
Rack1 Mediates the Interaction of P-Glycoprotein with Anxa2 and Regulates Migration and Invasion of Multidrug-Resistant Breast Cancer Cells

Abstract
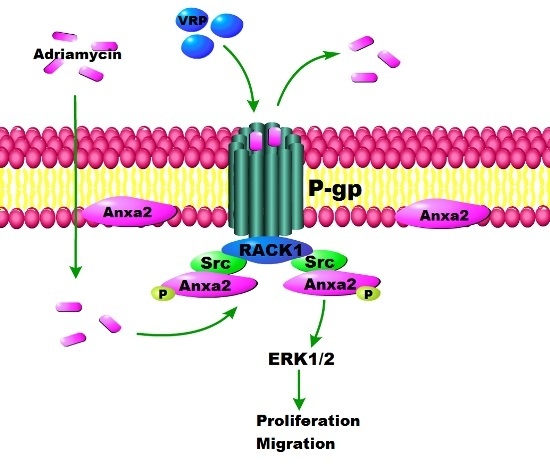
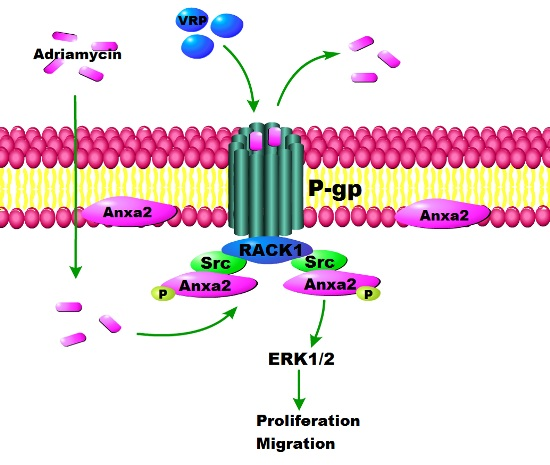
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
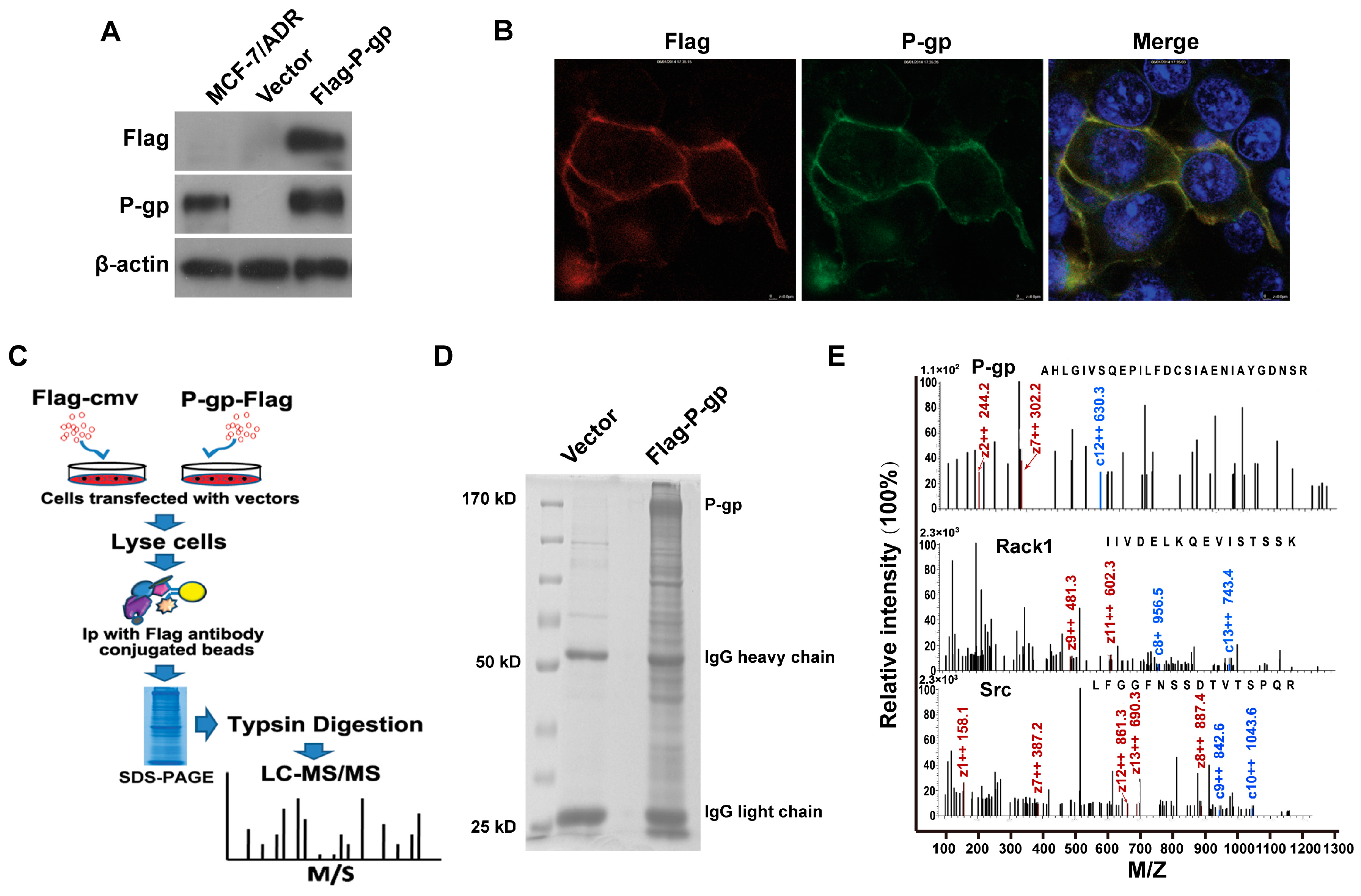
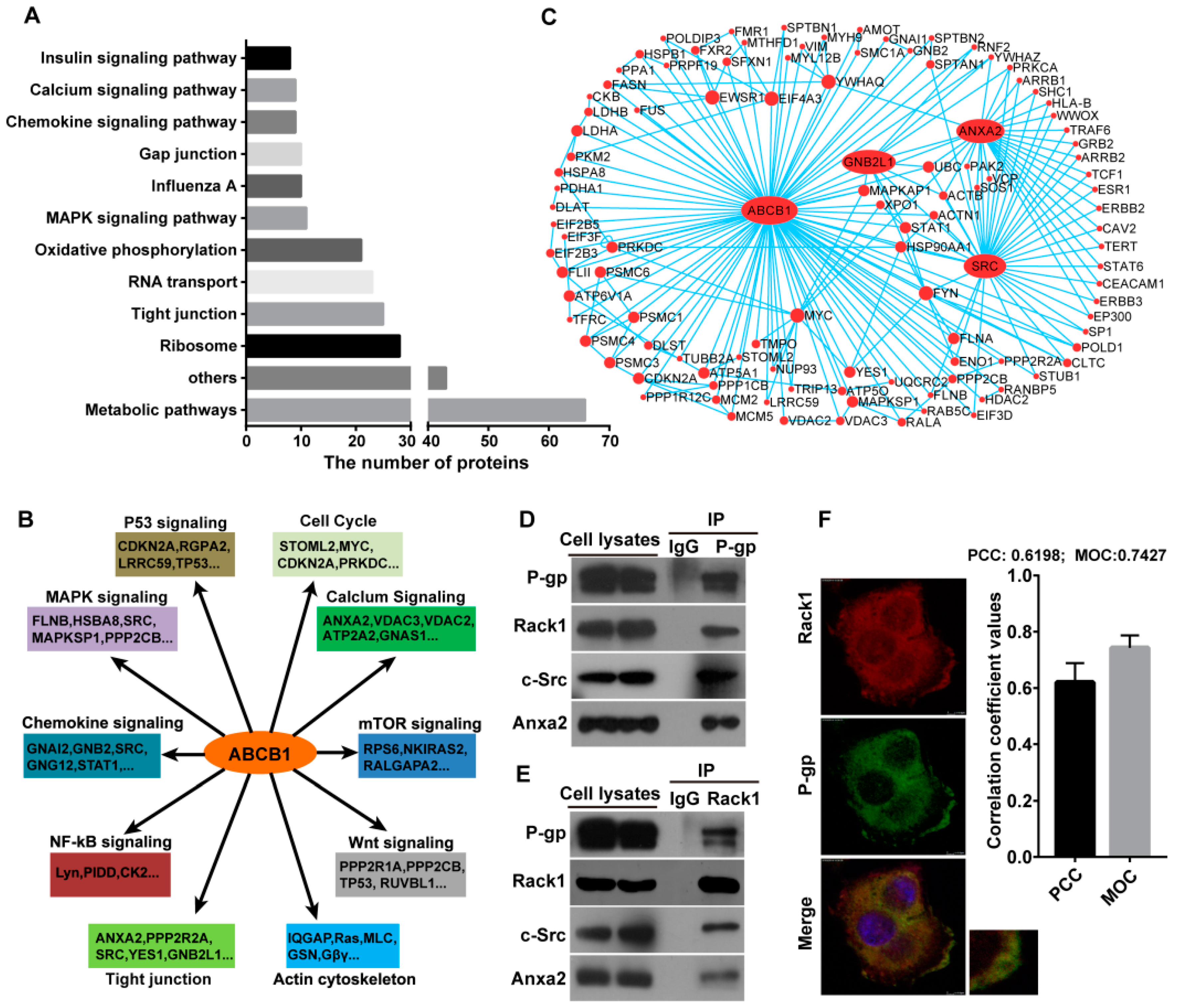
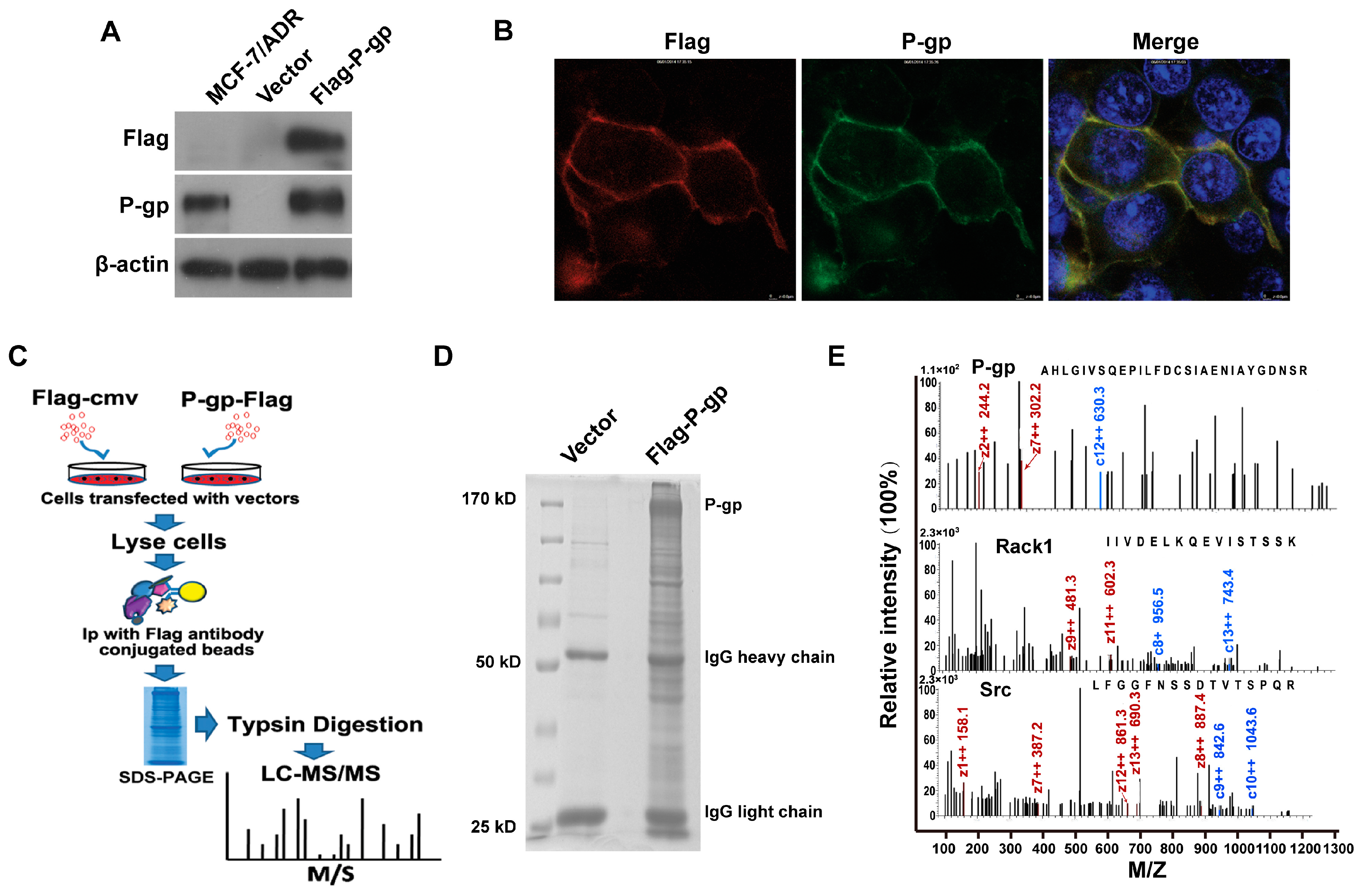
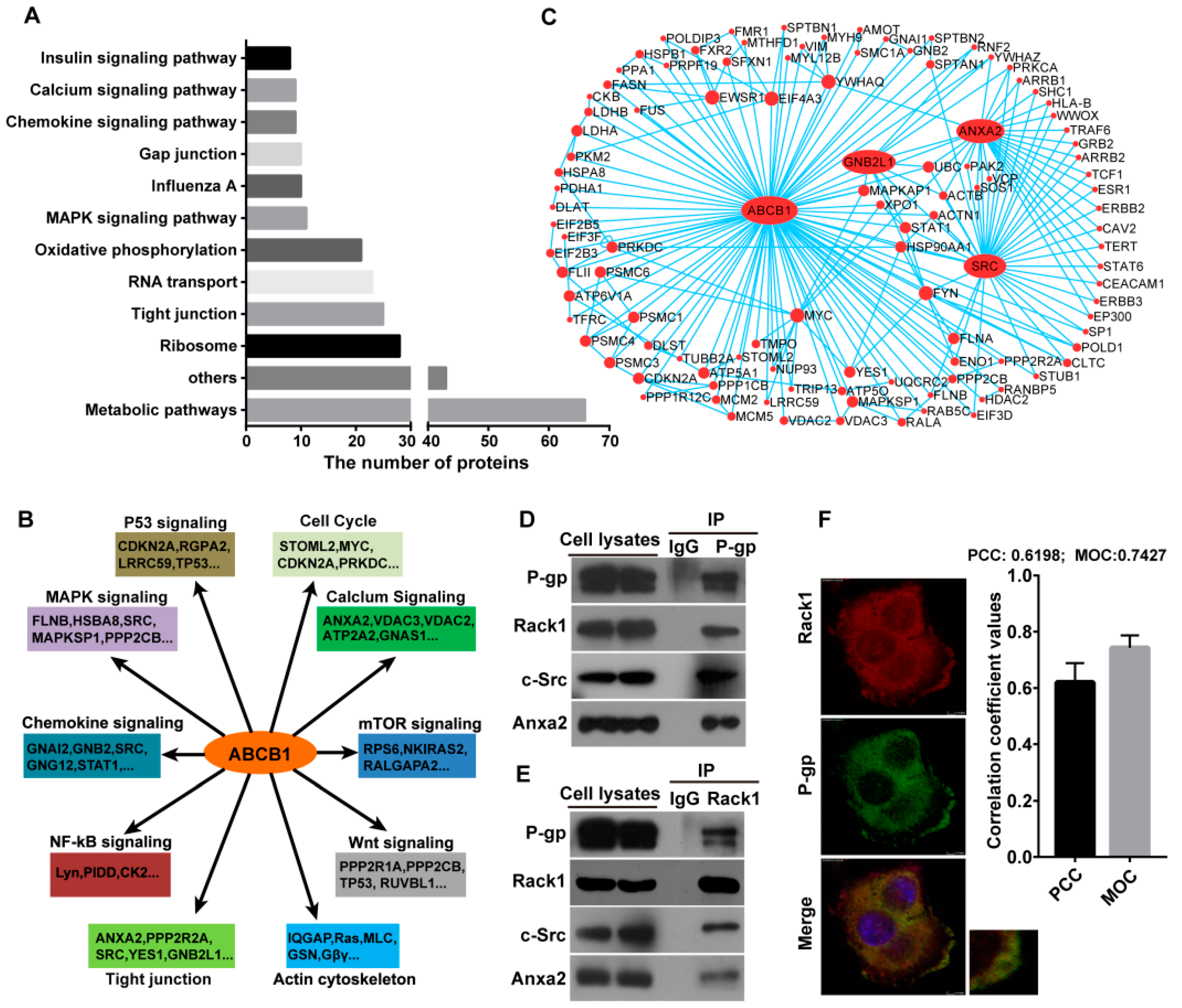
2.1. Identification of P-Glycoprotein Binding Proteins Using Interactome Analysis
2.2. Rack1 Is a Novel Binding Protein of P-Glycoprotein
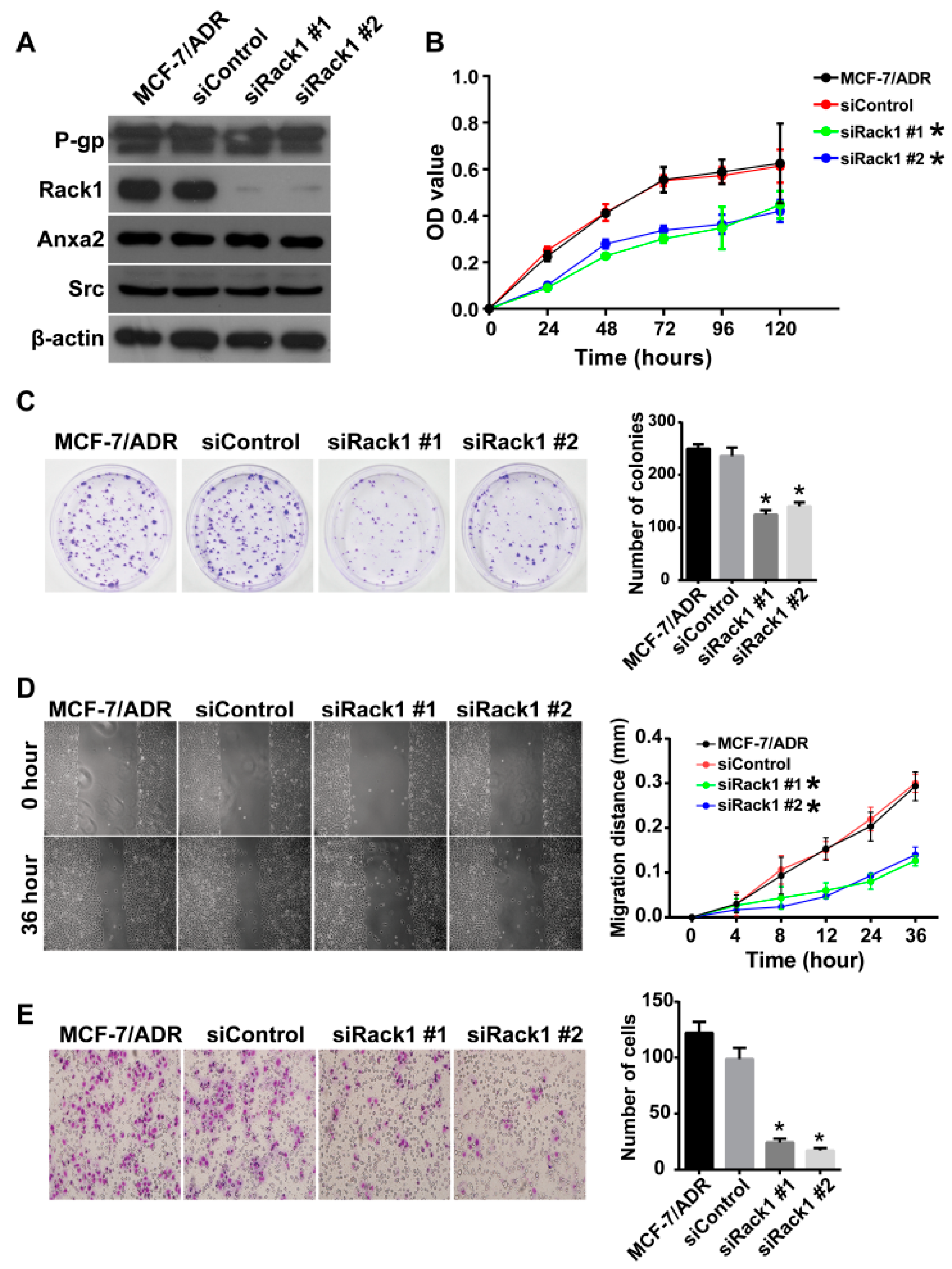
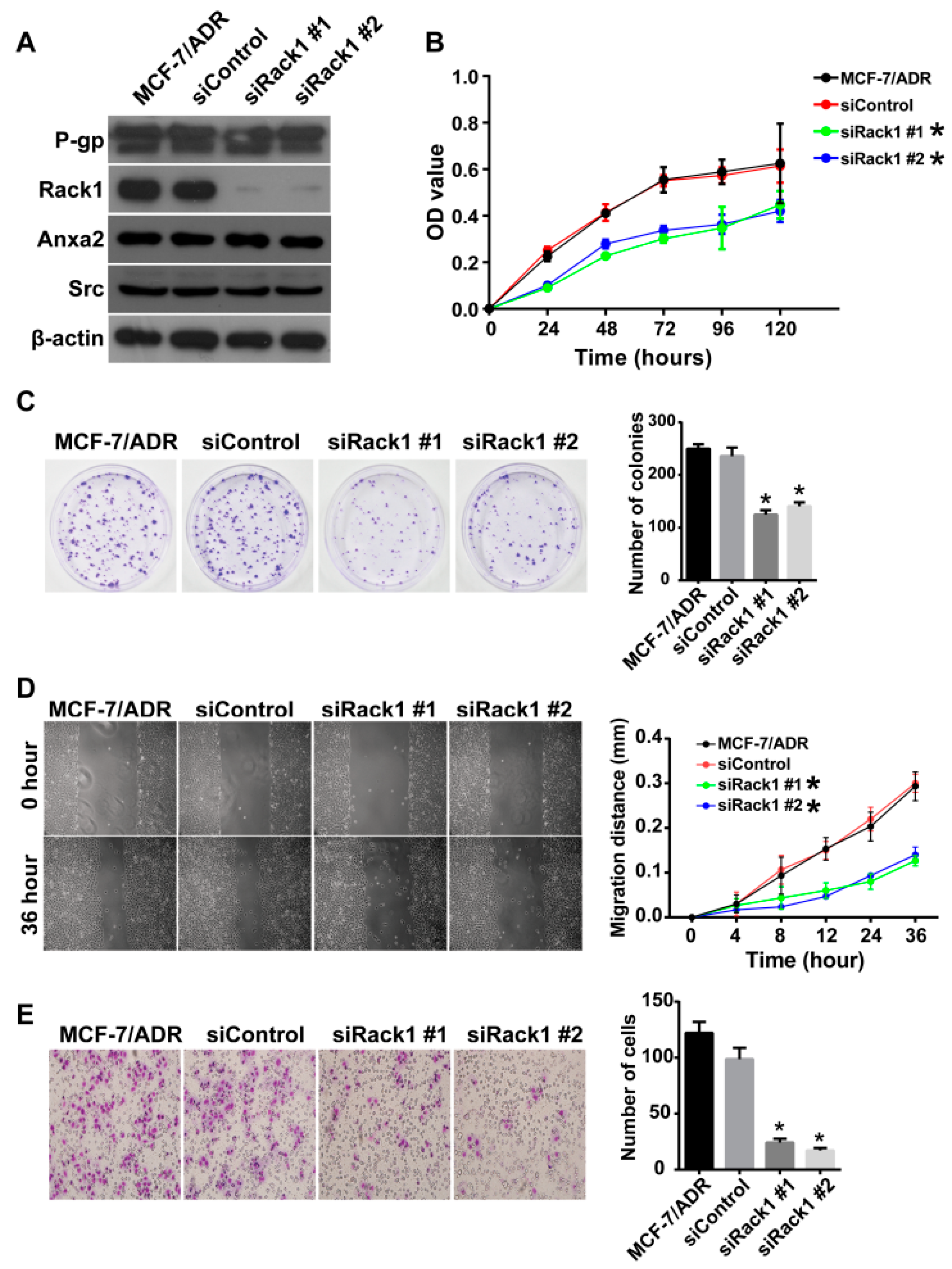
2.3. Knockdown of Rack1 Inhibits Proliferation, Migration and Invasiveness of Multidrug-Resistant Breast Cancer Cells
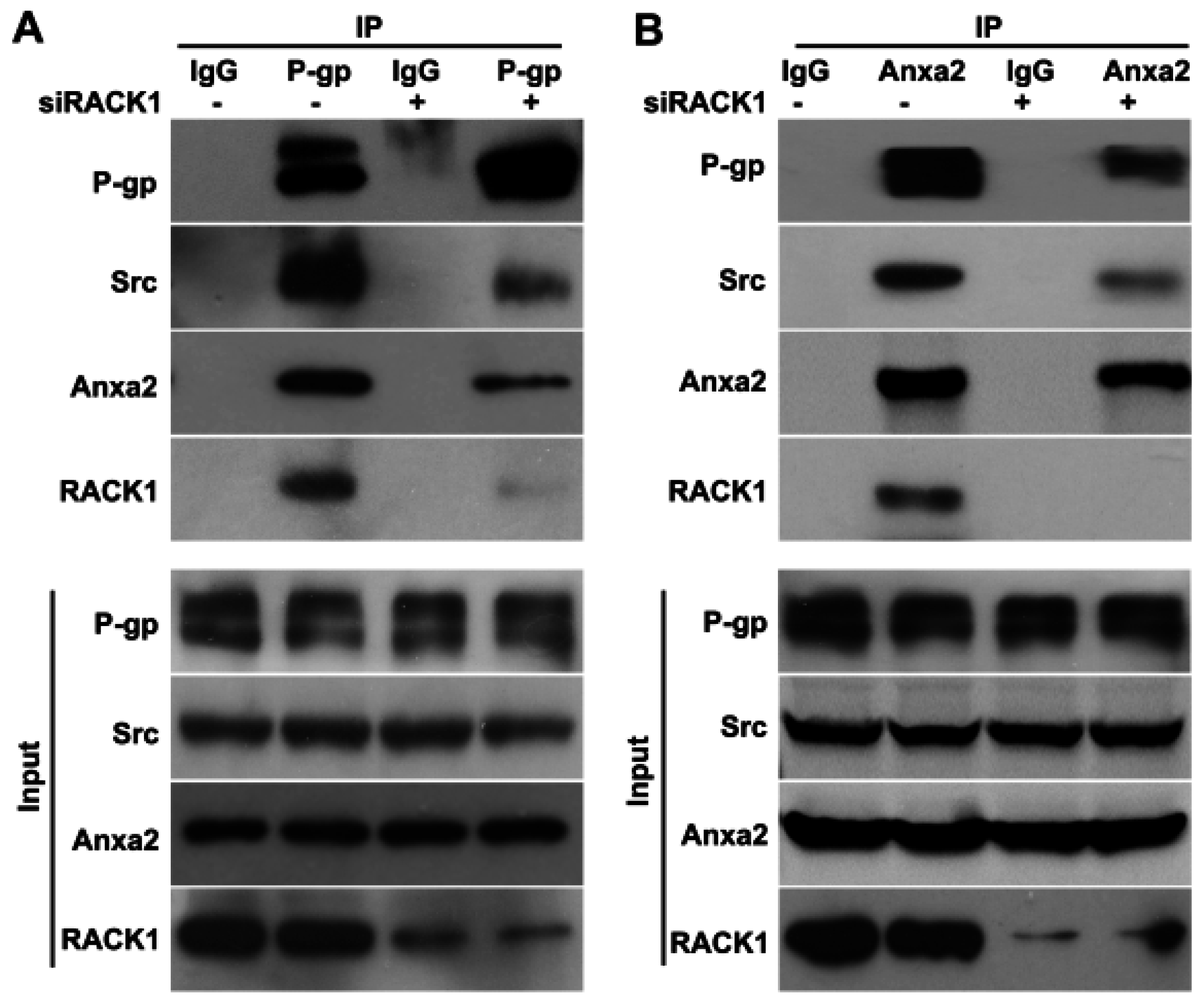
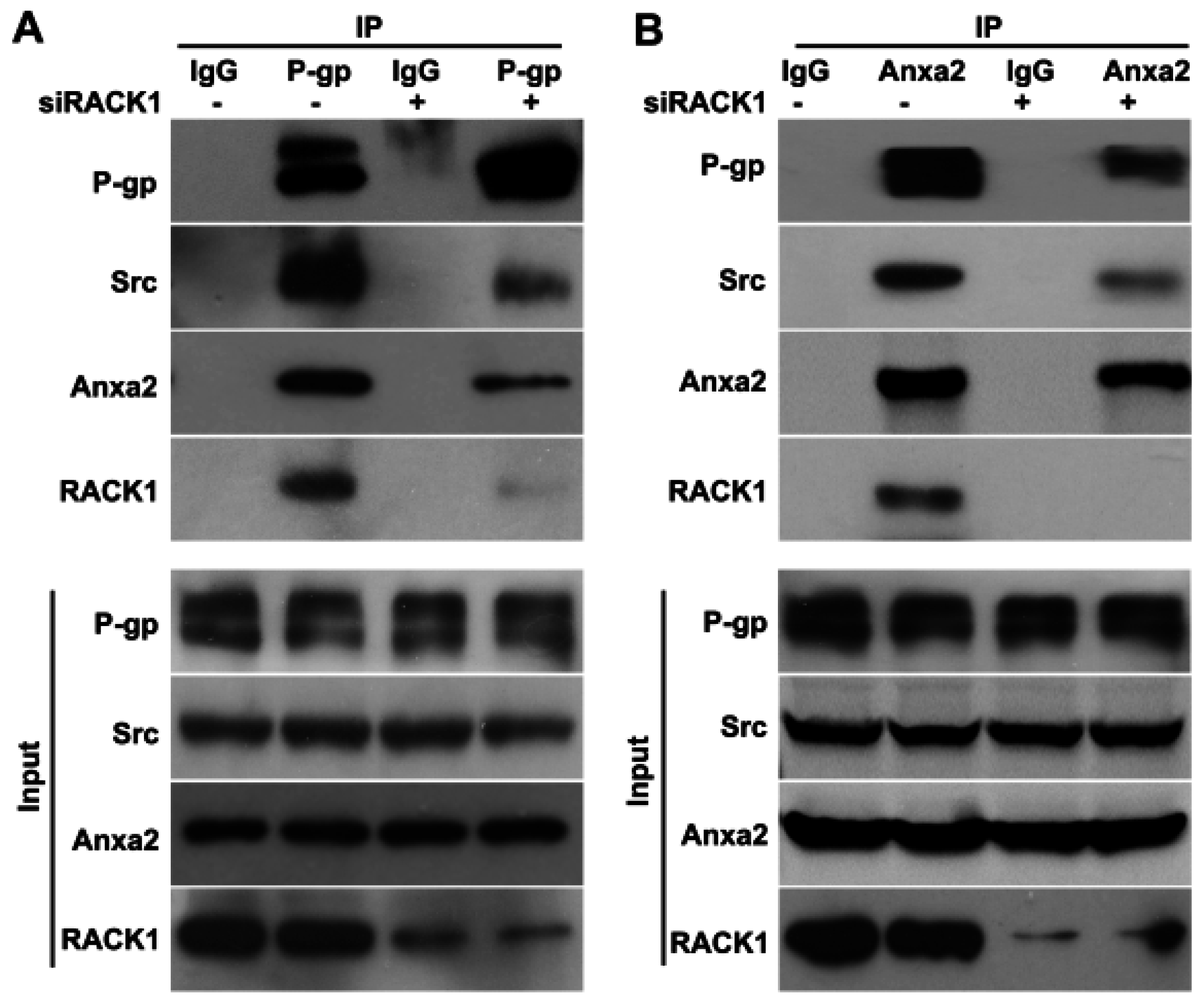
2.4. Knockdown of Rack1 Attenuates the Interaction of P-Glycoprotein with Anxa2 and Src
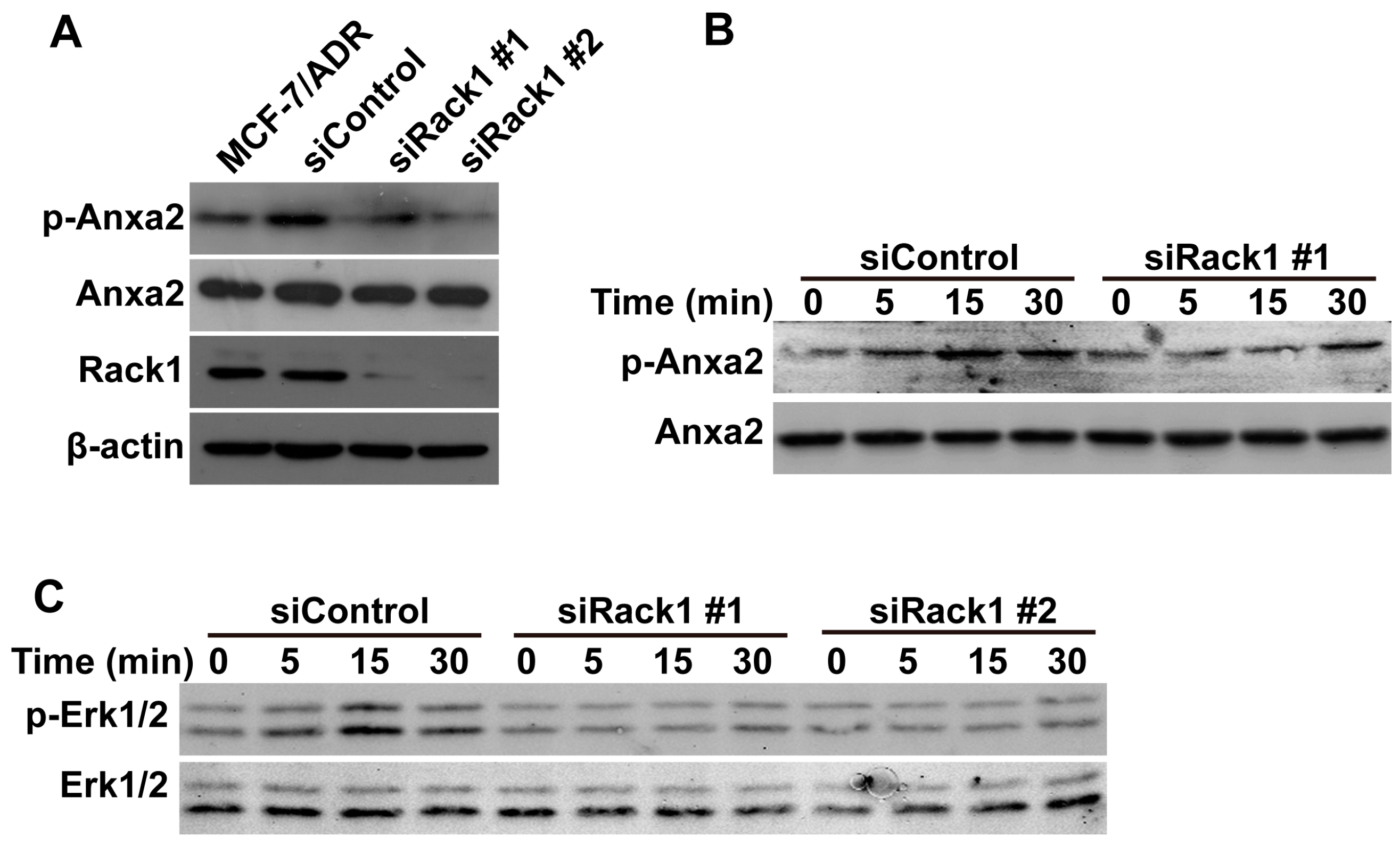
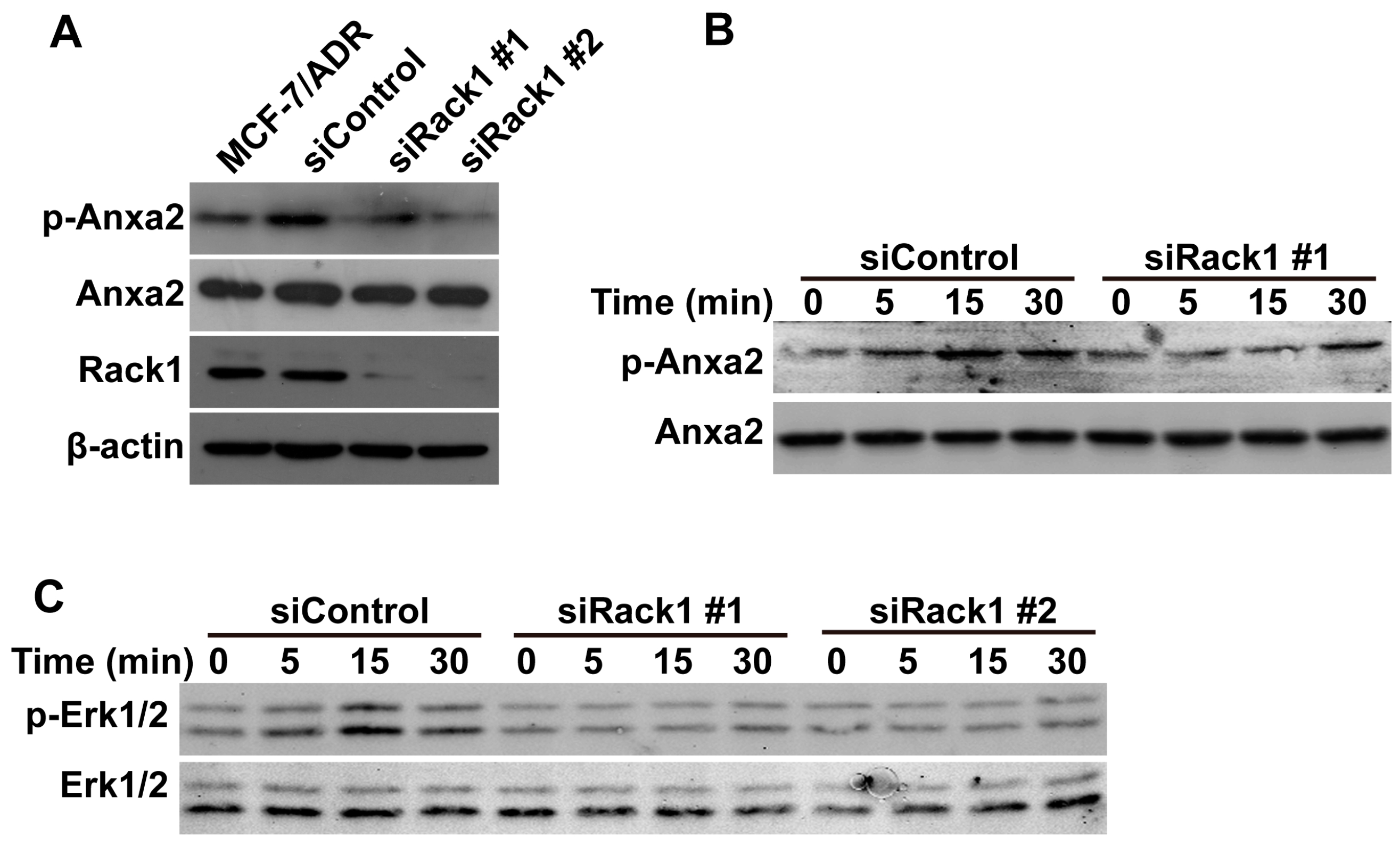
2.5. Knockdown of Rack1 Inhibits Anxa2 Phosphorylation and Adriamycin-Induced Erk1/2 Phosphorylation
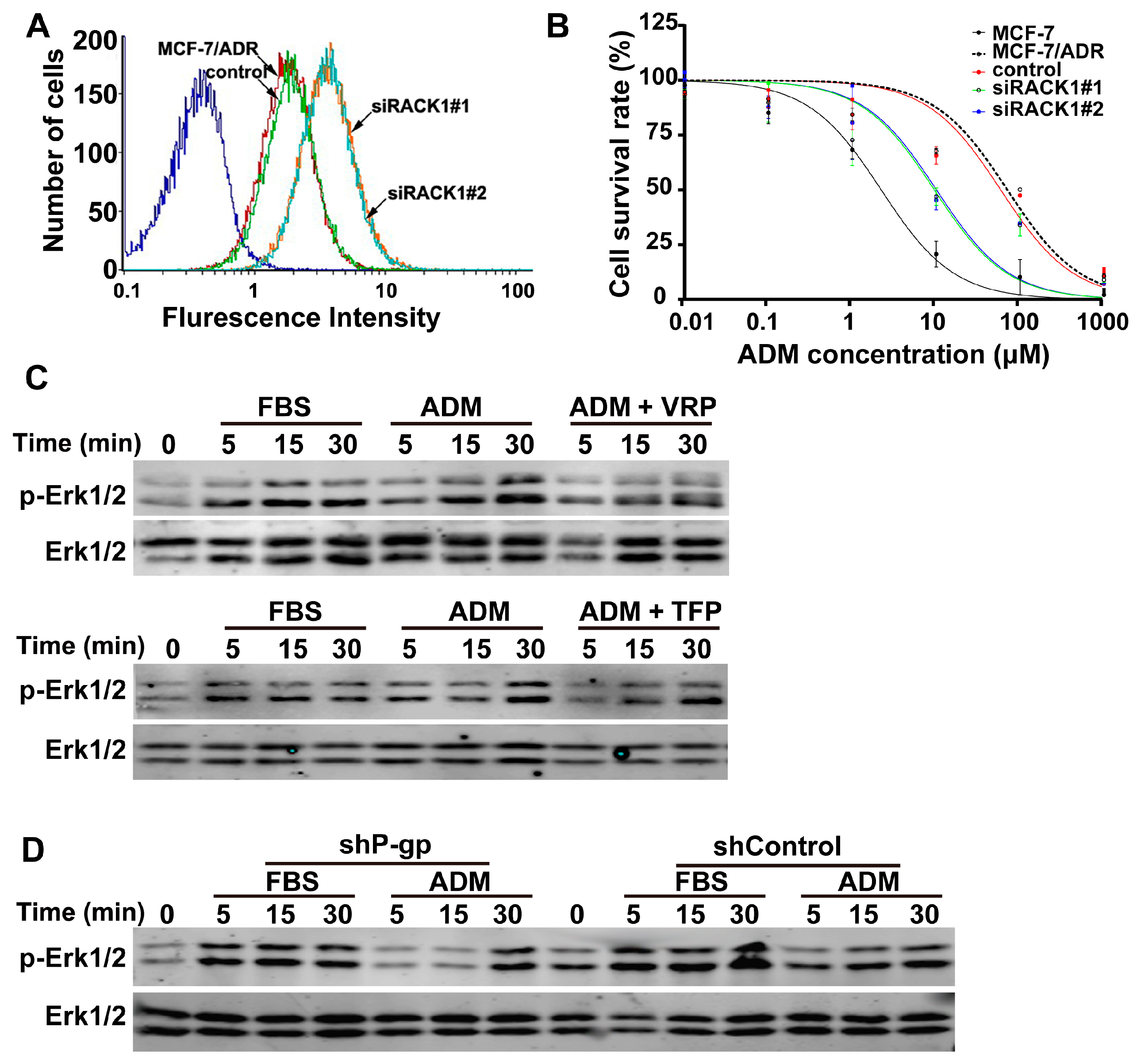
2.6. Knockdown of Rack1 Inhibited P-gp Activity, and P-gp Is Required for Adriamycin-Induced Erk1/2 Phosphorylation
3. Discussion
4. Materials and Methods
4.1. Cell Lines, Reagents and Antibodies
4.2. Vector Construction, siRNA and Plasmid Transfection
4.3. Western Blotting Analysis
4.4. Co-Immunoprecipitation Assay
4.5. In-Gel Trypsin Digestion, Protein Identification by LC-MS/MS and Data Analysis
4.6. Immunofluorescence Confocal Microscopy Analysis
4.7. Cell Viability Assay
4.8. Colony Formation Assay
4.9. IC50 Assay
4.10. Wound Healing Assay and in Vitro Cell Invasion Assay
4.11. Rh123 Efflux Assay
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wu, Q.; Yang, Z.; Nie, Y.; Shi, Y.; Fan, D. Multi-drug resistance in cancer chemotherapeutics: Mechanisms and lab approaches. Cancer Lett. 2014, 347, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.H.; Yu, A.M. ABC transporters in multidrug resistance and pharmacokinetics, and strategies for drug development. Curr. Pharm. Des. 2014, 20, 793–807. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, K.; Eno, M.L.; Ahn, E.H.; Shahzad, M.M.; Im, D.D.; Rosenshein, N.B.; Sood, A.K. Multidrug resistance gene (MDR-1) and risk of brain metastasis in epithelial ovarian, fallopian tube, and peritoneal cancer. Am. J. Clin. Oncol. 2011, 34, 488–493. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; Chen, H.; Madigan, M.C.; Cozzi, P.J.; Beretov, J.; Xiao, W.; Delprado, W.J.; Russell, P.J.; Li, Y. Co-expression of CD147 (EMMPRIN), CD44v3–10, MDR1 and monocarboxylate transporters is associated with prostate cancer drug resistance and progression. Br. J. Cancer 2010, 103, 1008–1018. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, J.I.; Haber, M.; Henderson, M.J.; Norris, M.D. ABC transporters in cancer: More than just drug efflux pumps. Nat. Rev. Cancer 2010, 10, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Iseri, O.D.; Kars, M.D.; Arpaci, F.; Atalay, C.; Pak, I.; Gunduz, U. Drug resistant MCF-7 cells exhibit epithelial-mesenchymal transition gene expression pattern. Biomed. Pharmacother. 2011, 65, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Zhang, H.; Wang, Z.; Yu, M.; Tian, R.; Ji, W.; Yang, Y.; Niu, R. P-glycoprotein associates with Anxa2 and promotes invasion in multidrug resistant breast cancer cells. Biochem. Pharmacol. 2014, 87, 292–302. [Google Scholar] [CrossRef] [PubMed]
- Kajiyama, H.; Shibata, K.; Terauchi, M.; Yamashita, M.; Ino, K.; Nawa, A.; Kikkawa, F. Chemoresistance to paclitaxel induces epithelial-mesenchymal transition and enhances metastatic potential for epithelial ovarian carcinoma cells. Int. J. Oncol. 2007, 31, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Lopes, E.C.; Ernst, G.; Aulicino, P.; Vanzulli, S.; Garcia, M.; Alvarez, E.; Hajos, S.E. Dissimilar invasive and metastatic behavior of vincristine and doxorubicin-resistant cell lines derived from a murine T cell lymphoid leukemia. Clin. Exp. Metastasis 2002, 19, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.Q.; Wang, W.J.; Xu, J.D.; Cao, X.X.; Chen, Q.; Yang, J.M.; Xu, Z.D. Up-regulation of CD147 and matrix metalloproteinase-2, -9 induced by P-glycoprotein substrates in multidrug resistant breast cancer cells. Cancer Sci. 2007, 98, 1767–1774. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Hao, J.; Wang, L.; Li, Y. Coexpression of invasive markers (uPA, CD44) and multiple drug-resistance proteins (MDR1, MRP2) is correlated with epithelial ovarian cancer progression. Br. J. Cancer 2009, 101, 432–440. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.D.; Fan, F.; Camp, E.R.; van Buren, G.; Liu, W.; Somcio, R.; Gray, M.J.; Cheng, H.; Hoff, P.M.; Ellis, L.M. Chronic oxaliplatin resistance induces epithelial-to-mesenchymal transition in colorectal cancer cell lines. Clin. Cancer Res. 2006, 12, 4147–4153. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.S.; Chen, L.; Ding, W.X.; Li, K.; Wu, J.J. Elevated expression of both MDR1 and MMP-2 genes in metastasized lymph node of invasive ductal breast cancer. Eur. Rev. Med. Pharmacol. Sci. 2012, 16, 2037–2043. [Google Scholar] [PubMed]
- Hoffmann, A.C.; Wild, P.; Leicht, C.; Bertz, S.; Danenberg, K.D.; Danenberg, P.V.; Stohr, R.; Stockle, M.; Lehmann, J.; Schuler, M.; et al. MDR1 and ERCC1 expression predict outcome of patients with locally advanced bladder cancer receiving adjuvant chemotherapy. Neoplasia 2010, 12, 628–636. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, K.; Maishi, N.; Ohga, N.; Hida, Y.; Ohba, Y.; Alam, M.T.; Kawamoto, T.; Ohmura, H.; Yamada, K.; Torii, C.; et al. Inhibition of multidrug transporter in tumor endothelial cells enhances antiangiogenic effects of low-dose metronomic paclitaxel. Am. J. Pathol. 2015, 185, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Barakat, S.; Turcotte, S.; Demeule, M.; Lachambre, M.P.; Regina, A.; Baggetto, L.G.; Beliveau, R. Regulation of brain endothelial cells migration and angiogenesis by P-glycoprotein/caveolin-1 interaction. Biochem. Biophys. Res. Commun. 2008, 372, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.; Fojo, T.; Bates, S. Tumour stem cells and drug resistance. Nat. Rev. Cancer 2005, 5, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Landreville, S.; Agapova, O.A.; Kneass, Z.T.; Salesse, C.; Harbour, J.W. ABCB1 identifies a subpopulation of uveal melanoma cells with high metastatic propensity. Pigment Cell Melanoma Res. 2011, 24, 430–437. [Google Scholar] [CrossRef] [PubMed]
- Walsh, N.; Kennedy, S.; Larkin, A.M.; Tryfonopoulos, D.; Eustace, A.J.; Mahgoub, T.; Conway, C.; Oglesby, I.; Collins, D.; Ballot, J.; et al. Membrane transport proteins in human melanoma: Associations with tumour aggressiveness and metastasis. Br. J. Cancer 2010, 102, 1157–1162. [Google Scholar] [CrossRef] [PubMed]
- Miletti-Gonzalez, K.E.; Chen, S.; Muthukumaran, N.; Saglimbeni, G.N.; Wu, X.; Yang, J.; Apolito, K.; Shih, W.J.; Hait, W.N.; Rodriguez-Rodriguez, L. The CD44 receptor interacts with P-glycoprotein to promote cell migration and invasion in cancer. Cancer Res. 2005, 65, 6660–6667. [Google Scholar] [CrossRef] [PubMed]
- Colone, M.; Calcabrini, A.; Toccacieli, L.; Bozzuto, G.; Stringaro, A.; Gentile, M.; Cianfriglia, M.; Ciervo, A.; Caraglia, M.; Budillon, A.; et al. The multidrug transporter P-glycoprotein: A mediator of melanoma invasion? J. Investig. Dermatol. 2008, 128, 957–971. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Liu, Y.; Deng, C.; Han, R.; Jia, Y.; Liu, S.; Jiang, Z.; Cao, X.; He, L.; Zhang, Q. Enhanced invasiveness in multidrug resistant leukemic cells is associated with overexpression of P-glycoprotein and cellular inhibitor of apoptosis protein. Leuk. Lymphoma 2011, 52, 1302–1311. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.; Chamberlain, M.; McLaren, A.; Goh, L.; Duncan, I.; Wolf, C.R. Cross-talk between signalling pathways and the multidrug resistant protein MDR-1. Br. J. Cancer 2001, 85, 1175–1184. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.Q.; Wang, W.J.; Xu, J.D.; Cao, X.X.; Chen, Q.; Yang, J.M.; Xu, Z.D. Involvement of CD147 in regulation of multidrug resistance to P-gp substrate drugs and in vitro invasion in breast cancer cells. Cancer Sci. 2007, 98, 1064–1069. [Google Scholar] [CrossRef] [PubMed]
- Pawson, T.; Nash, P. Protein-protein interactions define specificity in signal transduction. Genes Dev. 2000, 14, 1027–1047. [Google Scholar] [PubMed]
- Burack, W.R.; Shaw, A.S. Signal transduction: Hanging on a scaffold. Curr. Opin. Cell Biol. 2000, 12, 211–216. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Su, L.; Mruk, D.D.; Lui, W.Y.; Lee, W.M.; Cheng, C.Y. P-glycoprotein regulates blood-testis barrier dynamics via its effects on the occludin/zonula occludens 1 (ZO-1) protein complex mediated by focal adhesion kinase (FAK). Proc. Natl. Acad. Sci. USA 2011, 108, 19623–19628. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Cabrera, R.M.; Li, Y.; Miller, D.S.; Finnell, R.H. Functional regulation of P-glycoprotein at the blood-brain barrier in proton-coupled folate transporter (PCFT) mutant mice. FASEB J. 2013, 27, 1167–1175. [Google Scholar] [CrossRef] [PubMed]
- Sulova, Z.; Seres, M.; Barancik, M.; Gibalova, L.; Uhrik, B.; Polekova, L.; Breier, A. Does any relationship exist between P-glycoprotein-mediated multidrug resistance and intracellular calcium homeostasis. Gen. Physiol. Biophys. 2009, 28, F89–F95. [Google Scholar] [PubMed]
- Riganti, C.; Doublier, S.; Viarisio, D.; Miraglia, E.; Pescarmona, G.; Ghigo, D.; Bosia, A. Artemisinin induces doxorubicin resistance in human colon cancer cells via calcium-dependent activation of HIF-1α and P-glycoprotein overexpression. Br. J. Pharmacol. 2009, 156, 1054–1066. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Liu, Y.; Wang, Z.; Sun, X.; Yuan, J.; Wang, T.; Tian, R.; Ji, W.; Yu, M.; Zhao, Y.; et al. A novel Anxa2-interacting protein Ebp1 inhibits cancer proliferation and invasion by suppressing Anxa2 protein level. Mol. Cell. Endocrinol. 2015, 411, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Chang, B.Y.; Harte, R.A.; Cartwright, C.A. Rack1: A novel substrate for the Src protein-tyrosine kinase. Oncogene 2002, 21, 7619–7629. [Google Scholar] [CrossRef] [PubMed]
- Li, J.J.; Xie, D. RACK1, a versatile hub in cancer. Oncogene 2015, 34, 1890–1898. [Google Scholar] [CrossRef] [PubMed]
- Berns, H.; Humar, R.; Hengerer, B.; Kiefer, F.N.; Battegay, E.J. RACK1 is up-regulated in angiogenesis and human carcinomas. FASEB J. 2000, 14, 2549–2558. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Min, L.; Wang, X.; Zhao, J.; Chen, H.; Qin, J.; Chen, W.; Shen, Z.; Tang, Z.; Gan, Q.; et al. Loss of RACK1 promotes metastasis of gastric cancer by inducing a miR-302c/IL8 signaling loop. Cancer Res. 2015, 75, 3832–3841. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.X.; Xu, J.D.; Xu, J.W.; Liu, X.L.; Cheng, Y.Y.; Wang, W.J.; Li, Q.Q.; Chen, Q.; Xu, Z.D.; Liu, X.P. RACK1 promotes breast carcinoma proliferation and invasion/metastasis in vitro and in vivo. Breast Cancer Res. Treat. 2010, 123, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.X.; Xu, J.D.; Xu, J.W.; Liu, X.L.; Cheng, Y.Y.; Li, Q.Q.; Xu, Z.D.; Liu, X.P. RACK1 promotes breast carcinoma migration/metastasis via activation of the Rhoa/Rho kinase pathway. Breast Cancer Res. Treat. 2011, 126, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.R.; Ron, D.; Kiely, P.A. RACK1, a multifaceted scaffolding protein: Structure and function. Cell Commun. Signal. 2011, 9, 22. [Google Scholar] [CrossRef] [PubMed]
- McCahill, A.; Warwicker, J.; Bolger, G.B.; Houslay, M.D.; Yarwood, S.J. The RACK1 scaffold protein: A dynamic cog in cell response mechanisms. Mol. Pharmacol. 2002, 62, 1261–1273. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Foley, K.; Huang, L.; Leubner, A.; Mo, G.; Olino, K.; Edil, B.H.; Mizuma, M.; Sharma, R.; Le, D.T.; et al. Tyrosine 23 phosphorylation-dependent cell-surface localization of annexin A2 is required for invasion and metastases of pancreatic cancer. PLoS ONE 2011, 6, e19390. [Google Scholar] [CrossRef] [PubMed]
- Spijkers-Hagelstein, J.A.; Mimoso Pinhancos, S.; Schneider, P.; Pieters, R.; Stam, R.W. Src kinase-induced phosphorylation of annexin A2 mediates glucocorticoid resistance in MLL-rearranged infant acute lymphoblastic leukemia. Leukemia 2013, 27, 1063–1071. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liu, S.; Guo, C.; Zong, J.; Sun, M.Z. The association of annexin A2 and cancers. Clin. Transl. Oncol. 2012, 14, 634–640. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Zhang, L.; Zhang, B.; Wei, X.; Yang, Y.; Qi, R.Z.; Ying, G.; Zhang, N.; Niu, R. Anxa2 plays a critical role in enhanced invasiveness of the multidrug resistant human breast cancer cells. J. Proteome Res. 2009, 8, 5041–5047. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.X.; Xu, J.D.; Liu, X.L.; Xu, J.W.; Wang, W.J.; Li, Q.Q.; Chen, Q.; Xu, Z.D.; Liu, X.P. RACK1: A superior independent predictor for poor clinical outcome in breast cancer. Int. J. Cancer 2010, 127, 1172–1179. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.Z.; Yao, F.; Li, J.J.; Mao, Z.F.; Hu, P.T.; Long, L.Y.; Li, G.; Ji, X.D.; Shi, S.; Guan, D.X.; et al. RACK1 suppresses gastric tumorigenesis by stabilizing the β-catenin destruction complex. Gastroenterology 2012, 142, 812–823. [Google Scholar] [CrossRef] [PubMed]
- Nagashio, R.; Sato, Y.; Matsumoto, T.; Kageyama, T.; Satoh, Y.; Shinichiro, R.; Masuda, N.; Goshima, N.; Jiang, S.X.; Okayasu, I. Expression of RACK1 is a novel biomarker in pulmonary adenocarcinomas. Lung Cancer 2010, 69, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Meng, J.; Du, Y.; Huang, Y.; Jin, Y.; Zhang, J.; Wang, B.; Zhang, Y.; Sun, M.; Tang, J. RACK1 promotes the proliferation, migration and invasion capacity of mouse hepatocellular carcinoma cell line in vitro probably by PI3K/Rac1 signaling pathway. Biomed. Pharmacother. 2013, 67, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Wang, W.; Wang, J.; Feng, J.; Wang, Q.; Jin, J.; Lv, M.; Li, X.; Li, Y.; Ma, Y.; et al. Receptor for activated C kinase 1 promotes hepatocellular carcinoma growth by enhancing mitogen-activated protein kinase kinase 7 activity. Hepatology 2013, 57, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Guo, Y.; Feng, X.; Wang, Z.; Wang, Y.; Deng, P.; Zhang, D.; Wang, R.; Xie, L.; Xu, X.; et al. Receptor for activated C kinase 1 (RACK1): A regulator for migration and invasion in oral squamous cell carcinoma cells. J. Cancer Res. Clin. Oncol. 2012, 138, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, G.; Cartwright, C.A. RACK1 promotes epithelial cell-cell adhesion by regulating E-cadherin endocytosis. Oncogene 2012, 31, 376–389. [Google Scholar] [CrossRef] [PubMed]
- Daverey, A.; Drain, A.P.; Kidambi, S. Physical intimacy of breast cancer cells with mesenchymal stem cells elicits trastuzumab resistance through Src activation. Sci. Rep. 2015, 5, 13744. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Huang, W.C.; Li, P.; Guo, H.; Poh, S.B.; Brady, S.W.; Xiong, Y.; Tseng, L.M.; Li, S.H.; Ding, Z.; et al. Combating trastuzumab resistance by targeting Src, a common node downstream of multiple resistance pathways. Nat. Med. 2011, 17, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.V.; Gajowniczek, P.; Way, I.P.; Lee, D.Y.; Jiang, J.; Yuza, Y.; Classon, M.; Haber, D.A.; Settleman, J. A common signaling cascade may underlie “addiction” to the Src, BCR-ABL, and EGF receptor oncogenes. Cancer Cell 2006, 10, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Y.; Sun, L.; Hao, Y.; Wang, L.; Xu, J.; Zhang, W.; Xie, J.; Guo, L.; Zhou, L.; Yun, X.; et al. Ribosomal RACK1 promotes chemoresistance and growth in human hepatocellular carcinoma. J. Clin. Investig. 2012, 122, 2554–2566. [Google Scholar] [CrossRef] [PubMed]
- George, J.A.; Chen, T.; Taylor, C.C. Src tyrosine kinase and multidrug resistance protein-1 inhibitions act independently but cooperatively to restore paclitaxel sensitivity to paclitaxel-resistant ovarian cancer cells. Cancer Res. 2005, 65, 10381–10388. [Google Scholar] [CrossRef] [PubMed]
- Bird, R.J.; Baillie, G.S.; Yarwood, S.J. Interaction with receptor for activated C-kinase 1 (RACK1) sensitizes the phosphodiesterase PDE4D5 towards hydrolysis of cAMP and activation by protein kinase C. Biochem. J. 2010, 432, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Vomastek, T.; Iwanicki, M.P.; Schaeffer, H.J.; Tarcsafalvi, A.; Parsons, J.T.; Weber, M.J. RACK1 targets the extracellular signal-regulated kinase/mitogen-activated protein kinase pathway to link integrin engagement with focal adhesion disassembly and cell motility. Mol. Cell. Biol. 2007, 27, 8296–8305. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Mellor, J.; Wu, J.; DeLisi, C. VisANT: An online visualization and analysis tool for biological interaction data. BMC Bioinform. 2004, 5, 17. [Google Scholar] [CrossRef] [PubMed]

© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Y.; Wu, N.; Wang, Z.; Zhang, F.; Tian, R.; Ji, W.; Ren, X.; Niu, R. Rack1 Mediates the Interaction of P-Glycoprotein with Anxa2 and Regulates Migration and Invasion of Multidrug-Resistant Breast Cancer Cells. Int. J. Mol. Sci. 2016, 17, 1718. https://doi.org/10.3390/ijms17101718
Yang Y, Wu N, Wang Z, Zhang F, Tian R, Ji W, Ren X, Niu R. Rack1 Mediates the Interaction of P-Glycoprotein with Anxa2 and Regulates Migration and Invasion of Multidrug-Resistant Breast Cancer Cells. International Journal of Molecular Sciences. 2016; 17(10):1718. https://doi.org/10.3390/ijms17101718
Chicago/Turabian StyleYang, Yi, Na Wu, Zhiyong Wang, Fei Zhang, Ran Tian, Wei Ji, Xiubao Ren, and Ruifang Niu. 2016. "Rack1 Mediates the Interaction of P-Glycoprotein with Anxa2 and Regulates Migration and Invasion of Multidrug-Resistant Breast Cancer Cells" International Journal of Molecular Sciences 17, no. 10: 1718. https://doi.org/10.3390/ijms17101718
APA StyleYang, Y., Wu, N., Wang, Z., Zhang, F., Tian, R., Ji, W., Ren, X., & Niu, R. (2016). Rack1 Mediates the Interaction of P-Glycoprotein with Anxa2 and Regulates Migration and Invasion of Multidrug-Resistant Breast Cancer Cells. International Journal of Molecular Sciences, 17(10), 1718. https://doi.org/10.3390/ijms17101718