
Inhibition of NF-κB in Tumor Cells Exacerbates Immune Cell Activation Following Photodynamic Therapy

Abstract
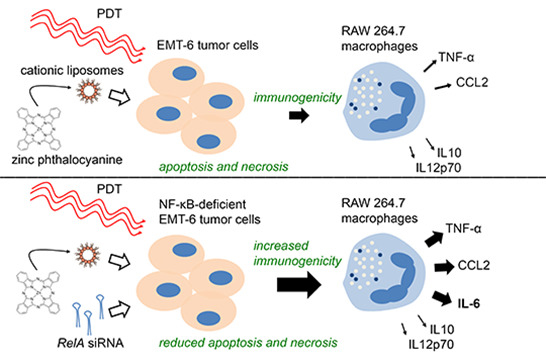
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
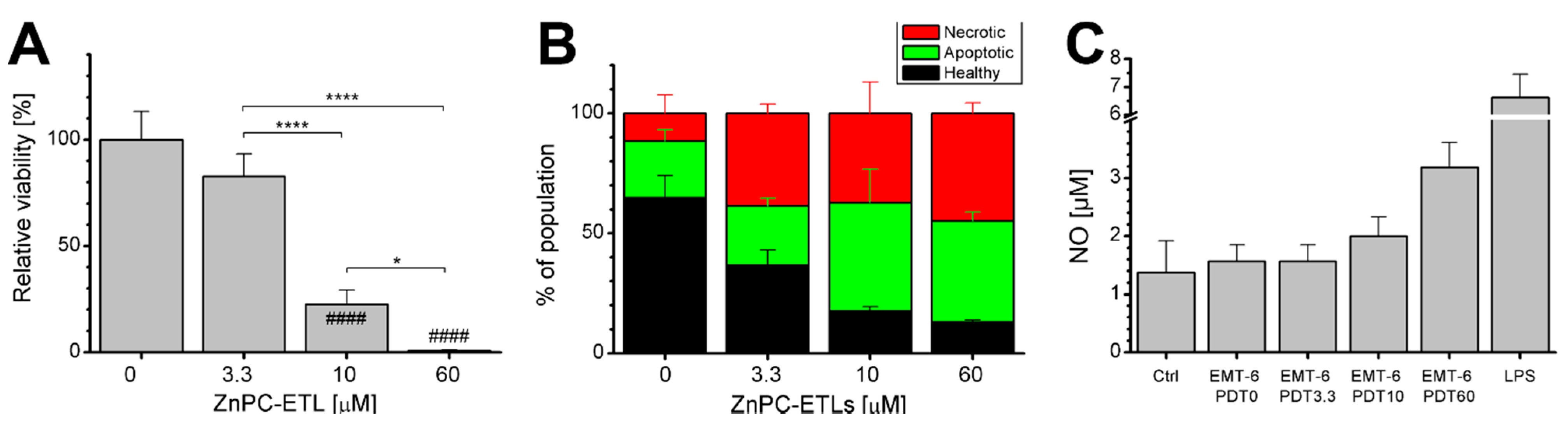
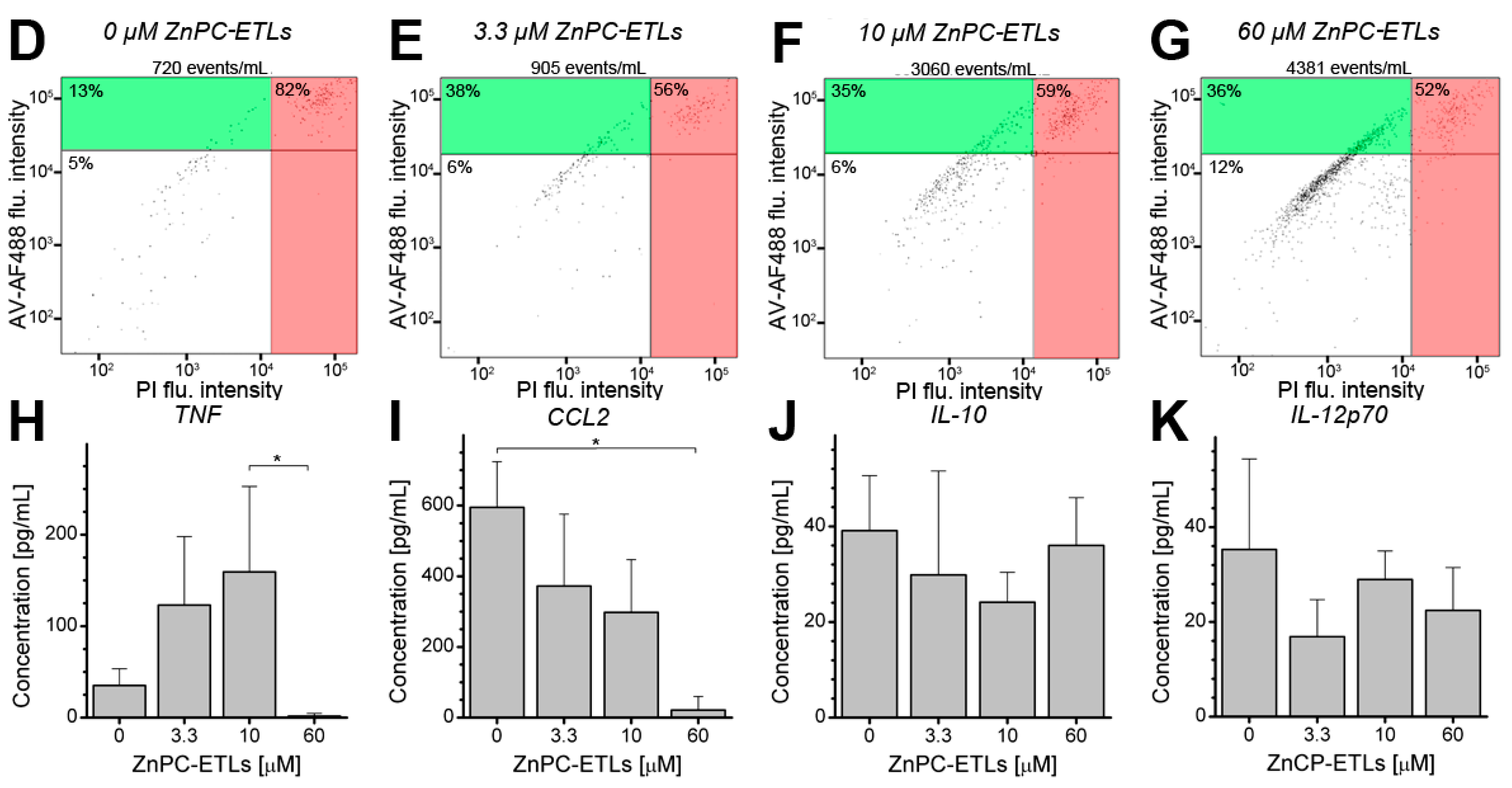
2.1. Photodynamic Therapy (PDT)-Killed EMT-6 Cells Are Immunogenic
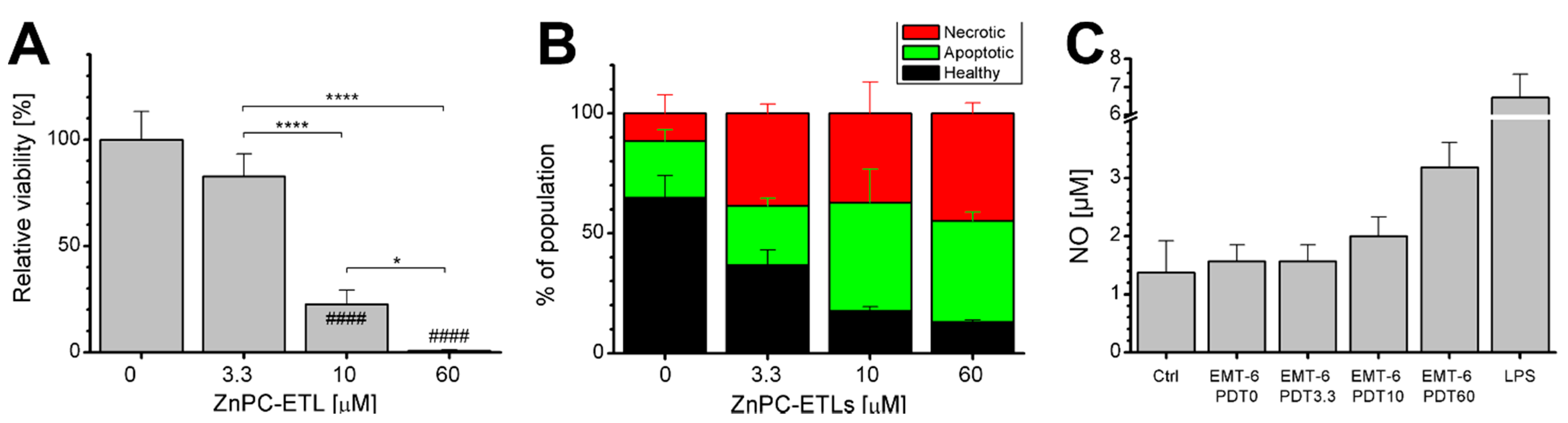
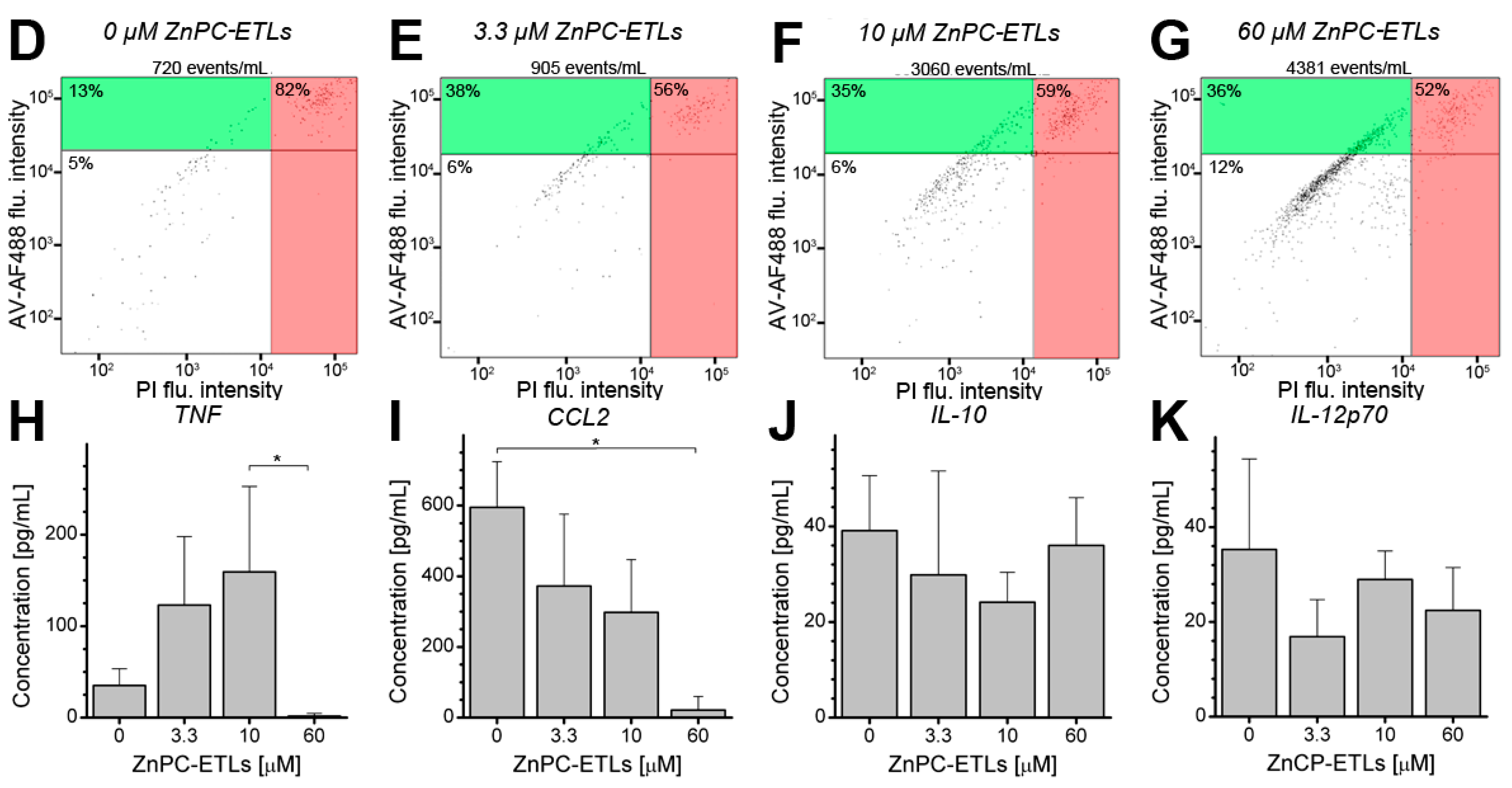
2.2. PDT-Killed EMT-6 Supernatant Contains Tumor Necrosis Factor α (TNF-α) and Chemokine C–C Motif Ligand 2 (CCL2)
2.3. Inhibition of Nuclear Factor κB (NF-κB) Reduces Cell Death
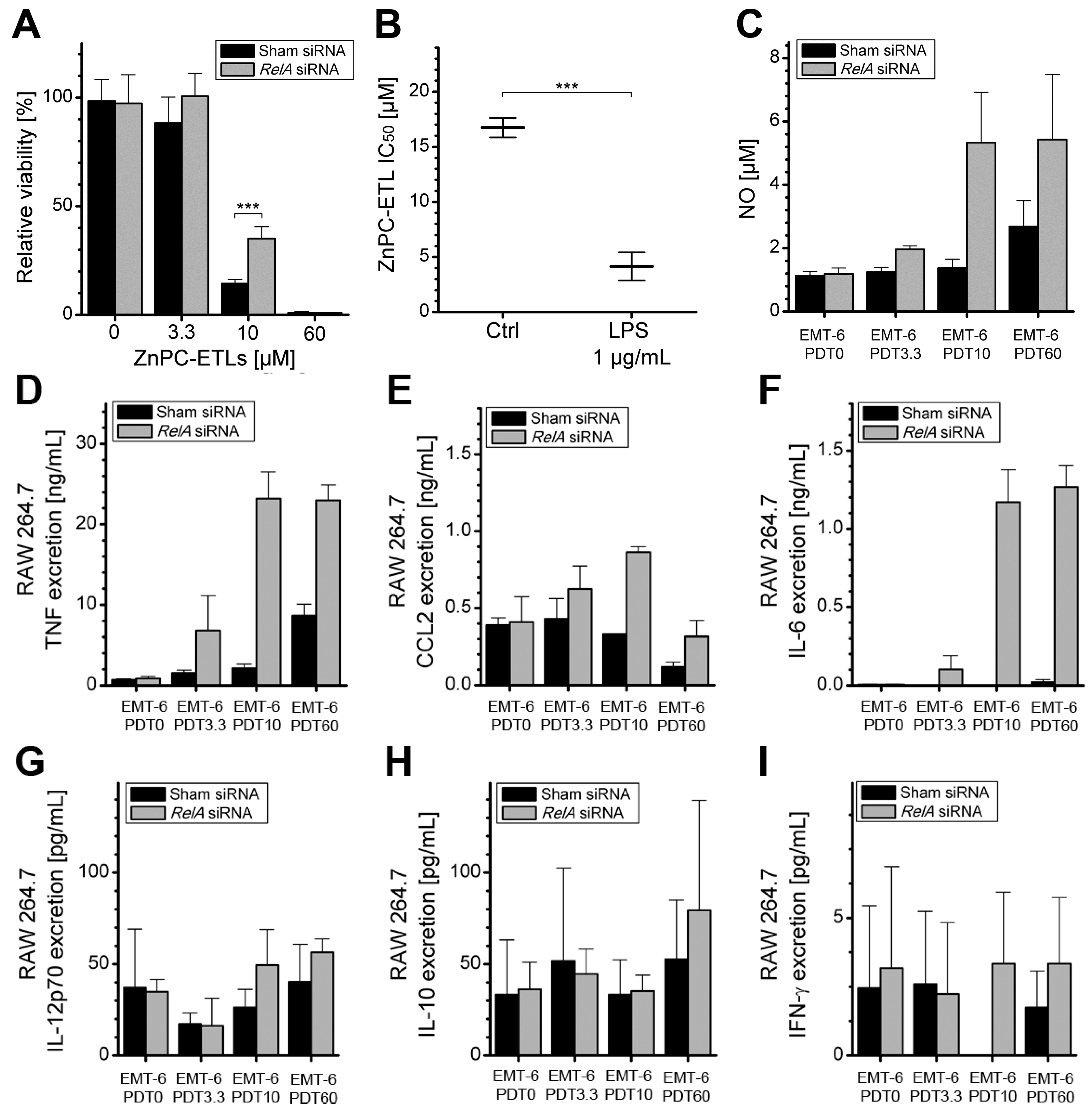
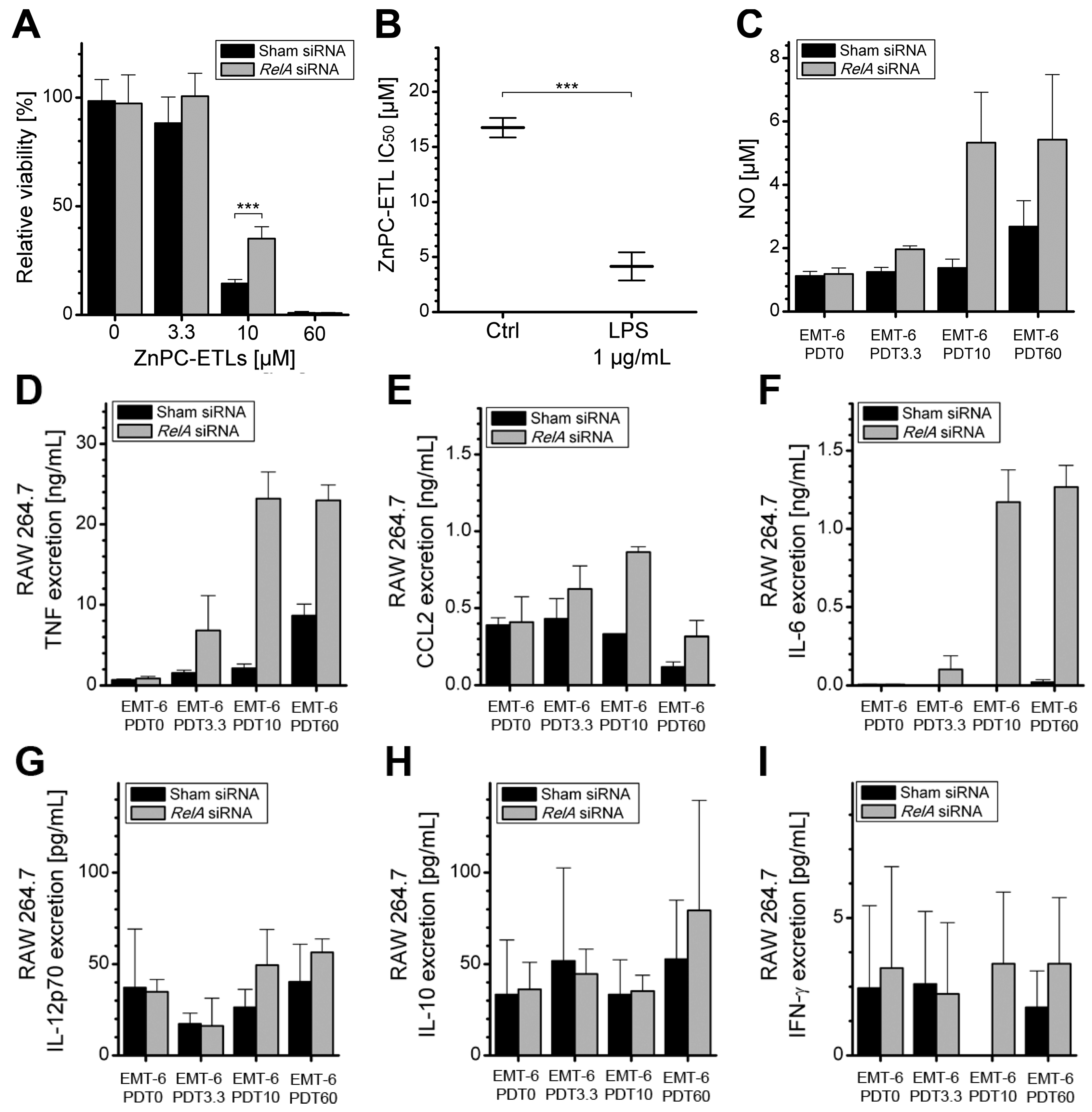
2.4. Increased Immunogenicity of PDT-Killed EMT-6-RelAkd Cells
3. Discussion
4. Experimental Section
4.1. Preparation of Zinc Phthalocyanine Liposomes
4.2. Cell Culture
4.3. Photodynamic Therapy
4.4. Stimulation of RAW 264.7 Macrophages
4.5. Cytokine Detection
4.6. Transfections
4.7. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Dolmans, D.E.J.G.J.; Fukumura, D.; Jain, R.K. Photodynamic therapy for cancer. Nat. Rev. Cancer 2003, 3, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Broekgaarden, M.; Weijer, R.; van Gulik, T.M.; Hamblin, M.R.; Heger, M. Tumor cell survival pathways activated by photodynamic therapy: A molecular basis for pharmacological inhibition strategies. Cancer Metastasis Rev. 2015, in press. [Google Scholar]
- Khan, S.A.; Thomas, H.C.; Davidson, B.R.; Taylor-Robinson, S.D. Cholangiocarcinoma. Lancet 2005, 366, 1303–1314. [Google Scholar] [CrossRef]
- O’Connor, A.E.; Gallagher, W.M.; Byrne, A.T. Porphyrin and nonporphyrin photosensitizers in oncology: Preclinical and clinical advances in photodynamic therapy. Photochem. Photobiol. 2009, 85, 1053–1074. [Google Scholar] [CrossRef] [PubMed]
- Weijer, R.; Broekgaarden, M.; Kos, M.; Vught, R.; Rauws, E.; van Gulik, T.M.; Heger, M. Enhancing photodynamic therapy of refractory solid cancers: Combining second generation photosensitizers with multi-targeted drug delivery. J. Photochem. Photobiol. C Photochem. Rev. 2015, 23, 103–131. [Google Scholar] [CrossRef]
- Broekgaarden, M.; de Kroon, A.I.P.M.; van Gulik, T.M.; Heger, M. Development and in vitro proof-of-concept of interstitially targeted zinc-phthalocyanine liposomes for photodynamic therapy. Curr. Med. Chem. 2013, 21, 377–391. [Google Scholar] [CrossRef]
- Rizvi, I.; Celli, J.P.; Evans, C.L.; Abu-Yousif, A.O.; Muzikansky, A.; Pogue, B.W.; Finkelstein, D.; Hasan, T. Synergistic enhancement of carboplatin efficacy with photodynamic therapy in a three-dimensional model for micrometastatic ovarian cancer. Cancer Res. 2010, 70, 9319–9328. [Google Scholar] [CrossRef] [PubMed]
- Broekgaarden, M.; Weijer, R.; van Wijk, A.C.; Cox, R.C.; Egmond, M.R.; Hoebe, R.; van Gulik, T.M.; Heger, M. Photodynamic therapy with liposomal zinc phthalocyanine and tirapazamine increases tumor cell death via DNA damage. J. Biomed. Nanotechnol. 2015. submitted. [Google Scholar]
- Coupienne, I.; Bontems, S.B.; Dewaele, M.; Rubio, N.; Habraken, Y.; Fulda, S.; Agostinis, P.; Piette, J. NF-κB inhibition improves the sensitivity of human glioblastoma cells to 5-aminolevulinic acid-based photodynamic therapy. Biochem. Pharmacol. 2011, 81, 606–616. [Google Scholar] [CrossRef] [PubMed]
- Matroule, J.Y.; Hellin, A.C.; Morliere, P.; Fabiano, A.S.; Santus, R.; Merville, M.P.; Piette, J. Role of nuclear factor-κB in colon cancer cell apoptosis mediated by aminopyropheophorbide photosensitization. Photochem. Photobiol. 1999, 70, 540–548. [Google Scholar] [CrossRef]
- Chen, H.M.; Liu, C.M.; Yang, H.; Chou, H.Y.; Chiang, C.P.; Kuo, M.Y. 5-Aminolevulinic acid induce apoptosis via NF-κB/JNK pathway in human oral cancer Ca9–22 cells. J. Oral Pathol. Med. 2011, 40, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.T.; Cummins, E.P. The role of NF-κB in hypoxia-induced gene expression. Ann. N. Y. Acad. Sci. 2009, 1177, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Cao, Y.; Greten, F.R.; Li, Z.-W. NF-κB in cancer: From innocent bystander to major culprit. Nat. Rev. Cancer 2002, 2, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Guttridge, D.C.; Albanese, C.; Reuther, J.Y.; Pestell, R.G.; Baldwin, A.S., Jr. NF-κB controls cell growth and differentiation through transcriptional regulation of cyclin D1. Mol. Cell. Biol. 1999, 19, 5785–5799. [Google Scholar] [PubMed]
- Schoonbroodt, S.; Piette, J. Oxidative stress interference with the nuclear factor-κB activation pathways. Biochem. Pharmacol. 2000, 60, 1075–1083. [Google Scholar] [CrossRef]
- Matroule, J.-Y.; Volanti, C.; Piette, J. NF-κB in photodynamic therapy: Discrepancies of a master regulator. Photochem. Photobiol. 2006, 82, 1241–1246. [Google Scholar] [CrossRef] [PubMed]
- Korbelik, M.; Cecic, I. Contribution of myeloid and lymphoid host cells to the curative outcome of mouse sarcoma treatment by photodynamic therapy. Cancer Lett. 1999, 137, 91–98. [Google Scholar] [CrossRef]
- Korbelik, M.; Krosl, G.; Krosl, J.; Dougherty, G.J. The role of host lymphoid populations in the response of mouse EMT6 tumor to photodynamic therapy. Cancer Res. 1996, 56, 5647–5652. [Google Scholar] [PubMed]
- Thong, P.S.-P.; Ong, K.W.; Goh, N.S.G.; Kho, K.W.; Manivasager, V.; Bhuvaneswari, R.; Olivo, M.; Soo, K.C. Photodynamic-therapy-activated immune response against distant untreated tumours in recurrent angiosarcoma. Lancet Oncol. 2007, 8, 950–952. [Google Scholar] [CrossRef]
- Kabingu, E.; Vaughan, L.; Owczarczak, B.; Ramsey, K.D.; Gollnick, S.O. CD8+ T cell-mediated control of distant tumours following local photodynamic therapy is independent of CD4+ T cells and dependent on natural killer cells. Br. J. Cancer 2007, 96, 1839–1848. [Google Scholar] [CrossRef] [PubMed]
- Matroule, J.Y.; Piette, J. Photosensitization and redox signaling. Antioxid. Redox Signal. 2000, 2, 301–315. [Google Scholar] [CrossRef] [PubMed]
- Piva, R.; Belardo, G.; Santoro, M.G. NF-κB: A stress-regulated switch for cell survival. Antioxid. Redox Signal. 2006, 8, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Castano, A.P.; Mroz, P.; Hamblin, M.R. Photodynamic therapy and anti-tumour immunity. Nat. Rev. Cancer 2006, 6, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Broekgaarden, M.; Weijer, R.; van Golen, R.F.; Maas, M.A.; van Wijk, A.C.; Furse, S.; van Gulik, T.M.; Heger, M. Development of tumor vascular endothelium targeted liposomes containing zinc phthalocyanine for application in photodynamic therapy. ACS Nano 2015. in preparation. [Google Scholar]
- Broekgaarden, M.; Weijer, R.; Krekorian, M.; van den IJssel, B.; Kos, M.; Alles, L.K.; van Wijk, A.C.; Bikadi, Z.; Hazai, E.; van Gulik, T.M.; et al. Inhibition of hypoxia inducible factor 1 with acriflavine sensitizes tumor cells to photodynamic therapy with zinc phthalocyanine-encapsulating cationic liposomes. Nano Res. 2015. submitted. [Google Scholar]
- Kloek, J.J.; Marechal, X.; Roelofsen, J.; Houtkooper, R.H.; van Kuilenburg, A.B.; Kulik, W.; Bezemer, R.; Neviere, R.; van Gulik, T.M.; Heger, M. Cholestasis is associated with hepatic microvascular dysfunction and aberrant energy metabolism before and during ischemia-reperfusion. Antioxid. Redox Signal. 2012, 17, 1109–1123. [Google Scholar] [CrossRef] [PubMed]
- Lyons, C.R.; Orloff, G.J.; Cunningham, J.M. Molecular cloning and functional expression of an inducible nitric oxide synthase from a murine macrophage cell line. J. Biol. Chem. 1992, 267, 6370–6374. [Google Scholar] [PubMed]
- Garg, A.D.; Krysko, D.V.; Vandenabeele, P.; Agostinis, P. DAMPs and PDT-mediated photo-oxidative stress: Exploring the unknown. Photochem. Photobiol. Sci. 2011, 10, 670–680. [Google Scholar] [CrossRef] [PubMed]
- Korbelik, M.; Hamblin, M.R. The impact of macrophage-cancer cell interaction on the efficacy of photodynamic therapy. Photochem. Photobiol. Sci. 2015, 14, 1403–1409. [Google Scholar] [CrossRef] [PubMed]
- Beg, A.A.; Baltimore, D. An essential role for NF-κB in preventing TNF-α-induced cell death. Science 1996, 274, 782–784. [Google Scholar] [CrossRef] [PubMed]
- Kundu, S.D.; Lee, C.; Billips, B.K.; Habermacher, G.M.; Zhang, Q.; Liu, V.; Wong, L.Y.; Klumpp, D.J.; Thumbikat, P. The toll-like receptor pathway: A novel mechanism of infection-induced carcinogenesis of prostate epithelial cells. Prostate 2008, 68, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Wijayanti, N.; Huber, S.; Samoylenko, A.; Kietzmann, T.; Immenschuh, S. Role of NF-κB and p38 MAP kinase signaling pathways in the lipopolysaccharide-dependent activation of heme oxygenase-1 gene expression. Antioxid. Redox Signal. 2004, 6, 802–810. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P. The TLR and IL-1 signaling network at a glance. J. Cell Sci. 2014, 127, 2383–2390. [Google Scholar] [CrossRef] [PubMed]
- Eigenbrod, T.; Park, J.H.; Harder, J.; Iwakura, Y.; Núñez, G. Cutting edge: Critical role for mesothelial cells in necrosis-induced inflammation through the recognition of IL-1α released from dying cells. J. Immunol. 2008, 181, 8194–8198. [Google Scholar] [CrossRef] [PubMed]
- Carr, M.W.; Roth, S.J.; Luther, E.; Rose, S.S.; Springer, T.A. Monocyte chemoattractant protein 1 acts as a T-lymphocyte chemoattractant. Proc. Natl. Acad. Sci. USA 1994, 91, 3652–3656. [Google Scholar] [CrossRef] [PubMed]
- Chu, W.M. Tumor necrosis factor. Cancer Lett. 2012, 328, 222–225. [Google Scholar] [CrossRef] [PubMed]
- Cecic, I.; Korbelik, M. Mediators of peripheral blood neutrophilia induced by photodynamic therapy of solid tumors. Cancer Lett. 2002, 183, 43–51. [Google Scholar] [CrossRef]
- Byun, J.Y.; Choi, H.Y.; Myung, K.B.; Choi, Y.W. Expression of IL-10, TGF-β1 and TNF-α in cultured keratinocytes (HaCaT cells) after IPL treatment or ALA-IPL photodynamic treatment. Ann. Dermatol. 2009, 21, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Shixiang, Y.; Xi, S.; Junliang, L.; Shanyi, Z.; Xingke, X.; Meiguang, Z.; Kai, W.; Fangcheng, L. Antitumor efficacy of a photodynamic therapy-generated dendritic cell glioma vaccine. Med. Oncol. 2011, 28, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F. Tumour necrosis factor and cancer. Nat. Rev. Cancer 2009, 9, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Ybanez, M.D.; Ahmadi, S.; Yeh, K.; Kaplowitz, N. Redox regulation of tumor necrosis factor signaling. Antioxid. Redox Signal. 2009, 11, 2245–2263. [Google Scholar] [CrossRef] [PubMed]
- Kawczyk-Krupka, A.; Bugaj, A.M.; Latos, W.; Wawrzyniec, K.; Oles, P.; Mertas, A.; Czuba, Z.; Krol, W.; Sieron-Stoltny, K.; Sieron, A. ALA-mediated photodynamic effect on apoptosis induction and secretion of macrophage migration inhibitory factor (MIF) and of monocyte chemotactic protein (MCP-1) by colon cancer cells in normoxia and in hypoxia-like conditions in vitro. Photodiagn. Photodyn. Ther. 2015, 12, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, M.R.; Merlino, G. The two faces of interferon-γ in cancer. Clin. Cancer Res. 2011, 17, 6118–6124. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Cheng, Y.; Lu, J.; Hu, R.; Wan, Q.; Feng, H. Photodynamic therapy boosts anti-glioma immunity in mice: A dependence on the activities of T cells and complement C3. J. Cell. Biochem 2011, 112, 3035–3043. [Google Scholar] [CrossRef] [PubMed]
- Trinchieri, G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat. Rev. Immunol. 2003, 3, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Gollnick, S.O.; Liu, X.; Owczarczak, B.; Musser, D.A.; Henderson, B.W. Altered expression of interleukin 6 and interleukin 10 as a result of photodynamic therapy in vivo. Cancer Res. 1997, 57, 3904–3909. [Google Scholar] [PubMed]
- Lin, W.W.; Karin, M. A cytokine-mediated link between innate immunity, inflammation, and cancer. J. Clin. Investig. 2007, 117, 1175–1183. [Google Scholar] [CrossRef] [PubMed]
- Panzarini, E.; Inguscio, V.; Tenuzzo, B.A.; Dini, L. In vitro and in vivo clearance of rose bengal acetate-photodynamic therapy-induced autophagic and apoptotic cells. Exp. Biol. Med. 2013, 238, 765–778. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; Krysko, D.V.; Verfaillie, T.; Kaczmarek, A.; Ferreira, G.B.; Marysael, T.; Rubio, N.; Firczuk, M.; Mathieu, C.; Roebroek, A.J.M.; et al. A novel pathway combining calreticulin exposure and ATP secretion in immunogenic cancer cell death. EMBO J. 2012, 31, 1062–1079. [Google Scholar] [CrossRef] [PubMed]
- Kick, G.; Messer, G.; Goetz, A.; Plewig, G.; Kind, P. Photodynamic therapy induces expression of interleukin 6 by activation of AP-1 but not NF-κB DNA binding. Cancer Res. 1995, 55, 2373–2379. [Google Scholar] [PubMed]
- Volanti, C.; Gloire, G.; Vanderplasschen, A.; Jacobs, N.; Habraken, Y.; Piette, J. Downregulation of ICAM-1 and VCAM-1 expression in endothelial cells treated by photodynamic therapy. Oncogene 2004, 23, 8649–8658. [Google Scholar] [CrossRef] [PubMed]
- Gollnick, S.O.; Evans, S.S.; Baumann, H.; Owczarczak, B.; Maier, P.; Vaughan, L.; Wang, W.C.; Unger, E.; Henderson, B.W. Role of cytokines in photodynamic therapy-induced local and systemic inflammation. Br. J. Cancer 2003, 88, 1772–1779. [Google Scholar] [CrossRef] [PubMed]
- De Vree, W.J.A.; Essers, M.C.; de Bruijn, H.S.; Star, W.M.; Koster, J.F.; Sluiter, W. Evidence for an important role of neutrophils in the efficacy of photodynamic therapy in vivo. Cancer Res. 1996, 56, 2908–2911. [Google Scholar]
- De Vree, W.J.A.; Essers, M.C.; Koster, J.F.; Sluiter, W. Role of interleukin 1 and granulocyte colony-stimulating factor in photofrin-based photodynamic therapy of rat rhabdomyosarcoma tumors. Cancer Res. 1997, 57, 2555–2558. [Google Scholar] [PubMed]
- Du, H.; Bay, B.-H.; Mahendran, R.; Olivo, M. Hypericin-mediated photodynamic therapy elicits differential interleukin-6 response in nasopharyngeal cancer. Cancer Lett. 2006, 235, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Heger, M.; van der Wal, A.C.; Storm, G.; van Gemert, M.J. Potential therapeutic benefits stemming from the thermal nature of irreversible electroporation of solid cancers. Hepatobiliary Pancreat. Dis. Int. 2015, 14, 331–333. [Google Scholar] [CrossRef]
- Wei, L.H.; Baumann, H.; Tracy, E.; Wang, Y.; Hutson, A.; Rose-John, S.; Henderson, B.W. Interleukin-6 trans signalling enhances photodynamic therapy by modulating cell cycling. Br. J. Cancer 2007, 97, 1513–1522. [Google Scholar] [CrossRef] [PubMed]
- Kushibiki, T.; Tajiri, T.; Tomioka, Y.; Awazu, K. Photodynamic therapy induces interleukin secretion from dendritic cells. Int. J. Clin. Exp. Med. 2010, 3, 110–114. [Google Scholar] [PubMed]
- Kim, R.; Emi, M.; Tanabe, K. Functional roles of immature dendritic cells in impaired immunity of solid tumour and their targeted strategies for provoking tumour immunity. Clin. Exp. Immunol. 2006, 146, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Demaria, S.; Santori, F.R.; Ng, B.; Liebes, L.; Formenti, S.C.; Vukmanovic, S. Select forms of tumor cell apoptosis induce dendritic cell maturation. J. Leukoc. Biol. 2005, 77, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Feng, F.; Wang, Y.-C.; Hu, X.-B.; Liu, X.-W.; Ji, G.; Chen, Y.-R.; Wang, L.; He, F.; Dou, G.-R.; Liang, L.; et al. The transcription factor RBP-J-mediated signaling is essential for dendritic cells to evoke efficient anti-tumor immune responses in mice. Mol. Cancer 2010, 9, 90–90. [Google Scholar] [CrossRef] [PubMed]
- Brackett, C.M.; Owczarczak, B.; Ramsey, K.; Maier, P.G.; Gollnick, S.O. IL-6 potentiates tumor resistance to photodynamic therapy (PDT). Lasers Surg. Med. 2011, 43, 676–685. [Google Scholar] [PubMed]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef] [PubMed]
- Hatzi, E.; Murphy, C.; Zoephel, A.; Rasmussen, H.; Morbidelli, L.; Ahorn, H.; Kunisada, K.; Tontsch, U.; Klenk, M.; Yamauchi-Takihara, K.; et al. N-myc oncogene overexpression down-regulates IL-6; evidence that IL-6 inhibits angiogenesis and suppresses neuroblastoma tumor growth. Oncogene 2002, 21, 3552–3561. [Google Scholar] [CrossRef] [PubMed]
- Usuda, J.; Okunaka, T.; Furukawa, K.; Tsuchida, T.; Kuroiwa, Y.; Ohe, Y.; Saijo, N.; Nishio, K.; Konaka, C.; Kato, H. Increased cytotoxic effects of photodynamic therapy in IL-6 gene transfected cells via enhanced apoptosis. Int. J. Cancer 2001, 93, 475–480. [Google Scholar] [CrossRef] [PubMed]
- Thurston, G.; McLean, J.W.; Rizen, M.; Baluk, P.; Haskell, A.; Murphy, T.J.; Hanahan, D.; McDonald, D.M. Cationic liposomes target angiogenic endothelial cells in tumors and chronic inflammation in mice. J. Clin. Investig. 1998, 101, 1401–1413. [Google Scholar] [CrossRef] [PubMed]
- Fingar, V.H. Vascular effects of photodynamic therapy. J. Clin. Laser Med. Surg. 1996, 14, 323–328. [Google Scholar] [PubMed]
- De Vries, E.M.; Kwakkel, J.; Eggels, L.; Kalsbeek, A.; Barrett, P.; Fliers, E.; Boelen, A. NFκB signaling is sssential for the lipopolysaccharide-induced increase of type 2 seiodinase in tanycytes. Endocrinology 2014, 155, 2000–2008. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Broekgaarden, M.; Kos, M.; Jurg, F.A.; Van Beek, A.A.; Van Gulik, T.M.; Heger, M. Inhibition of NF-κB in Tumor Cells Exacerbates Immune Cell Activation Following Photodynamic Therapy. Int. J. Mol. Sci. 2015, 16, 19960-19977. https://doi.org/10.3390/ijms160819960
Broekgaarden M, Kos M, Jurg FA, Van Beek AA, Van Gulik TM, Heger M. Inhibition of NF-κB in Tumor Cells Exacerbates Immune Cell Activation Following Photodynamic Therapy. International Journal of Molecular Sciences. 2015; 16(8):19960-19977. https://doi.org/10.3390/ijms160819960
Chicago/Turabian StyleBroekgaarden, Mans, Milan Kos, Freek A. Jurg, Adriaan A. Van Beek, Thomas M. Van Gulik, and Michal Heger. 2015. "Inhibition of NF-κB in Tumor Cells Exacerbates Immune Cell Activation Following Photodynamic Therapy" International Journal of Molecular Sciences 16, no. 8: 19960-19977. https://doi.org/10.3390/ijms160819960
APA StyleBroekgaarden, M., Kos, M., Jurg, F. A., Van Beek, A. A., Van Gulik, T. M., & Heger, M. (2015). Inhibition of NF-κB in Tumor Cells Exacerbates Immune Cell Activation Following Photodynamic Therapy. International Journal of Molecular Sciences, 16(8), 19960-19977. https://doi.org/10.3390/ijms160819960