mice | Delayed citalopram reversed the behavioral phenotype blocked the degeneration of dopaminergic midbrain neurons, and attenuated striatal atrophy after 4 months (p < 0.05). |
| Natalizumab | ||||
| Becker et al. [213] | 2001 | Rats underwent 3 h of MCAO followed by 45 h of reperfusion. 2 h after ischemia, one group received anti-α4 integrin antibody intraperitoneally and another an isotype control antibody | Male Lewis rats | Neurological deficits were less frequent in treated rats at 24 (p < 0.01) and 48 h (p = 0.01) after ischemia. White blood cell count was higher in treated rats (p < 0.01) with a lymphocyte/monocyte predominance. Infarction volume was reduced in treated animals (p = 0.012). |
| Relton et al. [237] | 2001 | Rats underwent 1-h MCAO followed by 23-h reperfusion. 24 h before MCAO were injected intravenously with anti-α4 integrin antibody (2.5 mg/kg) or isotype control antibody | Male spontaneously hypertensive rats or Sprague-Dawley rats | Treated animals showed reduced total infarct volume (p < 0.05–0.01). Moreover, treatment reduced brain myeloperoxidase activity (p < 0.05). No significant difference in white blood cell count was observed. Leukocyte counts were elevated in TA-2-treated rats. |
| Liesz et al. [226] | 2011 | 24 h before or 3 h after ischemia, mice were administered 300 mg of CD49d-specific monoclonal antibody intraperitoneally after; control animals received rat IgG2b isotype control monoclonal antibody | Male mice C57BL/6J aged 10–12 weeks | VLA-4 blockade improved outcome after 7 days from MCAO via the inhibition of cerebral leukocyte invasion and neurotoxic cytokine production (p < 0.01). VLA-4 inhibition reduced the post-ischemic VCAM-1 up-regulation (p < 0.01). |
| Langhauser et al. [224] | 2014 | 24 h before or 3 h after cerebral ischemia (both tMCAO and pMCAO), mice were treated with 300 μg of a monoclonal antibody anti-CD49d | Male C57Bl/6 mice | VLA-4 blocking reduced T cell and neutrophil invasion after 5 days following MCAO and inhibited the up-regulation of VCAM-1 (p < 0.05). Anti-CD49d antibody could not influence stroke outcome positively, irrespective of the model or the time point investigated. |
| Neumann et al. [232] | 2015 | After focal cerebral ischemia was induced by pMCAO, anti-CD49d treatment was administered intravenously | LysM-eGFP mice | The systemic blockade of VLA-4 resulted in reduction of adherence of neutrophils (p < 0.05) and inhibition of their infiltration (p < 0.01) 24 h after focal ischemia. Moreover, anti-VLA-4 treatment improved neurological outcome and reduced infarct volume at day 3 after stroke (p < 0.05). |
| Llovera et al. [230] | 2015 | After cMCAO (for small lesions confined to the cortex) or fMCAO (for lesions in the cortex and subcortical structures) was assessed, anti-CD49d treatment was administered intraperitoneally 3 h after stroke induction | 315 male C57BL/6J mice | Anti-CD49d treatment reduced infarct volume (p < 0.05) and leukocytes invasion into the ischemic brain (p < 0.001) after 7 days from cMCAO (p < 0.05). After fMCAO, mice had fewer cerebral leukocytes than after cMCAO (p < 0.001), but anti-CD49d treatment did not affect leukocyte invasion after fMCAO. |
| Cyclosporine A | ||||
| Uchino et al. [243] | 1998 | CsA was given intraperitoneally daily for 1 week before and 1 week after forebrain ischemia of 7 or 10 min duration | Rats | Systemically administered CsA ameliorated the damage to the CA1 sector of the hippocampus due to transient ischemia (p < 0.001). |
| Cho et al. [217] | 2013 | Rats underwent MCAO and then randomly treated by intracarotid CsA 10 mg/kg 20 min before MCAO (pre-treatment group); intracarotid CsA 10 mg/kg immediately after reperfusion (post-treatment); and intracarotid saline immediately after reperfusion | 27 Sprague-Dawley rats | On day 1, a significant reduction of infarct size in the pre-treatment group compared to the post-treatment (p < 0.004) was evaluated. A significant reduction of microglial cell count in the pre-treatment group compared to either saline or post-treatment groups was found (p < 0.001). |
| Yu et al. [250] | 2004 | Rats underwent MCAO then were randomly treated with either: low dose CsA, MP, low dose CsA plus MP, high dose CsA, or vehicle | Adult Sprague-Dawley rats | Animals receiving high dose CsA alone exhibited a minor motor asymmetry and less neurologic deficits 3 days after stroke (p < 0.0001) as well as those receiving low dose CsA and MP treatment but only on day 1 post-stroke (p <0.005). Animals receiving high dose CsA alone exhibited significantly (p < 0.0001). |
| Yuen et al. [253] | 2011 | Rats were equally divided into sham control, intraperitoneal physiological saline (at 0.5/24/48 h after stroke), CsA (20 mg/kg at 0.5/24 h intraperitoneally), EPO (5000 IU/kg at 0.5/24/48 h, subcutaneously), combined CsA and EPO after occlusion of distal left internal carotid artery | 50 adult-male Sprague-Dawley rats | On day 21, improvement in neurological function was found in CsA and EPO group (p < 0.05) and was higher when the combined treatment was administered (p < 0.004). Attenuation of inflammatory response, apoptosis, and oxidative stress was found with combined therapy with CsA and EPO (p < 0.05). |
| Edaravone | ||||
| Fujiwara et al. [221] | 2016 | Before 90-min MCAO followed by reperfusion, rats were randomly assigned to intravenous vehicle or intravenous edaravone 3 mg/kg | Male Sprague-Dawley rats | Edaravone decreased infarct volume and edema formation and IL-1β and MMP-9 levels 3 h after ischemia levels (p < 0.05). Edaravone was shown to reduce levels of many other pro-inflammatory cytokines. |
| Yamashita et al. [248] | 2015 | Thrombolysis was evaluated by using a He-Ne-laser-induced thrombosis model in mesenteric microvessels. 3 experimental groups (placebo, alteplase 0.6 mg/kg, alteplase 0.6 mg/kg + edaravone 10.5 mg/kg) | Male Wistar–ST rats | In the alteplase group, thrombus volume decreased (p < 0.01) after 20 min. In the alteplase+edaravone group, thrombus volume was more evident (p < 0.001). |
| Wu et al. [245] | 2014 | Rats were subjected to tMCAO and then administered edaravone 2.4 mg/kg; a subset of these animals were administered both edaravone 2.4 mg/kg and borneol 0.6 mg/kg | Sprague-Dawley rats | Edaravone was demonstrated to scavenge free radicals. Edavarone and borneol reduced the infarct area (p < 0.001) and the effect was increased when drugs were administered synergistically (p < 0.001). |
| Study | Year | Treatment | Sample Size | Outcome |
|---|---|---|---|---|
| IL-1Ra | ||||
| Emsley et al. [283] | 2005 | Within 6 h of the stroke onset, patients were randomized to rhIL-1ra (intravenously by a 100 mg loading dose over 60 s, followed by a 2 mg/kg/h infusion over 72 h.) or placebo. | 34 patients (17 rhIL-1Ra, 17 placebo) | Peripheral total white blood cell and neutrophil count, CRP, and IL-6 and neutrophil counts were lower in the rhIL-1ra-treated were lower in the treated group. The drug was safe and well tolerated. |
| Smith et al. [294] | 2012 | Blood samples prior to treatment initiation, at 24 h and 5 to 7 days. LPS stimulation was made to assess cytokine production by leukocytes. | 34 patients (17 rhIL-1Ra, 17 placebo) | Induction of TNF-α (p < 0.001), IL-1β (p < 0.005), IL-6, IL-8, and IL-10 (p < 0.02) by LPS was reduced in patients at admission. At 24 h, for patients treated with IL-1Ra, induction of TNF-α, IL-6 and IL-10 was greater than in the placebo group (p < 0.05). At 5 to 7 day, TNF-α and IL-1β induction remained suppressed only in the placebo group (p < 0.05). Plasma cortisol concentrations were elevated at admission in patients compared to controls but decreased at 24 h in treated patients (p < 0.05) and inversely correlated (p < 0.001) with either TNF-α or IL-1β induction at admission. |
| Statins | ||||
| Scandinavian Simvastatin Survival Study (4S) [277] | 1994 | Patients with angina pectoris or previous MI and serum cholesterol 5.5–8.0 mmol/L on a lipid-lowering diet were randomized to double-blind treatment with simvastatin or placebo. | 4444 patients (2221 simvastatin, 2223 placebo) | Over 5.4 years, simvastatin improved lipid profile, with few adverse effects. The relative risk of death in the simvastatin group was 0.70 (95% CI 0.58–0.85, p = 0.0003). In a post hoc analysis, simvastatin was demonstrated to reduce by 30% the rate of strokes and transient ischemic attacks. |
| Plehn et al. [290] | 1999 | Enrolled patients: 21–75 years old who had experienced a myocardial infarction within the past 3 to 20 months, total cholesterol <240 mg/dL, LDL cholesterol between 115 and 174 mg/dL, and fasting triglycerides <350 mg/dL during 4 weeks of treatment. | 4159 patients (2081 pravastatin 40 mg daily and 2078 placebo) | Compared with placebo, pravastatin lowered total and LDL cholesterol, and triglycerides by 20%, 32%, and 14%, respectively. A total of 128 strokes (52 on pravastatin, 76 on placebo) and 216 strokes or TIAs (92 on pravastatin, 124 on placebo) were observed, representing a 32% reduction (95% CI, 4%–52%, p = 0.03) in all-cause stroke and 27% reduction in stroke or TIA (95% CI, 4%–44%, p = 0.02). No increase in hemorrhagic stroke with pravastatin was found. |
| Montaner et al. [288] | 2008 | Simvastatin (40 mg/day for the first week followed by a dose of 20 mg/day until day 90) or placebo were given at 3–12 h from symptom onset. | 60 patients (30 simvastatin, 30 placebo) | Simvastatin-treated group presented greater improvements at several time points (p = 0.01). Simvastatin treatment and low temperatures were the only independent predictors of a great improvement by day 90 (OR 10.3, CI 2.05–52.2, p = 0.005 and OR 0.13, CI 0.02–0.70, p = 0.017, respectively). |
| Sever et al. [292] | 2003 | Hypertensive patients aged 40–79 years with at least 3 other cardiovascular risk factors. | 10305 (5168 atorvastatin 10 mg daily and 5137 placebo) | Treatment was stopped after a median follow-up of 3.3 years. In the atorvastatin group, less primary events occurred (HR 0.64, 95% CI 0.50–0.83, p = 0.0005), especially in the first year of follow-up. Fatal and non-fatal stroke (p = 0.024), total cardiovascular events and total coronary events (p = 0.0005) were also lowered. |
| Amarenco et al. [278] | 2006 | Patients with previous stroke or TIA within one to six months, LDL cholesterol levels of 100 to 190 mg/dL, and no known coronary heart disease. | 4731 patients (2365 atorvastatin 80 mg daily and 2366 placebo) | During 4.9 years, 265 patients under atorvastatin and 311 under placebo had a fatal or non-fatal stroke (5-year absolute reduction in risk, 2.2%; adjusted HR 0.84, 95% CI, 0.71–0.99, p = 0.03; unadjusted p = 0.05). The 5-year absolute reduction in the risk of major cardiovascular events was 3.5% (HR, 0.80, 95% CI, 0.69–0.92, p = 0.002). No difference in mortality rate was seen. |
| Shepherd et al. [293] | 2002 | Patients aged 70–82 years with a history of or risk factors for vascular disease. | 5804 patients (2891 pravastatin 40 mg daily and 2913 placebo) | Pravastatin lowered LDL cholesterol and reduced the incidence of the primary endpoint (HR 0.85, 95% CI 0.74–0.97, p = 0.014). Coronary heart disease death and non-fatal MI risk was also reduced (p = 0.006). Stroke risk was unaffected (p = 0.8) as well as for TIA (p = 0.051). New cancer diagnosis were more frequent in pravastatin group (p = 0.020). Mortality from coronary heart disease was lower in the pravastatin group (p = 0.043). No significant effect on cognitive function or disability was found. |
| Ridker et al. [291] | 2008 | Apparently healthy men and women with LDL cholesterol levels of less than 130 mg/dL and hs-CRP levels of 2.0 mg/L or higher. | 17802 patients (8901 rosuvastatin 20 mg daily and 8901 placebo) | Rosuvastatin reduced LDL cholesterol levels and hs-CRP levels. Rates of occurrence of the combined primary end point (MI, stroke, arterial revascularization, hospitalization for unstable angina, or death from cardiovascular causes) were 0.77 for rosuvastatin (HR 0.56, 95% CI: 0.46–0.69, p < 0.00001; HR for stroke 0.52, 95% CI 0.34–0.79, p = 0.002). |
| Donepezil | ||||
| Barrett et al. [280] | 2011 | Adults with ischemic stroke treated within 24 h after onset of symptoms. | 33 patients receiving donepezil 5 mg daily for 30 days followed by an increase to 10 mg/day for 60 days | 15 participants had a favorable clinical outcome (NIHSS score ≤1 at day 90) (p < 0.001). |
| Cyclosporine A | ||||
| Nighoghossian et al. [289] | 2015 | Patients aged 18–85 years with an anterior-circulation stroke and eligible for thrombolytic therapy and evaluation of infarct volume on MRI at 30 days. | 127 patients (61 CsA 2 mg/kg and 66 saline) | The reduction of infarct volume in CsA-treated patients was not significant (p = 0.18). In patients with proximal occlusion and effective recanalization, infarct volume decreased in CsA-treated group (p = 0.009). |
| Edaravone | ||||
| Edaravone Acute Infarction Study Group [282] | 2003 | Patients with acute ischemic stroke within 72 h from symptom onset. | 250 patients (125 edaravone 30 mg twice a day for 14 days and 125 placebo) | A significant improvement in functional outcome evaluated by the mRS was observed in the edaravone group (p = 0.039). |
| Kaste et al. [284] | 2013 | Patients with acute ischemic stroke within 24 h from stroke onset. | 36 patients (12 edaravone with loading dose 0.08 mg/kg + 0.2 mg/kg/h; 13 edaravone loading dose 0.16 mg/kg + 0.4 mg/kg/h; 11 placebo) | Both doses of the new formulation and dosing regimen were well tolerated and showed clinical improvement based on NIHSS score. |
| Takenaka et al. [295] | 2014 | Patients admitted to hospital for cerebral infarction within 3 h after the onset of infarction. | 48 patients (20 edaravone before rt-PA and 28 edaravone and rt-PA simultaneously) | NIHSS before rt-PA showed a statistically significant improvement after rt-PA administration (p < 0.001). The mRS at 90 days also improved. |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bonaventura, A.; Liberale, L.; Vecchié, A.; Casula, M.; Carbone, F.; Dallegri, F.; Montecucco, F. Update on Inflammatory Biomarkers and Treatments in Ischemic Stroke. Int. J. Mol. Sci. 2016, 17, 1967. https://doi.org/10.3390/ijms17121967
Bonaventura A, Liberale L, Vecchié A, Casula M, Carbone F, Dallegri F, Montecucco F. Update on Inflammatory Biomarkers and Treatments in Ischemic Stroke. International Journal of Molecular Sciences. 2016; 17(12):1967. https://doi.org/10.3390/ijms17121967
Chicago/Turabian StyleBonaventura, Aldo, Luca Liberale, Alessandra Vecchié, Matteo Casula, Federico Carbone, Franco Dallegri, and Fabrizio Montecucco. 2016. "Update on Inflammatory Biomarkers and Treatments in Ischemic Stroke" International Journal of Molecular Sciences 17, no. 12: 1967. https://doi.org/10.3390/ijms17121967
APA StyleBonaventura, A., Liberale, L., Vecchié, A., Casula, M., Carbone, F., Dallegri, F., & Montecucco, F. (2016). Update on Inflammatory Biomarkers and Treatments in Ischemic Stroke. International Journal of Molecular Sciences, 17(12), 1967. https://doi.org/10.3390/ijms17121967