
The Impact of Vitamin E and Other Fat-Soluble Vitamins on Alzheimer´s Disease



Abstract
:
1. Introduction
2. Vitamin A
3. Vitamin D
4. Vitamin E
5. Vitamin K
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Plassman, B.L.; Langa, K.M.; Fisher, G.G.; Heeringa, S.G.; Weir, D.R.; Ofstedal, M.B.; Burke, J.R.; Hurd, M.D.; Potter, G.G.; Rodgers, W.L.; et al. Prevalence of dementia in the united states: The aging, demographics, and memory study. Neuroepidemiology 2007, 29, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Alzheimer’s disease: Genes, proteins, and therapy. Physiol. Rev. 2001, 81, 741–766. [Google Scholar] [CrossRef] [PubMed]
- Ferri, C.P.; Prince, M.; Brayne, C.; Brodaty, H.; Fratiglioni, L.; Ganguli, M.; Hall, K.; Hasegawa, K.; Hendrie, H.; Huang, Y.; et al. Global prevalence of dementia: A delphi consensus study. Lancet 2005, 366, 2112–2117. [Google Scholar] [CrossRef]
- Prince, M.; Wimo, A.; Guerchet, M.; Ali, G.-C.; Wu, Y.T.; Prina, M.; Alzheimers Disease International. World Alzheimer Report; Alzheimers Disease International: London, UK, 2015. [Google Scholar]
- Scheff, S.W.; Price, D.A. Synapse loss in the temporal lobe in Alzheimer’s disease. Ann. Neurol. 1993, 33, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Isla, T.; Price, J.L.; McKeel, D.W., Jr.; Morris, J.C.; Growdon, J.H.; Hyman, B.T. Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer’s disease. J. Neurosci. 1996, 16, 4491–4500. [Google Scholar] [PubMed]
- Dickerson, B.C.; Goncharova, I.; Sullivan, M.P.; Forchetti, C.; Wilson, R.S.; Bennett, D.A.; Beckett, L.A.; deToledo-Morrell, L. Mri-derived entorhinal and hippocampal atrophy in incipient and very mild Alzheimer’s disease. Neurobiol. Aging 2001, 22, 747–754. [Google Scholar] [CrossRef]
- Mouton, P.R.; Martin, L.J.; Calhoun, M.E.; Dal Forno, G.; Price, D.L. Cognitive decline strongly correlates with cortical atrophy in Alzheimer’s dementia. Neurobiol. Aging 1998, 19, 371–377. [Google Scholar] [CrossRef]
- Selkoe, D.J. Cell biology of protein misfolding: The examples of Alzheimer’s and Parkinson’s diseases. Nat. Cell Biol. 2004, 6, 1054–1061. [Google Scholar] [CrossRef] [PubMed]
- Masters, C.L.; Simms, G.; Weinman, N.A.; Multhaup, G.; McDonald, B.L.; Beyreuther, K. Amyloid plaque core protein in Alzheimer disease and down syndrome. Proc. Natl. Acad. Sci. USA 1985, 82, 4245–4249. [Google Scholar] [CrossRef] [PubMed]
- Grundke-Iqbal, I.; Iqbal, K.; Quinlan, M.; Tung, Y.C.; Zaidi, M.S.; Wisniewski, H.M. Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J. Biol. Chem. 1986, 261, 6084–6089. [Google Scholar] [PubMed]
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 1984, 120, 885–890. [Google Scholar] [CrossRef]
- Dyrks, T.; Weidemann, A.; Multhaup, G.; Salbaum, J.M.; Lemaire, H.G.; Kang, J.; Muller-Hill, B.; Masters, C.L.; Beyreuther, K. Identification, transmembrane orientation and biogenesis of the amyloid A4 precursor of Alzheimer’s disease. EMBO J. 1988, 7, 949–957. [Google Scholar] [PubMed]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Iwatsubo, T.; Odaka, A.; Suzuki, N.; Mizusawa, H.; Nukina, N.; Ihara, Y. Visualization of Aβ42(43) and Aβ40 in senile plaques with end-specific Aβ monoclonals: Evidence that an initially deposited species is Aβ42(43). Neuron 1994, 13, 45–53. [Google Scholar] [CrossRef]
- Tamaoka, A.; Sawamura, N.; Odaka, A.; Suzuki, N.; Mizusawa, H.; Shoji, S.; Mori, H. Amyloid β protein 1–42/43 (Aβ 1–42/43) in cerebellar diffuse plaques: Enzyme-linked immunosorbent assay and immunocytochemical study. Brain Res. 1995, 679, 151–156. [Google Scholar] [CrossRef]
- Grimm, M.O.; Hartmann, T. Recent understanding of the molecular mechanisms of Alzheimer’s disease. J. Addict. Res. Ther. 2012, S5, 004. [Google Scholar] [CrossRef]
- Drachman, D.A. The amyloid hypothesis, time to move on: Amyloid is the downstream result, not cause, of Alzheimer’s disease. Alzheimer’s Dement. 2014, 10, 372–380. [Google Scholar] [CrossRef] [PubMed]
- Herrup, K. The case for rejecting the amyloid cascade hypothesis. Nat. Neurosci. 2015, 18, 794–799. [Google Scholar] [CrossRef] [PubMed]
- Grimm, M.O.; Mett, J.; Stahlmann, C.P.; Haupenthal, V.J.; Zimmer, V.C.; Hartmann, T. Neprilysin and abeta clearance: Impact of the APP intracellular domain in NEP regulation and implications in Alzheimer’s disease. Front. Aging Neurosci. 2013, 5, 98. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.S.; Ye, J.; Rawson, R.B.; Goldstein, J.L. Regulated intramembrane proteolysis: A control mechanism conserved from bacteria to humans. Cell 2000, 100, 391–398. [Google Scholar] [CrossRef]
- Grimm, M.O.; Grimm, H.S.; Tomic, I.; Beyreuther, K.; Hartmann, T.; Bergmann, C. Independent inhibition of Alzheimer disease β- and γ-secretase cleavage by lowered cholesterol levels. J. Biol. Chem. 2008, 283, 11302–11311. [Google Scholar] [CrossRef] [PubMed]
- Grimm, M.O.; Kuchenbecker, J.; Grosgen, S.; Burg, V.K.; Hundsdorfer, B.; Rothhaar, T.L.; Friess, P.; de Wilde, M.C.; Broersen, L.M.; Penke, B.; et al. Docosahexaenoic acid reduces amyloid β production via multiple pleiotropic mechanisms. J. Biol. Chem. 2011, 286, 14028–14039. [Google Scholar] [CrossRef] [PubMed]
- Grimm, M.O.; Grimm, H.S.; Patzold, A.J.; Zinser, E.G.; Halonen, R.; Duering, M.; Tschape, J.A.; de Strooper, B.; Muller, U.; Shen, J.; et al. Regulation of cholesterol and sphingomyelin metabolism by amyloid-β and presenilin. Nat. Cell Biol. 2005, 7, 1118–1123. [Google Scholar] [CrossRef] [PubMed]
- Grimm, M.O.; Zimmer, V.C.; Lehmann, J.; Grimm, H.S.; Hartmann, T. The impact of cholesterol, DHA, and sphingolipids on Alzheimer’s disease. BioMed Res. Int. 2013, 2013, 814390. [Google Scholar] [CrossRef] [PubMed]
- Grimm, M.O.; Zinser, E.G.; Grosgen, S.; Hundsdorfer, B.; Rothhaar, T.L.; Burg, V.K.; Kaestner, L.; Bayer, T.A.; Lipp, P.; Muller, U.; et al. Amyloid precursor protein (APP) mediated regulation of ganglioside homeostasis linking Alzheimer’s disease pathology with ganglioside metabolism. PLoS ONE 2012, 7, e34095. [Google Scholar] [CrossRef] [PubMed]
- Rothhaar, T.L.; Grosgen, S.; Haupenthal, V.J.; Burg, V.K.; Hundsdorfer, B.; Mett, J.; Riemenschneider, M.; Grimm, H.S.; Hartmann, T.; Grimm, M.O. Plasmalogens inhibit APP processing by directly affecting γ-secretase activity in Alzheimer’s disease. Sci. World J. 2012, 2012, 141240. [Google Scholar] [CrossRef] [PubMed]
- Grimm, M.O.; Rothhaar, T.L.; Grosgen, S.; Burg, V.K.; Hundsdorfer, B.; Haupenthal, V.J.; Friess, P.; Kins, S.; Grimm, H.S.; Hartmann, T. Trans fatty acids enhance amyloidogenic processing of the Alzheimer amyloid precursor protein (APP). J. Nutr. Biochem. 2012, 23, 1214–1223. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, T.; Kuchenbecker, J.; Grimm, M.O. Alzheimer’s disease: The lipid connection. J. Neurochem. 2007, 103 (Suppl. S1), 159–170. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Zhao, B.; Ratka, A. Oxidative stress and β-amyloid protein in Alzheimer’s disease. Neuromol. Med. 2011, 13, 223–250. [Google Scholar] [CrossRef] [PubMed]
- Grimm, M.O.; Haupenthal, V.J.; Mett, J.; Stahlmann, C.P.; Blumel, T.; Mylonas, N.T.; Endres, K.; Grimm, H.S.; Hartmann, T. Oxidized docosahexaenoic acid species and lipid peroxidation products increase amyloidogenic amyloid precursor protein processing. Neurodegener. Dis. 2016, 16, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Mohajeri, M.H.; Troesch, B.; Weber, P. Inadequate supply of vitamins and DHA in the elderly: Implications for brain aging and Alzheimer-type dementia. Nutrition 2015, 31, 261–275. [Google Scholar] [CrossRef] [PubMed]
- Lane, M.A.; Bailey, S.J. Role of retinoid signalling in the adult brain. Prog. Neurobiol. 2005, 75, 275–293. [Google Scholar] [CrossRef] [PubMed]
- Sodhi, R.K.; Singh, N. Retinoids as potential targets for Alzheimer’s disease. Pharmacol. Biochem. Behav. 2014, 120, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, P.; Polidori, M.C.; Metastasio, A.; Mariani, E.; Mattioli, P.; Cherubini, A.; Catani, M.; Cecchetti, R.; Senin, U.; Mecocci, P. Plasma antioxidants are similarly depleted in mild cognitive impairment and in Alzheimer’s disease. Neurobiol. Aging 2003, 24, 915–919. [Google Scholar] [CrossRef]
- Bourdel-Marchasson, I.; Delmas-Beauvieux, M.C.; Peuchant, E.; Richard-Harston, S.; Decamps, A.; Reignier, B.; Emeriau, J.P.; Rainfray, M. Antioxidant defences and oxidative stress markers in erythrocytes and plasma from normally nourished elderly Alzheimer patients. Age Ageing 2001, 30, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Jimenez, F.J.; Molina, J.A.; de Bustos, F.; Orti-Pareja, M.; Benito-Leon, J.; Tallon-Barranco, A.; Gasalla, T.; Porta, J.; Arenas, J. Serum levels of β-carotene, α-carotene and vitamin A in patients with Alzheimer’s disease. Eur. J. Neurol. 1999, 6, 495–497. [Google Scholar] [CrossRef] [PubMed]
- Perrig, W.J.; Perrig, P.; Stahelin, H.B. The relation between antioxidants and memory performance in the old and very old. J. Am. Geriatr. Soc. 1997, 45, 718–724. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, M.B.; Clarke, E.; Hobbs, C.; Malmqvist, T.; Deacon, R.; Jack, J.; Corcoran, J.P. Amyloid β inhibits retinoic acid synthesis exacerbating Alzheimer disease pathology which can be attenuated by an retinoic acid receptor α agonist. Eur. J. Neurosci. 2013, 37, 1182–1192. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, J.P.; So, P.L.; Maden, M. Disruption of the retinoid signalling pathway causes a deposition of amyloid β in the adult rat brain. Eur. J. Neurosci. 2004, 20, 896–902. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, S.; Grimm, M.O.; Stahlmann, C.; Hartmann, T.; Shudo, K.; Tomita, T.; Endres, K. Rescue of hypovitaminosis a induces non-amyloidogenic amyloid precursor protein (APP) processing. Curr. Alzheimer Res. 2016, 13, 1277–1289. [Google Scholar] [CrossRef]
- Ding, Y.; Qiao, A.; Wang, Z.; Goodwin, J.S.; Lee, E.S.; Block, M.L.; Allsbrook, M.; McDonald, M.P.; Fan, G.H. Retinoic acid attenuates β-amyloid deposition and rescues memory deficits in an Alzheimer’s disease transgenic mouse model. J. Neurosci. 2008, 28, 11622–11634. [Google Scholar] [CrossRef] [PubMed]
- Sodhi, R.K.; Singh, N. All-trans retinoic acid rescues memory deficits and neuropathological changes in mouse model of streptozotocin-induced dementia of Alzheimer’s type. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2013, 40, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Watamura, N.; Toba, J.; Yoshii, A.; Nikkuni, M.; Ohshima, T. Colocalization of phosphorylated forms of WAVE1, CRMP2, and tau in Alzheimer’s disease model mice: Involvement of Cdk5 phosphorylation and the effect of ATRA treatment. J. Neurosci. Res. 2016, 94, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, C.I.; Goncalves, M.B.; Clarke, E.; Dogruel, M.; Kalindjian, S.B.; Thomas, S.A.; Maden, M.; Corcoran, J.P. Retinoic acid receptor-α signalling antagonizes both intracellular and extracellular amyloid-β production and prevents neuronal cell death caused by amyloid-β. Eur. J. Neurosci. 2010, 32, 1246–1255. [Google Scholar] [CrossRef] [PubMed]
- Melino, G.; Draoui, M.; Bernardini, S.; Bellincampi, L.; Reichert, U.; Cohen, P. Regulation by retinoic acid of insulin-degrading enzyme and of a related endoprotease in human neuroblastoma cell lines. Cell Growth Differ. Mol. Biol. J. Am. Assoc. Cancer Res. 1996, 7, 787–796. [Google Scholar]
- Satoh, J.; Kuroda, Y. Amyloid precursor protein β-secretase (BACE) mRNA expression in human neural cell lines following induction of neuronal differentiation and exposure to cytokines and growth factors. Neuropathology 2000, 20, 289–296. [Google Scholar] [CrossRef]
- Hong, C.S.; Caromile, L.; Nomata, Y.; Mori, H.; Bredesen, D.E.; Koo, E.H. Contrasting role of presenilin-1 and presenilin-2 in neuronal differentiation in vitro. J. Neurosci. 1999, 19, 637–643. [Google Scholar]
- Yang, Y.; Quitschke, W.W.; Brewer, G.J. Upregulation of amyloid precursor protein gene promoter in rat primary hippocampal neurons by phorbol ester, IL-1 and retinoic acid, but not by reactive oxygen species. Mol. Brain Res. 1998, 60, 40–49. [Google Scholar] [CrossRef]
- Koryakina, A.; Aeberhard, J.; Kiefer, S.; Hamburger, M.; Kuenzi, P. Regulation of secretases by all-trans-retinoic acid. FEBS J. 2009, 276, 2645–2655. [Google Scholar] [CrossRef] [PubMed]
- Prinzen, C.; Muller, U.; Endres, K.; Fahrenholz, F.; Postina, R. Genomic structure and functional characterization of the human ADAM 10 promoter. FASEB J. 2005, 19, 1522–1524. [Google Scholar] [PubMed]
- Ono, K.; Yoshiike, Y.; Takashima, A.; Hasegawa, K.; Naiki, H.; Yamada, M. Vitamin A exhibits potent antiamyloidogenic and fibril-destabilizing effects in vitro. Exp. Neurol. 2004, 189, 380–392. [Google Scholar] [CrossRef] [PubMed]
- Takasaki, J.; Ono, K.; Yoshiike, Y.; Hirohata, M.; Ikeda, T.; Morinaga, A.; Takashima, A.; Yamada, M. Vitamin A has anti-oligomerization effects on amyloid-β in vitro. J. Alzheimer’s Dis. 2011, 27, 271–280. [Google Scholar]
- Annweiler, C.; Beauchet, O. Vitamin D-mentia: Randomized clinical trials should be the next step. Neuroepidemiology 2011, 37, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Khemka, V.K.; Ganguly, A.; Roy, D.; Ganguly, U.; Chakrabarti, S. Vitamin D and Alzheimer’s disease: Neurocognition to therapeutics. Int. J. Alzheimer’s Dis. 2015, 2015, 192747. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Asoh, T.; Oizumi, K. High prevalence of vitamin D deficiency and reduced bone mass in elderly women with Alzheimer’s disease. Bone 1998, 23, 555–557. [Google Scholar] [CrossRef]
- Kipen, E.; Helme, R.D.; Wark, J.D.; Flicker, L. Bone density, vitamin D nutrition, and parathyroid hormone levels in women with dementia. J. Am. Geriatr. Soc. 1995, 43, 1088–1091. [Google Scholar] [CrossRef] [PubMed]
- Llewellyn, D.J.; Lang, I.A.; Langa, K.M.; Muniz-Terrera, G.; Phillips, C.L.; Cherubini, A.; Ferrucci, L.; Melzer, D. Vitamin D and risk of cognitive decline in elderly persons. Arch. Intern. Med. 2010, 170, 1135–1141. [Google Scholar] [CrossRef] [PubMed]
- Balion, C.; Griffith, L.E.; Strifler, L.; Henderson, M.; Patterson, C.; Heckman, G.; Llewellyn, D.J.; Raina, P. Vitamin D, cognition, and dementia: A systematic review and meta-analysis. Neurology 2012, 79, 1397–1405. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Ji, H.F. Vitamin D deficiency is associated with increased risk of Alzheimer’s disease and dementia: Evidence from meta-analysis. Nutr. J. 2015, 14, 76. [Google Scholar] [CrossRef] [PubMed]
- Littlejohns, T.J.; Henley, W.E.; Lang, I.A.; Annweiler, C.; Beauchet, O.; Chaves, P.H.; Fried, L.; Kestenbaum, B.R.; Kuller, L.H.; Langa, K.M.; et al. Vitamin D and the risk of dementia and Alzheimer disease. Neurology 2014, 83, 920–928. [Google Scholar] [CrossRef] [PubMed]
- Llewellyn, D.J.; Lang, I.A.; Langa, K.M.; Melzer, D. Vitamin D and cognitive impairment in the elderly U.S. Population. J. Gerontol. Ser. A: Biol. Sci. Med. Sci. 2011, 66, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Przybelski, R.J.; Binkley, N.C. Is vitamin D important for preserving cognition? A positive correlation of serum 25-hydroxyvitamin D concentration with cognitive function. Arch. Biochem. Biophys. 2007, 460, 202–205. [Google Scholar] [CrossRef] [PubMed]
- Hooshmand, B.; Lokk, J.; Solomon, A.; Mangialasche, F.; Miralbell, J.; Spulber, G.; Annerbo, S.; Andreasen, N.; Winblad, B.; Cedazo-Minguez, A.; et al. Vitamin D in relation to cognitive impairment, cerebrospinal fluid biomarkers, and brain volumes. J. Gerontol. Ser A Biol. Sci. Med. Sci. 2014, 69, 1132–1138. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, D.J.; Refsum, H.; Warden, D.R.; Medway, C.; Wilcock, G.K.; Smith, A.D. The vitamin D receptor gene is associated with Alzheimer’s disease. Neurosci. Lett. 2011, 504, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Kim, J.H.; Song, G.G. Vitamin D receptor polymorphisms and susceptibility to Parkinson’s disease and Alzheimer’s disease: A meta-analysis. Neurol. Sci. 2014, 35, 1947–1953. [Google Scholar] [CrossRef] [PubMed]
- Gezen-Ak, D.; Dursun, E.; Ertan, T.; Hanagasi, H.; Gurvit, H.; Emre, M.; Eker, E.; Ozturk, M.; Engin, F.; Yilmazer, S. Association between vitamin D receptor gene polymorphism and Alzheimer’s disease. Tohoku J. Exp. Med. 2007, 212, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Hara, K.; van Baaren, J.M.; Price, J.C.; Beecham, G.W.; Gallins, P.J.; Whitehead, P.L.; Wang, G.; Lu, C.; Slifer, M.A.; et al. Vitamin D receptor and Alzheimer’s disease: A genetic and functional study. Neurobiol. Aging 2012, 33, 1841–1849. [Google Scholar] [CrossRef] [PubMed]
- Annweiler, C.; Fantino, B.; Parot-Schinkel, E.; Thiery, S.; Gautier, J.; Beauchet, O. Alzheimer’s disease—Input of vitamin D with memantine assay (AD-IDEA trial): Study protocol for a randomized controlled trial. Trials 2011, 12, 230. [Google Scholar] [CrossRef] [PubMed]
- Annweiler, C.; Brugg, B.; Peyrin, J.M.; Bartha, R.; Beauchet, O. Combination of memantine and vitamin D prevents axon degeneration induced by amyloid-β and glutamate. Neurobiol. Aging 2014, 35, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Annweiler, C.; Herrmann, F.R.; Fantino, B.; Brugg, B.; Beauchet, O. Effectiveness of the combination of memantine plus vitamin D on cognition in patients with Alzheimer disease: A pre-post pilot study. Cogn. Behav. Neurol. 2012, 25, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Stein, M.S.; Scherer, S.C.; Ladd, K.S.; Harrison, L.C. A randomized controlled trial of high-dose vitamin D2 followed by intranasal insulin in Alzheimer’s disease. J. Alzheimer’s Dis. 2011, 26, 477–484. [Google Scholar]
- Przybelski, R.; Agrawal, S.; Krueger, D.; Engelke, J.A.; Walbrun, F.; Binkley, N. Rapid correction of low vitamin D status in nursing home residents. Osteoporos. Int. 2008, 19, 1621–1628. [Google Scholar] [CrossRef] [PubMed]
- Keeney, J.T.; Butterfield, D.A. Vitamin D deficiency and Alzheimer disease: Common links. Neurobiol. Dis. 2015, 84, 84–98. [Google Scholar] [CrossRef] [PubMed]
- Durk, M.R.; Han, K.; Chow, E.C.; Ahrens, R.; Henderson, J.T.; Fraser, P.E.; Pang, K.S. 1α,25-dihydroxyvitamin D3 reduces cerebral amyloid-β accumulation and improves cognition in mouse models of Alzheimer’s disease. J. Neurosci. 2014, 34, 7091–7101. [Google Scholar] [CrossRef] [PubMed]
- Briones, T.L.; Darwish, H. Vitamin D mitigates age-related cognitive decline through the modulation of pro-inflammatory state and decrease in amyloid burden. J. Neuroinflamm. 2012, 9, 244. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Gattoni-Celli, M.; Zhu, H.; Bhat, N.R.; Sambamurti, K.; Gattoni-Celli, S.; Kindy, M.S. Vitamin D3-enriched diet correlates with a decrease of amyloid plaques in the brain of AβPP transgenic mice. J. Alzheimer’s Dis. 2011, 25, 295–307. [Google Scholar]
- Grimm, M.O.; Lehmann, J.; Mett, J.; Zimmer, V.C.; Grosgen, S.; Stahlmann, C.P.; Hundsdorfer, B.; Haupenthal, V.J.; Rothhaar, T.L.; Herr, C.; et al. Impact of vitamin D on amyloid precursor protein processing and amyloid-β peptide degradation in Alzheimer’s disease. Neurodegener. Dis. 2014, 13, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Taghizadeh, M.; Djazayery, A.; Salami, M.; Eshraghian, M.R.; Zavareh, S.A. Vitamin-D-free regimen intensifies the spatial learning deficit in Alzheimer’s disease. Int. J. Neurosci. 2011, 121, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Masoumi, A.; Goldenson, B.; Ghirmai, S.; Avagyan, H.; Zaghi, J.; Abel, K.; Zheng, X.; Espinosa-Jeffrey, A.; Mahanian, M.; Liu, P.T.; et al. 1α,25-Dihydroxyvitamin D3 interacts with curcuminoids to stimulate amyloid-β clearance by macrophages of Alzheimer’s disease patients. J. Alzheimer’s Dis. 2009, 17, 703–717. [Google Scholar]
- Ito, S.; Ohtsuki, S.; Nezu, Y.; Koitabashi, Y.; Murata, S.; Terasaki, T. 1α,25-Dihydroxyvitamin D3 enhances cerebral clearance of human amyloid-β peptide(1–40) from mouse brain across the blood-brain barrier. Fluids Barriers CNS 2011, 8, 20. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Xia, X.; Rui, Y.; Zhang, Z.; Qin, L.; Han, S.; Wan, Z. The combination of 1α,25dihydroxyvitaminD3 with resveratrol improves neuronal degeneration by regulating endoplasmic reticulum stress, insulin signaling and inhibiting tau hyperphosphorylation in SH-SY5Y cells. Food Chem. Toxicol. 2016, 93, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Brigelius-Flohe, R.; Traber, M.G. Vitamin E: Function and metabolism. FASEB J. 1999, 13, 1145–1155. [Google Scholar] [PubMed]
- Reiter, E.; Jiang, Q.; Christen, S. Anti-inflammatory properties of α- and γ-tocopherol. Mol. Asp. Med. 2007, 28, 668–691. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q. Natural forms of vitamin E: Metabolism, antioxidant, and anti-inflammatory activities and their role in disease prevention and therapy. Free Radic. Biol. Med. 2014, 72, 76–90. [Google Scholar] [CrossRef] [PubMed]
- Joshi, Y.B.; Pratico, D. Vitamin E in aging, dementia, and Alzheimer’s disease. BioFactors 2012, 38, 90–97. [Google Scholar] [CrossRef] [PubMed]
- La Fata, G.; Weber, P.; Mohajeri, M.H. Effects of vitamin E on cognitive performance during ageing and in Alzheimer’s disease. Nutrients 2014, 6, 5453–5472. [Google Scholar] [CrossRef] [PubMed]
- Mangialasche, F.; Xu, W.; Kivipelto, M.; Costanzi, E.; Ercolani, S.; Pigliautile, M.; Cecchetti, R.; Baglioni, M.; Simmons, A.; Soininen, H.; et al. Tocopherols and tocotrienols plasma levels are associated with cognitive impairment. Neurobiol. Aging 2012, 33, 2282–2290. [Google Scholar] [CrossRef] [PubMed]
- Lopes da Silva, S.; Vellas, B.; Elemans, S.; Luchsinger, J.; Kamphuis, P.; Yaffe, K.; Sijben, J.; Groenendijk, M.; Stijnen, T. Plasma nutrient status of patients with Alzheimer’s disease: Systematic review and meta-analysis. Alzheimer’s Dement. 2014, 10, 485–502. [Google Scholar] [CrossRef] [PubMed]
- Mangialasche, F.; Kivipelto, M.; Mecocci, P.; Rizzuto, D.; Palmer, K.; Winblad, B.; Fratiglioni, L. High plasma levels of vitamin E forms and reduced Alzheimer’s disease risk in advanced age. J. Alzheimer’s Dis. 2010, 20, 1029–1037. [Google Scholar]
- Li, F.J.; Shen, L.; Ji, H.F. Dietary intakes of vitamin E, vitamin C, and β-carotene and risk of Alzheimer’s disease: A meta-analysis. J. Alzheimer’s Dis. 2012, 31, 253–258. [Google Scholar]
- Morris, M.C.; Evans, D.A.; Tangney, C.C.; Bienias, J.L.; Wilson, R.S.; Aggarwal, N.T.; Scherr, P.A. Relation of the tocopherol forms to incident Alzheimer disease and to cognitive change. Am. J. Clin. Nutr. 2005, 81, 508–514. [Google Scholar] [PubMed]
- Morris, M.C.; Schneider, J.A.; Li, H.; Tangney, C.C.; Nag, S.; Bennett, D.A.; Honer, W.G.; Barnes, L.L. Brain tocopherols related to Alzheimer’s disease neuropathology in humans. Alzheimer’s Dement. 2015, 11, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Sano, M.; Ernesto, C.; Thomas, R.G.; Klauber, M.R.; Schafer, K.; Grundman, M.; Woodbury, P.; Growdon, J.; Cotman, C.W.; Pfeiffer, E.; et al. A controlled trial of selegiline, α-tocopherol, or both as treatment for Alzheimer’s disease. N. Engl. J. Med. 1997, 336, 1216–1222. [Google Scholar] [CrossRef] [PubMed]
- Dysken, M.W.; Sano, M.; Asthana, S.; Vertrees, J.E.; Pallaki, M.; Llorente, M.; Love, S.; Schellenberg, G.D.; McCarten, J.R.; Malphurs, J.; et al. Effect of vitamin E and memantine on functional decline in Alzheimer disease: The TEAM-AD va cooperative randomized trial. JAMA 2014, 311, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Petersen, R.C.; Thomas, R.G.; Grundman, M.; Bennett, D.; Doody, R.; Ferris, S.; Galasko, D.; Jin, S.; Kaye, J.; Levey, A.; et al. Vitamin E and donepezil for the treatment of mild cognitive impairment. N. Engl. J. Med. 2005, 352, 2379–2388. [Google Scholar] [CrossRef] [PubMed]
- Galasko, D.R.; Peskind, E.; Clark, C.M.; Quinn, J.F.; Ringman, J.M.; Jicha, G.A.; Cotman, C.; Cottrell, B.; Montine, T.J.; Thomas, R.G.; et al. Antioxidants for Alzheimer disease: A randomized clinical trial with cerebrospinal fluid biomarker measures. Arch. Neurol. 2012, 69, 836–841. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.R.; Pastor-Barriuso, R.; Dalal, D.; Riemersma, R.A.; Appel, L.J.; Guallar, E. Meta-analysis: High-dosage vitamin E supplementation may increase all-cause mortality. Ann. Intern. Med. 2005, 142, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Sung, S.; Yao, Y.; Uryu, K.; Yang, H.; Lee, V.M.; Trojanowski, J.Q.; Pratico, D. Early vitamin E supplementation in young but not aged mice reduces abeta levels and amyloid deposition in a transgenic model of Alzheimer’s disease. FASEB J. 2004, 18, 323–325. [Google Scholar] [PubMed]
- Sinha, M.; Bir, A.; Banerjee, A.; Bhowmick, P.; Chakrabarti, S. Multiple mechanisms of age-dependent accumulation of amyloid β protein in rat brain: Prevention by dietary supplementation with N-acetylcysteine, α-lipoic acid and α-tocopherol. Neurochem. Int. 2016, 95, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Thakurta, I.G.; Banerjee, P.; Bagh, M.B.; Ghosh, A.; Sahoo, A.; Chattopadhyay, S.; Chakrabarti, S. Combination of N-acetylcysteine, α-lipoic acid and α-tocopherol substantially prevents the brain synaptosomal alterations and memory and learning deficits of aged rats. Exp. Gerontol. 2014, 50, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.W.; Yang, S.G.; Liu, W.; Zhang, Y.X.; Xu, P.X.; Wang, T.; Ling, T.J.; Liu, R.T. α-Tocopherol quinine ameliorates spatial memory deficits by reducing β-amyloid oligomers, neuroinflammation and oxidative stress in transgenic mice with Alzheimer’s disease. Behav. Brain Res. 2016, 296, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Sandy, M.S.; di Monte, D.; Smith, M.T. Relationships between intracellular vitamin E, lipid peroxidation, and chemical toxicity in hepatocytes. Toxicol. Appl. Pharmacol. 1988, 93, 288–297. [Google Scholar] [CrossRef]
- Qi, X.L.; Xiu, J.; Shan, K.R.; Xiao, Y.; Gu, R.; Liu, R.Y.; Guan, Z.Z. Oxidative stress induced by β-amyloid peptide(1–42) is involved in the altered composition of cellular membrane lipids and the decreased expression of nicotinic receptors in human SH-SY5Y neuroblastoma cells. Neurochem. Int. 2005, 46, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Sun, Y.; Jiang, Z. Protective effects of vitamin E against oxidative damage induced by Aβ1–40Cu(II) complexes. Acta Biochim. Biophys. Sin. 2007, 39, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Shea, T.B.; Ortiz, D.; Nicolosi, R.J.; Kumar, R.; Watterson, A.C. Nanosphere-mediated delivery of vitamin E increases its efficacy against oxidative stress resulting from exposure to amyloid β. J. Alzheimer’s Dis. 2005, 7, 297–301. [Google Scholar]
- Nishida, Y.; Yokota, T.; Takahashi, T.; Uchihara, T.; Jishage, K.; Mizusawa, H. Deletion of vitamin E enhances phenotype of Alzheimer disease model mouse. Biochem. Biophys. Res. Commun. 2006, 350, 530–536. [Google Scholar] [CrossRef] [PubMed]
- Nishida, Y.; Ito, S.; Ohtsuki, S.; Yamamoto, N.; Takahashi, T.; Iwata, N.; Jishage, K.; Yamada, H.; Sasaguri, H.; Yokota, S.; et al. Depletion of vitamin E increases amyloid β accumulation by decreasing its clearances from brain and blood in a mouse model of Alzheimer disease. J. Biol. Chem. 2009, 284, 33400–33408. [Google Scholar] [CrossRef] [PubMed]
- Rota, C.; Rimbach, G.; Minihane, A.M.; Stoecklin, E.; Barella, L. Dietary vitamin E modulates differential gene expression in the rat hippocampus: Potential implications for its neuroprotective properties. Nutr. Neurosci. 2005, 8, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Grimm, M.O.; Stahlmann, C.P.; Mett, J.; Haupenthal, V.J.; Zimmer, V.C.; Lehmann, J.; Hundsdorfer, B.; Endres, K.; Grimm, H.S.; Hartmann, T. Vitamin E: Curse or benefit in Alzheimer’s disease? A systematic investigation of the impact of α-, γ- and δ-tocopherol on ass generation and degradation in neuroblastoma cells. J. Nutr. Health Aging 2015, 19, 646–656. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A. Amyloid β-peptide (1–42)-induced oxidative stress and neurotoxicity: Implications for neurodegeneration in Alzheimer’s disease brain. A review. Free Radic. Res. 2002, 36, 1307–1313. [Google Scholar] [CrossRef] [PubMed]
- Nohturfft, A.; Yabe, D.; Goldstein, J.L.; Brown, M.S.; Espenshade, P.J. Regulated step in cholesterol feedback localized to budding of SCAP from ER membranes. Cell 2000, 102, 315–323. [Google Scholar] [CrossRef]
- Sakai, J.; Duncan, E.A.; Rawson, R.B.; Hua, X.; Brown, M.S.; Goldstein, J.L. Sterol-regulated release of SREBP-2 from cell membranes requires two sequential cleavages, one within a transmembrane segment. Cell 1996, 85, 1037–1046. [Google Scholar] [CrossRef]
- Song, B.L.; DeBose-Boyd, R.A. Insig-dependent ubiquitination and degradation of 3-hydroxy-3-methylglutaryl coenzyme A reductase stimulated by δ- and γ-tocotrienols. J. Biol. Chem. 2006, 281, 25054–25061. [Google Scholar] [CrossRef] [PubMed]
- Valastyan, S.; Thakur, V.; Johnson, A.; Kumar, K.; Manor, D. Novel transcriptional activities of vitamin E: Inhibition of cholesterol biosynthesis. Biochemistry 2008, 47, 744–752. [Google Scholar] [CrossRef] [PubMed]
- Krycer, J.R.; Phan, L.; Brown, A.J. A key regulator of cholesterol homoeostasis, SREBP-2, can be targeted in prostate cancer cells with natural products. Biochem. J. 2012, 446, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Simons, M.; Keller, P.; de Strooper, B.; Beyreuther, K.; Dotti, C.G.; Simons, K. Cholesterol depletion inhibits the generation of β-amyloid in hippocampal neurons. Proc. Natl. Acad. Sci. USA 1998, 95, 6460–6464. [Google Scholar] [CrossRef] [PubMed]
- Fassbender, K.; Simons, M.; Bergmann, C.; Stroick, M.; Lutjohann, D.; Keller, P.; Runz, H.; Kuhl, S.; Bertsch, T.; von Bergmann, K.; et al. Simvastatin strongly reduces levels of Alzheimer’s disease β-amyloid peptides Aβ 42 and Aβ 40 in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2001, 98, 5856–5861. [Google Scholar] [CrossRef] [PubMed]
- Maulik, M.; Westaway, D.; Jhamandas, J.H.; Kar, S. Role of cholesterol in APP metabolism and its significance in Alzheimer’s disease pathogenesis. Mol. Neurobiol. 2013, 47, 37–63. [Google Scholar] [CrossRef] [PubMed]
- Sparks, D.L.; Scheff, S.W.; Hunsaker, J.C., III; Liu, H.; Landers, T.; Gross, D.R. Induction of Alzheimer-like β-amyloid immunoreactivity in the brains of rabbits with dietary cholesterol. Exp. Neurol. 1994, 126, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Refolo, L.M.; Malester, B.; LaFrancois, J.; Bryant-Thomas, T.; Wang, R.; Tint, G.S.; Sambamurti, K.; Duff, K.; Pappolla, M.A. Hypercholesterolemia accelerates the Alzheimer’s amyloid pathology in a transgenic mouse model. Neurobiol. Dis. 2000, 7, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Osenkowski, P.; Ye, W.; Wang, R.; Wolfe, M.S.; Selkoe, D.J. Direct and potent regulation of γ-secretase by its lipid microenvironment. J. Biol. Chem. 2008, 283, 22529–22540. [Google Scholar] [CrossRef] [PubMed]
- Kalvodova, L.; Kahya, N.; Schwille, P.; Ehehalt, R.; Verkade, P.; Drechsel, D.; Simons, K. Lipids as modulators of proteolytic activity of bace: Involvement of cholesterol, glycosphingolipids, and anionic phospholipids in vitro. J. Biol. Chem. 2005, 280, 36815–36823. [Google Scholar] [CrossRef] [PubMed]
- Hao, M.; Mukherjee, S.; Maxfield, F.R. Cholesterol depletion induces large scale domain segregation in living cell membranes. Proc. Natl. Acad. Sci. USA 2001, 98, 13072–13077. [Google Scholar] [CrossRef] [PubMed]
- Hicks, D.A.; Nalivaeva, N.N.; Turner, A.J. Lipid rafts and Alzheimer’s disease: Protein-lipid interactions and perturbation of signaling. Front. Physiol. 2012, 3, 189. [Google Scholar] [CrossRef] [PubMed]
- Cossec, J.C.; Simon, A.; Marquer, C.; Moldrich, R.X.; Leterrier, C.; Rossier, J.; Duyckaerts, C.; Lenkei, Z.; Potier, M.C. Clathrin-dependent APP endocytosis and Aβ secretion are highly sensitive to the level of plasma membrane cholesterol. Biochim. Biophys. Acta 2010, 1801, 846–852. [Google Scholar] [CrossRef] [PubMed]
- Kojro, E.; Gimpl, G.; Lammich, S.; Marz, W.; Fahrenholz, F. Low cholesterol stimulates the nonamyloidogenic pathway by its effect on the α-secretase ADAM 10. Proc. Natl. Acad. Sci. USA 2001, 98, 5815–5820. [Google Scholar] [CrossRef] [PubMed]
- Schneider, A.; Schulz-Schaeffer, W.; Hartmann, T.; Schulz, J.B.; Simons, M. Cholesterol depletion reduces aggregation of amyloid-β peptide in hippocampal neurons. Neurobiol. Dis. 2006, 23, 573–577. [Google Scholar] [CrossRef] [PubMed]
- Ferrera, P.; Mercado-Gomez, O.; Silva-Aguilar, M.; Valverde, M.; Arias, C. Cholesterol potentiates β-amyloid-induced toxicity in human neuroblastoma cells: Involvement of oxidative stress. Neurochem. Res. 2008, 33, 1509–1517. [Google Scholar] [CrossRef] [PubMed]
- Abramov, A.Y.; Ionov, M.; Pavlov, E.; Duchen, M.R. Membrane cholesterol content plays a key role in the neurotoxicity of β-amyloid: Implications for Alzheimer’s disease. Aging Cell 2011, 10, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Tran, K.; Wong, J.T.; Lee, E.; Chan, A.C.; Choy, P.C. Vitamin E potentiates arachidonate release and phospholipase A2 activity in rat heart myoblastic cells. Biochem. J. 1996, 319 Pt 2, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Robinson, B.S.; Hii, C.S.; Poulos, A.; Ferrante, A. Activation of neutral sphingomyelinase in human neutrophils by polyunsaturated fatty acids. Immunology 1997, 91, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Toman, R.E.; Movsesyan, V.; Murthy, S.K.; Milstien, S.; Spiegel, S.; Faden, A.I. Ceramide-induced cell death in primary neuronal cultures: Upregulation of ceramide levels during neuronal apoptosis. J. Neurosci. Res. 2002, 68, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Dawson, G.; Goswami, R.; Kilkus, J.; Wiesner, D.; Dawson, S. The formation of ceramide from sphingomyelin is associated with cellular apoptosis. Acta Biochim. Pol. 1998, 45, 287–297. [Google Scholar] [PubMed]
- Puglielli, L.; Ellis, B.C.; Saunders, A.J.; Kovacs, D.M. Ceramide stabilizes β-site amyloid precursor protein-cleaving enzyme 1 and promotes amyloid β-peptide biogenesis. J. Biol. Chem. 2003, 278, 19777–19783. [Google Scholar] [CrossRef] [PubMed]
- Ko, M.H.; Puglielli, L. Two endoplasmic reticulum (ER)/ER Golgi intermediate compartment-based lysine acetyltransferases post-translationally regulate BACE1 levels. J. Biol. Chem. 2009, 284, 2482–2492. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Mejia, R.O.; Newman, J.W.; Toh, S.; Yu, G.Q.; Zhou, Y.; Halabisky, B.; Cisse, M.; Scearce-Levie, K.; Cheng, I.H.; Gan, L.; et al. Phospholipase A2 reduction ameliorates cognitive deficits in a mouse model of Alzheimer’s disease. Nat. Neurosci. 2008, 11, 1311–1318. [Google Scholar] [CrossRef] [PubMed]
- Giraldo, E.; Lloret, A.; Fuchsberger, T.; Vina, J. Aβ and tau toxicities in Alzheimer’s are linked via oxidative stress-induced p38 activation: Protective role of vitamin E. Redox Biol. 2014, 2, 873–877. [Google Scholar] [CrossRef] [PubMed]
- Ekinci, F.J.; Linsley, M.D.; Shea, T.B. β-Amyloid-induced calcium influx induces apoptosis in culture by oxidative stress rather than tau phosphorylation. Mol. Brain Res. 2000, 76, 389–395. [Google Scholar] [CrossRef]
- Nakashima, H.; Ishihara, T.; Yokota, O.; Terada, S.; Trojanowski, J.Q.; Lee, V.M.; Kuroda, S. Effects of α-tocopherol on an animal model of tauopathies. Free Radic. Biol. Med. 2004, 37, 176–186. [Google Scholar] [CrossRef] [PubMed]
- Dias-Santagata, D.; Fulga, T.A.; Duttaroy, A.; Feany, M.B. Oxidative stress mediates tau-induced neurodegeneration in drosophila. J. Clin. Investig. 2007, 117, 236–245. [Google Scholar] [CrossRef] [PubMed]
- DiNicolantonio, J.J.; Bhutani, J.; O’Keefe, J.H. The health benefits of vitamin K. Open Heart 2015, 2, e000300. [Google Scholar] [CrossRef] [PubMed]
- Ferland, G. Vitamin K and the nervous system: An overview of its actions. Adv. Nutr. 2012, 3, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Presse, N.; Shatenstein, B.; Kergoat, M.J.; Ferland, G. Low vitamin K intakes in community-dwelling elders at an early stage of Alzheimer’s disease. J. Am. Diet. Assoc. 2008, 108, 2095–2099. [Google Scholar] [CrossRef] [PubMed]
- Shatenstein, B.; Kergoat, M.J.; Reid, I. Poor nutrient intakes during 1-year follow-up with community-dwelling older adults with early-stage Alzheimer dementia compared to cognitively intact matched controls. J. Am. Diet. Assoc. 2007, 107, 2091–2099. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Honda, Y.; Hayashida, N.; Iwamoto, J.; Kanoko, T.; Satoh, K. Vitamin K deficiency and osteopenia in elderly women with Alzheimer’s disease. Arch. Phys. Med. Rehabil. 2005, 86, 576–581. [Google Scholar] [CrossRef] [PubMed]
- Saupe, J.; Shearer, M.J.; Kohlmeier, M. Phylloquinone transport and its influence on γ-carboxyglutamate residues of osteocalcin in patients on maintenance hemodialysis. Am. J. Clin. Nutr. 1993, 58, 204–208. [Google Scholar] [PubMed]
- Annweiler, C.; Ferland, G.; Barberger-Gateau, P.; Brangier, A.; Rolland, Y.; Beauchet, O. Vitamin K antagonists and cognitive impairment: Results from a cross-sectional pilot study among geriatric patients. J. Gerontol. Ser A Biol. Sci. Med. Sci. 2015, 70, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Carrie, I.; Belanger, E.; Portoukalian, J.; Rochford, J.; Ferland, G. Lifelong low-phylloquinone intake is associated with cognitive impairments in old rats. J. Nutr. 2011, 141, 1495–1501. [Google Scholar] [CrossRef] [PubMed]
- Huy, P.D.; Yu, Y.C.; Ngo, S.T.; Thao, T.V.; Chen, C.P.; Li, M.S.; Chen, Y.C. In silico and in vitro characterization of anti-amyloidogenic activity of vitamin K3 analogues for Alzheimer’s disease. Biochim. Biophys. Acta 2013, 1830, 2960–2969. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, K.S.; Lev, M. Warfarin administration reduces synthesis of sulfatides and other sphingolipids in mouse brain. J. Lipid Res. 1988, 29, 1475–1479. [Google Scholar] [PubMed]
- Sundaram, K.S.; Fan, J.H.; Engelke, J.A.; Foley, A.L.; Suttie, J.W.; Lev, M. Vitamin K status influences brain sulfatide metabolism in young mice and rats. J. Nutr. 1996, 126, 2746–2751. [Google Scholar] [PubMed]
- Han, X.; Holtzman, D.M.; McKeel, D.W., Jr.; Kelley, J.; Morris, J.C. Substantial sulfatide deficiency and ceramide elevation in very early Alzheimer’s disease: Potential role in disease pathogenesis. J. Neurochem. 2002, 82, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Bandaru, V.V.; Troncoso, J.; Wheeler, D.; Pletnikova, O.; Wang, J.; Conant, K.; Haughey, N.J. ApoE4 disrupts sterol and sphingolipid metabolism in Alzheimer’s but not normal brain. Neurobiol. Aging 2009, 30, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Wang, M.; Li, J.L.; Cairns, N.J.; Han, X. Specific changes of sulfatide levels in individuals with pre-clinical Alzheimer’s disease: An early event in disease pathogenesis. J. Neurochem. 2013, 127, 733–738. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Fagan, A.M.; Cheng, H.; Morris, J.C.; Xiong, C.; Holtzman, D.M. Cerebrospinal fluid sulfatide is decreased in subjects with incipient dementia. Ann. Neurol. 2003, 54, 115–119. [Google Scholar] [CrossRef] [PubMed]
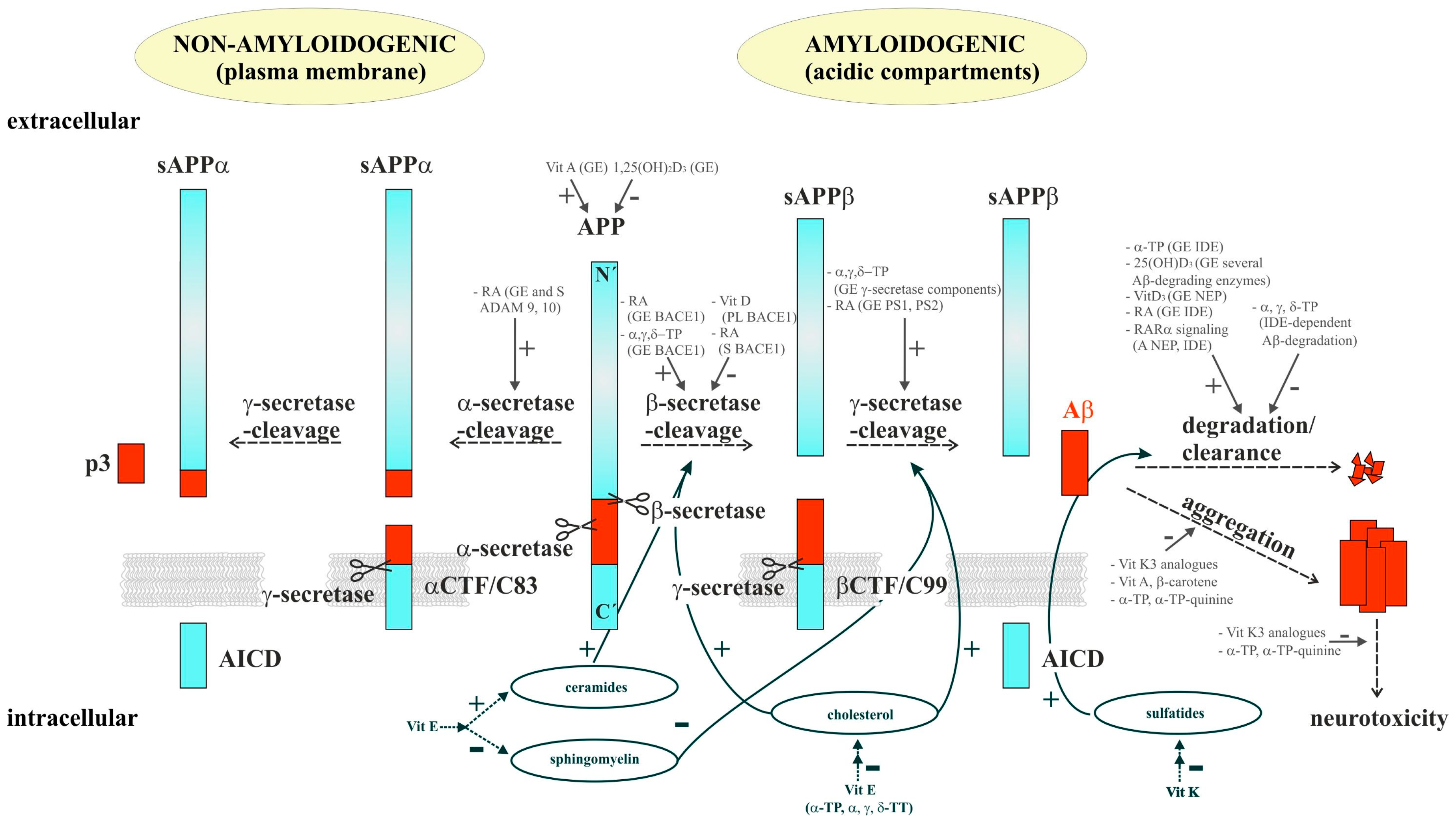

{kind=link}
{kind=link}
| Effect/Changes | Vitamin A | Vitamin D | Vitamin E | Vitamin K |
|---|---|---|---|---|
| Vitamin level in serum/plasma of AD/MCI patients | ↓ Vitamin A and provitamin A levels in serum/plasma of AD patients [36,37,38] | ↓ Vitamin D concentration in serum/plasma of patients suffering from all cause dementia and AD [33,57,58] | ↓ Vitamin E levels in plasma of patients suffering from AD or MCI [36,37,89] | ↓ Serum vitamin K concentration in persons suffering from AD [145,146,147] |
| Effect of serum/plasma vitamin level on cognition/AD risk in humans | ↑ β-carotene plasma levels →↑ Cognitive performances in the elderly [39] | ↓ Serum vitamin D levels →↑ Risk of cognitive decline and AD [59,60] ↑ 25-(OH)D3 plasma/serum levels →↑ Cognitive function, ↑ volumetric measures of brain structures affected by AD [64,65] | ↑ Plasma vitamin E levels/↑ intake of vitamin E or α-tocopherol equivalents →↓ Risk of AD [91,92,93] | |
| Effect of serum/plasma vitamin level/vitamin supplementation on AD progression in humans | Supplementation of vitamin D3 →↑ Cognition and memory in patients with moderate AD receiving memantine [70,72] | Inconsistent results [95,96,97,98] | Positive correlation between serum vitamin K level and cognition of AD patients [147,148] | |
| Effect of vitamin-supplementation on AD pathology/cognition in animal models | Treatment with retinoic acid →↓ Cerebral Aβ deposition, ↓ tau phosphorylation, ↑ cognitive functions [43,44] Treatment with all-trans retinoic acid →↓ Tau aggregation [45] | Vitamin D-enriched diet →↓ Brain Aβ burden, ↑ cognitive performances [76,77,78] | Vitamin E supplementation →↓ Cerebral Aβ-content in young APP-transgenic mice [100] →↓ Tau-induced neurodegeneration in Drosophila [142] α-tocopherol supplementation →↓ of age-related alterations in Aβ metabolism, ↓ deficits in learning and memory functions [101,102] →↓ Tau pathology [141] α-tocopherol quinine supplementation →↓ Memory impairment, ↓ cerebral levels of Aβ oligomers [103] | |
| Effect of vitamin-deficiency on AD pathology/cognition in animal models | Vitamin A deficiency →↑ cerebral Aβ deposition [41] →↑ cerebral Aβ production, ↓ cerebral sAPPα level [42] | Vitamin D deficiency →↑ Cerebral Aβ40 and Aβ42 levels [78,79] →↑ Spatial learning deficits [80] | α-tocopherol deficiency →↑ Cerebral Aβ-deposition [108] | Phylloquinone deficiency →↓ Cognitive functions in aged rats [150] |
| Effect of vitamin on AD-relevant molecular mechanisms | Retinoid acid →↑ Gene expression of APP, ADAM 9, ADAM10, BACE1, PS1, PS2, and IDE [47,48,49,50] →↑ Translocation of ADAM9, ADAM10, and BACE1 to the cellular membrane [51] vitamin A and β-carotene →↓ Aβ oligomerization, ↓ stability of Aβ fibrils [53,54] | Vitamin D →↑ Aβ clearance across the blood brain barrier, ↑ microglial Aβ-phagocytosis [77,81,82] →↑ NEP protein level, ↓ BACE1 protein level [77] →↓ Tau phosphorylation, ↓ Aβ-induced toxicity [83] 25-(OH)D3 →↑ Gene expression of Aβ-degrading enzymes [79] 1α,25-(OH)2D3 →↓ APP promoter activity [69] | α-, γ-, and δ-tocopherol →↑ Gene expression of the β- and γ-secretase components, ↓ Aβ-degradation [111] α-tocopherol, α-tocopherol quinine →↓ Aβaggregation, ↓ Aβ-induced toxicity [103,105,106,107] trolox (water-soluble vitamin E analog) →↓ Aβ-induced tau hyperphosphorylation [139] | Vitamin K3 analogues →↓ Aβ-aggregation, ↓ Aβ-induced toxicity [151] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grimm, M.O.W.; Mett, J.; Hartmann, T. The Impact of Vitamin E and Other Fat-Soluble Vitamins on Alzheimer´s Disease. Int. J. Mol. Sci. 2016, 17, 1785. https://doi.org/10.3390/ijms17111785
Grimm MOW, Mett J, Hartmann T. The Impact of Vitamin E and Other Fat-Soluble Vitamins on Alzheimer´s Disease. International Journal of Molecular Sciences. 2016; 17(11):1785. https://doi.org/10.3390/ijms17111785
Chicago/Turabian StyleGrimm, Marcus O. W., Janine Mett, and Tobias Hartmann. 2016. "The Impact of Vitamin E and Other Fat-Soluble Vitamins on Alzheimer´s Disease" International Journal of Molecular Sciences 17, no. 11: 1785. https://doi.org/10.3390/ijms17111785
APA StyleGrimm, M. O. W., Mett, J., & Hartmann, T. (2016). The Impact of Vitamin E and Other Fat-Soluble Vitamins on Alzheimer´s Disease. International Journal of Molecular Sciences, 17(11), 1785. https://doi.org/10.3390/ijms17111785