
Non-SMC Element 2 (NSMCE2) of the SMC5/6 Complex Helps to Resolve Topological Stress

Abstract
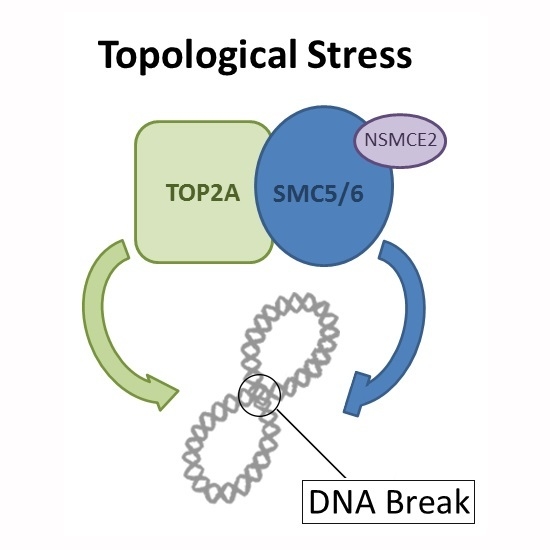
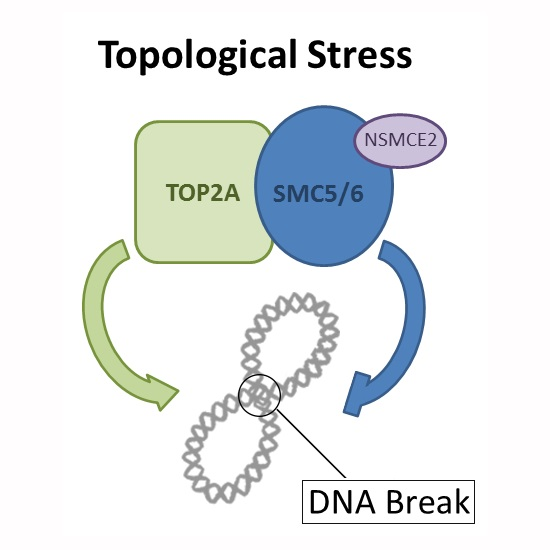
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
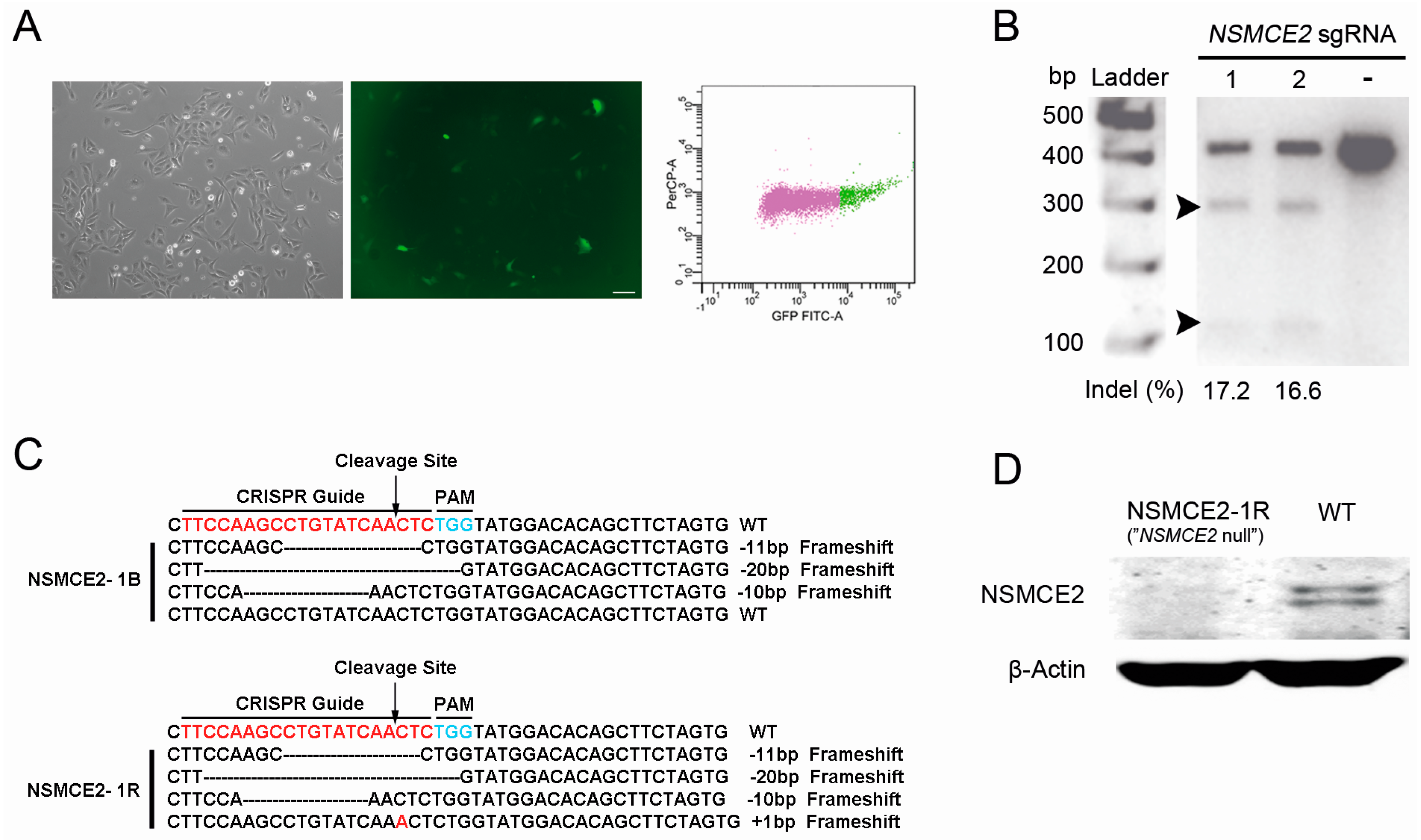
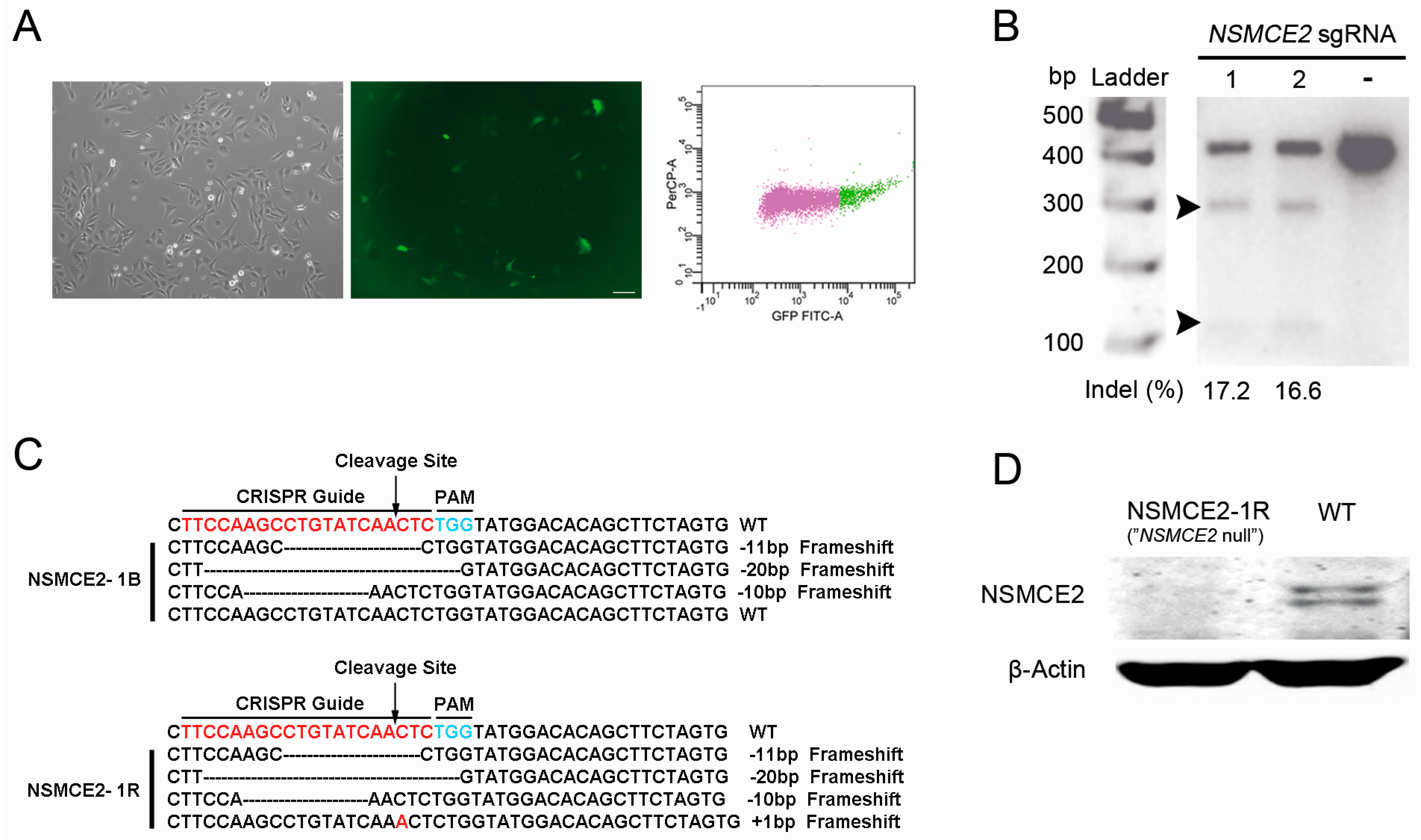
2.1. CRISPR-Cas9-Mediated Targeting of the SMC5/6 Complex
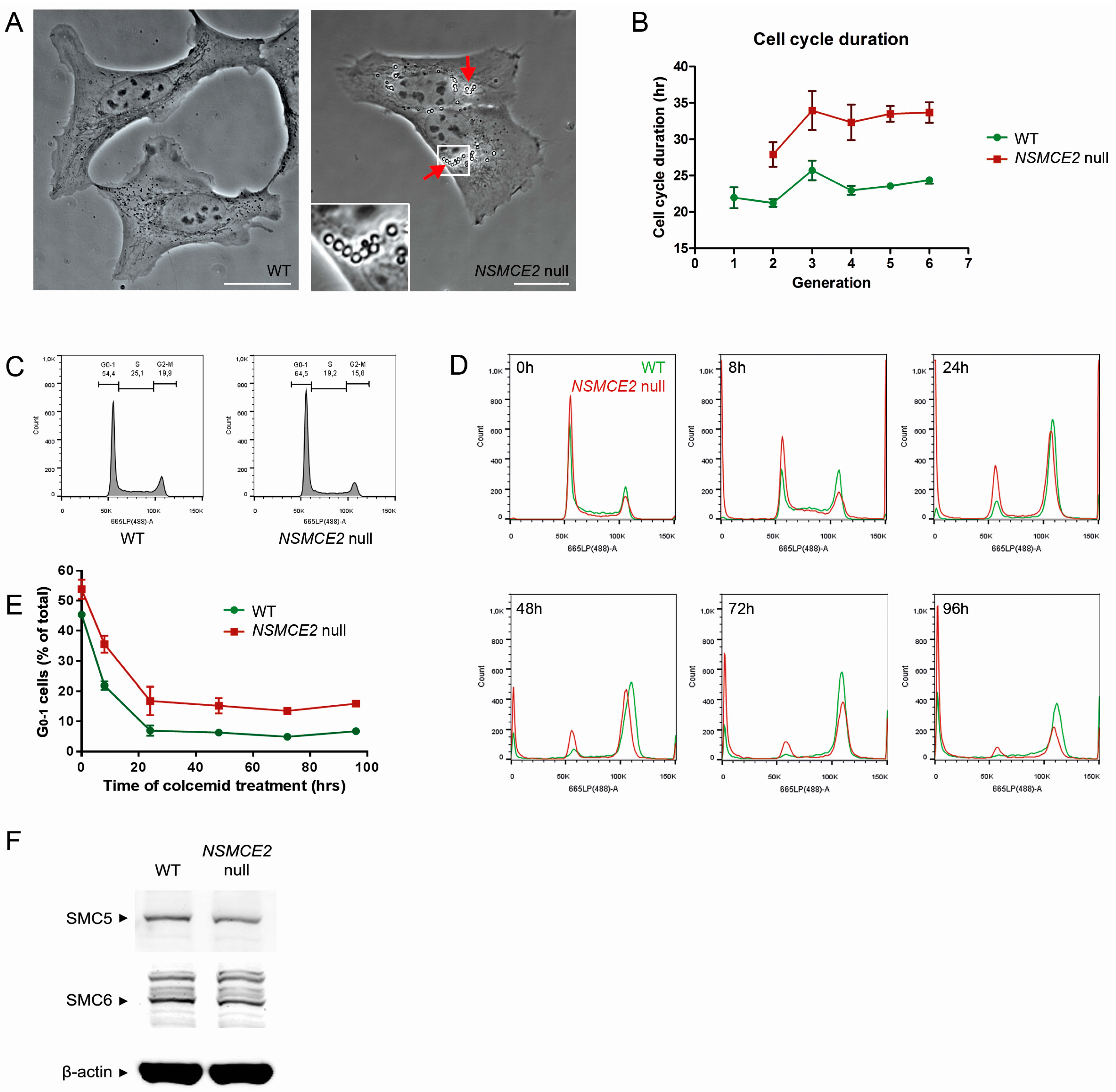
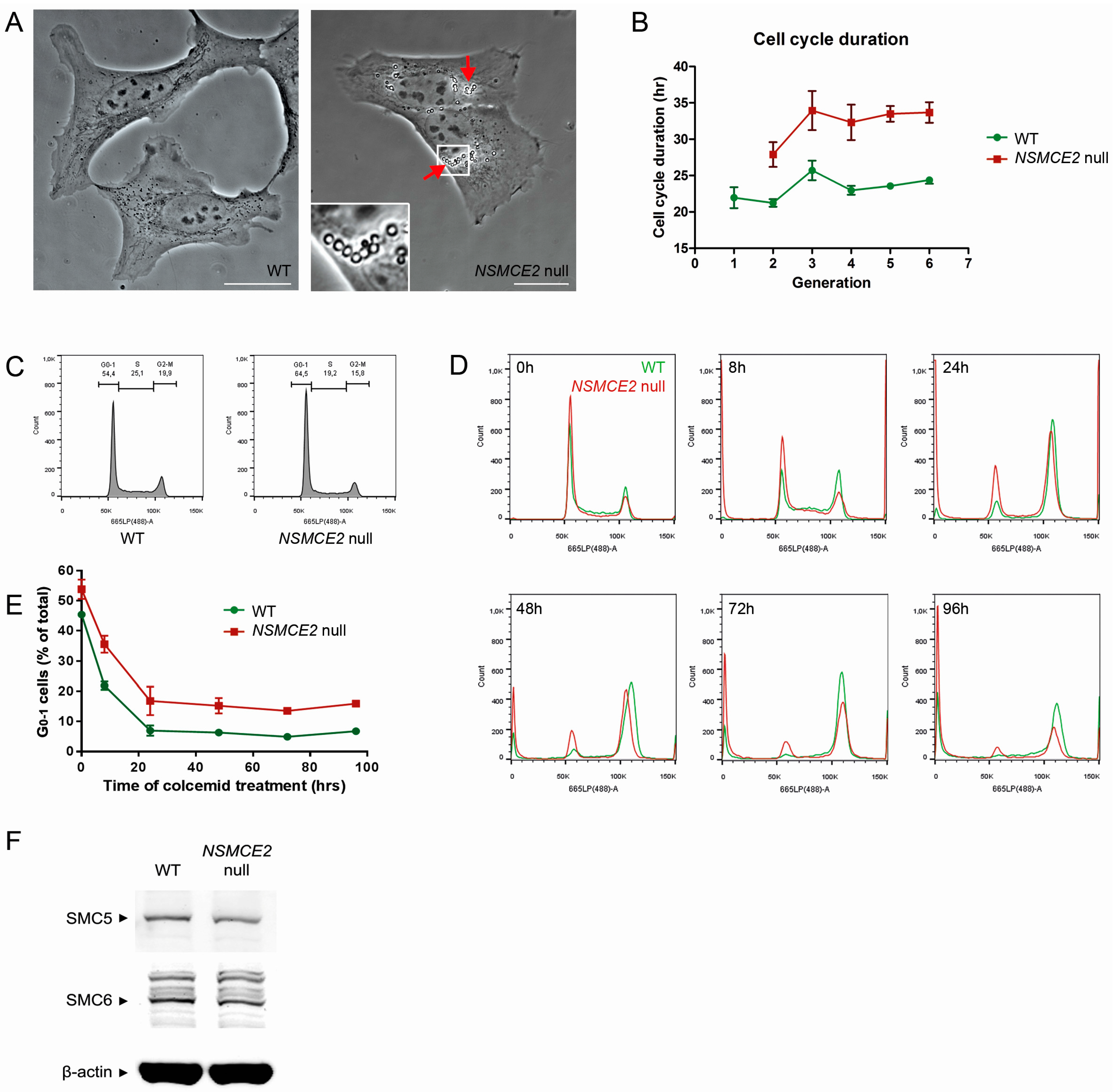
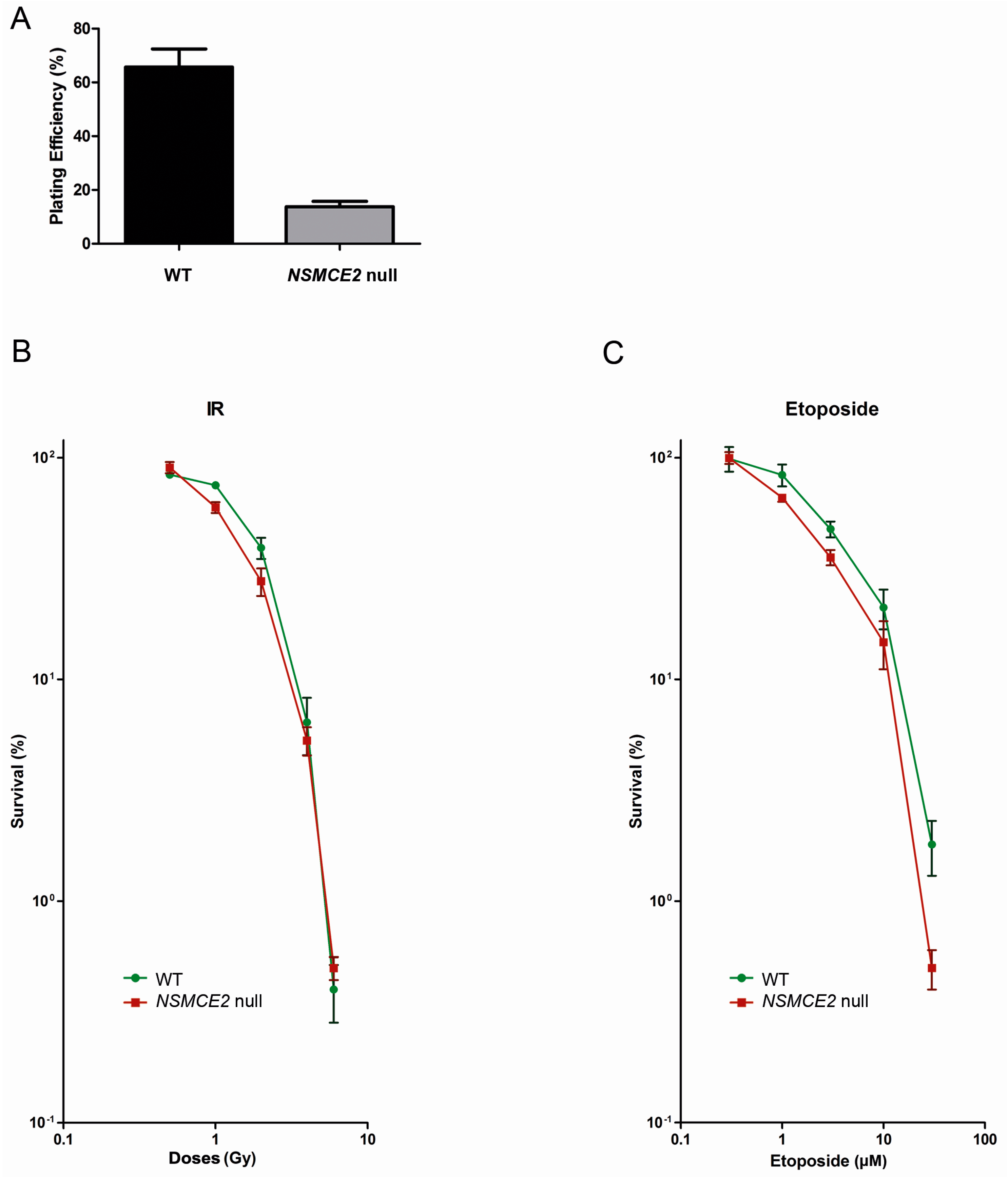
2.2. Characterization of NSMCE2 Null Cells
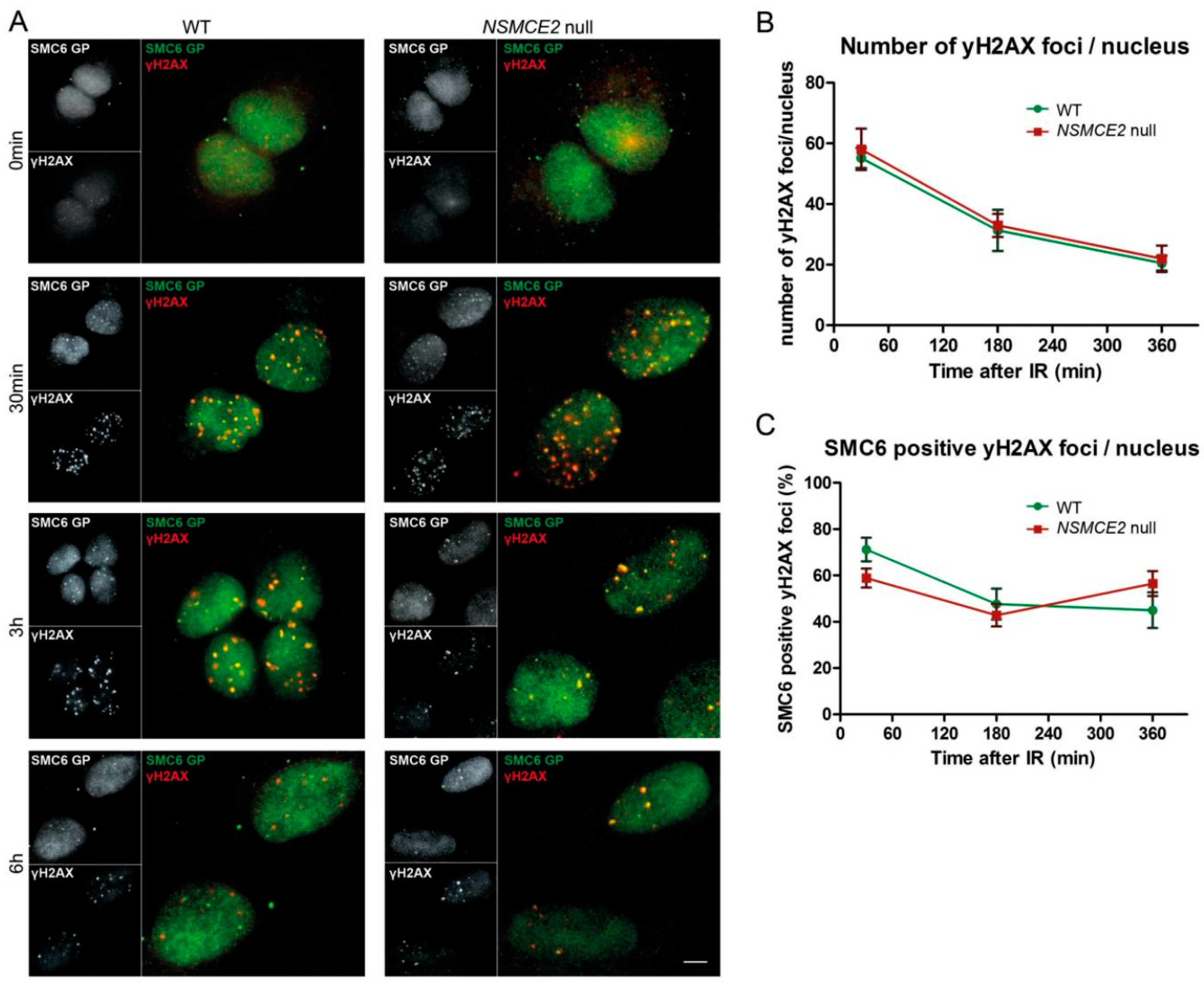
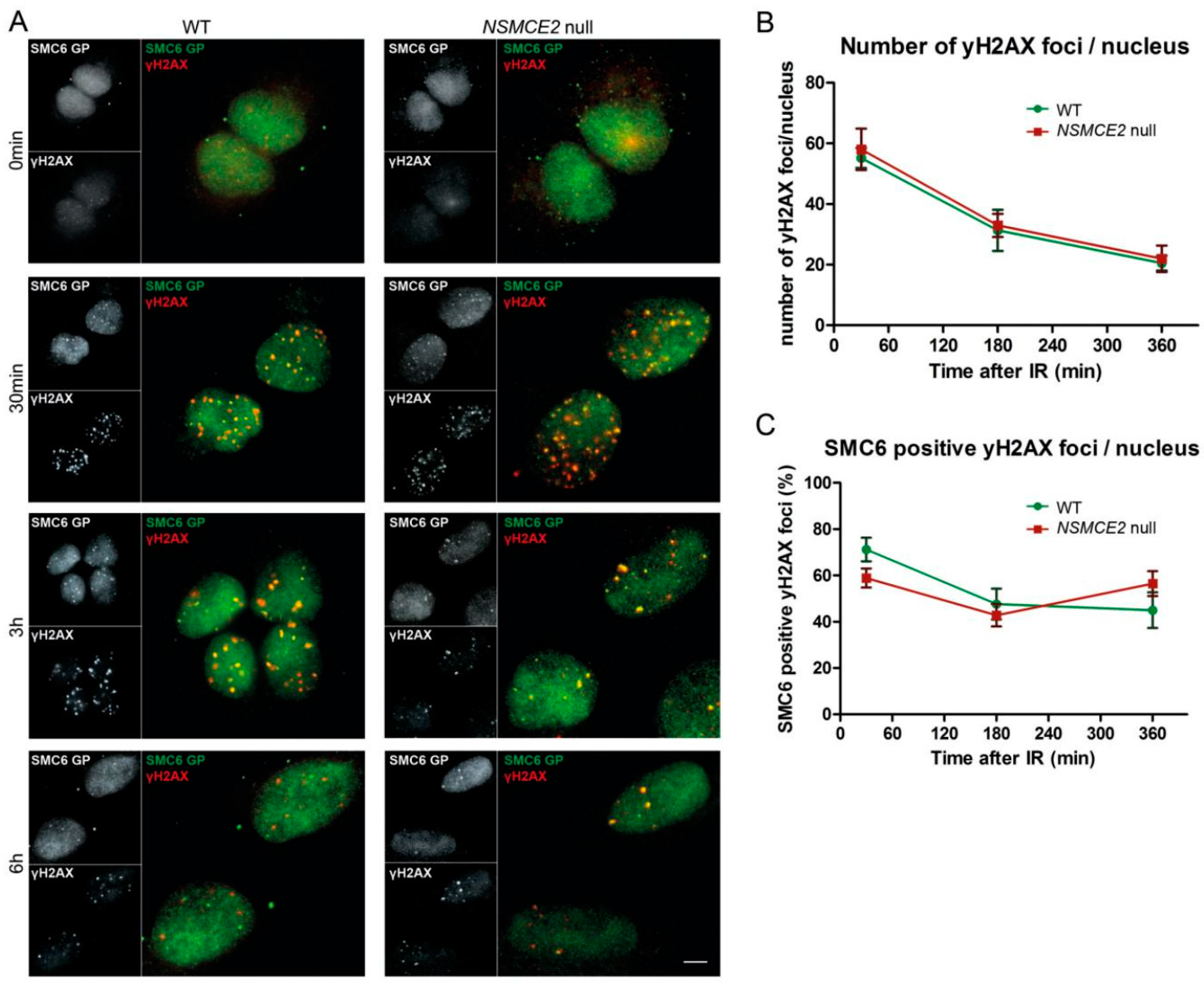
2.3. Irradiation-Induced SMC6 Foci Formation Occurs Independent of NSMCE2
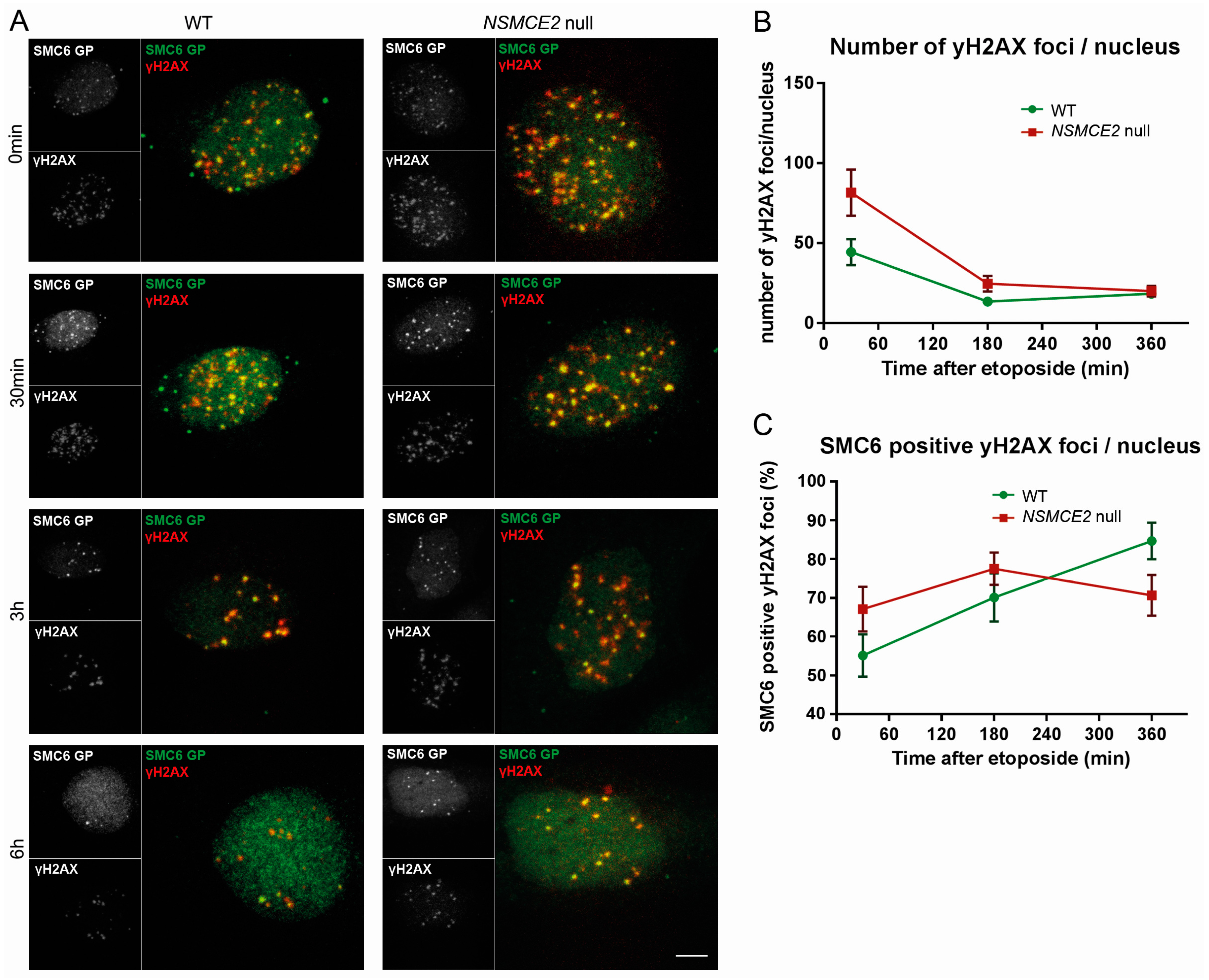
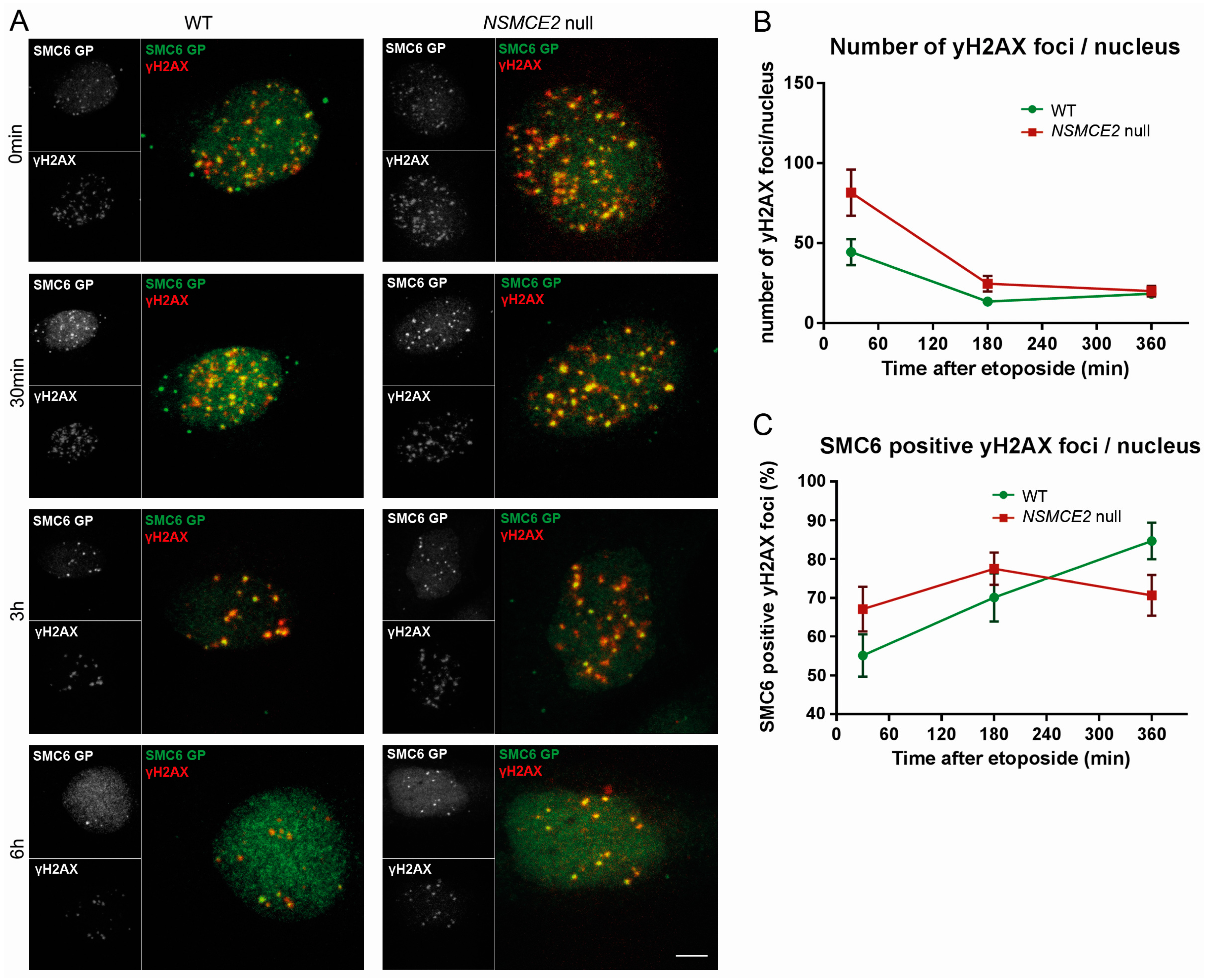
2.4. More Etoposide-Induced Double-Strand Break (DSB) Formation without NSMCE2
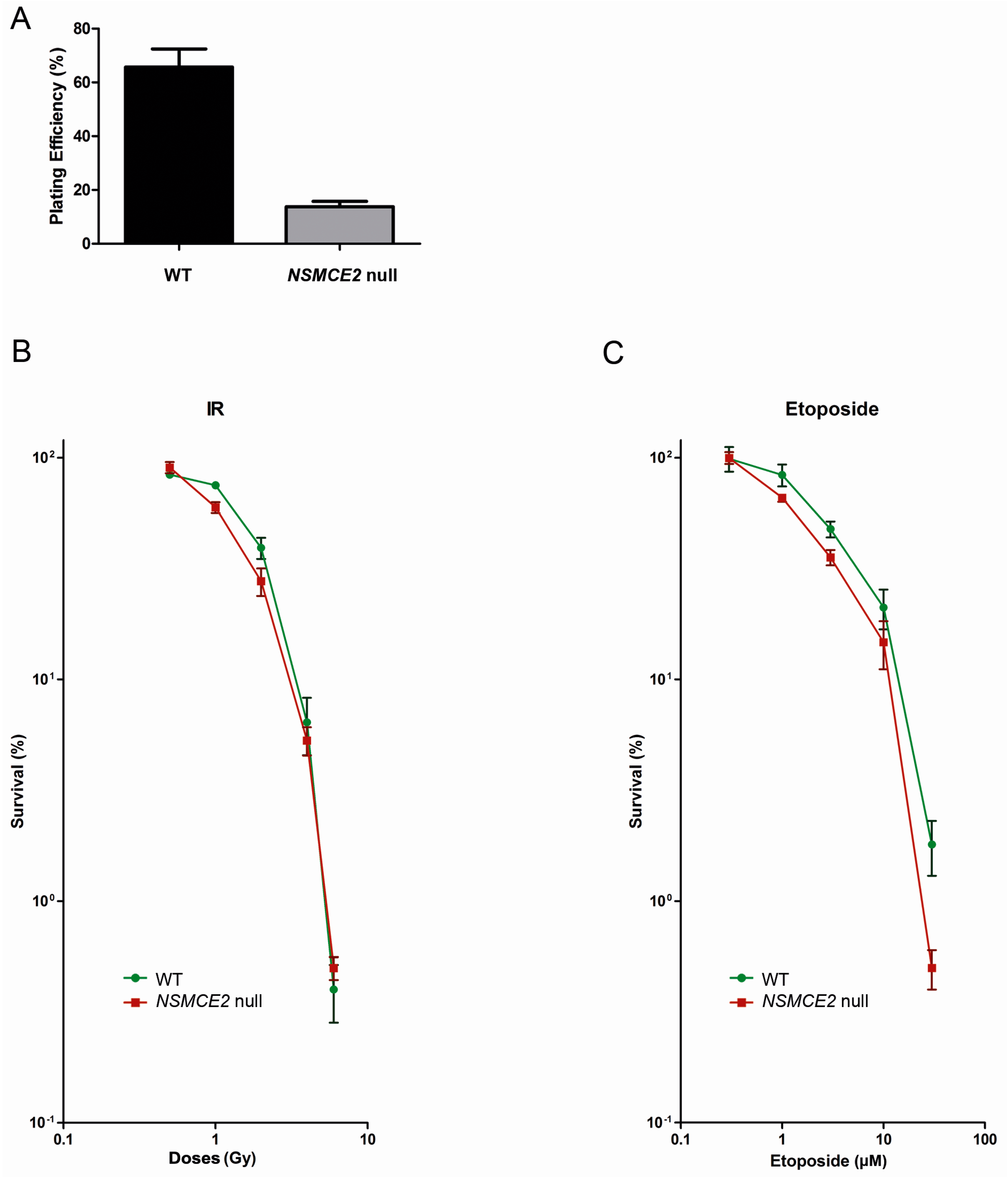
2.5. Absence of NSMCE2 Affects Survival upon Etoposide-Induced DSBs
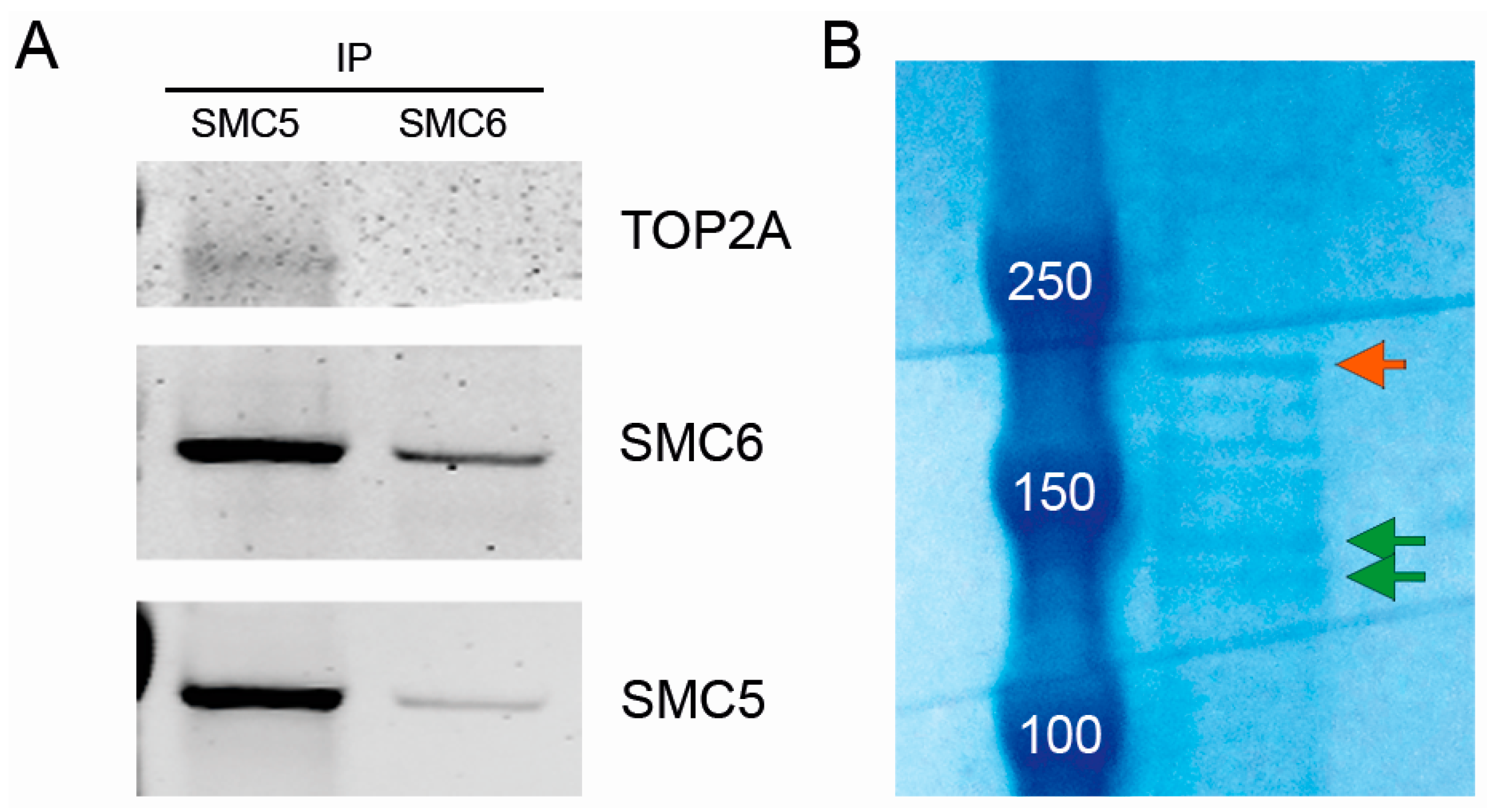
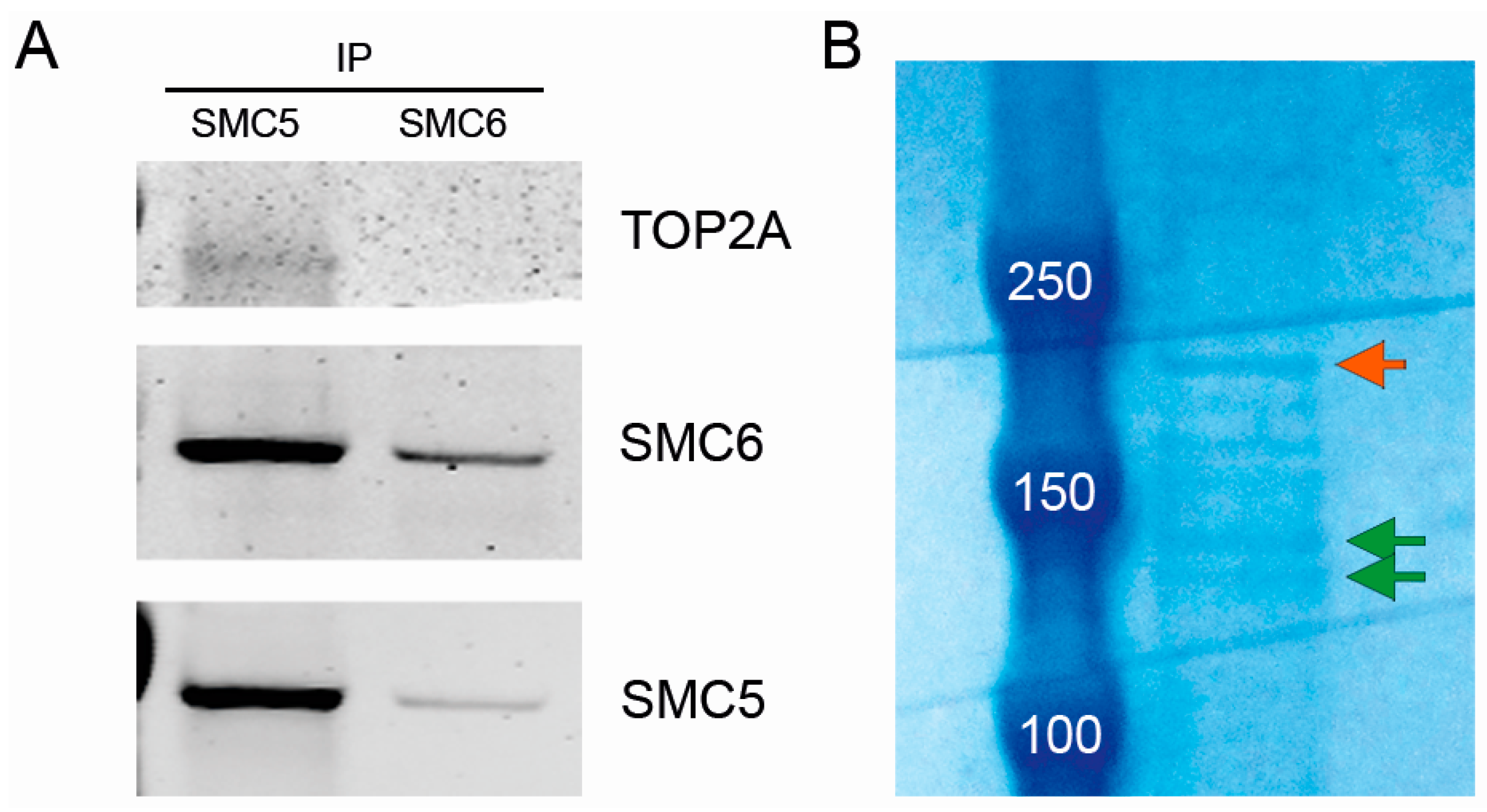
2.6. The SMC5/6 Complex Physically Interacts with Topoisomerase II α (TOP2A)
3. Discussion
4. Materials and Methods
4.1. Cells and Culture
4.2. Design of Single-Guide RNAs (sgRNAs) and Construction of the CRISPR-Cas9 Plasmids
4.3. Transfection of U2OS Cells with CRISPR Plasmids
4.4. Surveyor Assay for Verification of Genome Editing
4.5. Sequencing Analysis of the Targeting Site in NSMCE2
4.6. Off-Target Analysis in the NSMCE2-Devoid Cell Line
4.7. Live Cell Microscopy
4.8. Distribution of Cells over the Cell Cycle Phase
4.9. Ionizing Irradiation (IR)
4.10. Immunocytochemistry (ICC)
4.11. Quantitative Imaging
4.12. Clonogenic Assay
4.13. Protein Isolation
4.14. Western Blot Analysis
4.15. Immunoprecipitation (IP)
4.16. Mass Spectrometry
Supplementary Materials
Acknowledgments
Author Contribution
Conflicts of Interest
Abbreviations
| SMC | Structural Maintenance of Chromosomes |
| NSMCE | Non-SMC Element |
| DSB | DNA double-strand break |
| IR | Ionizing Radiation |
| TOP | Topoisomerase |
| SCI | Sister chromatid intertwining |
References
- Nasmyth, K.; Haering, C.H. The structure and function of SMC and kleisin complexes. Annu. Rev. Biochem. 2005, 74, 595–648. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.M.; Tedeschi, A.; Schmitz, J. The cohesin complex and its roles in chromosome biology. Genes Dev. 2008, 22, 3089–3114. [Google Scholar] [CrossRef] [PubMed]
- Hirano, T. At the heart of the chromosome: Smc proteins in action. Nat. Rev. 2006, 7, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Terret, M.E.; Sherwood, R.; Rahman, S.; Qin, J.; Jallepalli, P.V. Cohesin acetylation speeds the replication fork. Nature 2009, 462, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Wendt, K.S.; Peters, J.M. How cohesin and CTCF cooperate in regulating gene expression. Chromosome Res. 2009, 17, 201–214. [Google Scholar] [CrossRef] [PubMed]
- De Piccoli, G.; Cortes-Ledesma, F.; Ira, G.; Torres-Rosell, J.; Uhle, S.; Farmer, S.; Hwang, J.Y.; Machin, F.; Ceschia, A.; McAleenan, A.; et al. Smc5–Smc6 mediate DNA double-strand-break repair by promoting sister-chromatid recombination. Nat. Cell Biol. 2006, 8, 1032–1034. [Google Scholar] [CrossRef] [PubMed]
- Potts, P.R. The yin and yang of the MMS21-SMC5/6 SUMO ligase complex in homologous recombination. DNA Repair 2009, 8, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Yu, H. The SMC complexes in DNA damage response. Cell Biosci. 2012, 2, 5. [Google Scholar] [CrossRef] [PubMed]
- Verver, D.E.; Langedijk, N.S.; Jordan, P.W.; Repping, S.; Hamer, G. The SMC5/6 complex is involved in crucial processes during human spermatogenesis. Biol. Reprod. 2014, 91, 22. [Google Scholar] [CrossRef] [PubMed]
- Verver, D.E.; van Pelt, A.M.; Repping, S.; Hamer, G. Role for rodent Smc6 in pericentromeric heterochromatin domains during spermatogonial differentiation and meiosis. Cell Death Dis. 2013, 4, e749. [Google Scholar] [CrossRef] [PubMed]
- Verver, D.E.; Hwang, G.H.; Jordan, P.W.; Hamer, G. Resolving complex chromosome structures during meiosis: Versatile deployment of Smc5/6. Chromosoma 2016, 125, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Copsey, A.; Tang, S.; Jordan, P.W.; Blitzblau, H.G.; Newcombe, S.; Chan, A.C.; Newnham, L.; Li, Z.; Gray, S.; Herbert, A.D.; et al. Smc5/6 coordinates formation and resolution of joint molecules with chromosome morphology to ensure meiotic divisions. PLoS Genet. 2013, 9, e1004071. [Google Scholar] [CrossRef] [PubMed]
- Gomez, R.; Jordan, P.W.; Viera, A.; Alsheimer, M.; Fukuda, T.; Jessberger, R.; Llano, E.; Pendas, A.M.; Handel, M.A.; Suja, J.A. Dynamic localization of SMC5/6 complex proteins during mammalian meiosis and mitosis suggests functions in distinct chromosome processes. J. Cell Sci. 2013, 126, 4239–4252. [Google Scholar] [CrossRef] [PubMed]
- Bickel, J.S.; Chen, L.; Hayward, J.; Yeap, S.L.; Alkers, A.E.; Chan, R.C. Structural maintenance of chromosomes (SMC) proteins promote homolog-independent recombination repair in meiosis crucial for germ cell genomic stability. PLoS Genet. 2010, 6, e1001028. [Google Scholar] [CrossRef] [PubMed]
- Lilienthal, I.; Kanno, T.; Sjogren, C. Inhibition of the Smc5/6 complex during meiosis perturbs joint molecule formation and resolution without significantly changing crossover or non-crossover levels. PLoS Genet. 2013, 9, e1003898. [Google Scholar] [CrossRef] [PubMed]
- Xaver, M.; Huang, L.; Chen, D.; Klein, F. SMC5/6-MMS21 prevents and eliminates inappropriate recombination intermediates in meiosis. PLoS Genet. 2013, 9, e1004067. [Google Scholar] [CrossRef] [PubMed]
- Farmer, S.; San-Segundo, P.A.; Aragón, L. The Smc5–Smc6 complex is required to remove chromosome junctions in meiosis. PLoS ONE 2011, 6, e20948. [Google Scholar] [CrossRef] [PubMed]
- Torres-Rosell, J.; Sunjevaric, I.; de Piccoli, G.; Sacher, M.; Eckert-Boulet, N.; Reid, R.; Jentsch, S.; Rothstein, R.; Aragon, L.; Lisby, M. The Smc5–Smc6 complex and SUMO modification of Rad52 regulates recombinational repair at the ribosomal gene locus. Nat. Cell Biol. 2007, 9, 923–931. [Google Scholar] [CrossRef] [PubMed]
- Agostinho, A.; Meier, B.; Sonneville, R.; Jagut, M.; Woglar, A.; Blow, J.; Jantsch, V.; Gartner, A. Combinatorial regulation of meiotic holliday junction resolution in C. elegans by HIM-6 (BLM) helicase, SLX-4, and the SLX-1, MUS-81 and XPF-1 nucleases. PLoS Genet. 2013, 9, e1003591. [Google Scholar] [CrossRef]
- Hong, Y.; Sonneville, R.; Agostinho, A.; Meier, B.; Wang, B.; Blow, J.J.; Gartner, A. The SMC-5/6 complex and the HIM-6 (BLM) helicase synergistically promote meiotic recombination intermediate processing and chromosome maturation during caenorhabditis elegans meiosis. PLoS Genet. 2016, 12, e1005872. [Google Scholar] [CrossRef] [PubMed]
- O’Neil, N.J.; Martin, J.S.; Youds, J.L.; Ward, J.D.; Petalcorin, M.I.; Rose, A.M.; Boulton, S.J. Joint molecule resolution requires the redundant activities of MUS-81 and XPF-1 during Caenorhabditis elegans meiosis. PLoS Genet. 2013, 9, e1003582. [Google Scholar] [CrossRef] [PubMed]
- Wehrkamp-Richter, S.; Hyppa, R.W.; Prudden, J.; Smith, G.R.; Boddy, M.N. Meiotic DNA joint molecule resolution depends on Nse5–Nse6 of the Smc5–Smc6 holocomplex. Nucleic Acids Res. 2012, 40, 9633–9646. [Google Scholar] [CrossRef] [PubMed]
- Carter, S.D.; Sjogren, C. The SMC complexes, DNA and chromosome topology: Right or knot? Crit. Rev. Biochem. Mol. Biol. 2012, 47, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Jeppsson, K.; Kanno, T.; Shirahige, K.; Sjogren, C. The maintenance of chromosome structure: Positioning and functioning of smc complexes. Nat. Rev. 2012, 15, 601–614. [Google Scholar] [CrossRef] [PubMed]
- Langston, R.E.; Weinert, T. Nifty alleles, a plethora of interactions, and imagination advance understanding of Smc5/6′s roles with chromosomes. Mol. Cell 2015, 60, 832–833. [Google Scholar] [CrossRef] [PubMed]
- Ju, L.; Wing, J.; Taylor, E.; Brandt, R.; Slijepcevic, P.; Horsch, M.; Rathkolb, B.; Racz, I.; Becker, L.; Hans, W.; et al. SMC6 is an essential gene in mice, but a hypomorphic mutant in the atpase domain has a mild phenotype with a range of subtle abnormalities. DNA Repair 2013, 12, 356–366. [Google Scholar] [CrossRef] [PubMed]
- Jacome, A.; Gutierrez-Martinez, P.; Schiavoni, F.; Tenaglia, E.; Martinez, P.; Rodriguez-Acebes, S.; Lecona, E.; Murga, M.; Mendez, J.; Blasco, M.A.; et al. NSMCE2 suppresses cancer and aging in mice independently of its SUMO ligase activity. EMBO J. 2016, 34, 2604–2619. [Google Scholar] [CrossRef] [PubMed]
- Pryzhkova, M.V.; Jordan, P.W. Conditional mutation of Smc5 in mouse embryonic stem cells perturbs condensin localization and mitotic progression. J. Cell Sci. 2016, 129, 1619–1634. [Google Scholar] [CrossRef] [PubMed]
- Sluder, G. Role of spindle microtubules in the control of cell cycle timing. J. Cell Biol. 1979, 80, 674–691. [Google Scholar] [CrossRef] [PubMed]
- Andrews, E.A.; Palecek, J.; Sergeant, J.; Taylor, E.; Lehmann, A.R.; Watts, F.Z. Nse2, a component of the Smc5–6 complex, is a sumo ligase required for the response to DNA damage. Mol. Cell. Biol. 2005, 25, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Kliszczak, M.; Stephan, A.K.; Flanagan, A.M.; Morrison, C.G. SUMO ligase activity of vertebrate Mms21/Nse2 is required for efficient DNA repair but not for Smc5/6 complex stability. DNA Repair 2012, 11, 799–810. [Google Scholar] [CrossRef] [PubMed]
- McDonald, W.H.; Pavlova, Y.; Yates, J.R., 3rd; Boddy, M.N. Novel essential DNA repair proteins Nse1 and Nse2 are subunits of the fission yeast Smc5–Smc6 complex. J. Biol. Chem. 2003, 278, 45460–45467. [Google Scholar] [CrossRef] [PubMed]
- Rai, R.; Varma, S.P.; Shinde, N.; Ghosh, S.; Kumaran, S.P.; Skariah, G.; Laloraya, S. Small ubiquitin-related modifier ligase activity of Mms21 is required for maintenance of chromosome integrity during the unperturbed mitotic cell division cycle in saccharomyces cerevisiae. J. Biol. Chem. 2011, 286, 14516–14530. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Blobel, G. A SUMO ligase is part of a nuclear multiprotein complex that affects DNA repair and chromosomal organization. Proc. Natl. Acad. Sci. USA 2005, 102, 4777–4782. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.L.; Yang, L.; Rowe, T.C.; Halligan, B.D.; Tewey, K.M.; Liu, L.F. Nonintercalative antitumor drugs interfere with the breakage-reunion reaction of mammalian DNA topoisomerase II. J. Biol. Chem. 1984, 259, 13560–13566. [Google Scholar] [PubMed]
- Nitiss, J.L. DNA topoisomerase II and its growing repertoire of biological functions. Nat. Rev. Cancer 2009, 9, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.C. DNA topoisomerases. Annu. Rev. Biochem. 1996, 65, 635–692. [Google Scholar] [CrossRef] [PubMed]
- Franken, N.A.; Rodermond, H.M.; Stap, J.; Haveman, J.; van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef] [PubMed]
- Taylor, E.M.; Moghraby, J.S.; Lees, J.H.; Smit, B.; Moens, P.B.; Lehmann, A.R. Characterization of a novel human SMC heterodimer homologous to the Schizosaccharomyces pombe Rad18/Spr18 complex. Mol. Biol. Cell 2001, 12, 1583–1594. [Google Scholar] [CrossRef] [PubMed]
- Arimitsu, N.; Kogure, T.; Baba, T.; Nakao, K.; Hamamoto, H.; Sekimizu, K.; Yamamoto, A.; Nakanishi, H.; Taguchi, R.; Tagaya, M.; et al. p125/Sec23-interacting protein (Sec23ip) is required for spermiogenesis. FEBS Lett. 2011, 585, 2171–2176. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.; Gore, A.; Yan, W.; Abalde-Atristain, L.; Li, Z.; He, C.; Wang, Y.; Brodsky, R.A.; Zhang, K.; Cheng, L.; et al. Whole-genome sequencing analysis reveals high specificity of CRISPR/Cas9 and talen-based genome editing in human ipscs. Cell Stem Cell 2014, 15, 12–13. [Google Scholar] [CrossRef] [PubMed]
- Veres, A.; Gosis, B.S.; Ding, Q.; Collins, R.; Ragavendran, A.; Brand, H.; Erdin, S.; Cowan, C.A.; Talkowski, M.E.; Musunuru, K. Low incidence of off-target mutations in individual CRISPR-Cas9 and TALEN targeted human stem cell clones detected by whole-genome sequencing. Cell Stem Cell 2014, 15, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Ni, H.J.; Chang, Y.N.; Kao, P.H.; Chai, S.P.; Hsieh, Y.H.; Wang, D.H.; Fong, J.C. Depletion of SUMO ligase hMMS21 impairs G1 to S transition in MCF-7 breast cancer cells. Biochim. Biophys. Acta 2012, 1820, 1893–1900. [Google Scholar] [CrossRef] [PubMed]
- Pebernard, S.; McDonald, W.H.; Pavlova, Y.; Yates, J.R.; Boddy, M.N. Nse1, Nse2, and a novel subunit of the Smc5–Smc6 complex, Nse3, play a crucial role in meiosis. Mol. Biol. Cell 2004, 15, 4866–4876. [Google Scholar] [CrossRef] [PubMed]
- Raschle, M.; Smeenk, G.; Hansen, R.K.; Temu, T.; Oka, Y.; Hein, M.Y.; Nagaraj, N.; Long, D.T.; Walter, J.C.; Hofmann, K.; et al. DNA repair. Proteomics reveals dynamic assembly of repair complexes during bypass of DNA cross-links. Science 2015, 348, 1253671. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.C. Cellular roles of DNA topoisomerases: A molecular perspective. Nat. Rev. 2002, 3, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Harvey, S.H.; Sheedy, D.M.; Cuddihy, A.R.; O’Connell, M.J. Coordination of DNA damage responses via the Smc5/Smc6 complex. Mol. Cell. Biol. 2004, 24, 662–674. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Dulev, S.; Liu, X.; Hiller, N.J.; Zhao, X.; Strunnikov, A. Cooperation of sumoylated chromosomal proteins in rDNA maintenance. PLoS Genet. 2008, 4, e1000215. [Google Scholar] [CrossRef] [PubMed]
- Uemura, T.; Yanagida, M. Isolation of type I and II DNA topoisomerase mutants from fission yeast: Single and double mutants show different phenotypes in cell growth and chromatin organization. EMBO J. 1984, 3, 1737–1744. [Google Scholar] [PubMed]
- Verkade, H.M.; Bugg, S.J.; Lindsay, H.D.; Carr, A.M.; O’Connell, M.J. Rad18 is required for DNA repair and checkpoint responses in fission yeast. Mol. Biol. Cell 1999, 10, 2905–2918. [Google Scholar] [CrossRef] [PubMed]
- Kanno, T.; Berta, D.G.; Sjogren, C. The Smc5/6 complex is an ATP-dependent intermolecular DNA linker. Cell Rep. 2015, 12, 1471–1482. [Google Scholar] [CrossRef] [PubMed]
- Kegel, A.; Betts-Lindroos, H.; Kanno, T.; Jeppsson, K.; Strom, L.; Katou, Y.; Itoh, T.; Shirahige, K.; Sjogren, C. Chromosome length influences replication-induced topological stress. Nature 2011, 471, 392–396. [Google Scholar] [CrossRef] [PubMed]
- Lindroos, H.B.; Strom, L.; Itoh, T.; Katou, Y.; Shirahige, K.; Sjogren, C. Chromosomal association of the Smc5/6 complex reveals that it functions in differently regulated pathways. Mol. Cell 2006, 22, 755–767. [Google Scholar] [CrossRef] [PubMed]
- Menolfi, D.; Delamarre, A.; Lengronne, A.; Pasero, P.; Branzei, D. Essential roles of the Smc5/6 complex in replication through natural pausing sites and endogenous DNA damage tolerance. Mol. Cell 2015, 60, 835–846. [Google Scholar] [CrossRef] [PubMed]
- Bermejo, R.; Doksani, Y.; Capra, T.; Katou, Y.M.; Tanaka, H.; Shirahige, K.; Foiani, M. Top1- and Top2-mediated topological transitions at replication forks ensure fork progression and stability and prevent DNA damage checkpoint activation. Genes Dev. 2007, 21, 1921–1936. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.A.; Wang, J.C. Function of DNA topoisomerases as replication swivels in Saccharomyces cerevisiae. J. Mol. Biol. 1989, 208, 257–267. [Google Scholar] [CrossRef]
- Spell, R.M.; Holm, C. Nature and distribution of chromosomal intertwinings in Saccharomyces cerevisiae. Mol. Cell. Biol. 1994, 14, 1465–1476. [Google Scholar] [CrossRef] [PubMed]
- Bachant, J.; Alcasabas, A.; Blat, Y.; Kleckner, N.; Elledge, S.J. The SUMO-1 isopeptidase Smt4 is linked to centromeric cohesion through SUMO-1 modification of DNA topoisomerase II. Mol. Cell 2002, 9, 1169–1182. [Google Scholar] [CrossRef]
- Mao, Y.; Desai, S.D.; Liu, L.F. SUMO-1 conjugation to human DNA topoisomerase II isozymes. J. Biol. Chem. 2000, 275, 26066–26073. [Google Scholar] [CrossRef] [PubMed]
- Dawlaty, M.M.; Malureanu, L.; Jeganathan, K.B.; Kao, E.; Sustmann, C.; Tahk, S.; Shuai, K.; Grosschedl, R.; van Deursen, J.M. Resolution of sister centromeres requires RanBP2-mediated SUMOylation of topoisomerase IIalpha. Cell 2008, 133, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Ampatzidou, E.; Irmisch, A.; O’Connell, M.J.; Murray, J.M. Smc5/6 is required for repair at collapsed replication forks. Mol. Cell. Biol. 2006, 26, 9387–9401. [Google Scholar] [CrossRef] [PubMed]
- Miyabe, I.; Morishita, T.; Hishida, T.; Yonei, S.; Shinagawa, H. Rhp51-dependent recombination intermediates that do not generate checkpoint signal are accumulated in Schizosaccharomyces pombe rad60 and Smc5/6 mutants after release from replication arrest. Mol. Cell. Biol. 2006, 26, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [PubMed]
- Stax, M.J.; Mouser, E.E.; van Montfort, T.; Sanders, R.W.; de Vries, H.J.; Dekker, H.L.; Herrera, C.; Speijer, D.; Pollakis, G.; Paxton, W.A. Colorectal mucus binds DC-SIGN and inhibits HIV-1 trans-infection of CD4+ T-lymphocytes. PLoS ONE 2015, 10, e0122020. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Verver, D.E.; Zheng, Y.; Speijer, D.; Hoebe, R.; Dekker, H.L.; Repping, S.; Stap, J.; Hamer, G. Non-SMC Element 2 (NSMCE2) of the SMC5/6 Complex Helps to Resolve Topological Stress. Int. J. Mol. Sci. 2016, 17, 1782. https://doi.org/10.3390/ijms17111782
Verver DE, Zheng Y, Speijer D, Hoebe R, Dekker HL, Repping S, Stap J, Hamer G. Non-SMC Element 2 (NSMCE2) of the SMC5/6 Complex Helps to Resolve Topological Stress. International Journal of Molecular Sciences. 2016; 17(11):1782. https://doi.org/10.3390/ijms17111782
Chicago/Turabian StyleVerver, Dideke E., Yi Zheng, Dave Speijer, Ron Hoebe, Henk L. Dekker, Sjoerd Repping, Jan Stap, and Geert Hamer. 2016. "Non-SMC Element 2 (NSMCE2) of the SMC5/6 Complex Helps to Resolve Topological Stress" International Journal of Molecular Sciences 17, no. 11: 1782. https://doi.org/10.3390/ijms17111782
APA StyleVerver, D. E., Zheng, Y., Speijer, D., Hoebe, R., Dekker, H. L., Repping, S., Stap, J., & Hamer, G. (2016). Non-SMC Element 2 (NSMCE2) of the SMC5/6 Complex Helps to Resolve Topological Stress. International Journal of Molecular Sciences, 17(11), 1782. https://doi.org/10.3390/ijms17111782