
Mitochondrial Transcription Factor A and Mitochondrial Genome as Molecular Targets for Cisplatin-Based Cancer Chemotherapy

 ,
,
Abstract
: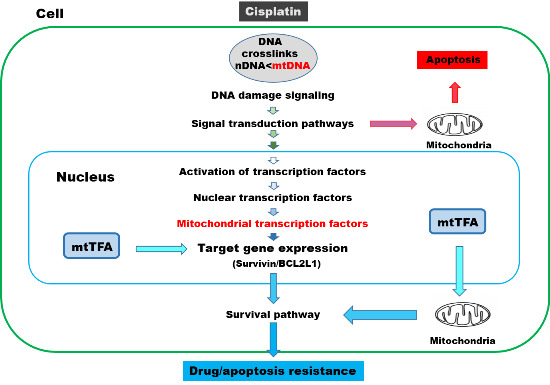
1. Introduction
2. Cisplatin and DNA
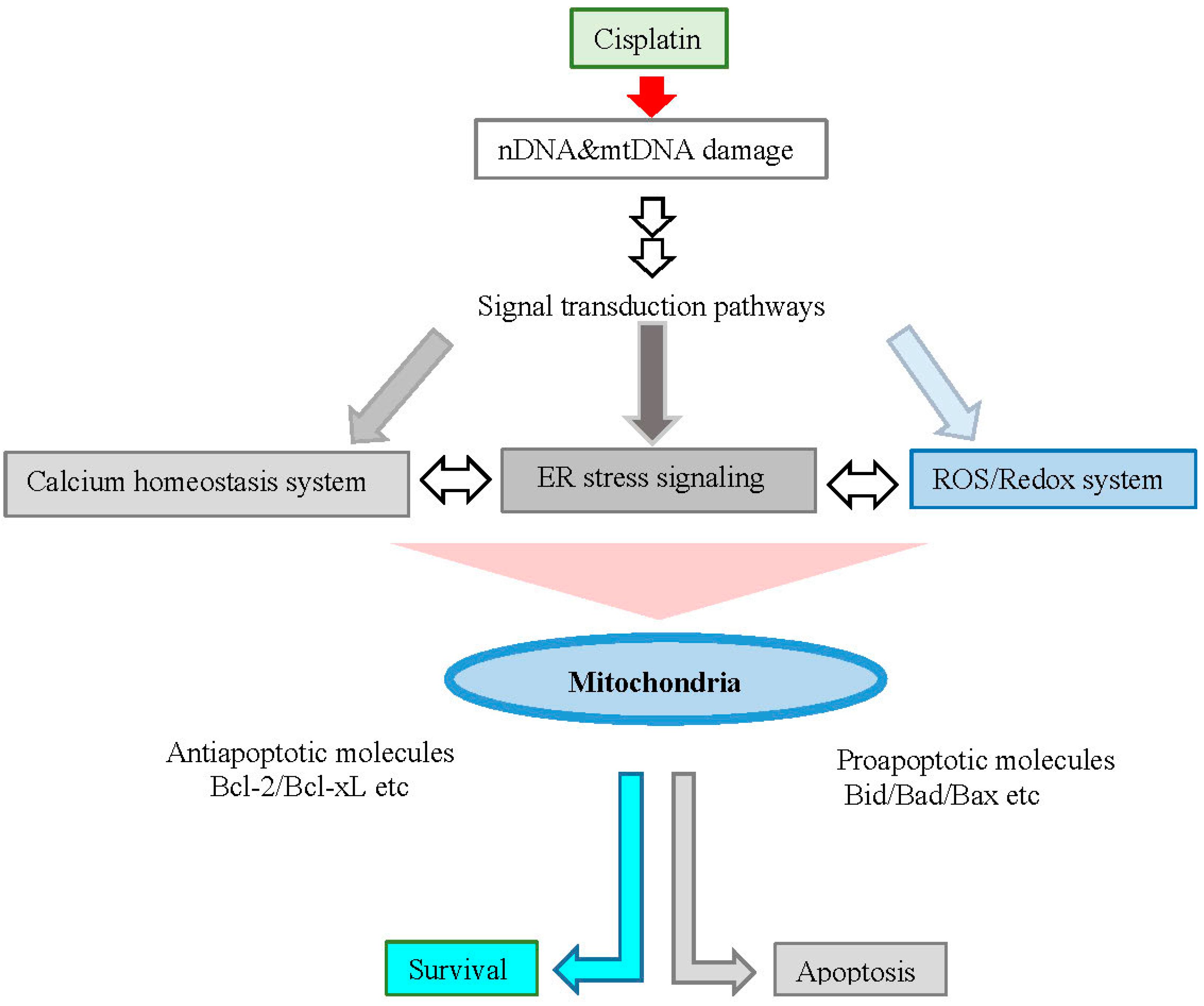
3. Calcium Ion, Endoplasmic Reticulum (ER) Stress, ROS/Redox System and Apoptosis
4. mtTFA and Cisplatin Resistance
5. Mitochondrial DNA as a Target for Cisplatin

{kind=link}
{kind=link}
{kind=link}
| Species | Total Number of mtDNA | GG | GGG | GGGG | GGGGG | |
|---|---|---|---|---|---|---|
| Human | 16,565 | L chain | 426 | 73 | 15 | 4 |
| H chain | 1772 | 624 | 224 | 69 | ||
| total | 2198 | 697 | 239 | 73 | ||
| Gorilla | 16,364 | L chain | 425 | 71 | 16 | 5 |
| H chain | 1712 | 596 | 216 | 72 | ||
| total | 2137 | 667 | 232 | 77 | ||
| Rat | 16,300 | L chain | 397 | 63 | 14 | 4 |
| H chain | 1299 | 377 | 99 | 32 | ||
| total | 1696 | 440 | 113 | 36 | ||
| Mouse | 16,300 | L chain | 397 | 58 | 11 | 3 |
| H chain | 1104 | 288 | 72 | 19 | ||
| total | 1501 | 346 | 83 | 22 | ||
| Xenopus | 17,553 | L chain | 445 | 72 | 12 | 1 |
| H chain | 1091 | 259 | 60 | 8 | ||
| total | 1536 | 331 | 72 | 9 | ||
| Drosophila | 16,019 | L chain | 599 | 230 | 16 | 1 |
| H chain | 770 | 358 | 78 | 30 | ||
| total | 1369 | 588 | 94 | 31 | ||
| Expectation | Double strand | 2071 | 518 | 129 | 32 | |
| Single strand | 1035 | 259 | 65 | 16 | ||
| Nuclear DNA | Double strand | 1919 ± 319 | 501 ± 77 | 118 ± 25 | 35 ± 14 |
6. Cellular Functions of mtTFA
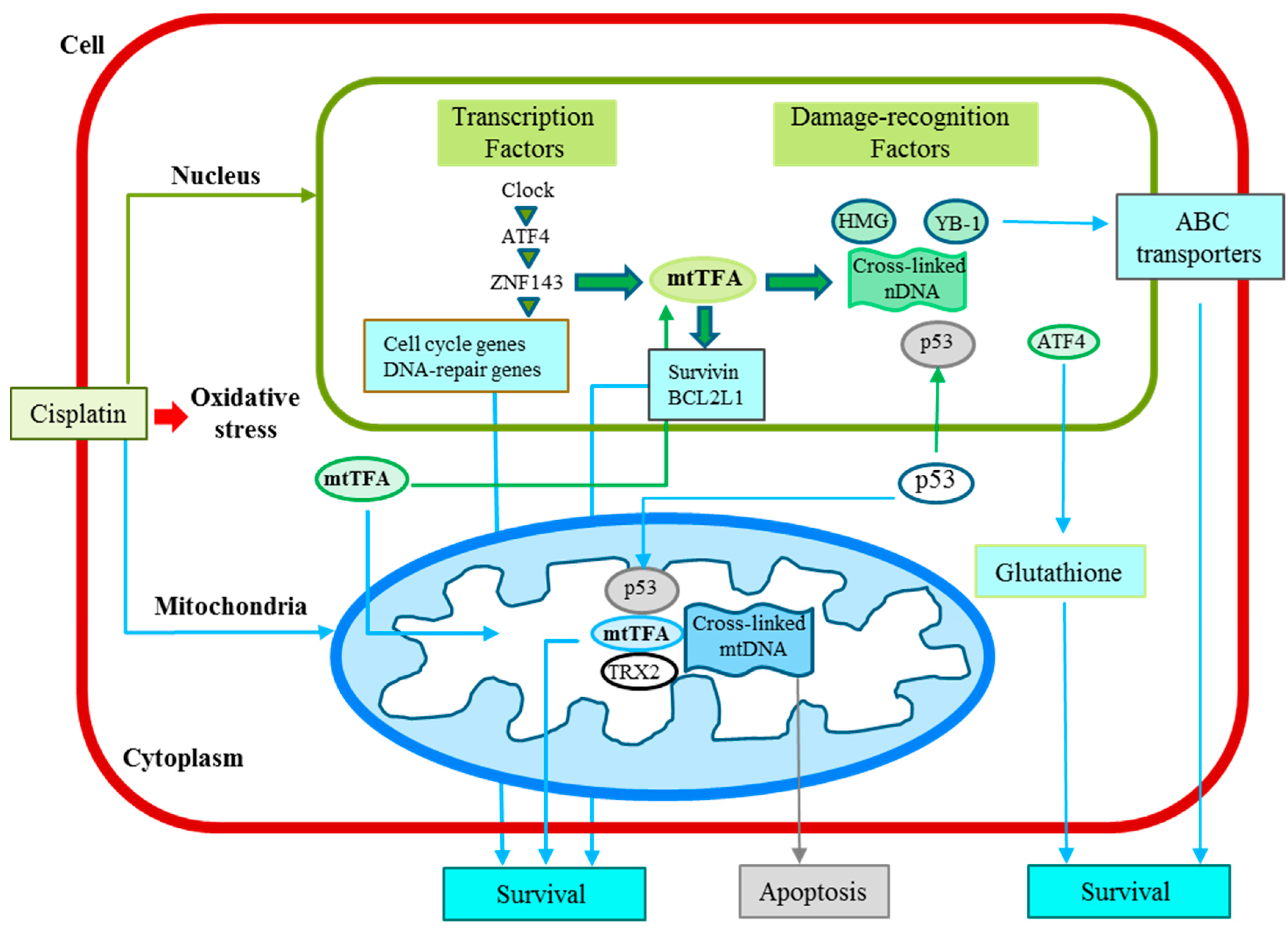
| Factors | Selected Target Genes | Selected Interacting Factors | Damage Recognition | Drug Resistance | Other Functions | References |
|---|---|---|---|---|---|---|
| mtTFA | Survivin BcLL2 | p53, TRX2 | + | + | Cell growth, anti-apoptosis | [54,55] |
| ZNF143 | mtTFA | p73 | + | + | Cell cycle, DNA repair | [35,57] |
| ATF4 | ZNF143 | + | Glutathione biosynthesis | [34,36] | ||
| Clock | ATF4, Tip60 | + | Circadian rhythm, DNA repair | [36,38] | ||
| YB-1 | MDR1 | p53, PCNA, Topo1 | + | + | Endothelial cell growth | [61,62,63] |
7. Clinical Implications of the mtTFA Expression in Tumors
| Tumor | No. of Cases | Malignant Characteristics | Reference |
|---|---|---|---|
| Serous ovarian | 60 | Poor prognosis, up-regulation of BCL2L1 expression | [64] |
| Pancreatic ductal adenocarcinoma | 70 | Poor prognosis, up-regulation of survivin expression | [65] |
| Colorectal | 105 | Poor prognosis | [66] |
| Metastatic colorectal | 59 | Poor clinical outcome with FOLFOX treatment | [67] |
| Endometrial | 245 | Invasion and metastasis, p53 mutation, poor prognosis | [68] |
8. Conclusions and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef] [PubMed]
- Guha, M.; Avadhani, N.G. Mitochondrial retrograde signaling at the crossroads of tumor bioenergetics, genetics and epigenetics. Mitochondrion 2013, 13, 577–591. [Google Scholar] [CrossRef] [PubMed]
- Igney, F.H.; Krammer, P.H. Death and anti-death: Tumour resistance to apoptosis. Nat. Rev. Cancer 2002, 2, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Holohan, C.; van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.M.; Lippard, S.J. Cisplatin: From DNA damage to cancer chemotherapy. Prog. Nucl. Acid Res. Mol. Biol. 2001, 67, 93–130. [Google Scholar]
- Wang, D.; Lippard, S.J. Cellular processing of platinum anticancer drugs. Nat. Rev. Drug Discov. 2005, 4, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Kelland, L. The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer 2007, 7, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Zamble, D.B.; Lippard, S.J. Cisplatin and DNA repair in cancer chemotherapy. Trends Biochem. Sci. 1995, 20, 435–439. [Google Scholar] [CrossRef]
- Correia, C.; Lee, S.H.; Meng, X.W.; Vincelette, N.D.; Knorr, K.L.; Ding, H.; Nowakowski, G.S.; Dai, H.; Kaufmann, S.H. Emerging understanding of Bcl-2 biology: Implications for neoplastic progression and treatment. Biochim. Biophys. Acta 2015, 1853, 1658–1671. [Google Scholar] [CrossRef] [PubMed]
- Frenzel, A.; Grespi, F.; Chmelewskij, W.; Villunger, A. Bcl2 family proteins in carcinogenesis and the treatment of cancer. Apoptosis 2009, 14, 584–596. [Google Scholar] [CrossRef] [PubMed]
- Lopez, J.; Tait, S.W. Mitochondrial apoptosis: Killing cancer using the enemy within. Br. J. Cancer 2015, 112, 957–962. [Google Scholar] [CrossRef] [PubMed]
- Orrenius, S.; Gogvadze, V.; Zhivotovsky, B. Calcium and mitochondria in the regulation of cell death. Biochem. Biophys. Res. Commun. 2015, 460, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Mandic, A.; Viktorsson, K.; Strandberg, L.; Heiden, T.; Hansson, J.; Linder, S.; Shoshan, M.C. Calpain-mediated Bid cleavage and calpain-independent Bak modulation: Two separate pathways in cisplatin-induced apoptosis. Mol. Cell. Biol. 2002, 22, 3003–3013. [Google Scholar] [CrossRef] [PubMed]
- Linder, S.; Shoshan, M.C. Lysosomes and endoplasmic reticulum: Targets for improved, selective anticancer therapy. Drug Resist. Updat. 2005, 8, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Wang, C.; Li, Z. A new strategy of promoting cisplatin chemotherapeutic efficiency by targeting endoplasmic reticulum stress. Mol. Clin. Oncol. 2014, 2, 3–7. [Google Scholar] [PubMed]
- Simran, S.S.; Paul, T.S. Mitochondrial ROS in cancer: Initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer 2014, 14, 709–721. [Google Scholar]
- Chirino, Y.I.; Pedraza, C.J. Role of oxidative and nitrosative stress in cisplatin-induced nephrotoxicity. Exp. Toxicol. Pathol. 2009, 61, 223–242. [Google Scholar] [CrossRef] [PubMed]
- Rybak, L.P.; Whitworth, C.A.; Mukherjea, D.; Ramkumar, V. Mechanisms of cisplatin-induced ototoxicity and prevention. Hear. Res. 2007, 226, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Torigoe, T.; Izumi, H.; Ishiguchi, H.; Yoshida, Y.; Tanabe, M.; Yoshida, T.; Igarashi, T.; Niina, I.; Wakasugi, T.; Imaizumi, T.; et al. Cisplatin resistance and transcription factors. Curr. Med. Chem. Anticancer Agents 2005, 5, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Kohno, K.; Uchiumi, T.; Niina, I.; Wakasugi, T.; Igarashi, T.; Momii, Y.; Yoshida, T.; Matsuo, K.; Miyamoto, N.; Izumi, H. Transcription factors and drug resistance. Eur. J. Cancer 2005, 41, 2577–2586. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Michels, J.; Brenner, C.; Szabadkai, G.; Harel-Bellan, A.; Castedo, M.; Kroemer, G. Systems biology of cisplatin resistance: Past, present and future. Cell Death Dis. 2014, 5, e1257. [Google Scholar] [CrossRef] [PubMed]
- Shen, D.W.; Pouliot, L.M.; Hall, M.D.; Gottesman, M.M. Cisplatin resistance: A cellular self-defense mechanism resulting from multiple epigenetic and genetic changes. Pharmacol. Rev. 2012, 64, 706–721. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular mechanisms of cisplatin resistance. Oncogene 2012, 31, 1869–1883. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.R.; Chua, K.N.; Sim, W.J.; Ng, H.C.; Bi, C.; Ho, J.; Nga, M.E.; Pang, Y.H.; Ong, W.R.; Soo, R.A.; et al. MEK Inhibition overcomes cisplatin resistance conferred by SOS/MAPK pathway activation in squamous cell carcinoma. Mol. Cancer Ther. 2015, 14, 1750–1760. [Google Scholar] [CrossRef] [PubMed]
- Crawford, N.; Chacko, A.D.; Savage, K.I.; McCoy, F.; Redmond, K.; Longley, D.B.; Fennell, D.A. Platinum resistant cancer cells conserve sensitivity to BH3 domains and obatoclax induced mitochondrial apoptosis. Apotosis 2011, 16, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.V.; Vanner, R.; Dirks, P.; Eaves, C.J. Cancer stem cells: An evolving concept. Nat. Rev. Cancer 2012, 12, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Visvader, J.E.; Lindeman, G.J. Cancer stem cells in solid tumours: Accumulating evidence and unresolved questions. Nat. Rev. Cancer 2008, 8, 755–768. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.; Fojo, T.; Bates, S. Tumour stem cells and drug resistance. Nat. Rev. Cancer 2005, 5, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Nagano, O.; Okazaki, S.; Saya, H. Redox regulation in stem-like cancer cells by CD44 variant isoforms. Oncogene 2013, 32, 5191–5198. [Google Scholar] [CrossRef] [PubMed]
- Song, I.S.; Jeong, J.Y.; Jeong, S.H.; Kim, H.K.; Ko, K.S.; Rhee, B.D.; Kim, N.; Han, J. Mitochondria as therapeutic targets for cancer stem cells. World J. Stem Cells 2015, 7, 418–427. [Google Scholar] [CrossRef] [PubMed]
- Kidani, A.; Izumi, H.; Yoshida, Y.; Kashiwagi, E.; Ohmori, H.; Tanaka, T.; Kuwano, M.; Kohno, K. Thioredoxin2 enhances the damaged DNA binding activity of mtTFA through direct interaction. Int. J. Oncol. 2009, 35, 1435–1440. [Google Scholar] [PubMed]
- Kohno, K.; Izumi, H.; Uchiumi, T.; Ashizuka, M.; Kuwano, M. The pleiotropic functions of the Y-box-binding protein, YB-1. Bioessays 2003, 25, 691–698. [Google Scholar] [CrossRef] [PubMed]
- Kashiwagi, E.; Izumi, H.; Yasuniwa, Y.; Baba, R.; Doi, Y.; Kidani, A.; Arao, T.; Nishio, K.; Naito, S.; Kohno, K. Enhanced expression of nuclear factor I/B in oxaliplatin-resistant human cancer cell lines. Cancer Sci. 2011, 102, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, M.; Izumi, H.; Ise, T.; Higuchi, S.; Yamori, T.; Yasumoto, K.; Kohno, K. Activating transcription factor 4 increases the cisplatin resistance of human cancer cell lines. Cancer Res. 2003, 63, 8592–8595. [Google Scholar] [PubMed]
- Wakasugi, T.; Izumi, H.; Uchiumi, T.; Suzuki, H.; Arao, T.; Nishio, K.; Kohno, K. ZNF143 interacts with p73 and is involved in cisplatin resistance through the transcriptional regulation of DNA repair genes. Oncogene 2007, 26, 5194–5203. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, T.; Izumi, H.; Uchiumi, T.; Nishio, K.; Arao, T.; Tanabe, M.; Uramoto, H.; Sugio, K.; Yasumoto, K.; Sasaguri, Y.; et al. Clock and ATF4 transcription system regulates drug resistance in human cancer cell lines. Oncogene 2007, 26, 4749–4760. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Bailey, S.M.; Udoh, U.S.; Young, M.E. Circadian regulation of metabolism. J. Endocrinol. 2014, 222, R75–R96. [Google Scholar] [CrossRef] [PubMed]
- Yasuniwa, Y.; Izumi, H.; Wang, K.Y.; Shimajiri, S.; Sasaguri, Y.; Kawai, K.; Kasai, H.; Shimada, T.; Miyake, K.; Kashiwagi, E.; et al. Circadian disruption accelerates tumor growth and angio/stromagenesis through a Wnt signaling pathway. PLoS ONE 2010, 5, e15330. [Google Scholar]
- Izumi, H.; Wang, K.Y.; Morimoto, Y.; Sasaguri, Y.; Kohno, K. Circadian disruption and cancer risk: A new concept of stromal niche (review). Int. J. Oncol. 2014, 44, 364–370. [Google Scholar] [PubMed]
- Guaragnella, N.; Giannattasio, S.; Moro, L. Mitochondrial dysfunction in cancer chemoresistance. Biochem. Pharmacol. 2014, 92, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, S.; Miyato, Y.; Shidara, Y.; Asoh, S.; Tokunaga, A.; Tajiri, T.; Ohta, S. Mutations in the mitochondrial genome confer resistance of cancer cells to anticancer drugs. Cancer Sci. 2009, 100, 1680–1687. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Jones, A.W.; Fassone, E.; Sweeney, M.G.; Lebiedzinska, M.; Suski, J.M.; Wieckowski, M.R.; Tajeddine, N.; Hargreaves, I.P.; Yasukawa, T.; et al. PGC-1β mediates adaptive chemoresistance associated with mitochondrial DNA mutations. Oncogene 2013, 32, 2592–2600. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Zheng, L.; Liu, W.; Wang, X.; Wang, Z.; Wang, Z.; French, A.J.; Kang, D.; Chen, L.; Thibodeau, S.N.; et al. Frequent truncating mutation of TFAM induces mitochondrial DNA depletion and apoptotic resistance in microsatellite-unstable colorectal cancer. Cancer Res. 2011, 71, 2978–2987. [Google Scholar] [CrossRef] [PubMed]
- Campbell, C.T.; Kolesar, J.E.; Kaufman, B.A. Mitochondrial transcription factor A regulates mitochondrial transcription initiation, DNA packaging, and genome copy number. Biochim. Biophys. Acta 2012, 1819, 921–929. [Google Scholar] [CrossRef] [PubMed]
- Olivero, O.A.; Semino, C.; Kassim, A.; Lopez-Larraza, D.M.; Poirier, M.C. Preferential binding of cisplatin to mitochondrial DNA of Chinese hamster ovary cells. Mutat. Res. 1995, 346, 221–230. [Google Scholar] [CrossRef]
- See comment in PubMed Commons below Olivero, O.A.; Chang, P.K.; Lopez-Larraza, D.M.; Semino-Mora, M.C.; Poirier, M.C. Preferential formation and decreased removal of cisplatin-DNA adducts in Chinese hamster ovary cell mitochondrial DNA as compared to nuclear DNA. Mutat. Res. 1997, 391, 79–86. [Google Scholar] [CrossRef]
- Murakami, T.; Shibuya, I.; Ise, T.; Chen, Z.S.; Akiyama, S.; Nakagawa, M.; Izumi, H.; Nakamura, T.; Matsuo, K.; Yamada, Y.; et al. Elevated expression of vacuolar proton pump genes and cellular pH in cisplatin resistance. Int. J. Cancer 2001, 93, 869–874. [Google Scholar] [CrossRef] [PubMed]
- Reshkin, S.J.; Greco, M.R.; Cardone, R.A. Role of pHi, and proton transporters in oncogene-driven neoplastic transformation. Philos. Trans. R. Soc. Lond. B 2014, 369, 20130100. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, Y.; Hoshino, S.; Izumi, H.; Kohno, K.; Yamashita, Y. New roles of mitochondrial transcription factor A in cancer. J. Phys. Chem. Biophys. 2011, 1, 101. [Google Scholar] [CrossRef]
- Han, B.; Izumi, H.; Yasuniwa, Y.; Akiyama, M.; Yamaguchi, T.; Fujimoto, N.; Matsumoto, T.; Wu, B.; Tanimoto, A.; Sasaguri, Y.; et al. Human mitochondrial transcription factor A functions in both nuclei and mitochondria and regulates cancer cell growth. Biochem. Biophys. Res. Commun. 2011, 408, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Altieri, D.C. Survivin, cancer networks and pathway-directed drug discovery. Nat. Rev. Cancer 2008, 8, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Cory, S.; Adams, J.M. The Bcl2 family: Regulators of the cellular life-or-death switch. Nat. Rev. Cancer 2002, 2, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Vaseva, A.V.; Moll, U.M. The mitochondrial p53 pathway. Biochim. Biophys. Acta 2009, 1787, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, Y.; Izumi, H.; Torigoe, T.; Ishiguchi, H.; Itoh, H.; Kang, D.; Kohno, K. P53 physically interacts with mitochondrial transcription factor A and differentially regulates binding to damaged DNA. Cancer Res. 2003, 63, 3729–3734. [Google Scholar] [PubMed]
- Wang, J.; Silva, J.P.; Gustafsson, C.M.; Rustin, P.; Larsson, N.G. Increased in vivo apoptosis in cells lacking mitochondrial DNA gene expression. Proc. Natl Acad. Sci. USA 2001, 98, 4038–4043. [Google Scholar] [CrossRef] [PubMed]
- Ishiguchi, H.; Izumi, H.; Torigoe, T.; Yoshida, Y.; Kubota, H.; Tsuji, S.; Kohno, K. ZNF143 activates gene expression in response to DNA damage and binds to cisplatin-modified DNA. Int. J. Cancer 2004, 111, 900–909. [Google Scholar] [CrossRef] [PubMed]
- Izumi, H.; Yasuniwa, Y.; Akiyama, M.; Yamaguchi, T.; Kuma, A.; Kitamura, N.; Kohno, K. Forced expression of ZNF143 restrains cancer cell growth. Cancers 2011, 3, 3909–3920. [Google Scholar] [CrossRef] [PubMed]
- Izumi, H.; Wakasugi, T.; Shimajiri, S.; Tanimoto, A.; Sasaguri, Y.; Kashiwagi, E.; Yasuniwa, Y.; Akiyama, M.; Han, B.; Wu, Y.; et al. Role of ZNF143 in tumor growth through transcriptional regulation of DNA replication and cell-cycle-associated genes. Cancer Sci. 2010, 101, 2538–2545. [Google Scholar] [CrossRef] [PubMed]
- Kawatsu, Y.; Kitada, S.; Uramoto, H.; Li, Z.; Takeda, T.; Kimura, T.; Horie, S.; Tanaka, F.; Sasaguri, Y.; Izumi, H.; et al. The combination of strong expression of ZNF143 and high MIB-1 labelling index independently predicts shorter disease-specific survival in lung adenocarcinoma. Br. J. Cancer. 2014, 110, 2583–2592. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Shimajiri, S.; Izumi, H.; Hirano, G.; Kashiwagi, E.; Yasuniwa, Y.; Wu, Y.; Han, B.; Akiyama, M.; Nishizawa, S.; et al. Y-box binding protein-1 is a novel molecular target for tumor vessels. Cancer Sci. 2010, 101, 1367–1373. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Wang, K.Y.; Li, Z.; Liu, Y.P.; Izumi, H.; Uramoto, H.; Nakayama, Y.; Ito, K.I.; Kohno, K. Y-box binding protein 1 enhances DNA topoisomerase 1 activity and sensitivity to camptothecin via direct interaction. J. Exp. Clin. Cancer Res. 2014, 33, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Wang, K.Y.; Li, Z.; Liu, Y.P.; Izumi, H.; Yamada, S.; Uramoto, H.; Nakayama, Y.; Ito, K.I.; Kohno, K. Y-box binding protein 1 expression in gastric cancer subtypes and association with cancer neovasculature. Clin. Transl. Oncol. 2015, 17, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Kurita, T.; Izumi, H.; Kagami, S.; Kawagoe, T.; Toki, N.; Matsuura, Y.; Hachisuga, T.; Kohno, K. Mitochondrial transcription factor A regulates BCL2L1 gene expression and is a prognostic factor in serous ovarian cancer. Cancer Sci. 2012, 103, 239–444. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Kitada, S.; Uramoto, H.; Li, Z.; Kawatsu, Y.; Takeda, T.; Horie, S.; Nabeshima, A.; Noguchi, H.; Sasaguri, Y.; et al. The combination of strong immunohistochemical mtTFA expression and a high survivin index predicts a shorter disease-specific survival in pancreatic ductal adenocarcinoma. Histol. Histopathol. 2015, 30, 193–204. [Google Scholar] [PubMed]
- Nakayama, Y.; Yamauchi, M.; Minagawa, N.; Torigoe, T.; Izumi, H.; Kohno, K.; Yamaguchi, K. Clinical significance of mitochondrial transcription factor A expression in patients with colorectal cancer. Oncol. Rep. 2012, 27, 1325–1330. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, Y.; Hasegawa, J.; Nezu, R.; Kim, Y.K.; Hirota, M.; Kawano, K.; Izumi, H.; Kohno, K. Clinical usefulness of mitochondrial transcription factor A expression as a predictive marker in colorectal cancer patients treated with FOLFOX. Cancer Sci. 2011, 102, 578–582. [Google Scholar] [CrossRef] [PubMed]
- Toki, N.; Kagami, S.; Kurita, T.; Kawagoe, T.; Matsuura, Y.; Hachisuga, T.; Matsuyama, A.; Hashimoto, H.; Izumi, H.; Kohno, K. Expression of mitochondrial transcription factor A in endometrial carcinomas: Clinicopathologic correlations and prognostic significance. Virchows Arch. 2010, 456, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Burnett, J.C.; Rossi, J.J. RNA-based therapeutics: Current progress and future prospects. Chem. Biol. 2012, 19, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Pirogova, E.; Istivan, T.; Gan, E.; Cosic, I. Advances in methods for therapeutic peptide discovery, design and development. Curr. Pharm. Biotechnol. 2011, 12, 1117–1127. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kohno, K.; Wang, K.-Y.; Takahashi, M.; Kurita, T.; Yoshida, Y.; Hirakawa, M.; Harada, Y.; Kuma, A.; Izumi, H.; Matsumoto, S. Mitochondrial Transcription Factor A and Mitochondrial Genome as Molecular Targets for Cisplatin-Based Cancer Chemotherapy. Int. J. Mol. Sci. 2015, 16, 19836-19850. https://doi.org/10.3390/ijms160819836
Kohno K, Wang K-Y, Takahashi M, Kurita T, Yoshida Y, Hirakawa M, Harada Y, Kuma A, Izumi H, Matsumoto S. Mitochondrial Transcription Factor A and Mitochondrial Genome as Molecular Targets for Cisplatin-Based Cancer Chemotherapy. International Journal of Molecular Sciences. 2015; 16(8):19836-19850. https://doi.org/10.3390/ijms160819836
Chicago/Turabian StyleKohno, Kimitoshi, Ke-Yong Wang, Mayu Takahashi, Tomoko Kurita, Yoichiro Yoshida, Masakazu Hirakawa, Yoshikazu Harada, Akihiro Kuma, Hiroto Izumi, and Shinji Matsumoto. 2015. "Mitochondrial Transcription Factor A and Mitochondrial Genome as Molecular Targets for Cisplatin-Based Cancer Chemotherapy" International Journal of Molecular Sciences 16, no. 8: 19836-19850. https://doi.org/10.3390/ijms160819836
APA StyleKohno, K., Wang, K.-Y., Takahashi, M., Kurita, T., Yoshida, Y., Hirakawa, M., Harada, Y., Kuma, A., Izumi, H., & Matsumoto, S. (2015). Mitochondrial Transcription Factor A and Mitochondrial Genome as Molecular Targets for Cisplatin-Based Cancer Chemotherapy. International Journal of Molecular Sciences, 16(8), 19836-19850. https://doi.org/10.3390/ijms160819836