
Statin Therapy and the Development of Cerebral Amyloid Angiopathy—A Rodent in Vivo Approach

 ,
,
Abstract
:
1. Introduction
2. Results
2.1. Influence of Statin Therapy on Mortality
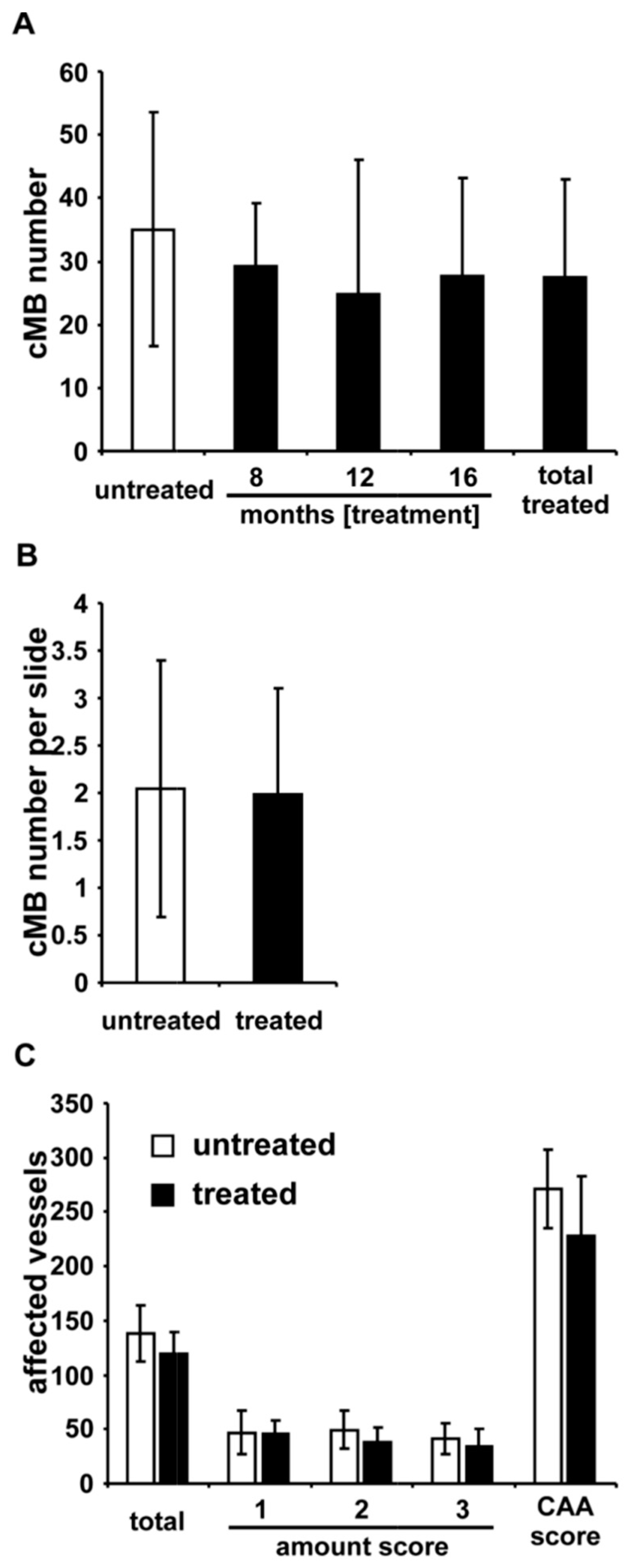
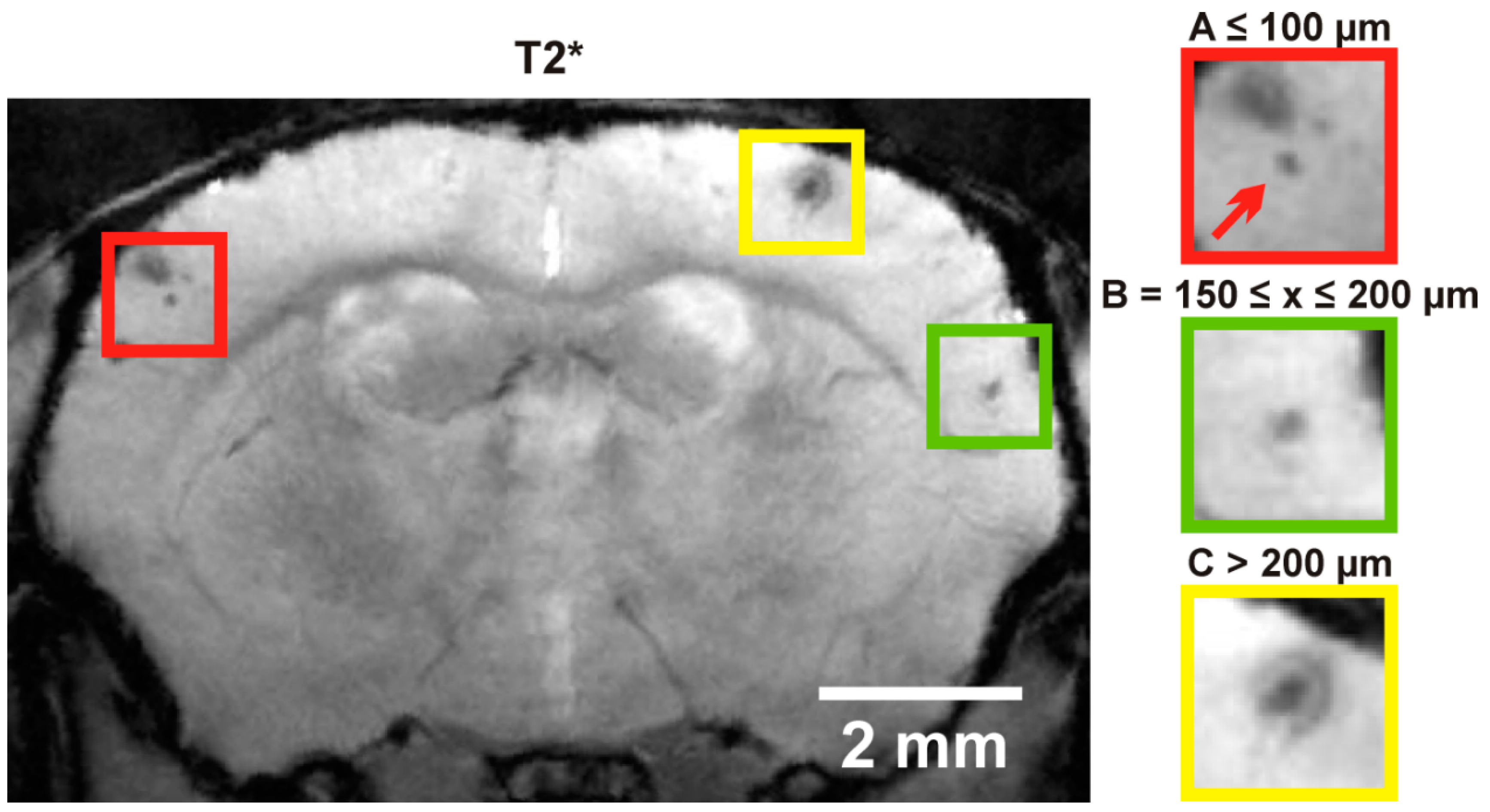
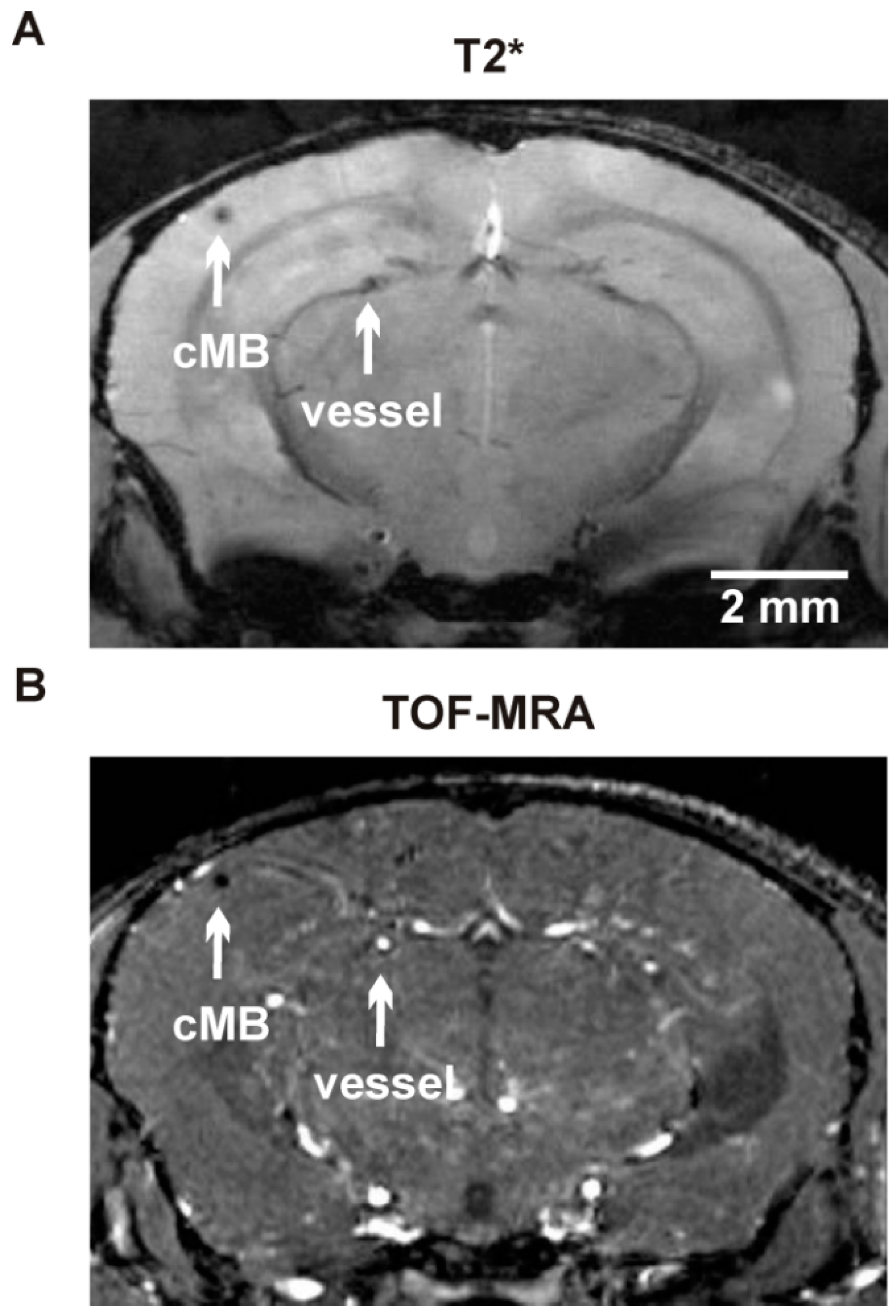
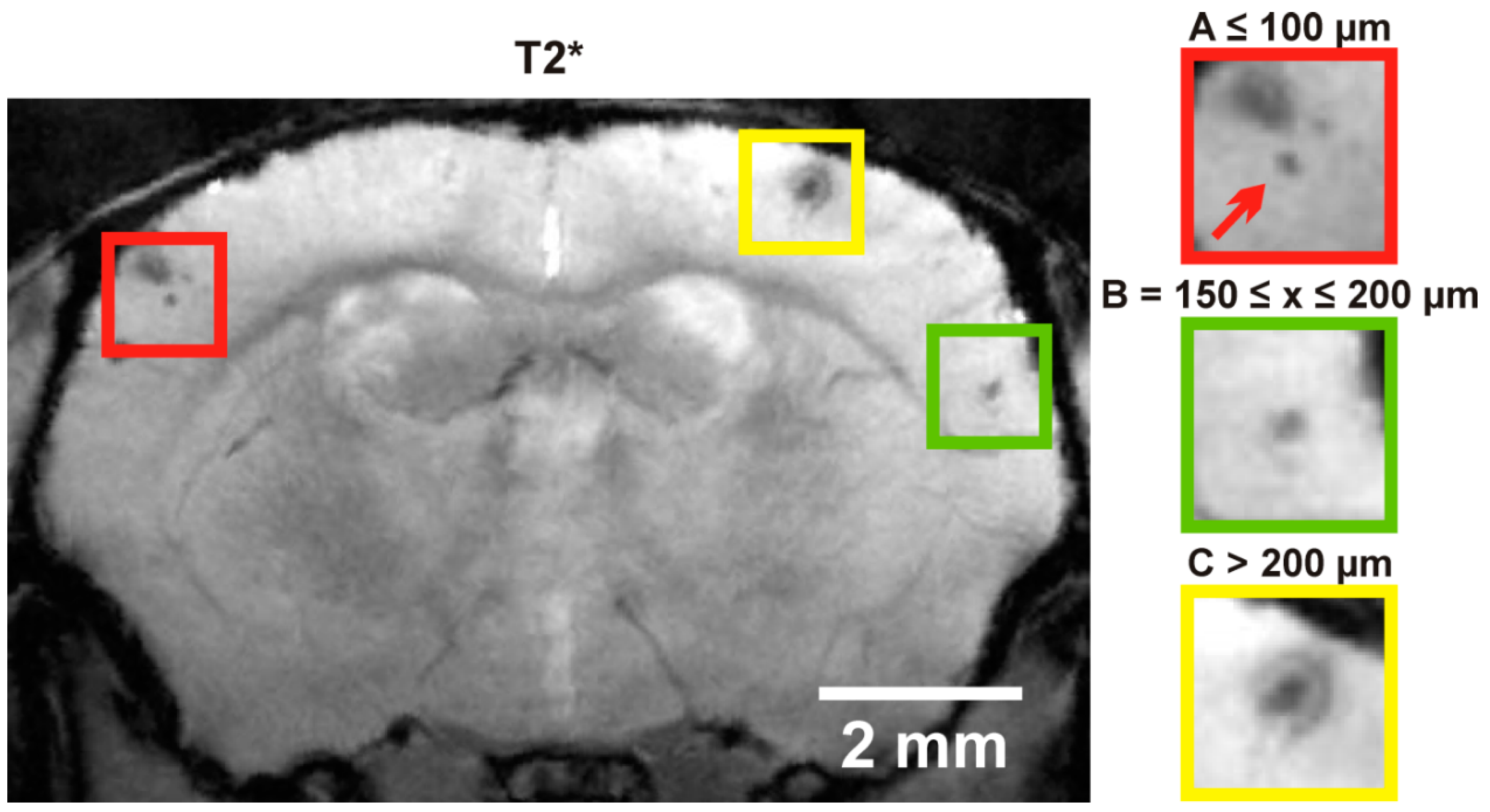
2.2. Cerebral Microbleeds
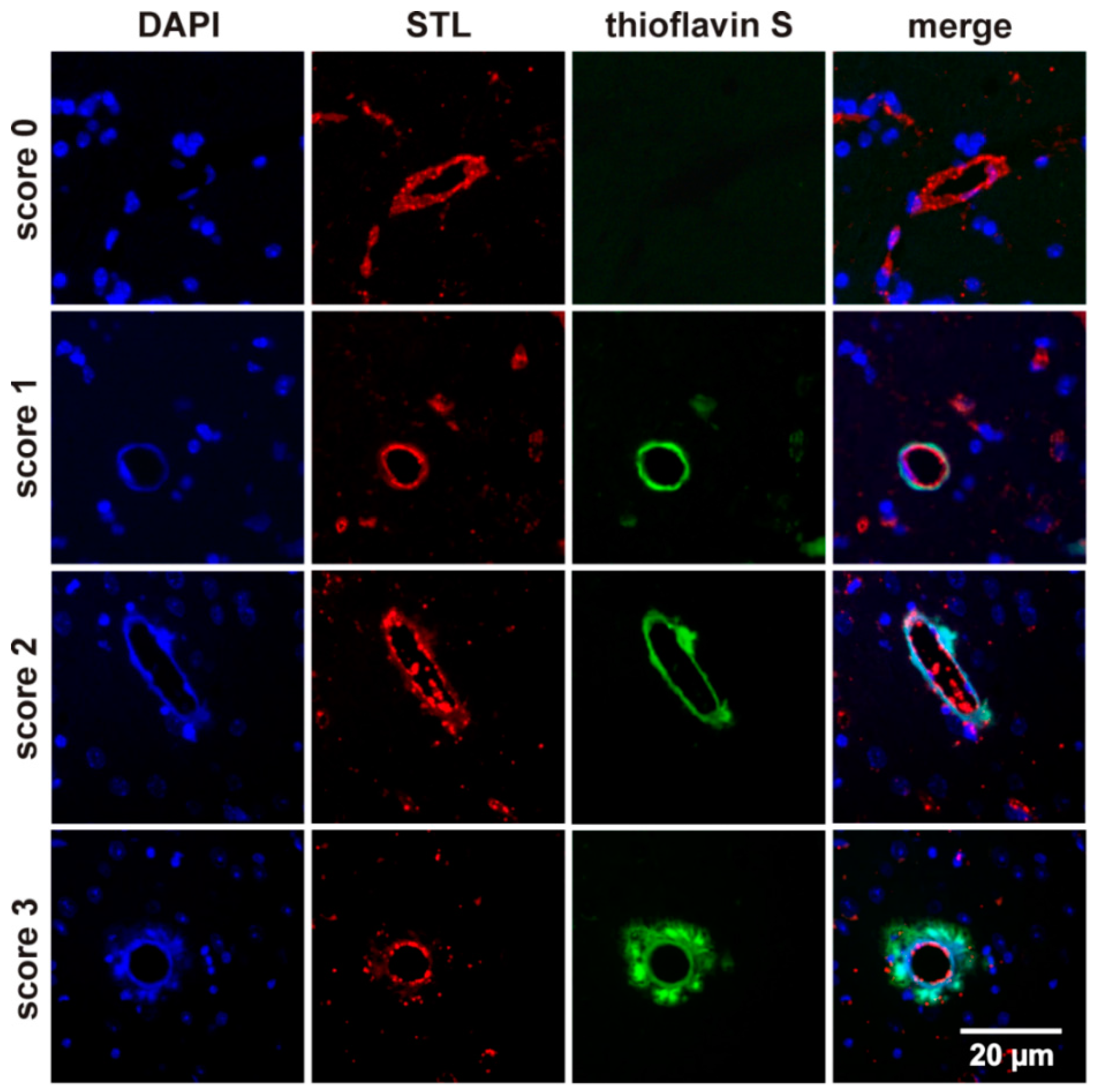
2.3. Vascular Amyloid β (Aβ) Deposition

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Mice (n) | Total cMBs | p-Value | cMBs ≤ 100 µm | p-Value | cMBs 150–200 µm | p-Value | cMBs > 200 µm | p-Value |
|---|---|---|---|---|---|---|---|---|---|
| Control group | 16 | 35 ± 18.5 | - | 17.2 ± 9.7 | - | 12.8 ± 6.9 | - | 5.1 ± 5.4 | - |
| 8 months of treatment | 6 | 29.3 ± 9.8 | p = 0.49 | 16.3 ± 6.4 | p = 0.84 | 9.8 ± 4.0 | p = 0.34 | 3.0 ± 2.1 | p = 0.38 |
| 12 months of treatment | 7 | 24.9 ± 21.3 | p = 0.26 | 12.9 ± 9.6 | p = 0.33 | 9.3 ± 10.3 | p = 0.34 | 2.7 ± 2.1 | p = 0.28 |
| 16 months of treatment | 13 | 27.8 ± 15.4 | p = 0.27 | 13.7 ± 9.4 | p = 0.34 | 10.2 ± 4.7 | p = 0.26 | 3.9 ± 2.8 | p = 0.5 |
| pooled treatment group | 26 | 27.4 ± 15.6 | p = 0.16 | 14.1 ± 8.6 | p = 0.29 | 9.9 ± 6.3 | p = 0.17 | 3.4 ± 2.5 | p = 0.26 |
3. Discussion
4. Materials and Methods
4.1. Animals
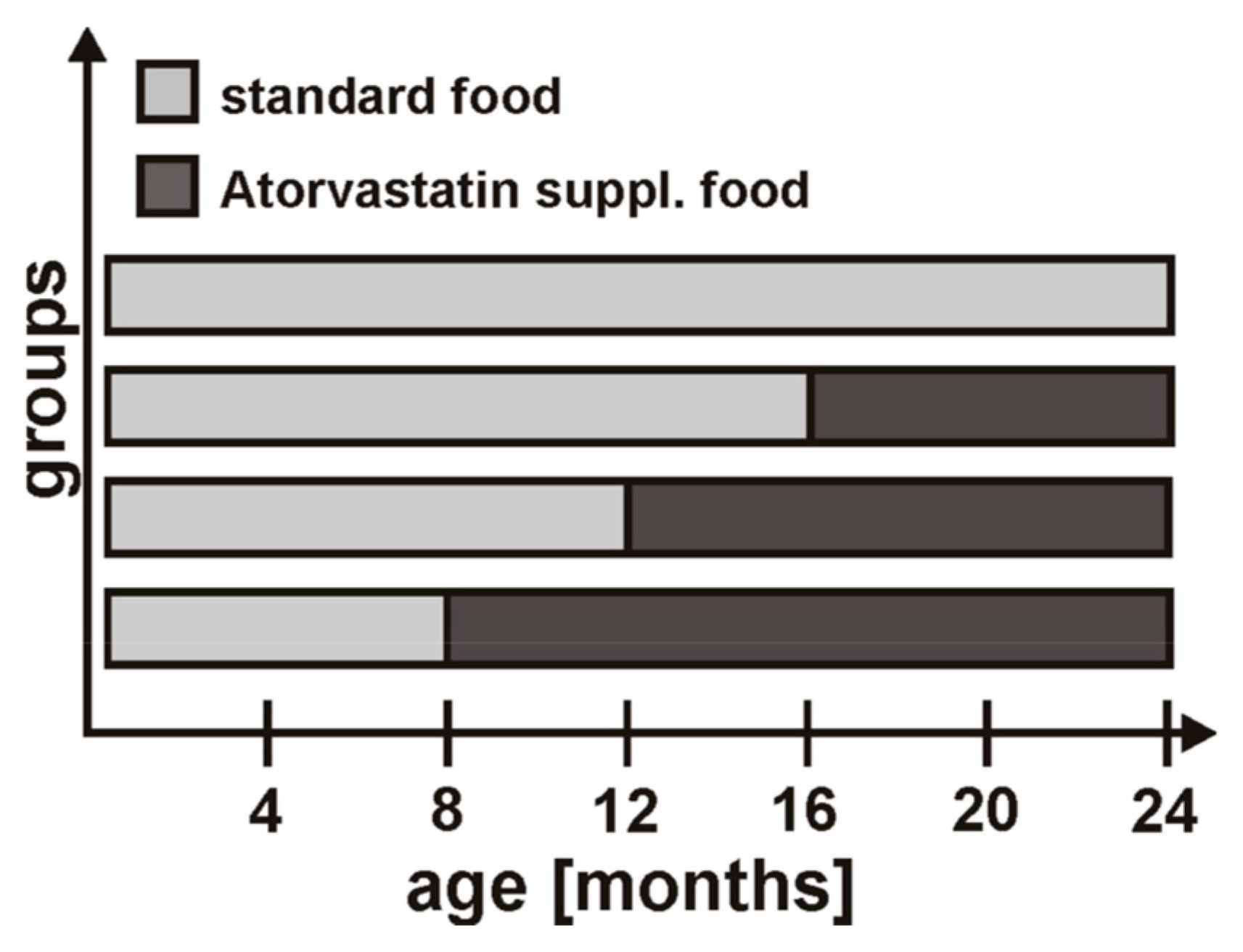
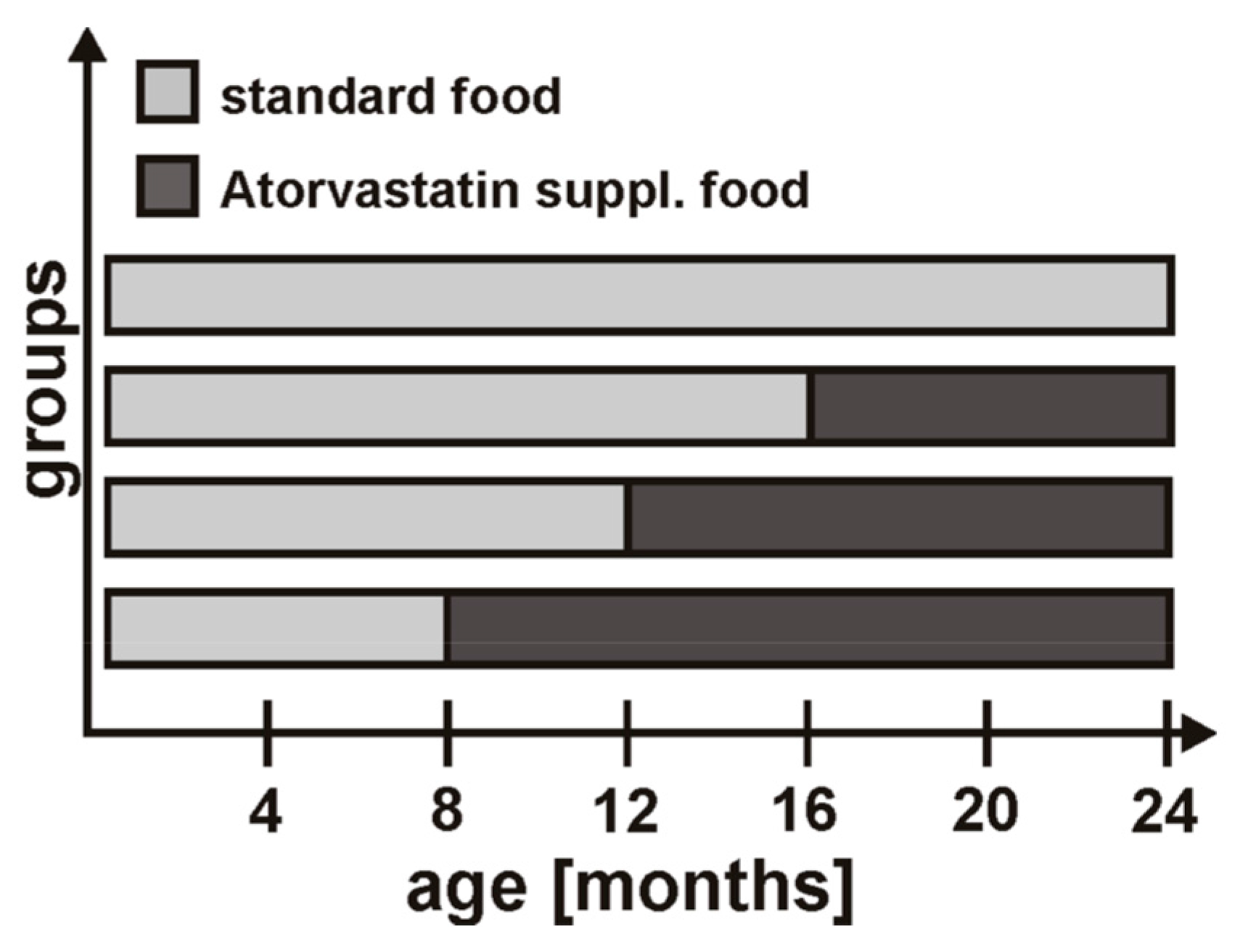
4.2. Statin Treatment
4.3. Magnetic Resonance Imaging (MRI) Protocol and Analysis
4.4. Histology
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Conflicts of Interest
Abbreviations
References
- Scholz, W. Studien zur Pathologie der Hirngefäβe. II. Die drusige Entartung der Hirnarterien und-capillaren (eine Form seniler Gefäβerkrankung). Z. Ges. Neurol. Psychiat. 1938, 162, 694–715. [Google Scholar] [CrossRef]
- Vinters, H.V. Cerebral amyloid angiopathy. A critical review. Stroke 1987, 18, 311–324. [Google Scholar] [CrossRef] [PubMed]
- Mandybur, T.I. The incidence of cerebral amyloid angiopathy in Alzheimer’s disease. Neurology 1975, 25, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Coria, F.; Castano, E.M.; Frangione, B. Brain amyloid in normal aging and cerebral amyloid angiopathy is antigenically related to Alzheimer’s disease β-protein. Am. J. Pathol. 1987, 129, 422–428. [Google Scholar] [PubMed]
- Revesz, T.; Holton, J.L.; Lashley, T.; Plant, G.; Rostagno, A.; Ghiso, J.; Frangione, B. Sporadic and familial cerebral amyloid angiopathies. Brain Pathol. 2002, 12, 343–357. [Google Scholar] [CrossRef] [PubMed]
- Keage, H.A.; Carare, R.O.; Friedland, R.P.; Ince, P.G.; Love, S.; Nicoll, J.A.; Wharton, S.B.; Weller, R.O.; Brayne, C. Population studies of sporadic cerebral amyloid angiopathy and dementia: A systematic review. BMC Neurol. 2009, 9. [Google Scholar] [CrossRef] [PubMed]
- Schlote, W. Die Amyloidnatur der kongophilen, drusigen Entartung der Hirnarterien (Scholz) im Senium. Acta Neuropathol. 1965, 4, 449–468. [Google Scholar] [CrossRef]
- Viswanathan, A.; Greenberg, S.M. Cerebral amyloid angiopathy in the elderly. Ann. Neurol. 2011, 70, 871–880. [Google Scholar] [CrossRef] [PubMed]
- Divry, P. De l’amyloidose vasculaire cérébrale et méningée (méningopathie amyloide) dans la démence sénile. J. Belge Neurol. Psychiat. 1941/42, 41/42, 141–158. [Google Scholar]
- Pfeifer, L.A.; White, L.R.; Ross, G.W.; Petrovitch, H.; Launer, L.J. Cerebral amyloid angiopathy and cognitive function: the HAAS autopsy study. Neurology 2002, 58, 1629–1634. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.E.; Greenberg, S.M. β-Amyloid, blood vessels, and brain function. Stroke 2009, 40, 2601–2606. [Google Scholar] [CrossRef] [PubMed]
- Kimberly, W.T.; Gilson, A.; Rost, N.S.; Rosand, J.; Viswanathan, A.; Smith, E.E.; Greenberg, S.M. Silent ischemic infarcts are associated with hemorrhage burden in cerebral amyloid angiopathy. Neurology 2009, 72, 1230–1235. [Google Scholar] [CrossRef] [PubMed]
- Linn, J.; Halpin, A.; Demaerel, P.; Ruhland, J.; Giese, A.D.; Dichgans, M.; van Buchem, M.A.; Bruckmann, H.; Greenberg, S.M. Prevalence of superficial siderosis in patients with cerebral amyloid angiopathy. Neurology 2010, 74, 1346–1350. [Google Scholar] [CrossRef] [PubMed]
- Charidimou, A.; Jager, R.H.; Fox, Z.; Peeters, A.; Vandermeeren, Y.; Laloux, P.; Baron, J.C.; Werring, D.J. Prevalence and mechanisms of cortical superficial siderosis in cerebral amyloid angiopathy. Neurology 2013, 81, 626–632. [Google Scholar] [PubMed]
- Schrag, M.; McAuley, G.; Pomakian, J.; Jiffry, A.; Tung, S.; Mueller, C.; Vinters, H.V.; Haacke, E.M.; Holshouser, B.; Kido, D.; et al. Correlation of hypointensities in susceptibility-weighted images to tissue histology in dementia patients with cerebral amyloid angiopathy: A postmortem MRI study. Acta Neuropathol. 2010, 119, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Dierksen, G.A.; Skehan, M.E.; Khan, M.A.; Jeng, J.; Nandigam, R.N.; Becker, J.A.; Kumar, A.; Neal, K.L.; Betensky, R.A.; Frosch, M.P.; et al. Spatial relation between microbleeds and amyloid deposits in amyloid angiopathy. Ann. Neurol. 2010, 68, 545–548. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Seo, S.W.; Kim, C.; Kim, G.H.; Noh, H.J.; Kim, S.T.; Kwak, K.C.; Yoon, U.; Lee, J.M.; Lee, J.W.; et al. Pathogenesis of cerebral microbleeds: In vivo imaging of amyloid and subcortical ischemic small vessel disease in 226 individuals with cognitive impairment. Ann. Neurol. 2013, 73, 584–593. [Google Scholar] [CrossRef] [PubMed]
- Mendoza-Oliva, A.; Zepeda, A.; Arias, C. The complex actions of statins in brain and their relevance for Alzheimer’s disease treatment: An analytical review. Curr. Alzheimer Res. 2014, 11, 817–833. [Google Scholar] [CrossRef] [PubMed]
- Giannopoulos, S.; Katsanos, A.H.; Tsivgoulis, G.; Marshall, R.S. Statins and cerebral hemodynamics. J. Cereb. Blood Flow Metab. 2012, 32, 1973–1976. [Google Scholar] [CrossRef] [PubMed]
- Winkler, D.T.; Bondolfi, L.; Herzig, M.C.; Jann, L.; Calhoun, M.E.; Wiederhold, K.H.; Tolnay, M.; Staufenbiel, M.; Jucker, M. Spontaneous hemorrhagic stroke in a mouse model of cerebral amyloid angiopathy. J. Neurosci. 2001, 21, 1619–1627. [Google Scholar] [PubMed]
- Greenberg, S.M.; Al-Shahi Salman, R.; Biessels, G.J.; van Buchem, M.; Cordonnier, C.; Lee, J.M.; Montaner, J.; Schneider, J.A.; Smith, E.E.; Vernooij, M.; et al. Outcome markers for clinical trials in cerebral amyloid angiopathy. Lancet Neurol. 2014, 13, 419–428. [Google Scholar] [CrossRef]
- Garcia-Alloza, M.; Prada, C.; Lattarulo, C.; Fine, S.; Borrelli, L.A.; Betensky, R.; Greenberg, S.M.; Frosch, M.P.; Bacskai, B.J. Matrix metalloproteinase inhibition reduces oxidative stress associated with cerebral amyloid angiopathy in vivo in transgenic mice. J. Neurochem. 2009, 109, 1636–1647. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, J.R. Statins in endothelial signaling and activation. Antioxid. Redox Signal. 2009, 11, 811–821. [Google Scholar] [CrossRef] [PubMed]
- Reuter, B.; Rodemer, C.; Grudzenski, S.; Meairs, S.; Bugert, P.; Hennerici, M.G.; Fatar, M. Effect of simvastatin on MMPs and TIMPs in human brain endothelial cells and experimental stroke. Transl. Stroke Res. 2015, 6, 156–159. [Google Scholar] [PubMed]
- Kunte, H.; Amberger, N.; Busch, M.A.; Ruckert, R.I.; Meiners, S.; Harms, L. Markers of instability in high-risk carotid plaques are reduced by statins. J. Vasc. Surg. 2008, 47, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.A.; O’Regan, D.J.; Ball, S.G.; Porter, K.E. Simvastatin inhibits MMP-9 secretion from human saphenous vein smooth muscle cells by inhibiting the RhoA/ROCK pathway and reducing MMP-9 mRNA levels. FASEB J. 2005, 19, 804–806. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Yin, K.; Hsin, I.; Chen, S.; Fryer, J.D.; Holtzman, D.M.; Hsu, C.Y.; Xu, J. Matrix metalloproteinase-9 in cerebral-amyloid-angiopathy-related hemorrhage. J. Neurol. Sci. 2005, 229–230, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Andras, I.E.; Rha, G.; Huang, W.; Eum, S.; Couraud, P.O.; Romero, I.A.; Hennig, B.; Toborek, M. Simvastatin protects against amyloid β and HIV-1 Tat-induced promoter activities of inflammatory genes in brain endothelial cells. Mol. Pharmacol. 2008, 73, 1424–1433. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Yin, K.J.; Hsin, I.; Chen, S.; Fryer, J.D.; Holtzman, D.M.; Hsu, C.Y.; Xu, J. Matrix metalloproteinase-9 and spontaneous hemorrhage in an animal model of cerebral amyloid angiopathy. Ann. Neurol. 2003, 54, 379–382. [Google Scholar] [CrossRef] [PubMed]
- Wolozin, B.; Manger, J.; Bryant, R.; Cordy, J.; Green, R.C.; McKee, A. Re-assessing the relationship between cholesterol, statins and Alzheimer’s disease. Acta Neurol. Scand. Suppl. 2006, 185, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Li, P.; Wang, J.; Qing, X.; Li, W. The interaction of amyloid β and the receptor for advanced glycation endproducts induces matrix metalloproteinase-2 expression in brain endothelial cells. Cell. Mol. Neurobiol. 2012, 32, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Piazza, F.; Greenberg, S.M.; Savoiardo, M.; Gardinetti, M.; Chiapparini, L.; Raicher, I.; Nitrini, R.; Sakaguchi, H.; Brioschi, M.; Billo, G.; et al. Anti-amyloid β autoantibodies in cerebral amyloid angiopathy-related inflammation: Implications for amyloid-modifying therapies. Ann. Neurol. 2013, 73, 449–458. [Google Scholar] [PubMed]
- Castro Caldas, A.; Silva, C.; Albuquerque, L.; Pimentel, J.; Silva, V.; Ferro, J.M. Cerebral amyloid angiopathy associated with inflammation: Report of 3 cases and systematic review. J. Stroke Cerebrovasc. Dis. 2015, 24, 2039–2048. [Google Scholar] [CrossRef] [PubMed]
- Auriel, E.; Charidimou, A.; Gurol, M.E.; Ni, J.; van Etten, E.S.; Martinez-Ramirez, S.; Boulouis, G.; Piazza, F.; DiFrancesco, J.C.; Frosch, M.P.; et al. Validation of clinicoradiological criteria for the diagnosis of cerebral amyloid angiopathy-related inflammation. JAMA Neurol. 2015. [Google Scholar] [CrossRef]
- Tong, X.K.; Lecrux, C.; Rosa-Neto, P.; Hamel, E. Age-dependent rescue by simvastatin of Alzheimer’s disease cerebrovascular and memory deficits. J. Neurosci. 2012, 32, 4705–4715. [Google Scholar] [PubMed]
- Undas, A.; Brummel-Ziedins, K.E.; Mann, K.G. Statins and blood coagulation. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, L.B.; Amarenco, P.; Szarek, M.; Callahan, A., 3rd; Hennerici, M.; Sillesen, H.; Zivin, J.A.; Welch, K.M.; Investigators, S. Hemorrhagic stroke in the stroke prevention by aggressive reduction in cholesterol levels study. Neurology 2008, 70 Pt 2, 2364–2370. [Google Scholar] [CrossRef] [PubMed]
- Haussen, D.C.; Henninger, N.; Kumar, S.; Selim, M. Statin use and microbleeds in patients with spontaneous intracerebral hemorrhage. Stroke 2012, 43, 2677–2681. [Google Scholar] [CrossRef] [PubMed]
- Romero, J.R.; Preis, S.R.; Beiser, A.; DeCarli, C.; Viswanathan, A.; Martinez-Ramirez, S.; Kase, C.S.; Wolf, P.A.; Seshadri, S. Risk factors, stroke prevention treatments, and prevalence of cerebral microbleeds in the Framingham Heart Study. Stroke 2014, 45, 1492–1494. [Google Scholar] [PubMed]
- Kurata, T.; Miyazaki, K.; Kozuki, M.; Panin, V.L.; Morimoto, N.; Ohta, Y.; Nagai, M.; Ikeda, Y.; Matsuura, T.; Abe, K. Atorvastatin and pitavastatin improve cognitive function and reduce senile plaque and phosphorylated tau in aged APP mice. Brain Res. 2011, 1371, 161–170. [Google Scholar] [PubMed]
- Reuter, B.; Grudzenski, S.; Chatzikonstantinou, E.; Meairs, S.; Ebert, A.; Heiler, P.; Schad, L.R.; Staufenbiel, M.; Hennerici, M.G.; Fatar, M. Thrombolysis in experimental cerebral amyloid angiopathy and the risk of secondary intracerebral hemorrhage. Stroke 2014, 45, 2411–2416. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Himmelreich, U.; Hoehn, M. Stem cell labeling for magnetic resonance imaging. Minim Invasive Ther. Allied. Technol. 2008, 17, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Fazekas, F.; Kleinert, R.; Roob, G.; Kleinert, G.; Kapeller, P.; Schmidt, R.; Hartung, H.P. Histopathologic analysis of foci of signal loss on gradient-echo T2*-weighted MR images in patients with spontaneous intracerebral hemorrhage: Evidence of microangiopathy-related microbleeds. AJNR. Am. J. Neuroradiol. 1999, 20, 637–642. [Google Scholar] [PubMed]
- Guntern, R.; Bouras, C.; Hof, P.R.; Vallet, P.G. An improved thioflavine S method for staining neurofibrillary tangles and senile plaques in Alzheimer’s disease. Experientia 1992, 48, 8–10. [Google Scholar] [CrossRef] [PubMed]
- Hartig, W.; Reichenbach, A.; Voigt, C.; Boltze, J.; Bulavina, L.; Schuhmann, M.U.; Seeger, J.; Schusser, G.F.; Freytag, C.; Grosche, J. Triple fluorescence labelling of neuronal, glial and vascular markers revealing pathological alterations in various animal models. J. Chem. Neuroanat. 2009, 37, 128–138. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reuter, B.; Venus, A.; Grudzenski, S.; Heiler, P.; Schad, L.; Staufenbiel, M.; Hennerici, M.G.; Fatar, M. Statin Therapy and the Development of Cerebral Amyloid Angiopathy—A Rodent in Vivo Approach. Int. J. Mol. Sci. 2016, 17, 126. https://doi.org/10.3390/ijms17010126
Reuter B, Venus A, Grudzenski S, Heiler P, Schad L, Staufenbiel M, Hennerici MG, Fatar M. Statin Therapy and the Development of Cerebral Amyloid Angiopathy—A Rodent in Vivo Approach. International Journal of Molecular Sciences. 2016; 17(1):126. https://doi.org/10.3390/ijms17010126
Chicago/Turabian StyleReuter, Björn, Alexander Venus, Saskia Grudzenski, Patrick Heiler, Lothar Schad, Matthias Staufenbiel, Michael G. Hennerici, and Marc Fatar. 2016. "Statin Therapy and the Development of Cerebral Amyloid Angiopathy—A Rodent in Vivo Approach" International Journal of Molecular Sciences 17, no. 1: 126. https://doi.org/10.3390/ijms17010126
APA StyleReuter, B., Venus, A., Grudzenski, S., Heiler, P., Schad, L., Staufenbiel, M., Hennerici, M. G., & Fatar, M. (2016). Statin Therapy and the Development of Cerebral Amyloid Angiopathy—A Rodent in Vivo Approach. International Journal of Molecular Sciences, 17(1), 126. https://doi.org/10.3390/ijms17010126