
In Silico Analysis of Correlations between Protein Disorder and Post-Translational Modifications in Algae

Abstract
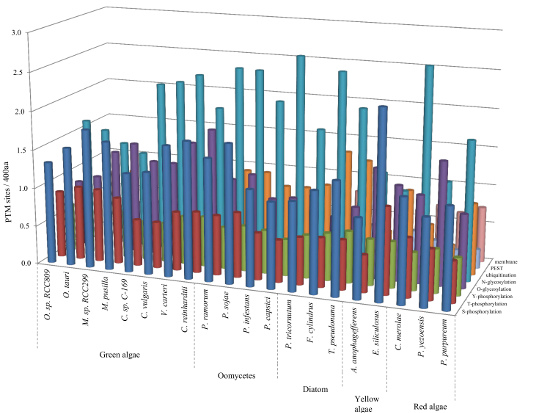
:
1. Introduction
2. Results
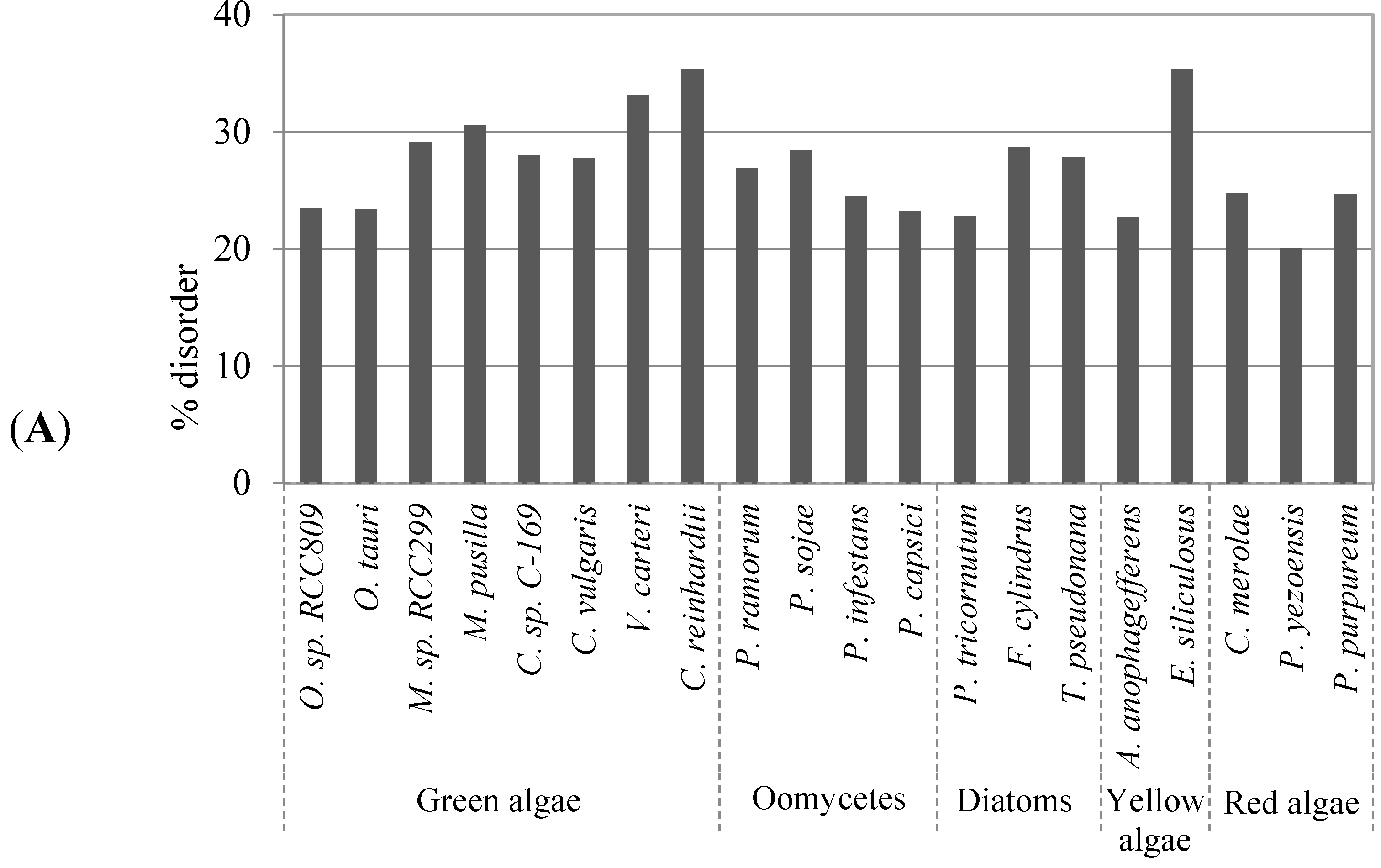
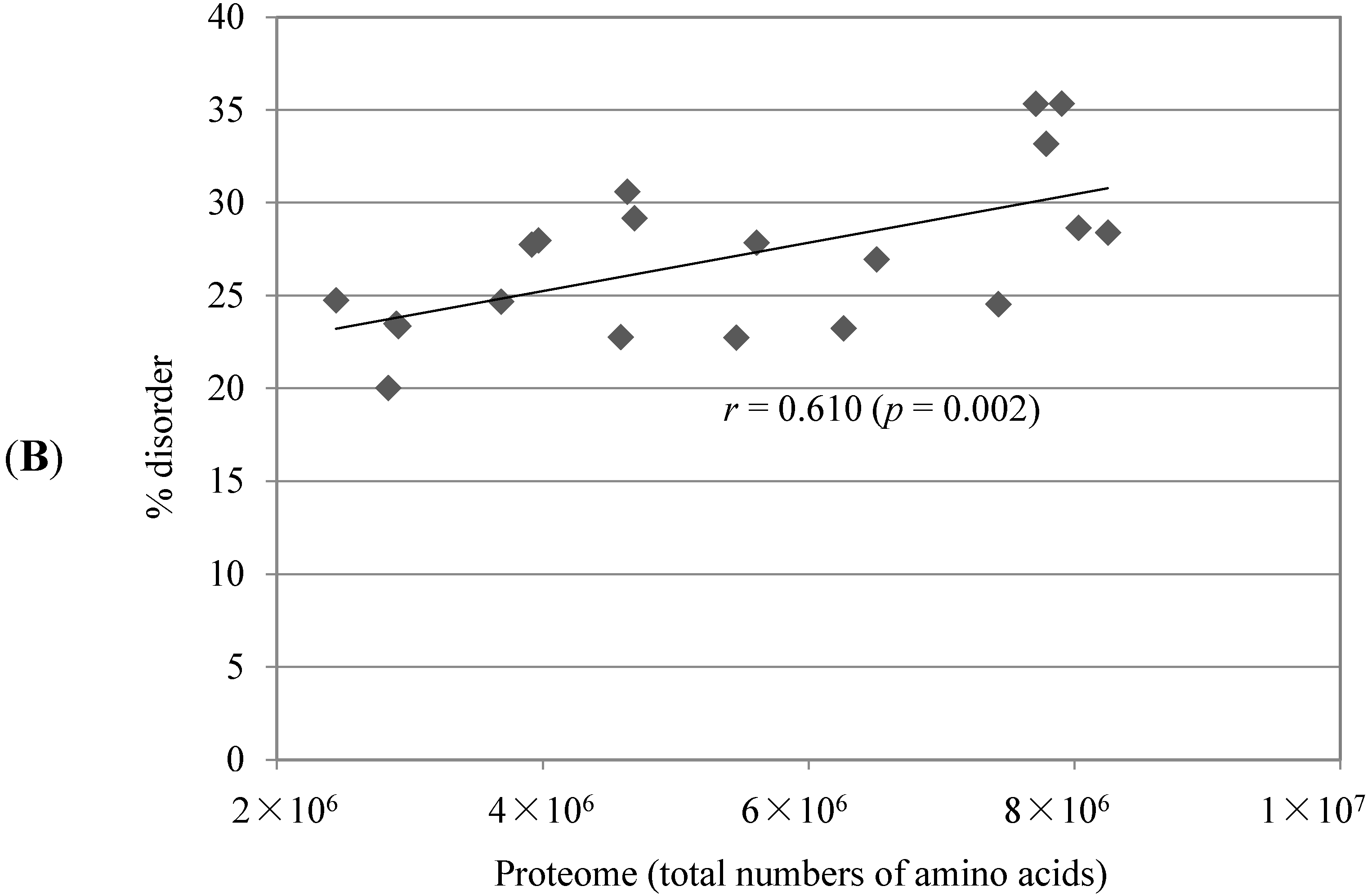
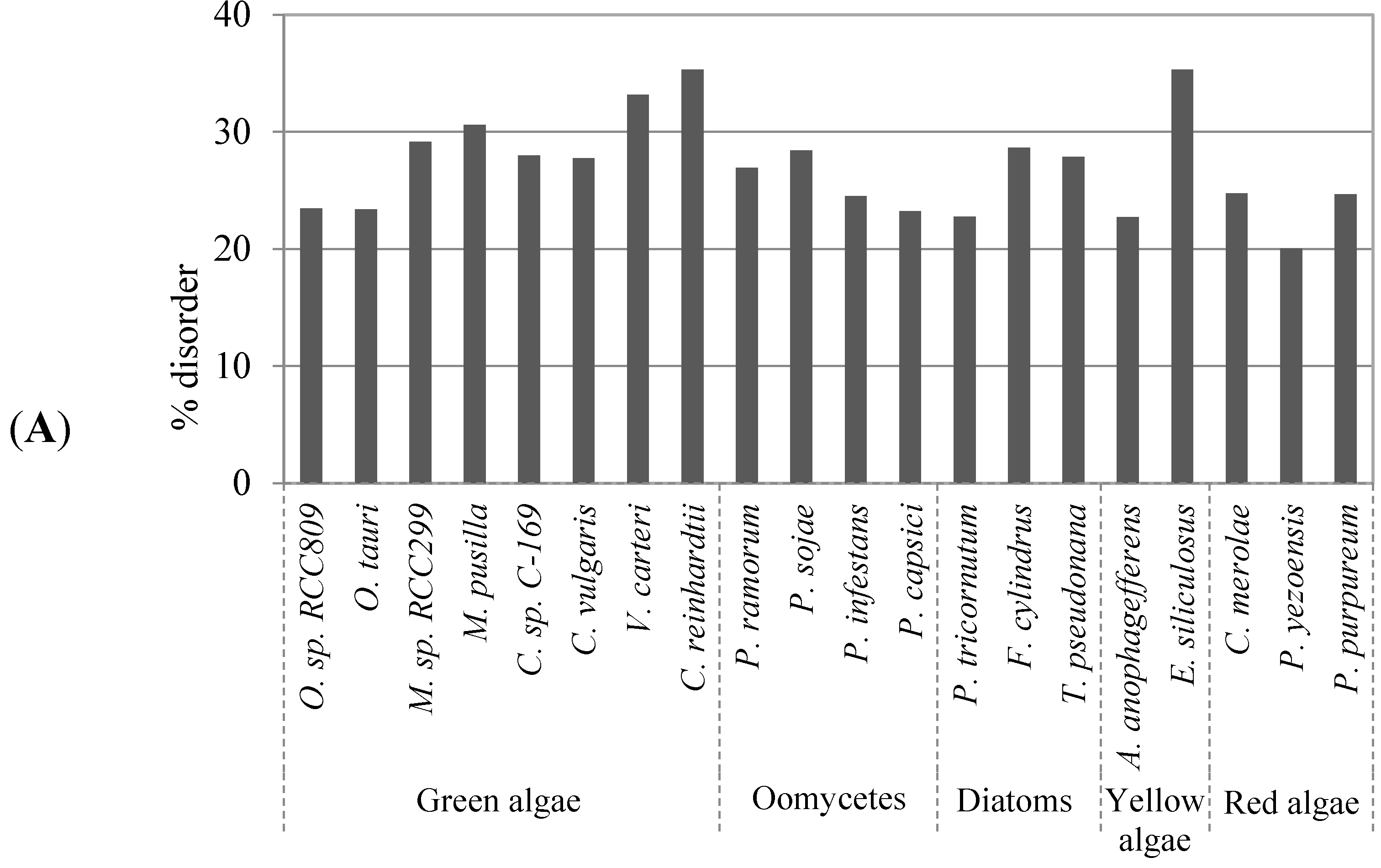
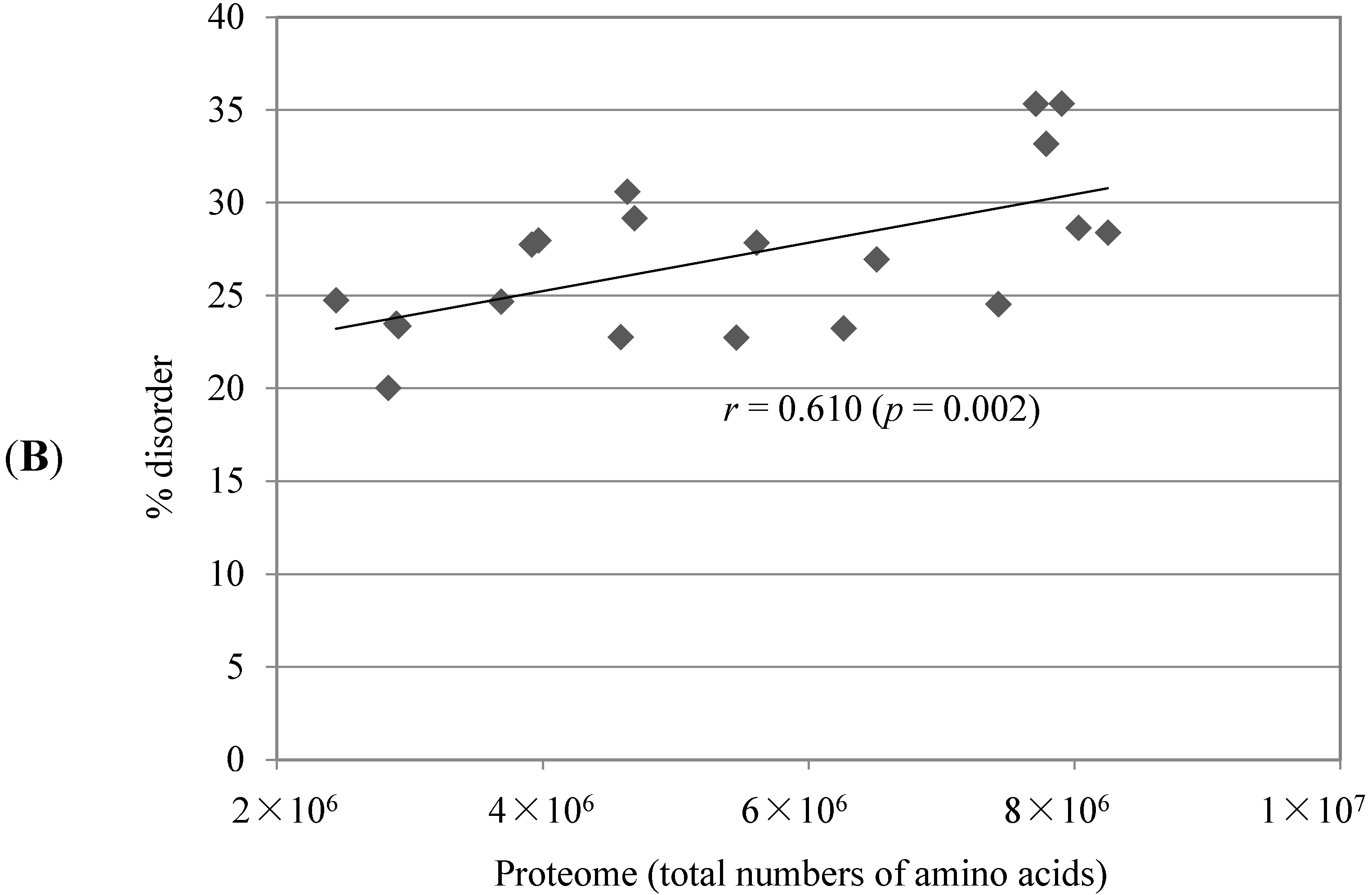
2.1. Comparison of Data Sets and Global Intrinsic Disorder in Algae Proteomes
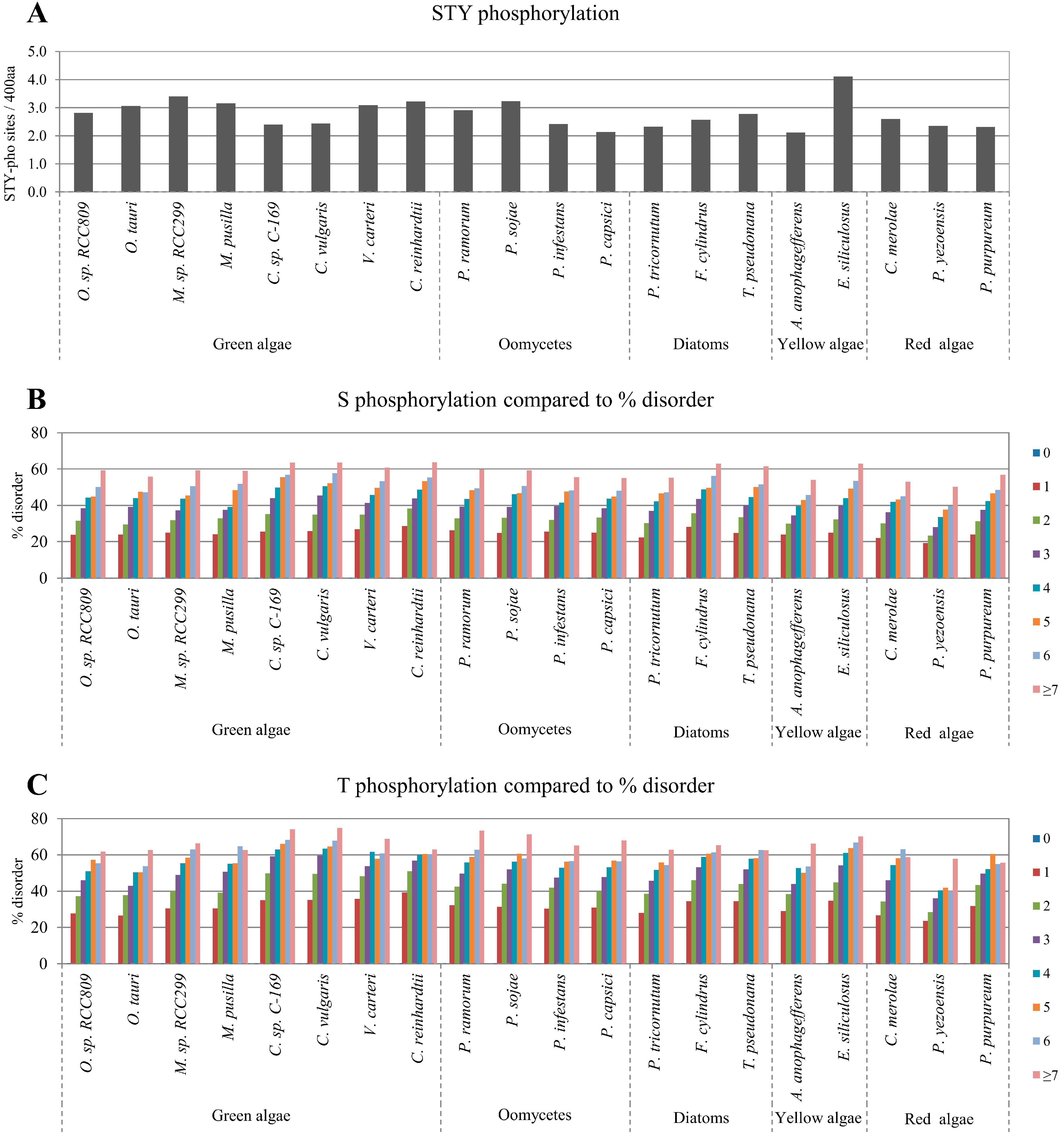
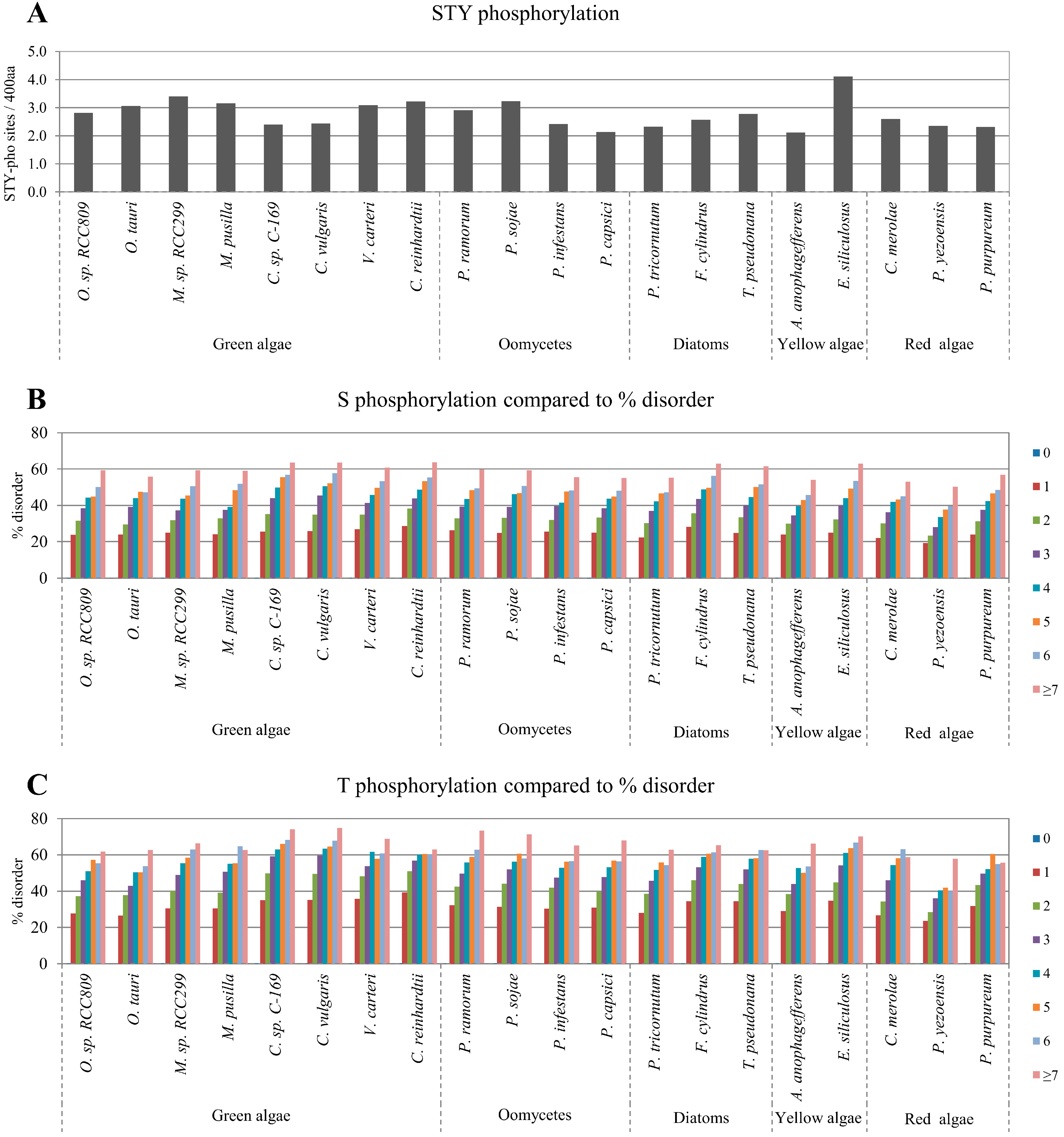
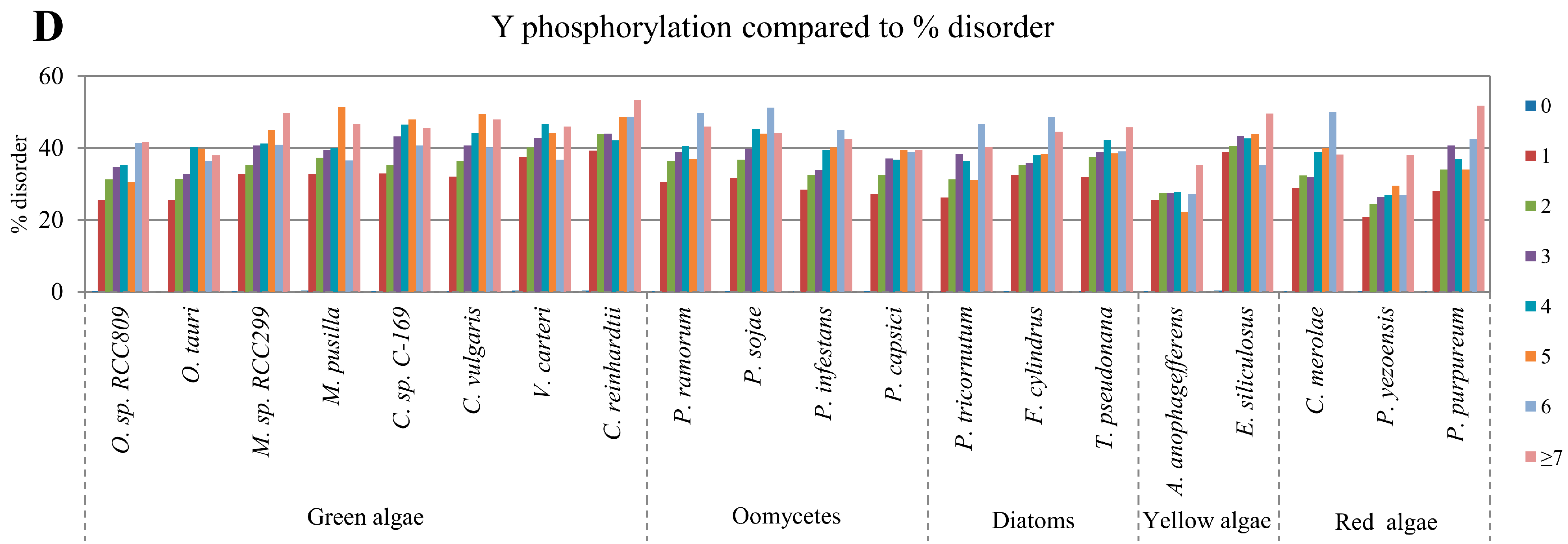
2.2. Correlation between Intrinsic Disorder and Phosphorylation
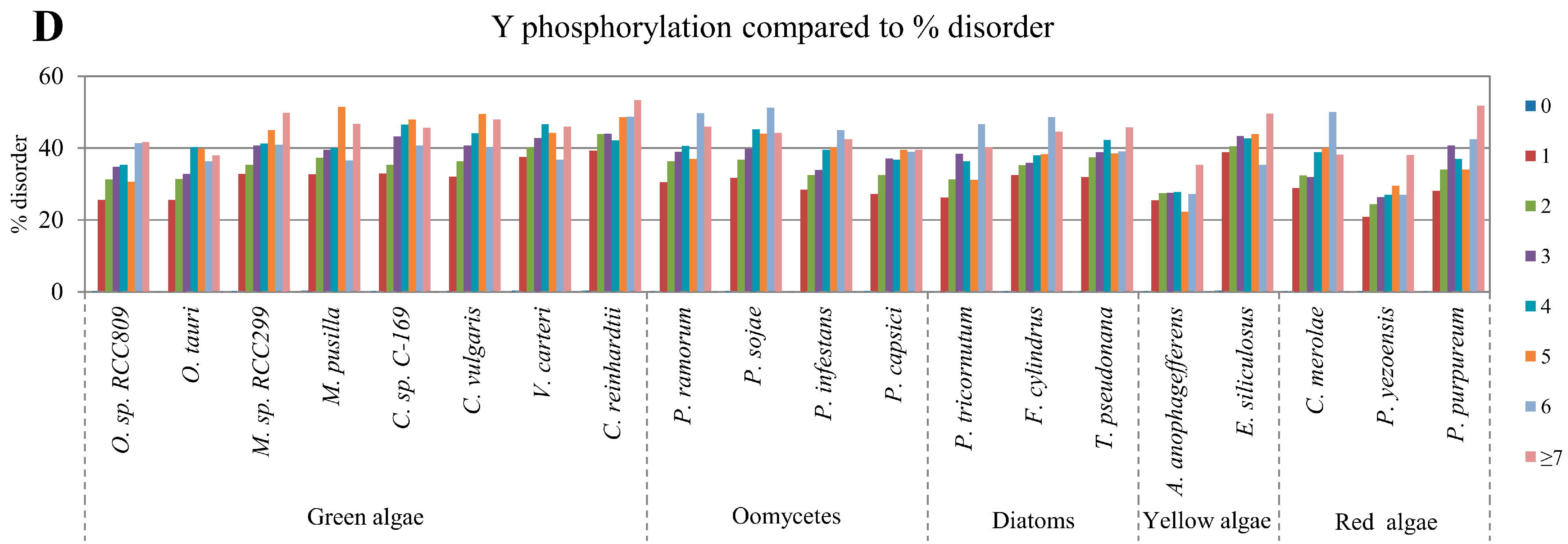

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
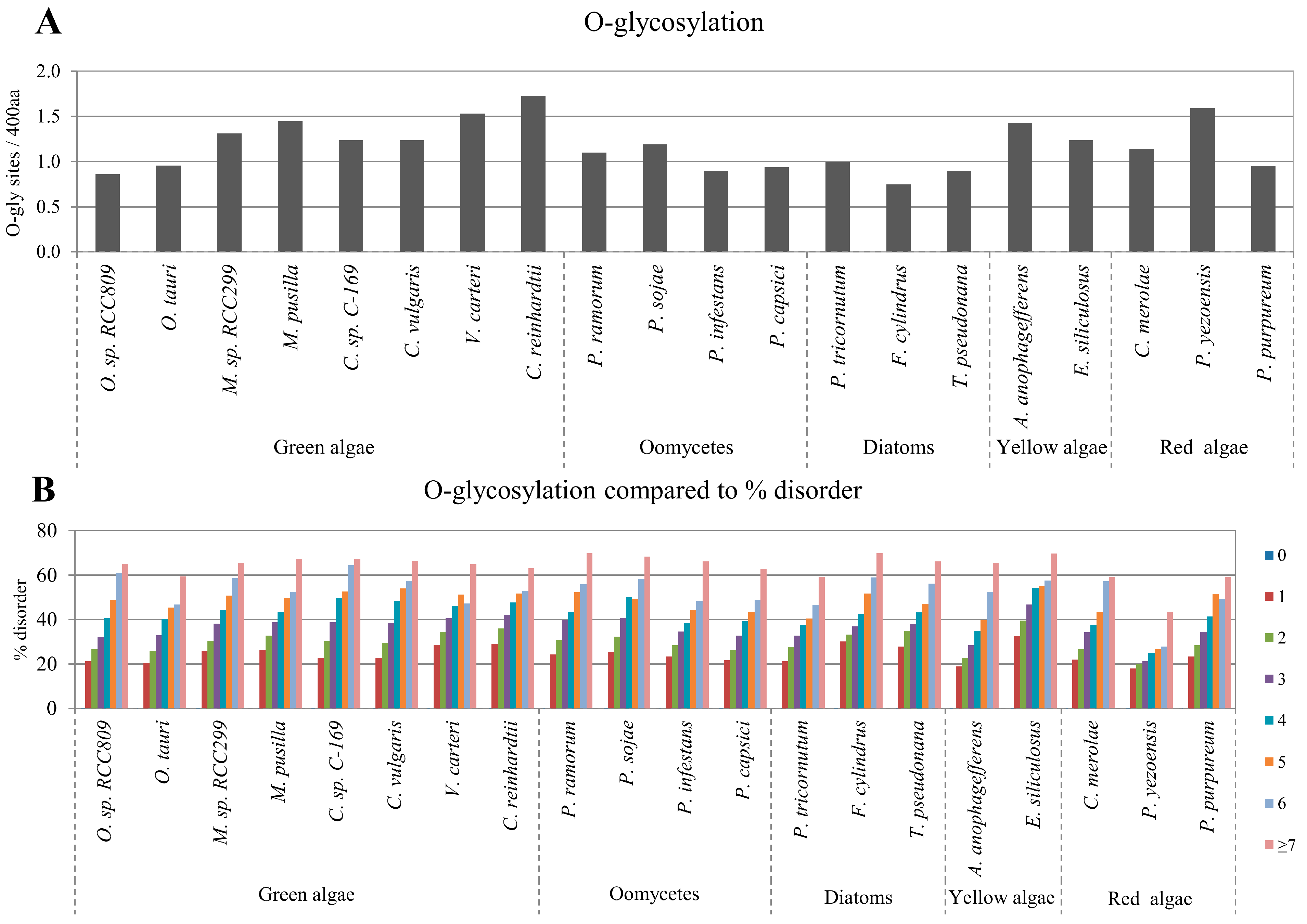
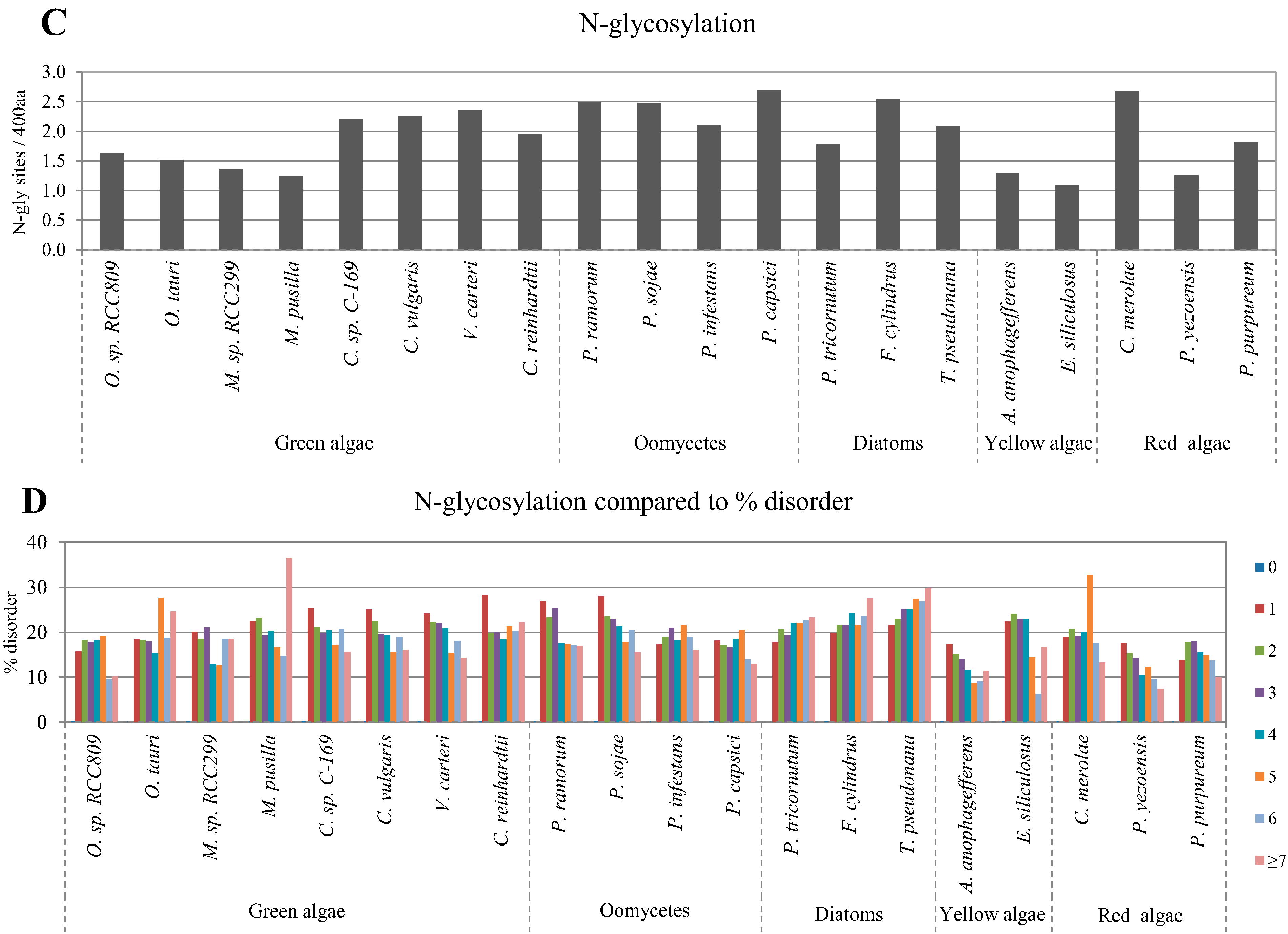
| Species | S-pho | T-pho | Y-pho | O-gly | N-gly | PEST | Ubi | TM | |
|---|---|---|---|---|---|---|---|---|---|
| Green algae | O. sp. RCC809 | 0.976 | 0.961 | 0.867 | 0.968 | −0.705 | 0.953 | 0.993 | −0.830 |
| 2 × 10−5 | 3 × 10−4 | 3 × 10−3 | 4 × 10−5 | 3 × 10−2 | 2 × 10−3 | 4 × 10−7 | 5 × 10−3 | ||
| 3 × 10−5 | 1 × 10−3 | 5 × 10−3 | 4 × 10−5 | 3 × 10−2 | 2 × 10−3 | 9 × 10−7 | 6 × 10−3 | ||
| O. tauri | 0.967 | 0.958 | 0.895 | 0.973 | −0.333 | 0.999 | 0.994 | −0.803 | |
| 4 × 10−5 | 3 × 10−4 | 1 × 10−3 | 3 × 10−5 | 2 × 10−1 | 6 × 10−7 | 3 × 10−7 | 8 × 10−3 | ||
| 4 × 10−5 | 1 × 10−3 | 3 × 10−3 | 3 × 10−5 | 2 × 10−1 | 1 × 10−5 | 7 × 10−7 | 9 × 10−3 | ||
| M. sp. RCC299 | 0.987 | 0.977 | 0.891 | 0.992 | −0.869 | 0.982 | 0.992 | −0.958 | |
| 3 × 10−6 | 8 × 10−5 | 1 × 10−3 | 5 × 10−7 | 3 × 10−3 | 2 × 10−4 | 8 × 10−7 | 9 × 10−5 | ||
| 2 × 10−5 | 5 × 10−4 | 3 × 10−3 | 3 × 10−6 | 4 × 10−3 | 4 × 10−4 | 2 × 10−6 | 2 × 10−4 | ||
| M. pusilla | 0.985 | 0.979 | 0.701 | 0.992 | −0.970 | 0.994 | 0.990 | −0.976 | |
| 4 × 10−6 | 6 × 10−5 | 3 × 10−2 | 7 × 10−7 | 3 × 10−5 | 3 × 10−5 | 1 × 10−6 | 2 × 10−5 | ||
| 2 × 10−5 | 6 × 10−4 | 3 × 10−2 | 3 × 10−6 | 2 × 10−4 | 1 × 10−4 | 2 × 10−6 | 5 × 10−5 | ||
| C. sp. C-169 | 0.980 | 0.935 | 0.831 | 0.988 | −0.980 | 0.970 | 0.968 | −0.950 | |
| 1 × 10−5 | 1 × 10−3 | 5 × 10−3 | 2 × 10−6 | 1 × 10−5 | 7 × 10−4 | 4 × 10−5 | 2 × 10−4 | ||
| 3 × 10−5 | 1 × 10−3 | 8 × 10−3 | 5 × 10−6 | 2 × 10−4 | 9 × 10−4 | 5 × 10−5 | 3 × 10−4 | ||
| C. vulgaris | 0.974 | 0.929 | 0.846 | 0.986 | −0.967 | 0.971 | 0.976 | −0.966 | |
| 2 × 10−5 | 1 × 10−3 | 4 × 10−3 | 3 × 10−6 | 4 × 10−5 | 6 × 10−4 | 2 × 10−5 | 5 × 10−5 | ||
| 3 × 10−5 | 1 × 10−3 | 6 × 10−3 | 7 × 10−6 | 2 × 10−4 | 9 × 10−4 | 2 × 10−5 | 1 × 10−4 | ||
| V. carteri | 0.982 | 0.908 | 0.578 | 0.958 | −0.883 | 0.965 | 0.910 | −0.985 | |
| 7 × 10−6 | 2 × 10−3 | 7 × 10−2 | 9 × 10−5 | 2 × 10−3 | 9 × 10−4 | 8 × 10−4 | 4 × 10−6 | ||
| 2 × 10−5 | 2 × 10−3 | 7 × 10−2 | 9 × 10−5 | 4 × 10−3 | 1 × 10−3 | 8 × 10−4 | 3 × 10−5 | ||
| C. reinhardtii | 0.976 | 0.886 | 0.879 | 0.987 | −0.805 | 0.962 | 0.967 | −0.987 | |
| 2 × 10−5 | 4 × 10−3 | 2 × 10−3 | 2 × 10−6 | 8 × 10−3 | 1 × 10−3 | 4 × 10−5 | 3 × 10−6 | ||
| 3 × 10−5 | 4 × 10−3 | 4 × 10−3 | 5 × 10−6 | 1 × 10−2 | 1 × 10−3 | 5 × 10−5 | 3 × 10−5 | ||
| Oomycetes | P. ramorum | 0.974 | 0.967 | 0.903 | 0.991 | −0.971 | 0.995 | 0.995 | −0.941 |
| 2 × 10−5 | 2 × 10−4 | 1 × 10−3 | 1 × 10−6 | 3 × 10−5 | 2 × 10−5 | 1 × 10−7 | 2 × 10−4 | ||
| 3 × 10−5 | 9 × 10−4 | 3 × 10−3 | 3 × 10−6 | 3 × 10−4 | 9 × 10−5 | 6 × 10−7 | 3 × 10−4 | ||
| P. sojae | 0.975 | 0.932 | 0.976 | 0.985 | −0.890 | 0.996 | 0.994 | −0.948 | |
| 2 × 10−5 | 1 × 10−3 | 2 × 10−5 | 5 × 10−6 | 2 × 10−3 | 1 × 10−5 | 2 × 10−7 | 2 × 10−4 | ||
| 3 × 10−5 | 1 × 10−3 | 2 × 10−4 | 8 × 10−6 | 3 × 10−3 | 6 × 10−5 | 7 × 10−7 | 3 × 10−4 | ||
| P. infestans | 0.972 | 0.947 | 0.982 | 0.995 | −0.533 | 0.984 | 0.996 | −0.960 | |
| 3 × 10−5 | 6 × 10−4 | 8 × 10−6 | 1 × 10−7 | 9 × 10−2 | 2 × 10−4 | 7 × 10−8 | 8 × 10−5 | ||
| 3 × 10−5 | 1 × 10−3 | 2 × 10−4 | 1 × 10−6 | 1 × 10−1 | 4 × 10−4 | 5 × 10−7 | 2 × 10−4 | ||
| P. capsici | 0.971 | 0.953 | 0.923 | 0.990 | −0.462 | 0.966 | 0.995 | −0.943 | |
| 3 × 10−5 | 4 × 10−4 | 5 × 10−4 | 1 × 10−6 | 1 × 10−1 | 9 × 10−4 | 2 × 10−7 | 2 × 10−4 | ||
| 3 × 10−5 | 1 × 10−3 | 2 × 10−3 | 3 × 10−6 | 1 × 10−1 | 1 × 10−3 | 7 × 10−7 | 3 × 10−4 | ||
| Diatom | P. tricornutum | 0.977 | 0.950 | 0.849 | 0.996 | 0.900 | 0.998 | 0.996 | −0.935 |
| 1 × 10−5 | 5 × 10−4 | 4 × 10−3 | 6 × 10−8 | 1 × 10−3 | 2 × 10−6 | 9 × 10−8 | 3 × 10−4 | ||
| 3 × 10−5 | 1 × 10−3 | 6 × 10−3 | 1 × 10−6 | 3 × 10−3 | 2 × 10−5 | 4 × 10−7 | 4 × 10−4 | ||
| F. cylindrus | 0.977 | 0.940 | 0.927 | 0.982 | 0.919 | 0.972 | 0.998 | −0.977 | |
| 1 × 10−5 | 8 × 10−4 | 5 × 10−4 | 7 × 10−6 | 6 × 10−4 | 6 × 10−4 | 2 × 10−8 | 2 × 10−5 | ||
| 3 × 10−5 | 1 × 10−3 | 2 × 10−3 | 1 × 10−5 | 2 × 10−3 | 1 × 10−3 | 4 × 10−7 | 6 × 10−5 | ||
| T. pseudonana | 0.979 | 0.947 | 0.789 | 0.993 | 0.765 | 0.997 | 0.991 | −0.981 | |
| 1 × 10−5 | 6 × 10−4 | 1 × 10−2 | 5 × 10−7 | 1 × 10−2 | 7 × 10−6 | 9 × 10−7 | 9 × 10−6 | ||
| 3 × 10−5 | 1 × 10−3 | 1 × 10−2 | 3 × 10−6 | 2 × 10−2 | 5 × 10−5 | 2 × 10−6 | 4 × 10−5 | ||
| Yellow algae | A. anophagefferens | 0.975 | 0.937 | 0.386 | 0.979 | −0.951 | 0.989 | 0.948 | −0.991 |
| 2 × 10−5 | 9 × 10−4 | 2 × 10−1 | 1 × 10−5 | 1 × 10−4 | 9 × 10−5 | 2 × 10−4 | 9 × 10−7 | ||
| 3 × 10−5 | 1 × 10−3 | 2 × 10−1 | 2 × 10−5 | 6 × 10−4 | 2 × 10−4 | 2 × 10−4 | 2 × 10−5 | ||
| E. siliculosus | 0.993 | 0.965 | 0.381 | 0.978 | −0.875 | 0.991 | 0.982 | −0.931 | |
| 5 × 10−7 | 2 × 10−4 | 2 × 10−1 | 1 × 10−5 | 2 × 10−3 | 6 × 10−5 | 7 × 10−6 | 4 × 10−4 | ||
| 5 × 10−6 | 8 × 10−4 | 2 × 10−1 | 2 × 10−5 | 4 × 10−3 | 2 × 10−4 | 1 × 10−5 | 5 × 10−4 | ||
| Red algae | C. merolae | 0.971 | 0.989 | 0.965 | 0.968 | −0.086 | 0.970 | 0.967 | −0.936 |
| 3 × 10−5 | 1 × 10−5 | 5 × 10−5 | 4 × 10−5 | 4 × 10−1 | 7 × 10−4 | 4 × 10−5 | 3 × 10−4 | ||
| 3 × 10−5 | 3 × 10−4 | 3 × 10−4 | 4 × 10−5 | 4 × 10−1 | 9 × 10−4 | 5 × 10−5 | 4 × 10−4 | ||
| P. yezoensis | 0.995 | 0.937 | 0.907 | 0.980 | −0.942 | 0.948 | 0.947 | −0.731 | |
| 1 × 10−7 | 9 × 10−4 | 9 × 10−4 | 1 × 10−5 | 2 × 10−4 | 2 × 10−3 | 2 × 10−4 | 2 × 10−2 | ||
| 2 × 10−6 | 1 × 10−3 | 3 × 10−3 | 2 × 10−5 | 8 × 10−4 | 2 × 10−3 | 2 × 10−4 | 2 × 10−2 | ||
| P. purpureum | 0.983 | 0.923 | 0.827 | 0.973 | −0.378 | 0.984 | 0.997 | −0.951 | |
| 6 × 10−6 | 1 × 10−3 | 6 × 10−3 | 2 × 10−5 | 2 × 10−1 | 2 × 10−4 | 2 × 10−8 | 1 × 10−4 | ||
| 2 × 10−5 | 2 × 10−3 | 8 × 10−3 | 3 × 10−5 | 2 × 10−1 | 4 × 10−4 | 2 × 10−7 | 3 × 10−4 | ||
2.3. Correlation between Intrinsic Disorder and Glycosylation
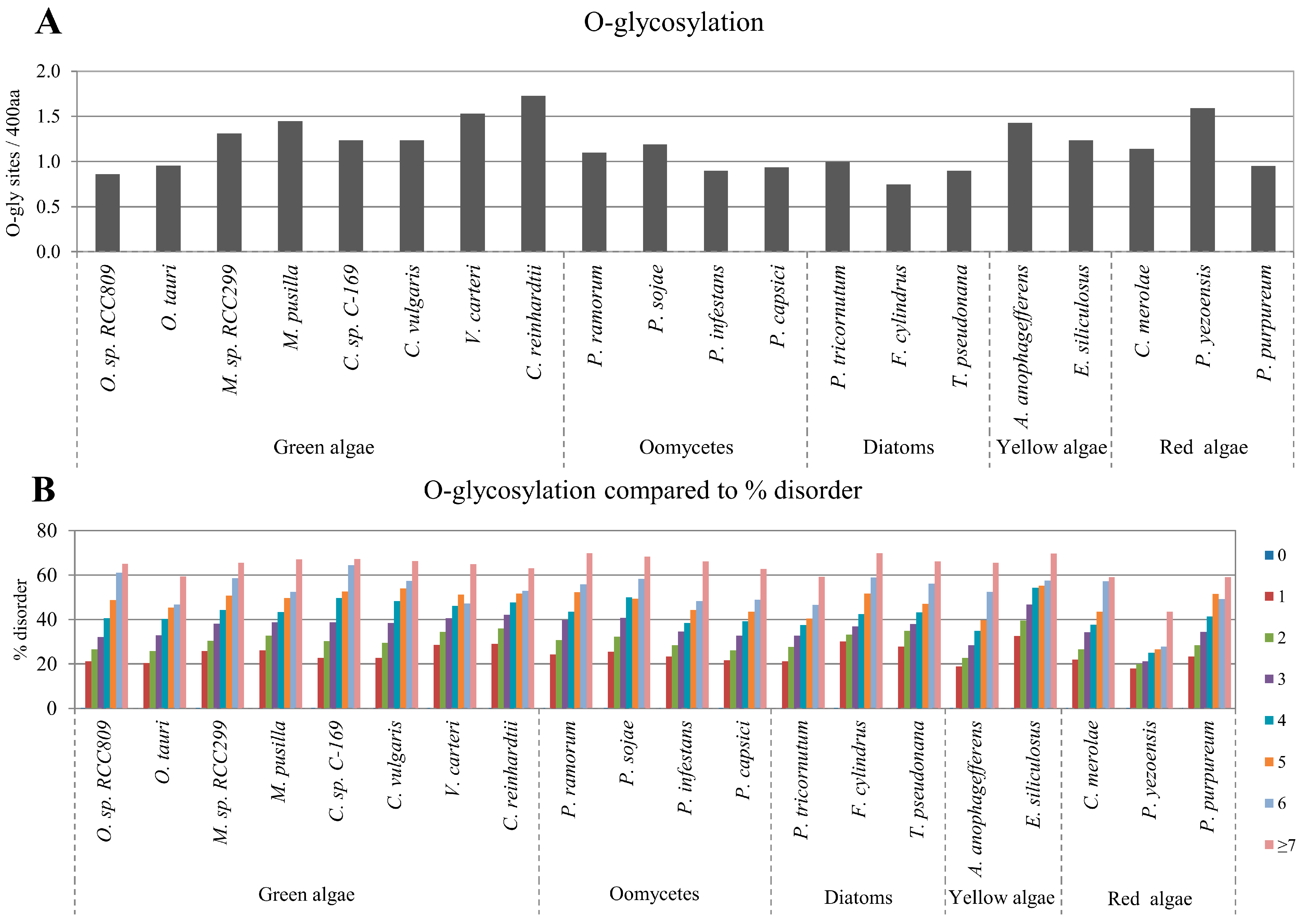
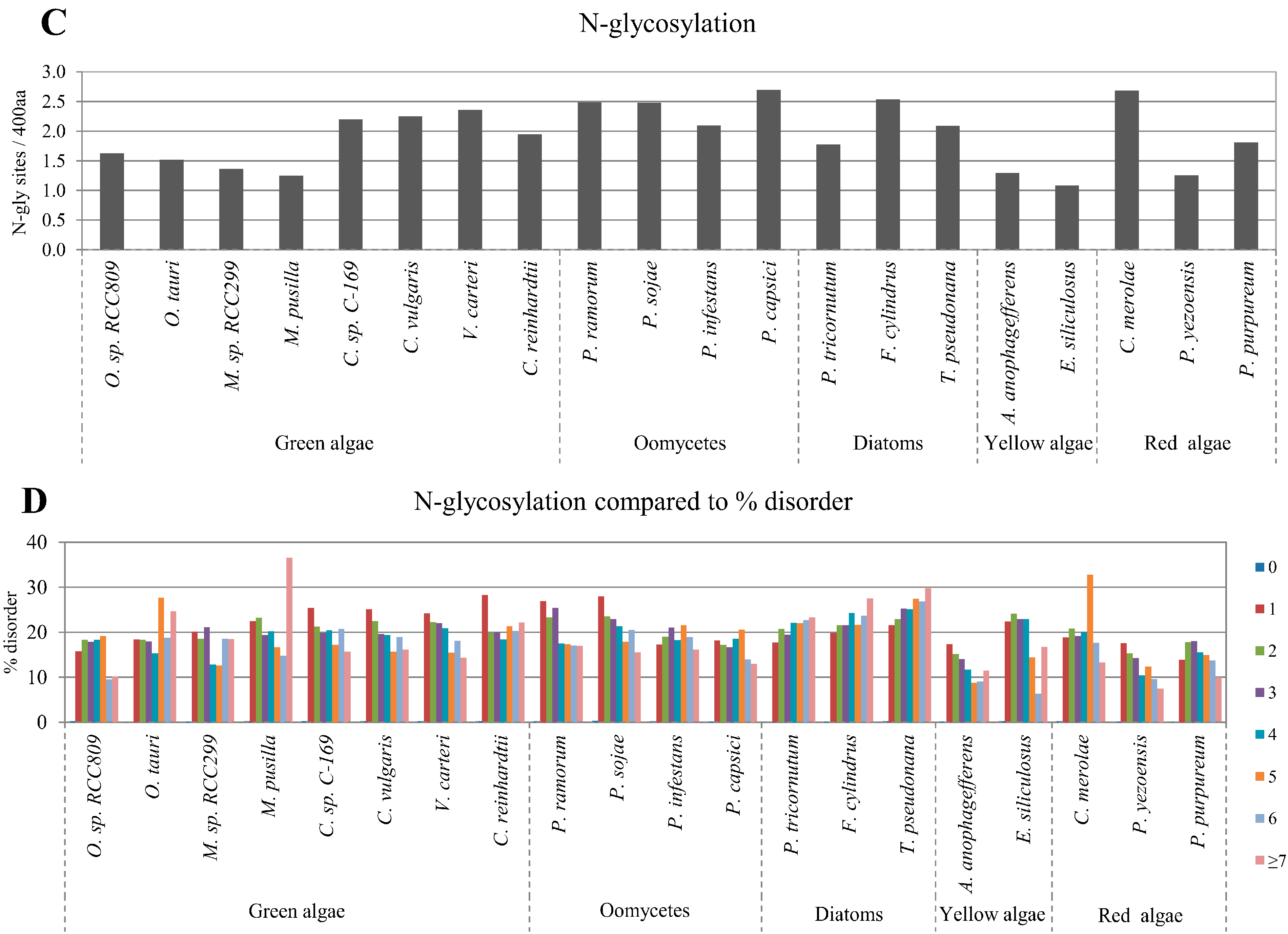
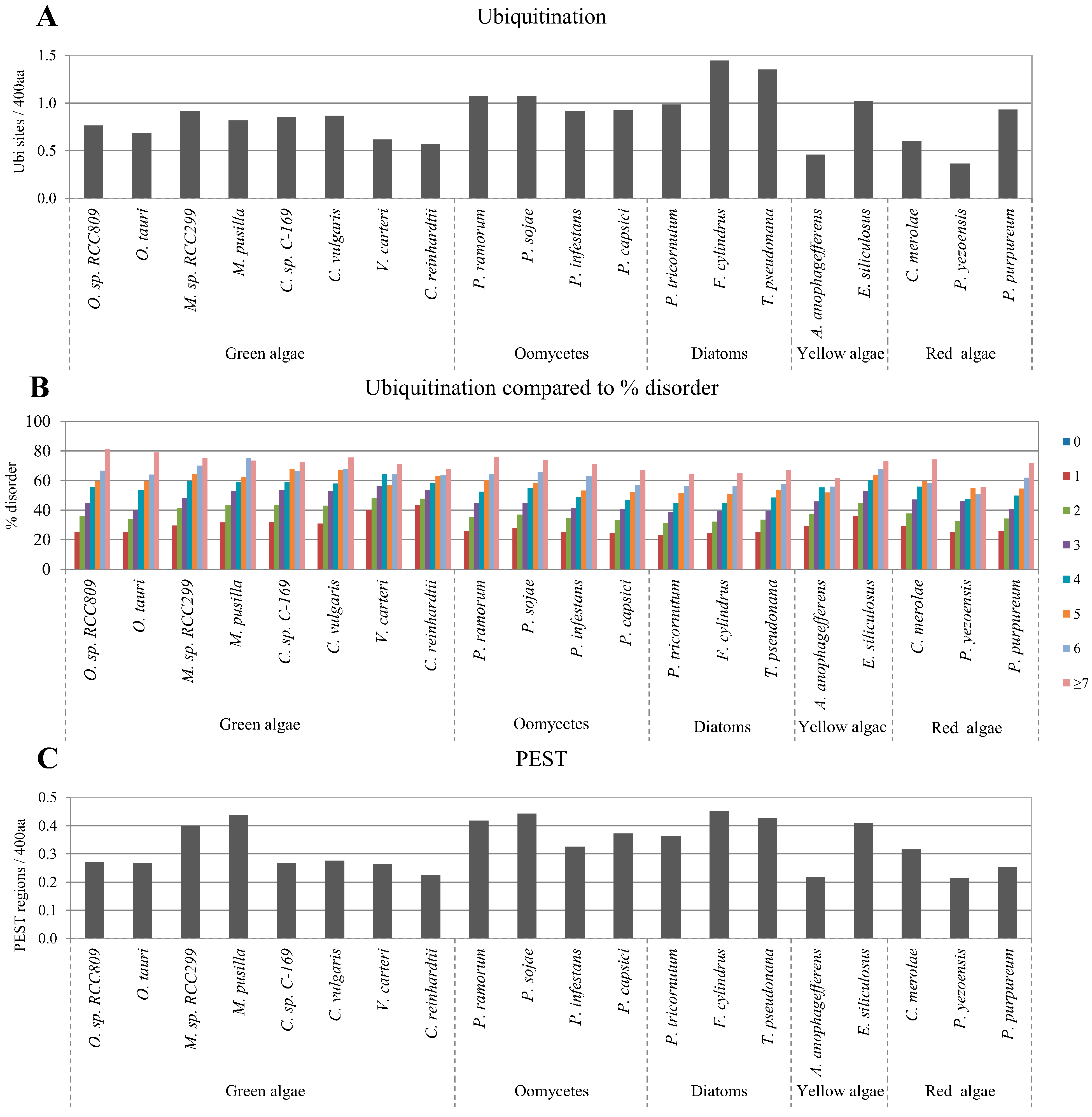
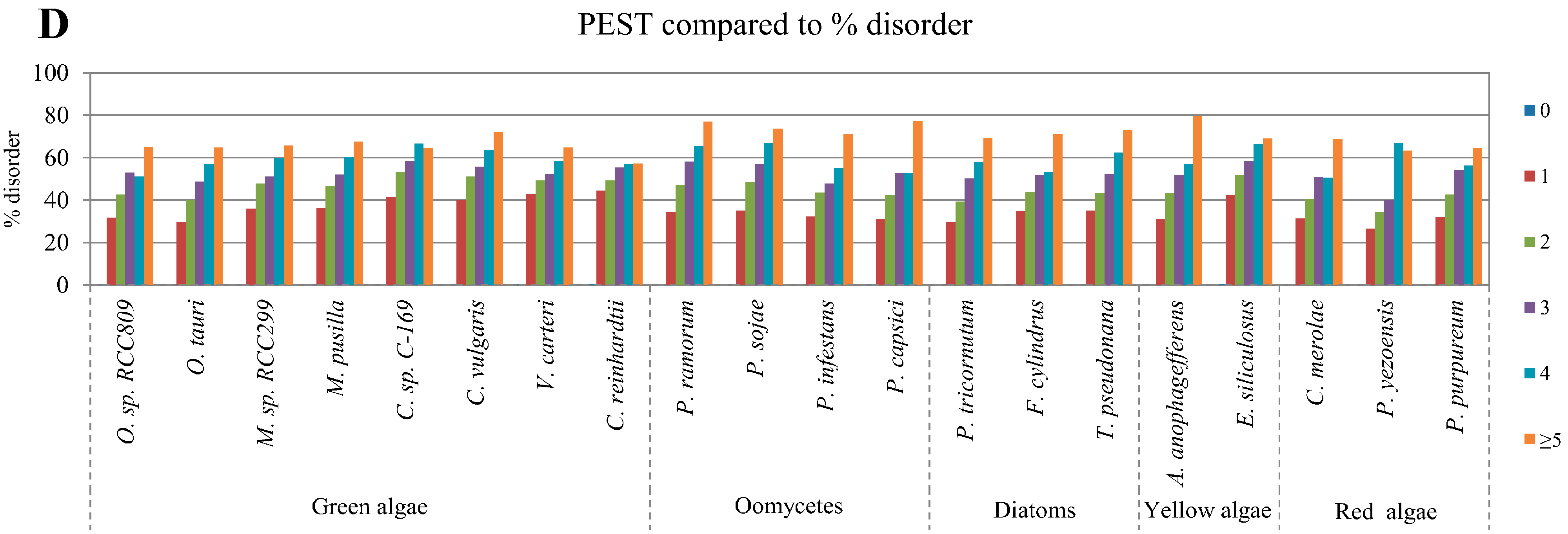
2.4. Correlation between Intrinsic Disorder and Ubiquitination and PEST (Region Enriched in Proline (P), Glutamic Acid (E), Serine (S), and Threonine (T) Residues)
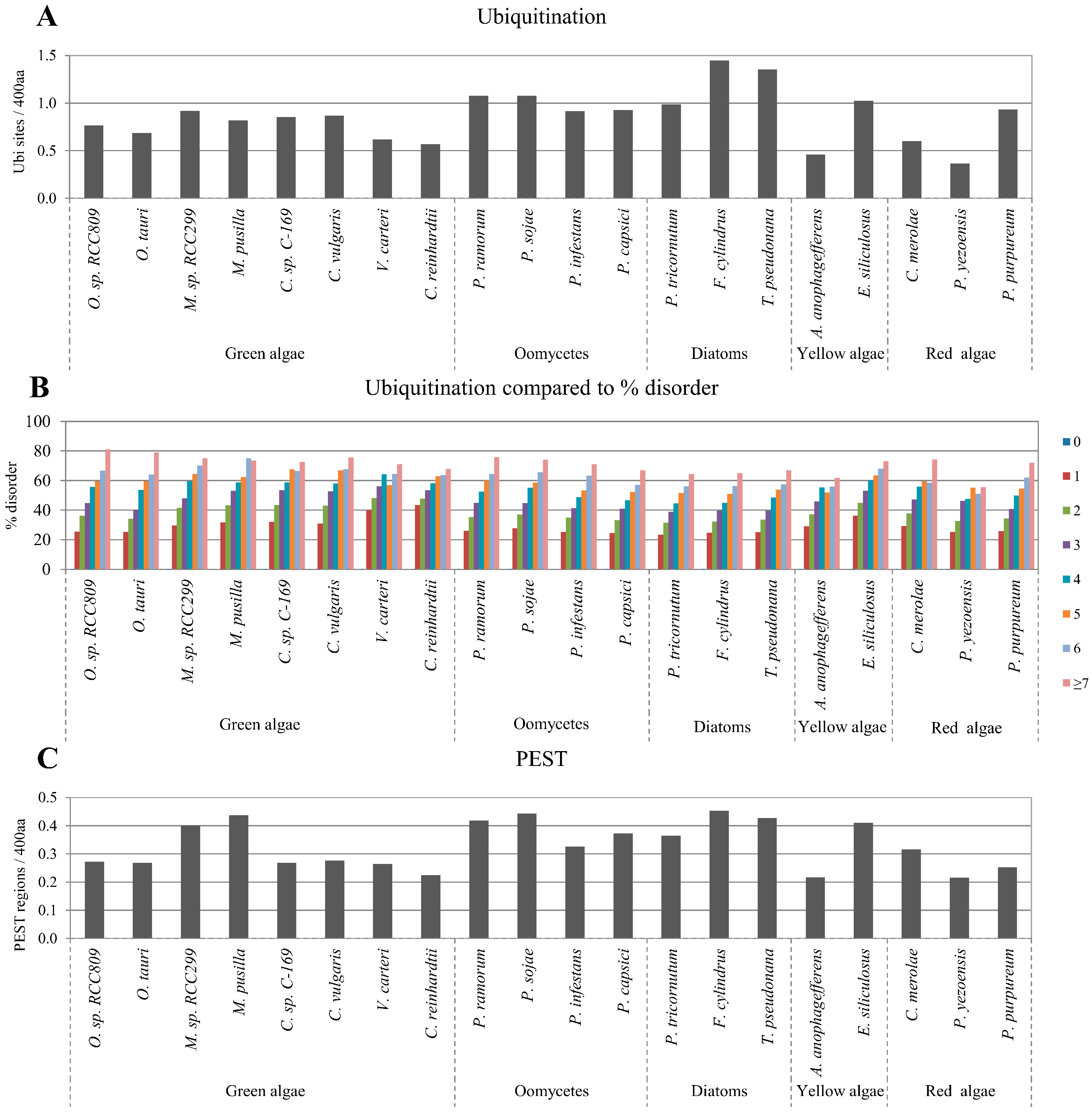
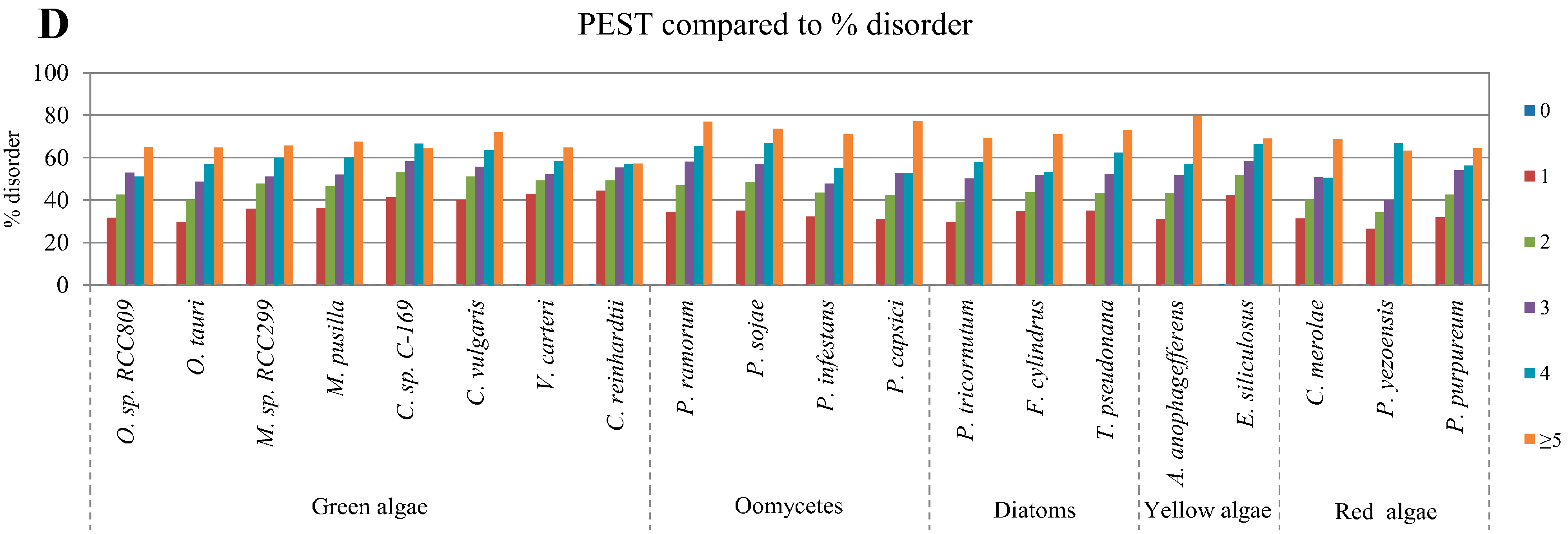
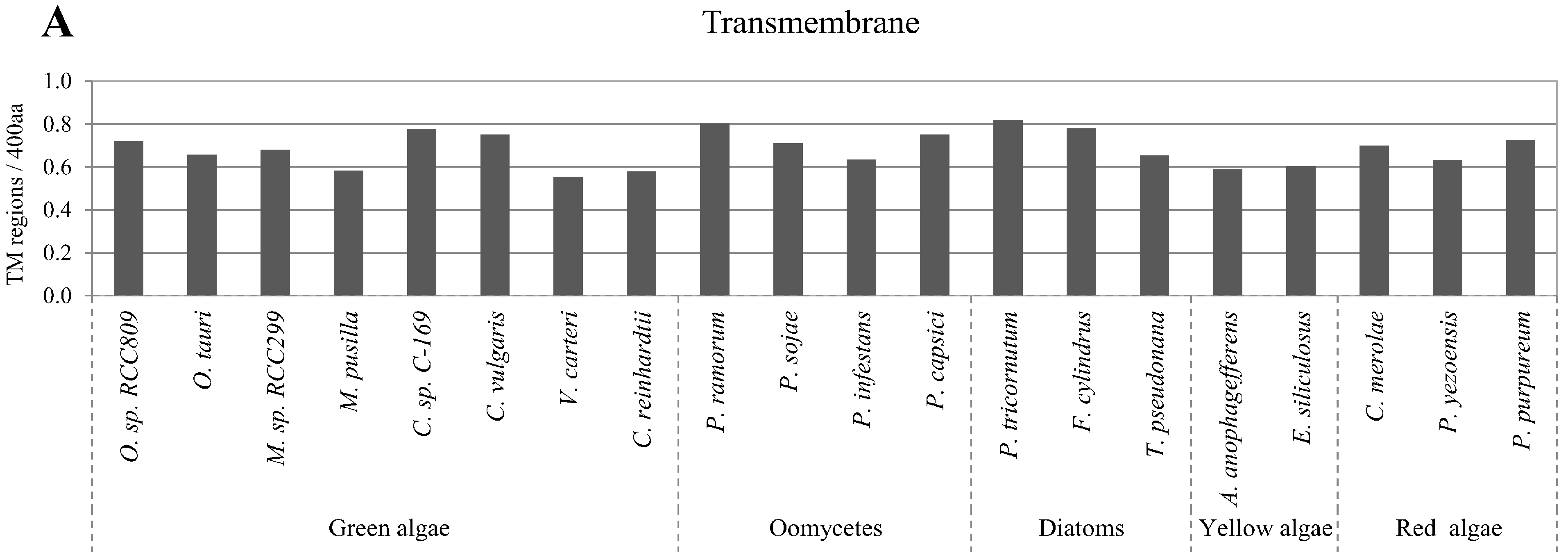
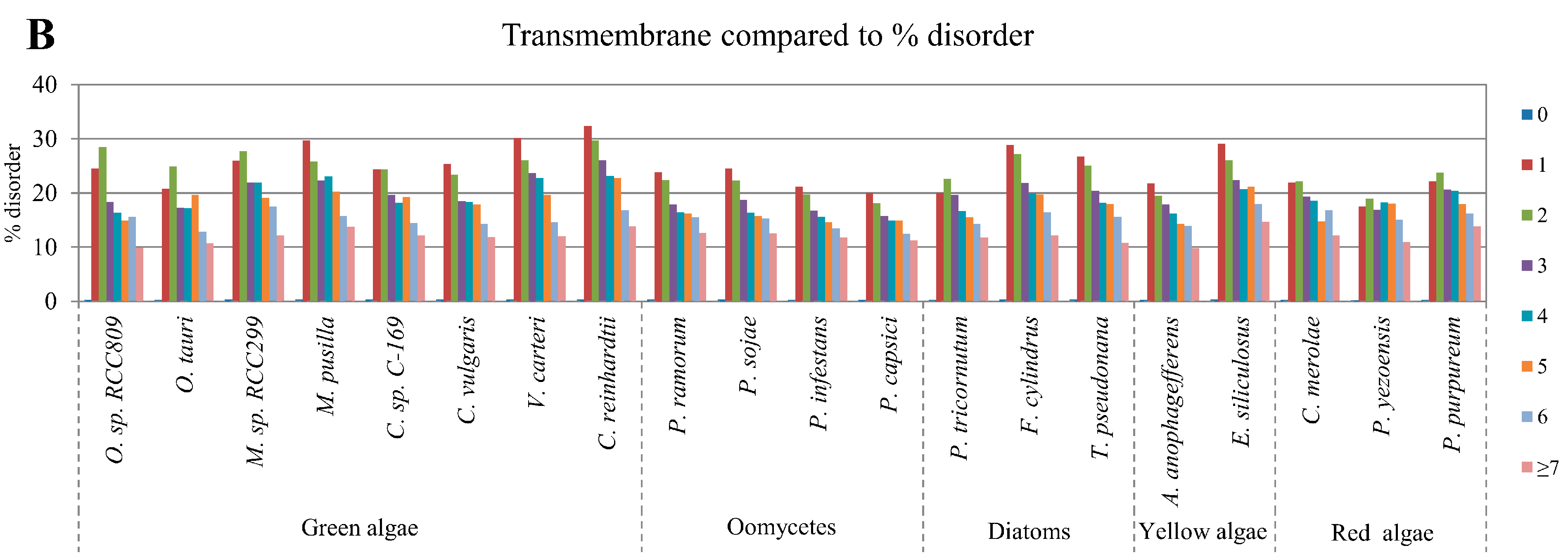
2.5. Correlation of Intrinsic Disorder with Transmembrane Helices
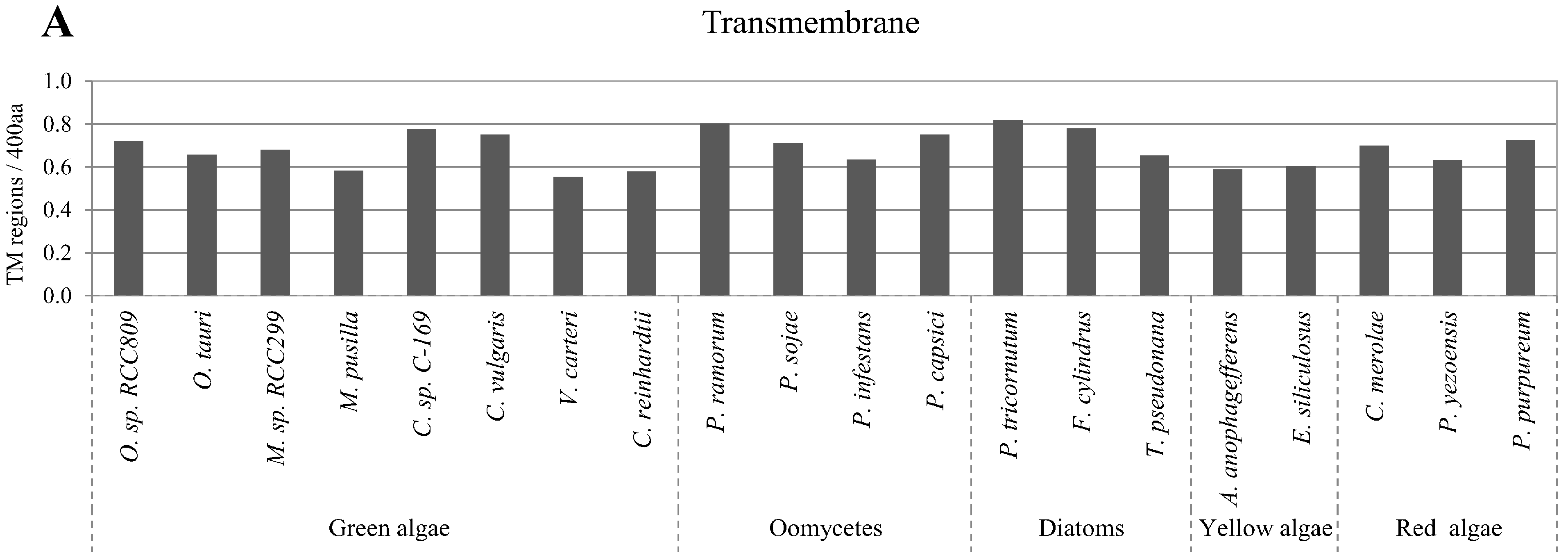
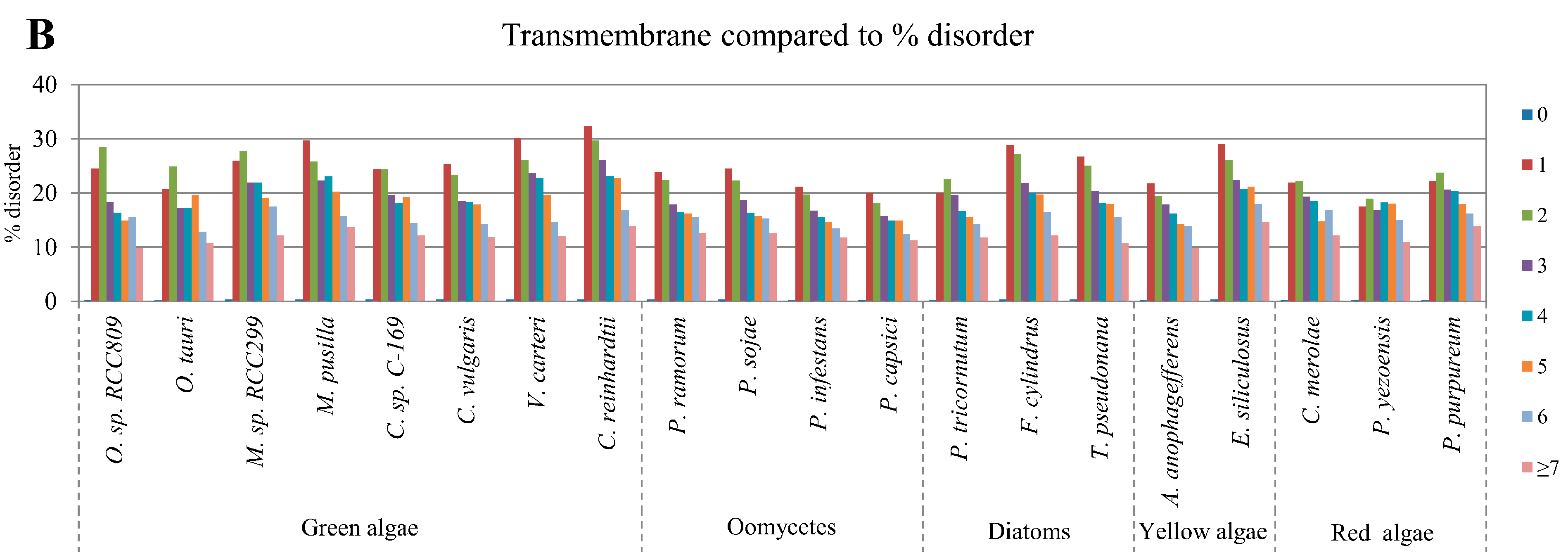
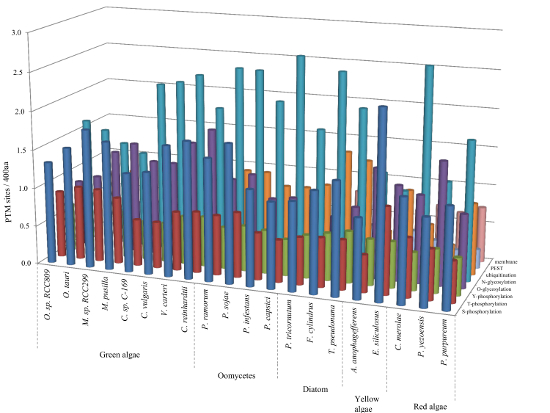
2.6. Relative Content of PTMs (Post-Translational Modifications) in Ordered and Disordered Regions of Algae Proteins
| Category | S-pho | T-pho | Y-pho | O-gly | N-gly | Ubi |
|---|---|---|---|---|---|---|
| Green algae | 15.1 | 8.4 | 1.1 | 3.2 | 0.9 | 11.0 |
| Oomycetes | 25.9 | 14.5 | 1.7 | 3.6 | 1.2 | 16.0 |
| Diatom | 19.8 | 10.3 | 1.3 | 2.9 | 1.9 | 11.9 |
| Yellow algae | 17.4 | 9.9 | 1.0 | 3.4 | 1.2 | 10.5 |
| Red algae | 19.2 | 11.3 | 1.6 | 2.6 | 1.2 | 16.6 |
| Total | 18.8 | 10.5 | 1.3 | 3.3 | 1.2 | 12.5 |
2.7. Relative Disordered Protein Content and the Number of PTMs (Post-Translational Modifications) in Specific and Common Protein Clusters of Algae Proteomes
| Category | Disorder | S-pho | T-pho | Y-pho | O-gly | N-gly | Ubi |
|---|---|---|---|---|---|---|---|
| Specific | 34% | 2.1 | 1.0 | 0.7 | 1.4 | 2.1 | 1.0 |
| Common | 20% | 0.6 | 0.3 | 0.5 | 0.8 | 1.3 | 0.6 |
| Ratio of s/c | 1.7 | 3.4 | 3.2 | 1.4 | 1.8 | 1.6 | 1.8 |
3. Discussion
4. Experimental Section
4.1. Data Sets
4.2. Prediction of Multiple Properties of Protein Sequences
4.3. Relative Content of PTMs in Ordered and Disordered Segments
4.4. Classification of Species-Specific Proteins and Common Proteins of Algae Proteomes
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Dunker, A.K.; Lawson, J.D.; Brown, C.J.; Williams, R.M.; Romero, P.; Oh, J.S.; Oldfield, C.J.; Campen, A.M.; Ratliff, C.R.; Hipps, K.W.; et al. Intrinsically disordered protein. J. Mol. Graph. Model. 2001, 19, 26–59. [Google Scholar] [CrossRef]
- Pancsa, R.; Tompa, P. Structural disorder in eukaryotes. PLoS ONE 2012, 7, e34687. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.J.; Sodhi, J.S.; McGuffin, L.J.; Buxton, B.F.; Jones, D.T. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J. Mol. Biol. 2004, 337, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.C.; Lu, X.; Ross, E.D.; Woody, R.W. Intrinsic protein disorder, amino acid composition, and histone terminal domains. J. Biol. Chem. 2006, 281, 1853–1856. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, I.; Nakajima, Y.; Ito, M.; Fukuchi, S.; Homma, K.; Nishikawa, K. Computational prediction of O-linked glycosylation sites that preferentially map on intrinsically disordered regions of extracellular proteins. Int. J. Mol. Sci. 2010, 11, 4992–5009. [Google Scholar] [CrossRef] [PubMed]
- Romero, P.; Obradovic, Z.; Li, X.H.; Garner, E.C.; Brown, C.J.; Dunker, A.K. Sequence complexity of disordered protein. Proteins 2001, 42, 38–48. [Google Scholar] [CrossRef]
- Shimizu, K.; Hirose, S.; Noguchi, T. POODLE-S: Web application for predicting protein disorder by using physicochemical features and reduced amino acid set of a position-specific scoring matrix. Bioinformatics 2007, 23, 2337–2338. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Gillespie, J.R.; Fink, A.L. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins 2000, 41, 415–427. [Google Scholar] [CrossRef]
- Uversky, V.N.; Oldfield, C.J.; Midic, U.; Xie, H.B.; Xue, B.; Vucetic, S.; Iakoucheva, L.M.; Obradovic, Z.; Dunker, A.K. Unfoldomics of human diseases: Linking protein intrinsic disorder with diseases. BMC Genom. 2009, 10, S7. [Google Scholar] [CrossRef] [PubMed]
- Bandaru, V.; Cooper, W.; Wallace, S.S.; Doublie, S. Overproduction, crystallization and preliminary crystallographic analysis of a novel human DNA-repair enzyme that recognizes oxidative DNA damage. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 1142–1144. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Wang, K.J.; Liu, Y.L.; Xue, B.; Uversky, V.N.; Dunker, A.K. Predicting intrinsic disorder in proteins: An overview. Cell Res. 2009, 19, 929–949. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Intrinsically disordered proteins from A to Z. Int. J. Biochem. Cell Biol. 2011, 43, 1090–1103. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Brown, C.J.; Lawson, J.D.; Iakoucheva, L.M.; Obradovic, Z. Intrinsic disorder and protein function. Biochemistry 2002, 41, 6573–6582. [Google Scholar] [CrossRef] [PubMed]
- van der Lee, R.; Buljan, M.; Lang, B.; Weatheritt, R.J.; Daughdrill, G.W.; Dunker, A.K.; Fuxreiter, M.; Gough, J.; Gsponer, J.; Jones, D.T.; et al. Classification of intrinsically disordered regions and proteins. Chem. Rev. 2014, 114, 6589–6631. [Google Scholar] [CrossRef] [PubMed]
- Karve, T.M.; Cheema, A.K. Small changes huge impact: The role of protein posttranslational modifications in cellular homeostasis and disease. J. Amino Acids 2011, 2011, ID207691. [Google Scholar] [CrossRef] [PubMed]
- Fukuchi, S.; Hosoda, K.; Homma, K.; Gojobori, T.; Nishikawa, K. Binary classification of protein molecules into intrinsically disordered and ordered segments. BMC Struct. Biol. 2011, 11, 29. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Thelen, J.J.; Dunker, A.K.; Xu, D. Musite, a tool for global prediction of general and kinase-specific phosphorylation sites. Mol. Cell. Proteom. 2010, 9, 2586–2600. [Google Scholar] [CrossRef] [PubMed]
- Iakoucheva, L.M.; Radivojac, P.; Brown, C.J.; O’Connor, T.R.; Sikes, J.G.; Obradovic, Z.; Dunker, A.K. The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res. 2004, 32, 1037–1049. [Google Scholar] [CrossRef] [PubMed]
- Yao, Q.; Gao, J.; Bollinger, C.; Thelen, J.J.; Xu, D. Predicting and analyzing protein phosphorylation sites in plants using musite. Front. Plant Sci. 2012, 3, 186. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Xu, D. Correlation between posttranslational modification and intrinsic disorder in protein. Pac. Symp. Biocomput. 2012, 17, 94–103. [Google Scholar]
- Kurotani, A.; Tokmakov, A.A.; Kuroda, Y.; Fukami, Y.; Shinozaki, K.; Sakurai, T. Correlations between predicted protein disorder and post-translational modifications in plants. Bioinformatics 2014, 30, 1095–1103. [Google Scholar] [CrossRef] [PubMed]
- Xue, B.; Dunker, A.K.; Uversky, V.N. Orderly order in protein intrinsic disorder distribution: Disorder in 3500 proteomes from viruses and the three domains of life. J. Biomol. Struct. Dyn. 2012, 30, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Pietrosemoli, N.; Garcia-Martin, J.A.; Solano, R.; Pazos, F. Genome-wide analysis of protein disorder in arabidopsis thaliana: Implications for plant environmental adaptation. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Umezawa, T.; Sugiyama, N.; Takahashi, F.; Anderson, J.C.; Ishihama, Y.; Peck, S.C.; Shinozaki, K. Genetics and phosphoproteomics reveal a protein phosphorylation network in the abscisic acid signaling pathway in Arabidopsis thaliana. Sci. Signal. 2013, 6, rs8. [Google Scholar] [CrossRef] [PubMed]
- The Gene Ontology Consortium. Gene Ontology Consortium: Going forward. Nucleic Acids Res. 2015, 43, D1049–D1056. [Google Scholar]
- Li, D.; Berardini, T.Z.; Muller, R.J.; Huala, E. Building an efficient curation workflow for the Arabidopsis literature corpus. Database 2012, 2012, bas047. [Google Scholar] [CrossRef] [PubMed]
- Kourmpetis, Y.A.; van Dijk, A.D.; van Ham, R.C.; ter Braak, C.J.F. Genome-wide computational function prediction of arabidopsis proteins by integration of multiple data sources. Plant Physiol. 2011, 155, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, K.; Kurotani, A.; Iida, K.; Kuromori, T.; Shinozaki, K.; Sakurai, T. RARGE II: An integrated phenotype database of Arabidopsis mutant traits using a controlled vocabulary. Plant Cell Physiol. 2013, 4, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kurotani, A.; Yamada, Y.; Shinozaki, K.; Kuroda, Y.; Sakurai, T. Plant-PrAS: A database of physicochemical and structural properties and novel functional regions in plant proteomes. Plant Cell Physiol. 2015, 56, e11. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Mackey, A.J.; Stoeckert, C.J.; Roos, D.S. OrthoMCL-DB: Querying a comprehensive multi-species collection of ortholog groups. Nucleic Acids Res. 2006, 34, D363–D368. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.J.; McGuffin, L.J.; Bryson, K.; Buxton, B.F.; Jones, D.T. The disopred server for the prediction of protein disorder. Bioinformatics 2004, 20, 2138–2139. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.R.; Thomson, R.; McNeil, P.; Esnouf, R.M. RONN: The bio-basis function neural network technique applied to the detection of natively disordered regions in proteins. Bioinformatics 2005, 21, 3369–3376. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate—A practical and powerful approach to multiple testing. J. Roy. Stat. Soc. B Met. 1995, 57, 289–300. [Google Scholar]
- Sanders, S.L.; Gentzsch, M.; Tanner, W.; Herskowitz, I. O-glycosylation of Axl2/Bud10p by Pmt4p is required for its stability, localization, and function in daughter cells. J. Cell Biol 1999, 145, 1177–1188. [Google Scholar] [CrossRef] [PubMed]
- Narhi, L.O.; Arakawa, T.; Aoki, K.H.; Elmore, R.; Rohde, M.F.; Boone, T.; Strickland, T.W. The effect of carbohydrate on the structure and stability of erythropoietin. J. Biol. Chem. 1991, 266, 23022–23026. [Google Scholar] [PubMed]
- Diaz, C.L.; Logman, T.J.J.; Stam, H.C.; Kijne, J.W. Sugar-binding activity of pea lectin expressed in white clover hairy roots. Plant Physiol. 1995, 109, 1167–1177. [Google Scholar] [PubMed]
- Webster, D.E.; Thomas, M.C. Post-translational modification of plant-made foreign proteins; glycosylation and beyond. Biotechnol. Adv. 2012, 30, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, K.K.; Bojsen, K.; Roepstorff, P.; Mikkelsen, J.D. A hydroxyproline-containing class-IV chitinase of sugar-beet is glycosylated with xylose. Plant Mol. Biol. 1994, 25, 241–257. [Google Scholar] [CrossRef] [PubMed]
- Steentoft, C.; Vakhrushev, S.Y.; Joshi, H.J.; Kong, Y.; Vester-Christensen, M.B.; Schjoldager, K.T.B.G.; Lavrsen, K.; Dabelsteen, S.; Pedersen, N.B.; Marcos-Silva, L.; et al. Precision mapping of the human O-GalNAc glycoproteome through simplecell technology. EMBO J. 2013, 32, 1478–1488. [Google Scholar] [CrossRef] [PubMed]
- Gomord, V.; Fitchette, A.C.; Menu-Bouaouiche, L.; Saint-Jore-Dupas, C.; Plasson, C.; Michaud, D.; Faye, L. Plant-specific glycosylation patterns in the context of therapeutic protein production. Plant Biotechnol. J. 2010, 8, 564–587. [Google Scholar] [CrossRef] [PubMed]
- Wilson, I.B.H. Glycosylation of proteins in plants and invertebrates. Curr. Opin. Struct. Biol. 2002, 12, 569–577. [Google Scholar] [CrossRef]
- Lam, P.V.; Goldman, R.; Karagiannis, K.; Narsule, T.; Simonyan, V.; Soika, V.; Mazumder, R. Structure-based comparative analysis and prediction of N-linked glycosylation sites in evolutionarily distant eukaryotes. Genom. Proteom. Bioinform. 2013, 11, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Stanley, P.; Schachter, H.; Taniguchi, N. N-glycans. In Essentials of Glycobiology, 2nd ed.; Varki, A., Cummings, R.D., Esko, J.D., Freeze, H.H., Stanley, P., Bertozzi, C.R., Hart, G.W., Etzler, M.E., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2009. [Google Scholar]
- Gupta, R.; Brunak, S. Prediction of glycosylation across the human proteome and the correlation to protein function. Pac. Symp. Biocomput. 2002, 322, 310–322. [Google Scholar]
- Chauhan, J.S.; Rao, A.; Raghava, G.P.S. In silico platform for prediction of N-, O- and C-glycosites in eukaryotic protein sequences. PLoS ONE 2013, 8, e67008. [Google Scholar] [CrossRef] [PubMed]
- Petersen, T.N.; Brunak, S.; von Heijne, G.; Nielsen, H. Signalp 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef] [PubMed]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Rechsteiner, M.; Rogers, S.W. Pest sequences and regulation by proteolysis. Trends Biochem. Sci. 1996, 21, 267–271. [Google Scholar] [CrossRef]
- Rogers, S.; Wells, R.; Rechsteiner, M. Amino-acid-sequences common to rapidly degraded proteins the PEST hypothesis. Science 1986, 234, 364–368. [Google Scholar] [CrossRef] [PubMed]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The european molecular biology open software suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Brunquell, J.; Yuan, J.; Erwin, A.; Westerheide, S.D.; Xue, B. DBC1/CCAR2 and CCAR1 are largely disordered proteins that have evolved from one common ancestor. Biomed. Res. Int. 2014, 2014, 418458. [Google Scholar] [CrossRef] [PubMed]
- Kim, P.M.; Sboner, A.; Xia, Y.; Gerstein, M. The role of disorder in interaction networks: A structural analysis. Mol. Syst. Biol. 2008, 4, 179. [Google Scholar] [CrossRef] [PubMed]
- Mosca, R.; Pache, R.A.; Aloy, P. The role of structural disorder in the rewiring of protein interactions through evolution. Mol. Cell. Proteom. 2012, 11, M111.014969. [Google Scholar] [CrossRef] [PubMed]
- Peng, K.; Radivojac, P.; Vucetic, S.; Dunker, A.K.; Obradovic, Z. Length-dependent prediction of protein intrinsic disorder. BMC Bioinform. 2006, 7, 208. [Google Scholar] [CrossRef] [PubMed]
- Burra, P.V.; Kalmar, L.; Tompa, P. Reduction in structural disorder and functional complexity in the thermal adaptation of prokaryotes. PLoS ONE 2010, 5, e12069. [Google Scholar] [CrossRef] [PubMed]
- Schad, E.; Tompa, P.; Hegyi, H. The relationship between proteome size, structural disorder and organism complexity. Genome Biol. 2011, 12, R120. [Google Scholar] [CrossRef] [PubMed]
- Oates, M.E.; Romero, P.; Ishida, T.; Ghalwash, M.; Mizianty, M.J.; Xue, B.; Dosztanyi, Z.; Uversky, V.N.; Obradovic, Z.; Kurgan, L.; et al. D2P2: Database of disordered protein predictions. Nucleic Acids Res. 2013, 41, D508–D516. [Google Scholar] [CrossRef] [PubMed]
- Pang, C.N.I.; Hayen, A.; Wilkins, M.R. Surface accessibility of protein post-translational modifications. J. Proteome Res. 2007, 6, 1833–1845. [Google Scholar] [CrossRef] [PubMed]
- Sirota, F.L.; Maurer-Stroh, S.; Eisenhaber, B.; Eisenhaber, F. Single-residue posttranslational modification sites at the N-terminus, C-terminus or in-between: To be or not to be exposed for enzyme access. Proteomics 2015, 15, 2525–2546. [Google Scholar] [CrossRef] [PubMed]
- Petrescu, A.J.; Milac, A.L.; Petrescu, S.M.; Dwek, R.A.; Wormald, M.R. Statistical analysis of the protein environment of N-glycosylation sites: Implications for occupancy, structure, and folding. Glycobiology 2004, 14, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Varki, A.; Esko, J.D.; Colley, K.J. Cellular organization of glycosylation. In Essentials of Glycobiology, 2nd ed.; Varki, A., Cummings, R.D., Esko, J.D., Freeze, H.H., Stanley, P., Bertozzi, C.R., Hart, G.W., Etzler, M.E., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2009. [Google Scholar]
- Wustman, B.A.; Gretz, M.R.; Hoagland, K.D. Extracellular matrix assembly in diatoms (Bacillariophyceae). I. A model of adhesives based on chemical characterization and localization of polysaccharides from the marine diatom achnanthes longipes and other diatoms. Plant Physiol. 1997, 113, 1059–1069. [Google Scholar] [PubMed]
- Belizario, J.E.; Alves, J.; Garay-Malpartida, M.; Occhiucci, J.M. Coupling caspase cleavage and proteasomal degradation of proteins carrying PEST motif. Curr. Protein Pept. Sci. 2008, 9, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Silman, I.; Uversky, V.N.; Sussman, J.L. Function and structure of inherently disordered proteins. Curr. Opin. Struct. Biol. 2008, 18, 756–764. [Google Scholar] [CrossRef] [PubMed]
- Tokmakov, A.A.; Kurotani, A.; Shirouzu, M.; Fukami, Y.; Yokoyama, S. Bioinformatics analysis and optimization of cell-free protein synthesis. Methods Mol. Biol. 2014, 1118, 17–33. [Google Scholar] [PubMed]
- Ba, A.N.N.; Yeh, B.J.; van Dyk, D.; Davidson, A.R.; Andrews, B.J.; Weiss, E.L.; Moses, A.M. Proteome-wide discovery of evolutionary conserved sequences in disordered regions. Sci. Signal. 2012, 5, rs1. [Google Scholar]
- Palenik, B.; Grimwood, J.; Aerts, A.; Rouze, P.; Salamov, A.; Putnam, N.; Dupont, C.; Jorgensen, R.; Derelle, E.; Rombauts, S.; et al. The tiny eukaryote ostreococcus provides genomic insights into the paradox of plankton speciation. Proc. Natl. Acad. Sci. USA 2007, 104, 7705–7710. [Google Scholar] [CrossRef] [PubMed]
- Worden, A.Z.; Lee, J.H.; Mock, T.; Rouze, P.; Simmons, M.P.; Aerts, A.L.; Allen, A.E.; Cuvelier, M.L.; Derelle, E.; Everett, M.V.; et al. Green evolution and dynamic adaptations revealed by genomes of the marine picoeukaryotes micromonas. Science 2009, 324, 268–272. [Google Scholar] [CrossRef] [PubMed]
- Blanc, G.; Agarkova, I.; Grimwood, J.; Kuo, A.; Brueggeman, A.; Dunigan, D.D.; Gurnon, J.; Ladunga, I.; Lindquist, E.; Lucas, S.; et al. The genome of the polar eukaryotic microalga Coccomyxa subellipsoidea reveals traits of cold adaptation. Genome Biol. 2012, 13, R39. [Google Scholar] [CrossRef] [PubMed]
- Wakasugi, T.; Nagai, T.; Kapoor, M.; Sugita, M.; Ito, M.; Ito, S.; Tsudzuki, J.; Nakashima, K.; Tsudzuki, T.; Suzuki, Y.; et al. Complete nucleotide sequence of the chloroplast genome from the green alga chlorella vulgaris: The existence of genes possibly involved in chloroplast division. Proc. Natl. Acad. Sci. USA 1997, 94, 5967–5972. [Google Scholar] [CrossRef] [PubMed]
- Prochnik, S.E.; Umen, J.; Nedelcu, A.M.; Hallmann, A.; Miller, S.M.; Nishii, I.; Ferris, P.; Kuo, A.; Mitros, T.; Fritz-Laylin, L.K.; et al. Genomic analysis of organismal complexity in the multicellular green alga Volvox carteri. Science 2010, 329, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Merchant, S.S.; Prochnik, S.E.; Vallon, O.; Harris, E.H.; Karpowicz, S.J.; Witman, G.B.; Terry, A.; Salamov, A.; Fritz-Laylin, L.K.; Marechal-Drouard, L.; et al. The chlamydomonas genome reveals the evolution of key animal and plant functions. Science 2007, 318, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Tyler, B.M.; Tripathy, S.; Zhang, X.M.; Dehal, P.; Jiang, R.H.Y.; Aerts, A.; Arredondo, F.D.; Baxter, L.; Bensasson, D.; Beynon, J.L.; et al. Phytophthora genome sequences uncover evolutionary origins and mechanisms of pathogenesis. Science 2006, 313, 1261–1266. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Kamoun, S.; Zody, M.C.; Jiang, R.H.Y.; Handsaker, R.E.; Cano, L.M.; Grabherr, M.; Kodira, C.D.; Raffaele, S.; Torto-Alalibo, T.; et al. Genome sequence and analysis of the irish potato famine pathogen Phytophthora infestans. Nature 2009, 461, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Lamour, K.H.; Mudge, J.; Gobena, D.; Hurtado-Gonzales, O.P.; Schmutz, J.; Kuo, A.; Miller, N.A.; Rice, B.J.; Raffaele, S.; Cano, L.M.; et al. Genome sequencing and mapping reveal loss of heterozygosity as a mechanism for rapid adaptation in the vegetable pathogen Phytophthora capsici. Mol. Plant Microbe Interact. 2012, 25, 1350–1360. [Google Scholar] [CrossRef] [PubMed]
- Bowler, C.; Allen, A.E.; Badger, J.H.; Grimwood, J.; Jabbari, K.; Kuo, A.; Maheswari, U.; Martens, C.; Maumus, F.; Otillar, R.P.; et al. The phaeodactylum genome reveals the evolutionary history of diatom genomes. Nature 2008, 456, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Armbrust, E.V.; Berges, J.A.; Bowler, C.; Green, B.R.; Martinez, D.; Putnam, N.H.; Zhou, S.G.; Allen, A.E.; Apt, K.E.; Bechner, M.; et al. The genome of the diatom Thalassiosira pseudonana: Ecology, evolution, and metabolism. Science 2004, 306, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Gobler, C.J.; Berry, D.L.; Dyhrman, S.T.; Wilhelm, S.W.; Salamov, A.; Lobanov, A.V.; Zhang, Y.; Collier, J.L.; Wurch, L.L.; Kustka, A.B.; et al. Niche of harmful alga Aureococcus anophagefferens revealed through ecogenomics. Proc. Natl. Acad. Sci. USA 2011, 108, 4352–4357. [Google Scholar] [CrossRef] [PubMed]
- Cock, J.M.; Sterck, L.; Rouze, P.; Scornet, D.; Allen, A.E.; Amoutzias, G.; Anthouard, V.; Artiguenave, F.; Aury, J.M.; Badger, J.H.; et al. The ectocarpus genome and the independent evolution of multicellularity in brown algae. Nature 2010, 465, 617–621. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, M.; Misumi, O.; Shin, I.T.; Maruyama, S.; Takahara, M.; Miyagishima, S.Y.; Mori, T.; Nishida, K.; Yagisawa, F.; Nishida, K.; et al. Genome sequence of the ultrasmall unicellular red alga Cyanidioschyzon merolae 10D. Nature 2004, 428, 653–657. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Sasaki, N.; Kobayashi, M.; Ojima, N.; Yasuike, M.; Shigenobu, Y.; Satomi, M.; Fukuma, Y.; Shiwaku, K.; Tsujimoto, A.; et al. The first symbiont-free genome sequence of marine red alga, Susabi-nori (Pyropia yezoensis). PLoS ONE 2013, 8, e57122. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, D.; Price, D.C.; Chan, C.X.; Qiu, H.; Rose, N.; Ball, S.; Weber, A.P.M.; Arias, M.C.; Henrissat, B.; Coutinho, P.M.; et al. Genome of the red alga Porphyridium purpureum. Nat. Commun. 2013, 4, 1941. [Google Scholar] [CrossRef] [PubMed]
- Goodstein, D.M.; Shu, S.Q.; Howson, R.; Neupane, R.; Hayes, R.D.; Fazo, J.; Mitros, T.; Dirks, W.; Hellsten, U.; Putnam, N.; et al. Phytozome: A comparative platform for green plant genomics. Nucleic Acids Res. 2012, 40, D1178–D1186. [Google Scholar] [CrossRef] [PubMed]
- Nordberg, H.; Cantor, M.; Dusheyko, S.; Hua, S.; Poliakov, A.; Shabalov, I.; Smirnova, T.; Grigoriev, I.V.; Dubchak, I. The genome portal of the department of energy joint genome institute: 2014 updates. Nucleic Acids Res. 2014, 42, D26–D31. [Google Scholar] [CrossRef] [PubMed]
- Rossman, A.Y.; Palm, M.E. Why are phytophthora and other oomycota not true fungi? Outlooks Pest Manag. 2006, 17, 217–219. [Google Scholar] [CrossRef]
- Radivojac, P.; Vacic, V.; Haynes, C.; Cocklin, R.R.; Mohan, A.; Heyen, J.W.; Goebl, M.G.; Iakoucheva, L.M. Identification, analysis, and prediction of protein ubiquitination sites. Proteins 2010, 78, 365–380. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Schneeberger, K.; Ossowski, S.; Gunther, T.; Bender, S.; Fitz, J.; Koenig, D.; Lanz, C.; Stegle, O.; Lippert, C.; et al. Whole-genome sequencing of multiple Arabidopsis thaliana populations. Nat. Genet. 2011, 43, 956–963. [Google Scholar] [CrossRef] [PubMed]
- Rafalski, A. Applications of single nucleotide polymorphisms in crop genetics. Curr. Opin. Plant Biol. 2002, 5, 94–100. [Google Scholar] [CrossRef]
- Sakurai, T.; Mochida, K.; Yoshida, T.; Akiyama, K.; Ishitani, M.; Seki, M.; Shinozaki, K. Genome-wide discovery and information resource development of DNA polymorphisms in cassava. PLoS ONE 2013, 8, e74056. [Google Scholar] [CrossRef] [PubMed]
- Bais, P.; Moon, S.M.; He, K.; Leitao, R.; Dreher, K.; Walk, T.; Sucaet, Y.; Barkan, L.; Wohlgemuth, G.; Roth, M.R.; et al. PlantMetabolomics.Org: A web portal for plant metabolomics experiments. Plant Physiol. 2010, 152, 1807–1816. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, K.; Chikayama, E.; Yuasa, H.; Shimada, Y.; Tohge, T.; Shinozaki, K.; Hirai, M.; Sakurai, T.; Kikuchi, J.; Saito, K. PRIMe: A web site that assembles tools for metabolomics and transcriptomics. In Silico Biol. 2008, 8, 339–345. [Google Scholar] [PubMed]
- Kudo, T.; Akiyama, K.; Kojima, M.; Makita, N.; Sakurai, T.; Sakakibara, H. UniVIO: A multiple omics database with hormonome and transcriptome data from rice. Plant Cell Physiol. 2013, 54, E9. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, T.; Yamada, T.; Sawada, Y.; Matsuda, F.; Akiyama, K.; Shinozaki, K.; Hirai, M.Y.; Saito, K. PRIMe update: Innovative content for plant metabolomics and integration of gene expression and metabolite accumulation. Plant Cell Physiol. 2013, 54, E5. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kurotani, A.; Sakurai, T. In Silico Analysis of Correlations between Protein Disorder and Post-Translational Modifications in Algae. Int. J. Mol. Sci. 2015, 16, 19812-19835. https://doi.org/10.3390/ijms160819812
Kurotani A, Sakurai T. In Silico Analysis of Correlations between Protein Disorder and Post-Translational Modifications in Algae. International Journal of Molecular Sciences. 2015; 16(8):19812-19835. https://doi.org/10.3390/ijms160819812
Chicago/Turabian StyleKurotani, Atsushi, and Tetsuya Sakurai. 2015. "In Silico Analysis of Correlations between Protein Disorder and Post-Translational Modifications in Algae" International Journal of Molecular Sciences 16, no. 8: 19812-19835. https://doi.org/10.3390/ijms160819812
APA StyleKurotani, A., & Sakurai, T. (2015). In Silico Analysis of Correlations between Protein Disorder and Post-Translational Modifications in Algae. International Journal of Molecular Sciences, 16(8), 19812-19835. https://doi.org/10.3390/ijms160819812