
Mitochondrial and Ubiquitin Proteasome System Dysfunction in Ageing and Disease: Two Sides of the Same Coin?


Abstract
:
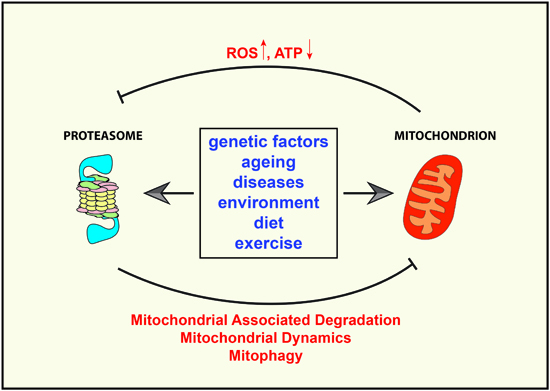{kind=link}
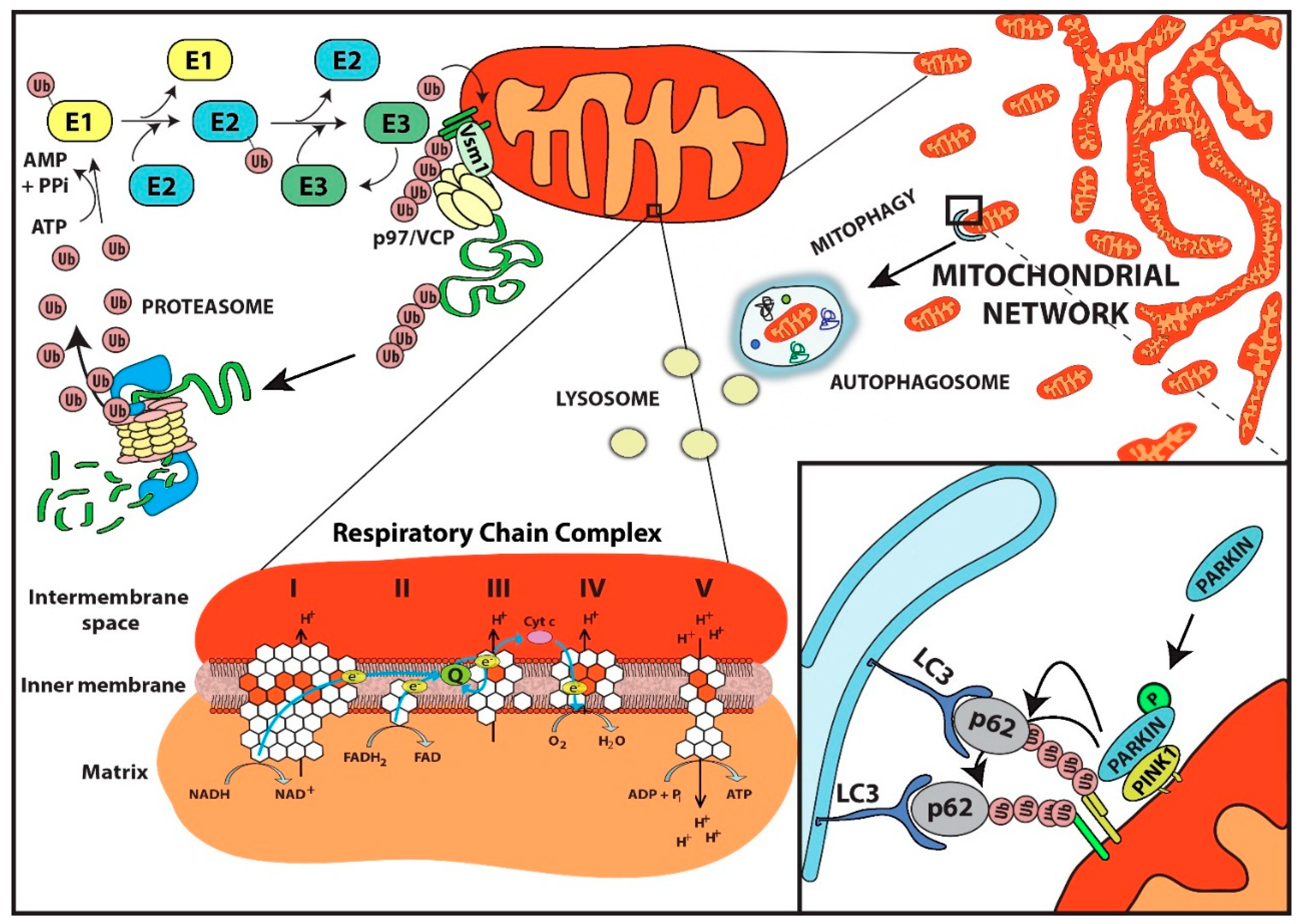{kind=link}
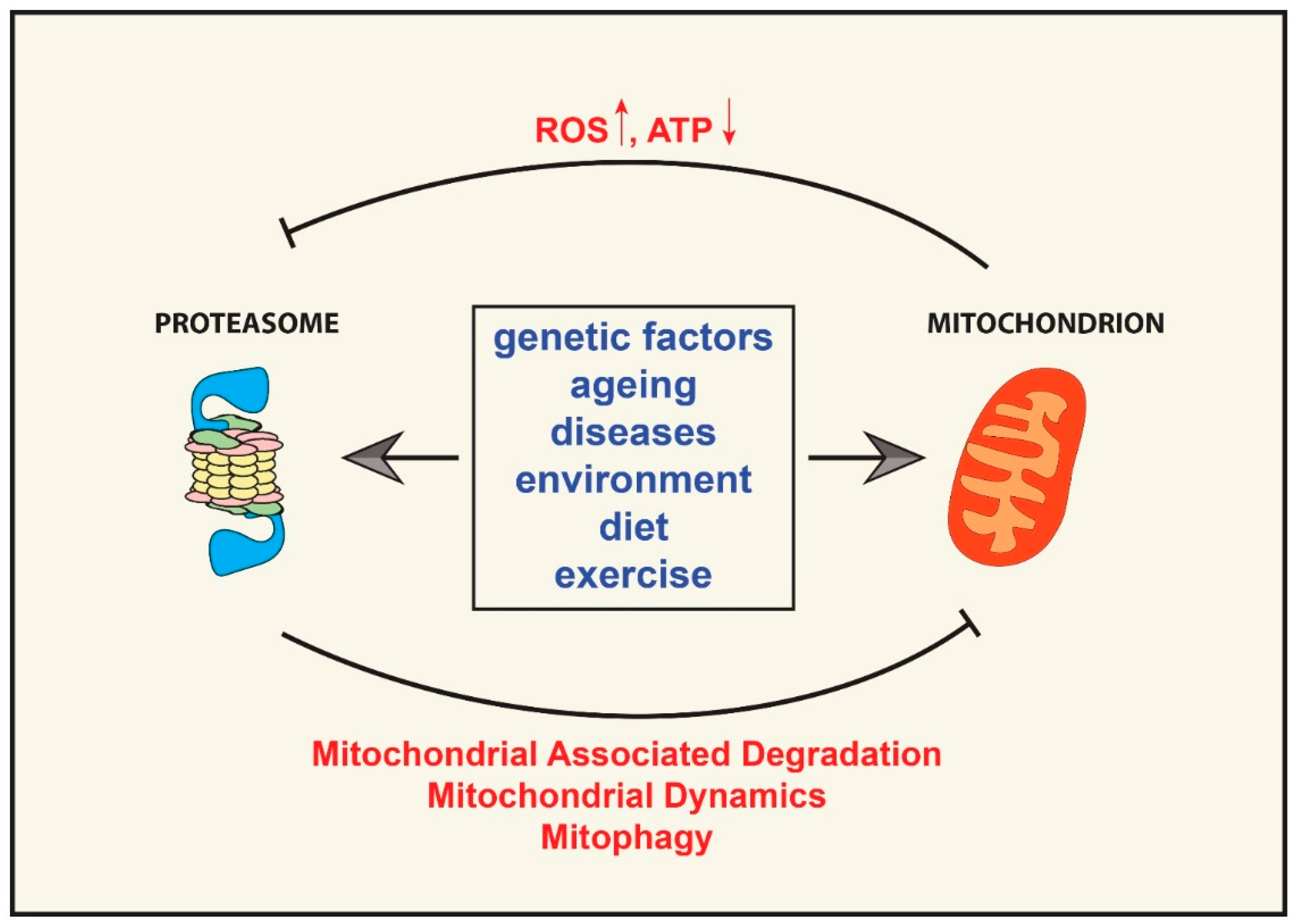{kind=link}
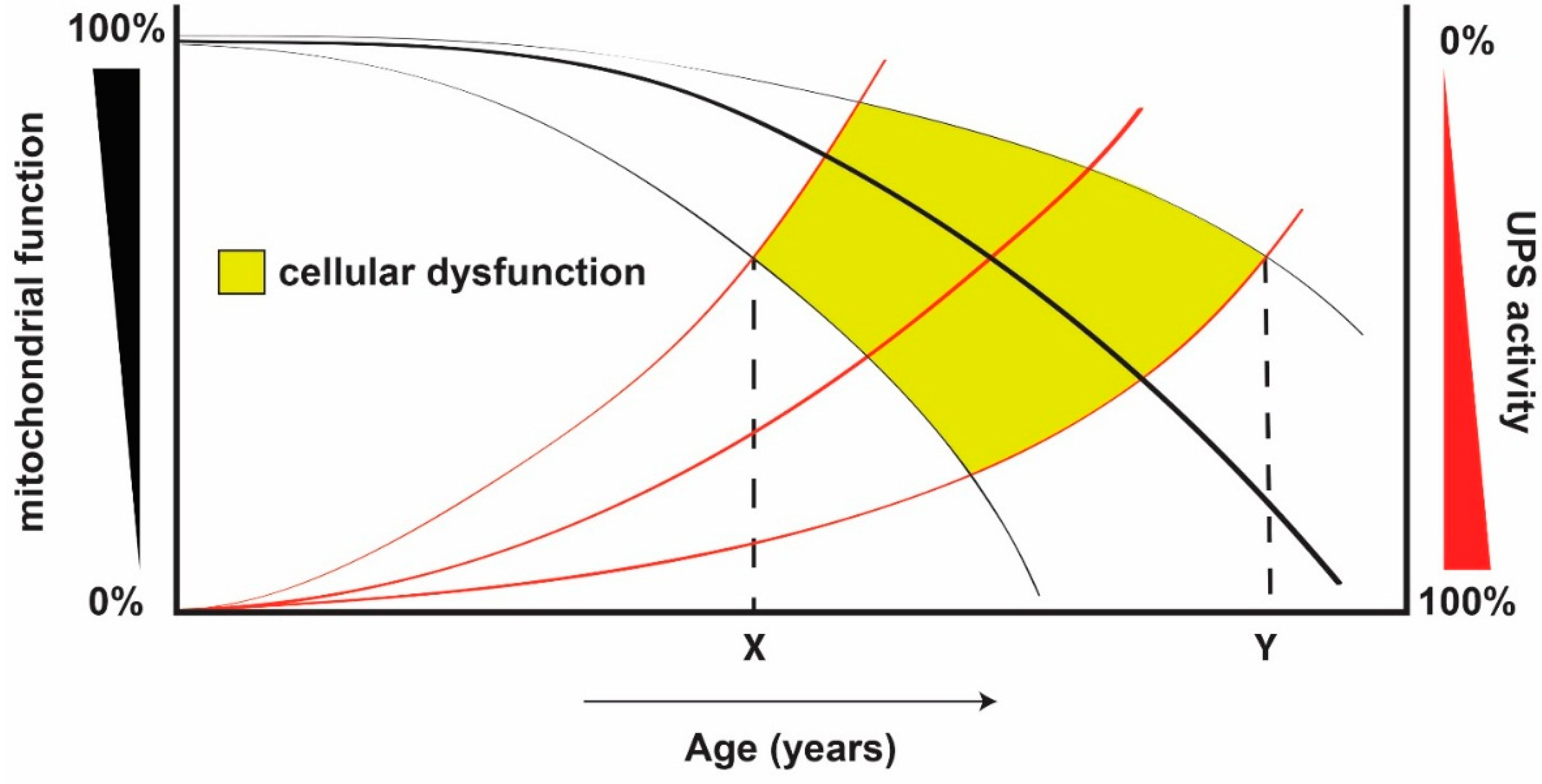{kind=link}
1. Introduction
2. The Ubiquitin Proteasome System
3. Mitochondria
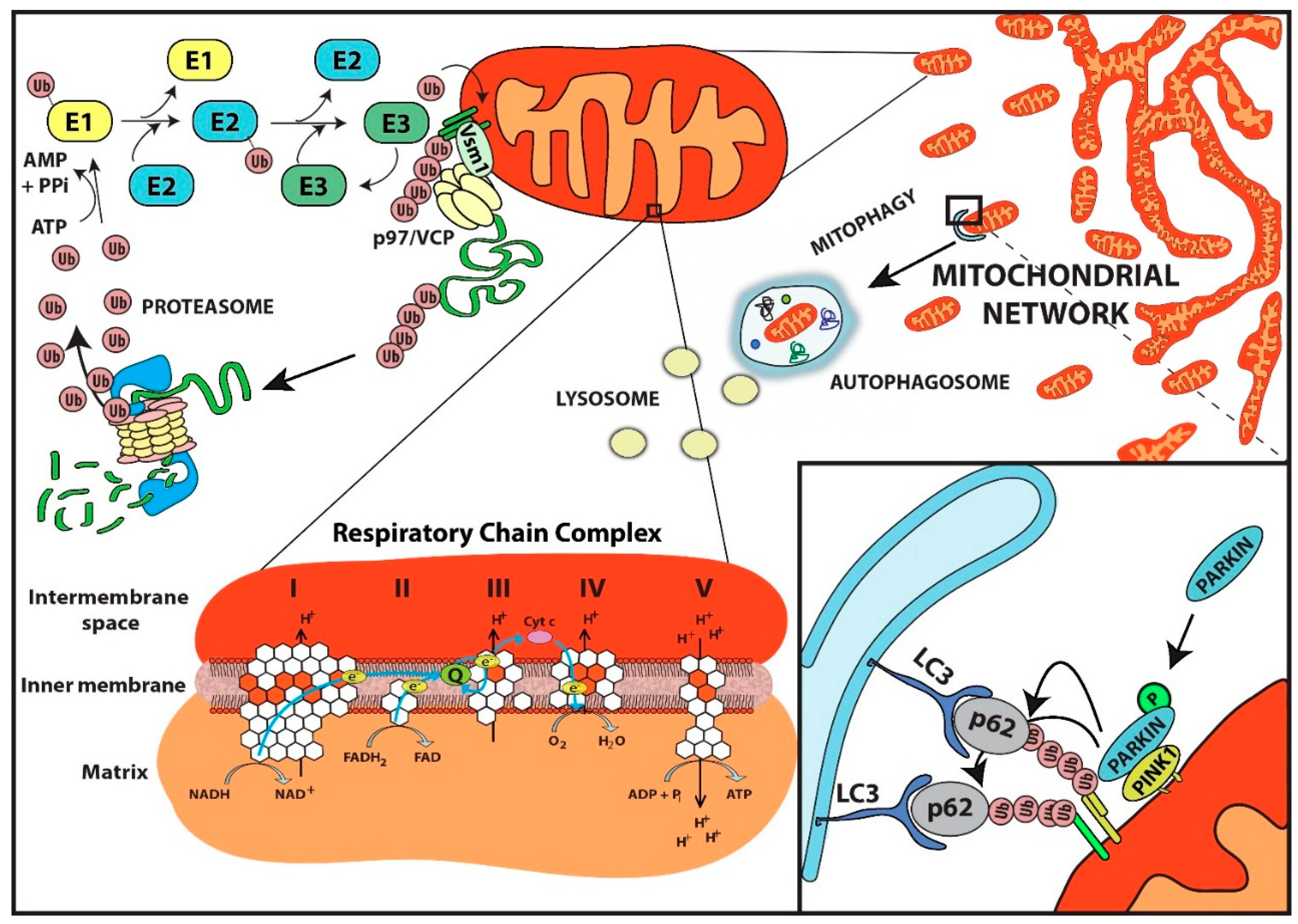
4. Role of the Ubiquitin Proteasome System in Mitochondrial Protein Quality Control
5. Effect of Mitochondrial Dysfunction on the Ubiquitin Proteasome System
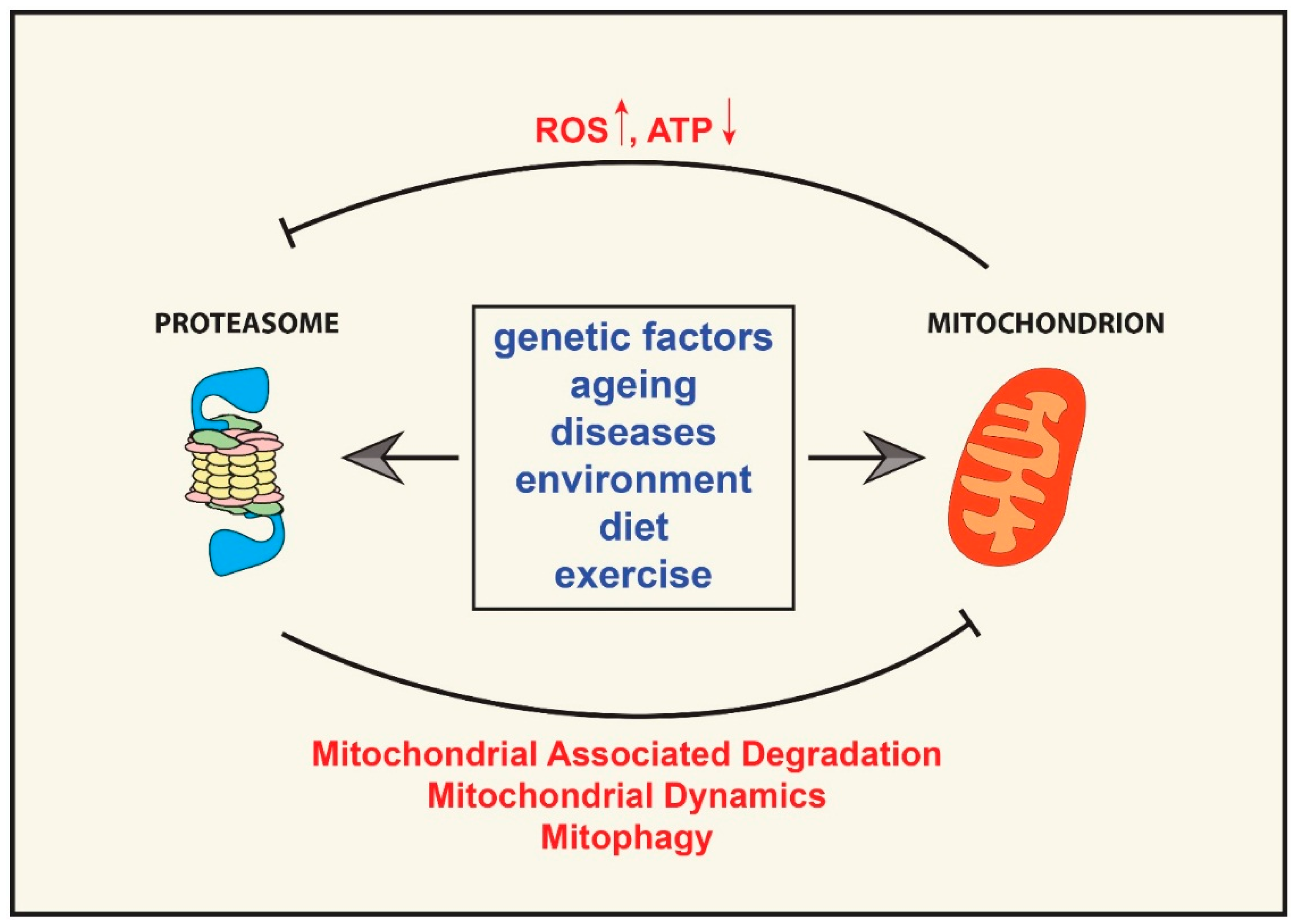
6. The “Mitochondrion—Ubiquitin Proteasome System Axis” in Ageing and Age-Related Diseases
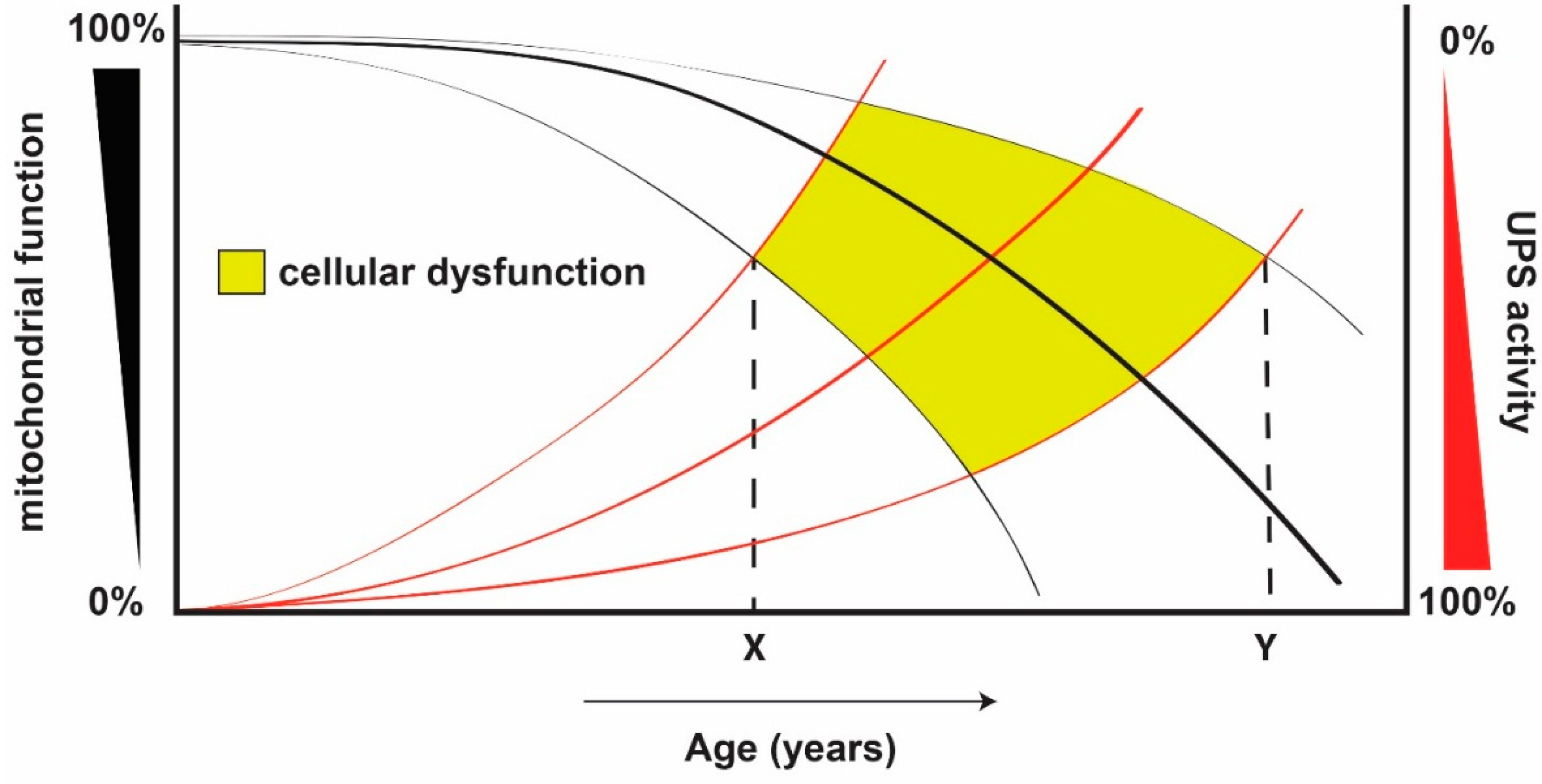
7. Conclusions and Future Prospects
Acknowledgments
Conflicts of Interest
Abbreviations
References
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, W.; Li, L.; Perry, G.; Lee, H.-G.; Zhu, X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. Acta 2014, 1842, 1240–1247. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, R.I. Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes Dev. 2008, 22, 1427–1438. [Google Scholar] [CrossRef] [PubMed]
- Keller, J.N.; Hanni, K.B.; Markesbery, W.R. Possible involvement of proteasome inhibition in aging: Implications for oxidative stress. Mech. Ageing Dev. 2000, 113, 61–70. [Google Scholar] [CrossRef]
- Gautier, C.A.; Corti, O.; Brice, A. Mitochondrial dysfunctions in Parkinson’s disease. Rev. Neurol. 2014, 170, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Büeler, H. Impaired mitochondrial dynamics and function in the pathogenesis of Parkinson’s disease. Exp. Neurol. 2009, 218, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Jung, T.; Catalgol, B.; Grune, T. The proteasomal system. Mol. Asp. Med. 2009, 30, 191–296. [Google Scholar] [CrossRef] [PubMed]
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef] [PubMed]
- Glickman, M.H.; Ciechanover, A. The ubiquitin-proteasome proteolytic pathway: Destruction for the sake of construction. Physiol. Rev. 2002, 82, 373–428. [Google Scholar] [CrossRef] [PubMed]
- Hochstrasser, M. Ubiquitin-dependent protein degradation. Annu. Rev. Genet. 1996, 30, 405–439. [Google Scholar] [CrossRef] [PubMed]
- Kim, A. A panoramic overview of mitochondria and mitochondrial redox biology. Toxicol. Res. 2014, 30, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.; Chan, D.C. Mitochondrial dynamics and inheritance during cell division, development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 634–646. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Galluzzi, L.; Kroemer, G. Cell biology. Metabolic control of cell death. Science 2014, 345, 1250256. [Google Scholar] [CrossRef] [PubMed]
- De Duve, C.; Pressman, B.C.; Gianetto, R.; Wattiaux, R.; Appelmans, F. Tissue fractionation studies. 6. Intracellular distribution patterns of enzymes in rat-liver tissue. Biochem. J. 1955, 60, 604–617. [Google Scholar] [CrossRef] [PubMed]
- Baraibar, M.A.; Friguet, B. Changes of the proteasomal system during the aging process. Prog. Mol. Biol. Transl. Sci. 2012, 109, 249–275. [Google Scholar]
- Kaushik, S.; Cuervo, A.M. Chaperone-mediated autophagy: A unique way to enter the lysosome world. Trends Cell Biol. 2012, 22, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [PubMed]
- Ciechanover, A. Proteolysis: From the lysosome to ubiquitin and the proteasome. Nat. Rev. Mol. Cell Biol. 2005, 6, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Pickart, C.M.; Eddins, M.J. Ubiquitin: Structures, functions, mechanisms. Biochim. Biophys. Acta 2004, 1695, 55–72. [Google Scholar] [CrossRef] [PubMed]
- Komander, D.; Rape, M. The ubiquitin code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef] [PubMed]
- Finley, D. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu. Rev. Biochem. 2009, 78, 477–513. [Google Scholar] [CrossRef] [PubMed]
- Pelzer, C.; Kassner, I.; Matentzoglu, K.; Singh, R.K.; Wollscheid, H.P.; Scheffner, M.; Schmidtke, G.; Groettrup, M. UBE1L2, a novel E1 enzyme specific for ubiquitin. J. Biol. Chem. 2007, 282, 23010–23014. [Google Scholar] [CrossRef] [PubMed]
- Metzger, M.B.; Hristova, V.A.; Weissman, A.M. HECT and RING finger families of E3 ubiquitin ligases at a glance. J. Cell Sci. 2012, 125, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Amerik, A.Y.; Hochstrasser, M. Mechanism and function of deubiquitinating enzymes. Biochim. Biophys. Acta 2004, 1695, 189–207. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Turcu, F.E.; Ventii, K.H.; Wilkinson, K.D. Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu. Rev. Biochem. 2009, 78, 363–397. [Google Scholar] [CrossRef] [PubMed]
- Jung, T.; Grune, T. Structure of the proteasome. Prog. Mol. Biol. Transl. Sci. 2012, 109, 1–39. [Google Scholar] [PubMed]
- Lander, G.C.; Estrin, E.; Matyskiela, M.E.; Bashore, C.; Nogales, E.; Martin, A. Complete subunit architecture of the proteasome regulatory particle. Nature 2012, 482, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Van Nocker, S.; Sadis, S.; Rubin, D.M.; Glickman, M.; Fu, H.; Coux, O.; Wefes, I.; Finley, D.; Vierstra, R.D. The multiubiquitin-chain-binding protein Mcb1 is a component of the 26S proteasome in Saccharomyces cerevisiae and plays a nonessential, substrate-specific role in protein turnover. Mol. Cell. Biol. 1996, 16, 6020–6028. [Google Scholar] [PubMed]
- Deveraux, Q.; Ustrell, V.; Pickart, C.; Rechsteiner, M. A 26 S protease subunit that binds ubiquitin conjugates. J. Biol. Chem. 1994, 269, 7059–7061. [Google Scholar] [PubMed]
- Husnjak, K.; Elsasser, S.; Zhang, N.; Chen, X.; Randles, L.; Shi, Y.; Hofmann, K.; Walters, K.J.; Finley, D.; Dikic, I. Proteasome subunit Rpn13 is a novel ubiquitin receptor. Nature 2008, 453, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Braun, B.C.; Glickman, M.; Kraft, R.; Dahlmann, B.; Kloetzel, P.M.; Finley, D.; Schmidt, M. The base of the proteasome regulatory particle exhibits chaperone-like activity. Nat. Cell Biol. 1999, 1, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.W.; Li, X.; Thompson, D.; Wooding, K.; Chang, T.; Tang, Z.; Yu, H.; Thomas, P.J.; DeMartino, G.N. ATP binding and ATP hydrolysis play distinct roles in the function of 26S proteasome. Mol. Cell 2006, 24, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Verma, R.; Aravind, L.; Oania, R.; McDonald, W.H.; Yates, J.R.; Koonin, E.V.; Deshaies, R.J. Role of Rpn11 metalloprotease in deubiquitination and degradation by the 26S proteasome. Science 2002, 298, 611–615. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.; Mentel, M.; van Hellemond, J.J.; Henze, K.; Woehle, C.; Gould, S.B.; Yu, R.Y.; van der Giezen, M.; Tielens, A.G.; Martin, W.F. Biochemistry and evolution of anaerobic energy metabolism in eukaryotes. Microbiol. Mol. Biol. Rev. 2012, 76, 444–495. [Google Scholar] [CrossRef] [PubMed]
- Rafelski, S.M. Mitochondrial network morphology: Building an integrative, geometrical view. BMC Biol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Dhingra, R.; Kirshenbaum, L.A. Regulation of mitochondrial dynamics and cell fate. Circ. J. 2014, 78, 803–810. [Google Scholar] [CrossRef] [PubMed]
- Van der Bliek, A.M.; Shen, Q.; Kawajiri, S. Mechanisms of mitochondrial fission and fusion. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, A.V.; Hermann, M.; Saks, V.; Hengster, P.; Margreiter, R. The cell-type specificity of mitochondrial dynamics. Int. J. Biochem. Cell Biol. 2009, 41, 1928–1939. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, A.V.; Margreiter, R. Heterogeneity of mitochondria and mitochondrial function within cells as another level of mitochondrial complexity. Int. J. Mol. Sci. 2009, 10, 1911–1929. [Google Scholar] [CrossRef] [PubMed]
- Pagliarini, D.J.; Calvo, S.E.; Chang, B.; Sheth, S.A.; Vafai, S.B.; Ong, S.E.; Walford, G.A.; Sugiana, C.; Boneh, A.; Chen, W.K. A mitochondrial protein compendium elucidates complex I disease biology. Cell 2008, 134, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Chacinska, A.; Koehler, C.M.; Milenkovic, D.; Lithgow, T.; Pfanner, N. Importing mitochondrial proteins: Machineries and mechanisms. Cell 2009, 138, 628–644. [Google Scholar] [CrossRef] [PubMed]
- Baker, M.J.; Frazier, A.E.; Gulbis, J.M.; Ryan, M.T. Mitochondrial protein-import machinery: Correlating structure with function. Trends Cell Biol. 2007, 17, 456–464. [Google Scholar] [CrossRef] [PubMed]
- Baker, M.J.; Palmer, C.S.; Stojanovski, D. Mitochondrial protein quality control in health and disease. Br. J. Pharmacol. 2014, 171, 1870–1889. [Google Scholar] [CrossRef] [PubMed]
- Baker, M.J.; Tatsuta, T.; Langer, T. Quality control of mitochondrial proteostasis. Cold Spring Harb. Perspect. Biol. 2011, 3. [Google Scholar] [CrossRef] [PubMed]
- Voos, W.; Ward, L.A.; Truscott, K.N. The role of AAA+ proteases in mitochondrial protein biogenesis, homeostasis and activity control. Subcell Biochem. 2013, 66, 223–263. [Google Scholar] [PubMed]
- Taylor, E.B.; Rutter, J. Mitochondrial quality control by the ubiquitin-proteasome system. Biochem. Soc. Trans. 2011, 39, 1509–1513. [Google Scholar] [CrossRef] [PubMed]
- Fritz, S.; Weinbach, N.; Westermann, B. Mdm30 is an F-box protein required for maintenance of fusion-competent mitochondria in yeast. Mol. Biol. Cell 2003, 14, 2303–2313. [Google Scholar] [CrossRef] [PubMed]
- Sickmann, A.; Reinders, J.; Wagner, Y.; Joppich, C.; Zahedi, R.; Meyer, H.E.; Schönfisch, B.; Perschil, I.; Chacinska, A.; Guiard, B. The proteome of Saccharomyces cerevisiae mitochondria. Proc. Natl. Acad. Sci. USA 2003, 100, 13207–13212. [Google Scholar] [CrossRef] [PubMed]
- Jeon, H.B.; Choi, E.S.; Yoon, J.H.; Hwang, J.H.; Chang, J.W.; Lee, E.K.; Choi, H.W.; Park, Z.Y.; Yoo, Y.J. A proteomics approach to identify the ubiquitinated proteins in mouse heart. Biochem. Biophys. Res. Commun. 2007, 357, 731–736. [Google Scholar] [CrossRef] [PubMed]
- Schubert, U.; Antón, L.C.; Gibbs, J.; Norbury, C.C.; Yewdell, J.W.; Bennink, J.R. Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature 2000, 404, 770–774. [Google Scholar] [PubMed]
- Chatenay-Lapointe, M.; Shadel, G.S. Stressed-out mitochondria get MAD. Cell Metab. 2010, 12, 559–560. [Google Scholar] [CrossRef] [PubMed]
- Heo, J.-M.; Livnat-Levanon, N.; Taylor, E.B.; Jones, K.T.; Dephoure, N.; Ring, J.; Xie, J.; Brodsky, J.L.; Madeo, F.; Gygi, S.P. A stress-responsive system for mitochondrial protein degradation. Mol. Cell 2010, 40, 465–480. [Google Scholar] [CrossRef] [PubMed]
- Neutzner, A.; Youle, R.J.; Karbowski, M. Outer mitochondrial membrane protein degradation by the proteasome. Novartis Found. Symp. 2007, 287, 4–14. [Google Scholar] [PubMed]
- Margineantu, D.H.; Emerson, C.B.; Diaz, D.; Hockenbery, D.M. Hsp90 inhibition decreases mitochondrial protein turnover. PLoS ONE 2007, 2, e1066. [Google Scholar] [CrossRef] [PubMed]
- Azzu, V.; Brand, M.D. Degradation of an intramitochondrial protein by the cytosolic proteasome. J. Cell Sci. 2010, 123, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Clarke, K.J.; Adams, A.E.; Manzke, L.H.; Pearson, T.W.; Borchers, C.H.; Porter, R.K. A role for ubiquitinylation and the cytosolic proteasome in turnover of mitochondrial uncoupling protein 1 (UCP1). Biochim. Biophys. Acta 2012, 1817, 1759–1767. [Google Scholar] [CrossRef] [PubMed]
- Radke, S.; Chander, H.; Schäfer, P.; Meiss, G.; Krüger, R.; Schulz, J.B.; Germain, D. Mitochondrial protein quality control by the proteasome involves ubiquitination and the protease Omi. J. Biol. Chem. 2008, 283, 12681–12685. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Peng, G.; Wang, Y.; Fang, S.; Karbowski, M. The AAA-ATPase p97 is essential for outer mitochondrial membrane protein turnover. Mol. Biol. Cell. 2011, 22, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Braun, R.J.; Sommer, C.; Leibiger, C.; Gentier, R.J.G.; Dumit, V.I.; Paduch, K.; Eisenberg, T.; Habernig, L.; Trausinger, G.; Magnes, C. Accumulation of basic amino acids at mitochondria dictates the cytotoxicity of aberrant ubiquitin. Cell Rep. 2015. [Google Scholar] [CrossRef] [PubMed]
- Bartolome, F.; Wu, H.C.; Burchell, V.S.; Preza, E.; Wray, S.; Mahoney, C.J.; Fox, N.C.; Calvo, A.; Canosa, A.; Moglia, C.; et al. Pathogenic VCP mutations induce mitochondrial uncoupling and reduced ATP levels. Neuron 2013, 78, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Braun, R.J.; Zischka, H.; Madeo, F.; Eisenberg, T.; Wissing, S.; Büttner, S.; Engelhardt, S.M.; Büringer, D.; Ueffing, M. Crucial mitochondrial impairment upon CDC48 mutation in apoptotic yeast. J. Biol. Chem. 2006, 281, 25757–25767. [Google Scholar] [CrossRef] [PubMed]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [PubMed]
- Koyano, F.; Matsuda, N. Molecular mechanisms underlying PINK1 and Parkin catalyzed ubiquitylation of substrates on damaged mitochondria. Biochim. Biophys. Acta 2015. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, D.M.; Lissounov, A.; Brzovic, P.S.; Klevit, R.E. UBCH7 reactivity profile reveals parkin and HHARI to be RING/HECT hybrids. Nature 2011, 474, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.S.; Saiki, S.; Kawajiri, S.; Sato, F.; Kimura, M.; et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Kane, L.A.; Lazarou, M.; Fogel, A.I.; Li, Y.; Yamano, K.; Sarraf, S.A.; Banerjee, S.; Youle, R.J. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J. Cell Biol. 2014, 205, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Koyano, F.; Okatsu, K.; Kosako, H.; Tamura, Y.; Go, E.; Kimura, M.; Kimura, Y.; Tsuchiya, H.; Yoshihara, H.; Hirokawa, T.; et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 2014, 510, 162–166. [Google Scholar] [PubMed]
- Kazlauskaite, A.; Kondapalli, C.; Gourlay, R.; Campbell, D.G.; Ritorto, M.S.; Hofmann, K.; Alessi, D.R.; Knebel, A.; Trost, M.; Muqit, M.M.K. Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser65. Biochem. J. 2014, 460, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Geisler, S.; Holmström, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010, 12, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Glauser, L.; Sonnay, S.; Stafa, K.; Moore, D.J. Parkin promotes the ubiquitination and degradation of the mitochondrial fusion factor mitofusin 1. J. Neurochem. 2011, 118, 636–645. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.; Tanaka, A.; Suen, D.-F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Sarraf, S.A.; Raman, M.; Guarani-Pereira, V.; Sowa, M.E.; Huttlin, E.L.; Gygi, S.P.; Harper, J.W. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature 2013, 496, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Rana, A.; Rera, M.; Walker, D.W. Parkin overexpression during aging reduces proteotoxicity, alters mitochondrial dynamics, and extends lifespan. Proc. Natl. Acad. Sci. USA 2013, 110, 8638–8643. [Google Scholar] [CrossRef] [PubMed]
- Melrose, H.L.; Lincoln, S.J.; Tyndall, G.M.; Farrer, M.J. Parkinson’s disease: A rethink of rodent models. Exp. Brain Res. 2006, 173, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Sterky, F.H.; Lee, S.; Wibom, R.; Olson, L.; Larsson, N.G. Impaired mitochondrial transport and Parkin-independent degeneration of respiratory chain-deficient dopamine neurons in vivo. Proc. Natl. Acad. Sci. USA 2011, 108, 12937–12942. [Google Scholar] [CrossRef] [PubMed]
- Nagashima, S.; Tokuyama, T.; Yonashiro, R.; Inatome, R.; Yanagi, S. Roles of mitochondrial ubiquitin ligase MITOL/MARCH5 in mitochondrial dynamics and diseases. J. Biochem. 2014, 155, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Karbowski, M.; Neutzner, A.; Youle, R.J. The mitochondrial E3 ubiquitin ligase MARCH5 is required for Drp1 dependent mitochondrial division. J. Cell Biol. 2007, 178, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Yonashiro, R.; Ishido, S.; Kyo, S.; Fukuda, T.; Goto, E.; Matsuki, Y.; Ohmura-Hoshino, M.; Sada, K.; Hotta, H.; Yamamura, H.; et al. A novel mitochondrial ubiquitin ligase plays a critical role in mitochondrial dynamics. EMBO J. 2006, 25, 3618–3626. [Google Scholar] [CrossRef] [PubMed]
- Yonashiro, R.; Sugiura, A.; Miyachi, M.; Fukuda, T.; Matsushita, N.; Inatome, R.; Ogata, Y.; Suzuki, T.; Dohmae, N.; Yanagi, S. Mitochondrial ubiquitin ligase MITOL ubiquitinates mutant SOD1 and attenuates mutant SOD1-induced reactive oxygen species generation. Mol. Biol. Cell 2009, 20, 4524–4530. [Google Scholar] [CrossRef] [PubMed]
- Durcan, T.M.; Kontogiannea, M.; Thorarinsdottir, T.; Fallon, L.; Williams, A.J.; Djarmati, A.; Fantaneanu, T.; Paulson, H.L.; Fon, E.A. The Machado–Joseph disease-associated mutant form of ataxin-3 regulates parkin ubiquitination and stability. Hum. Mol. Genet. 2011, 20, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Wang, H.; Perry, S.W.; Figueiredo-Pereira, M.E. Negative regulation of 26S proteasome stability via calpain-mediated cleavage of Rpn10 upon mitochondrial dysfunction in neurons. J. Biol. Chem. 2013, 288, 12161–12174. [Google Scholar] [CrossRef] [PubMed]
- Livnat-Levanon, N.; Kevei, É.; Kleifeld, O.; Krutauz, D.; Segref, A.; Rinaldi, T.; Erpapazoglou, Z.; Cohen, M.; Reis, N.; Hoppe, T.; et al. Reversible 26S proteasome disassembly upon mitochondrial stress. Cell Rep. 2014, 7, 1371–1380. [Google Scholar] [CrossRef] [PubMed]
- Segref, A.; Kevei, É.; Pokrzywa, W.; Schmeisser, K.; Mansfeld, J.; Livnat-Levanon, N.; Ensenauer, R.; Glickman, M.H.; Ristow, M.; Hoppe, T. Pathogenesis of human mitochondrial diseases is modulated by reduced activity of the ubiquitin/proteasome system. Cell Metab. 2014, 19, 642–652. [Google Scholar] [CrossRef] [PubMed]
- Grune, T.; Reinheckel, T.; Davies, K.J. Degradation of oxidized proteins in K562 human hematopoietic cells by proteasome. J. Biol. Chem. 1996, 271, 15504–15509. [Google Scholar] [PubMed]
- Grune, T.; Merker, K.; Sandig, G.; Davies, K.J.A. Selective degradation of oxidatively modified protein substrates by the proteasome. Biochem. Biophys. Res. Commun. 2003, 305, 709–718. [Google Scholar] [CrossRef]
- Shringarpure, R.; Grune, T.; Mehlhase, J.; Davies, K.J.A. Ubiquitin conjugation is not required for the degradation of oxidized proteins by proteasome. J. Biol. Chem. 2003, 278, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Hershko, A.; Heller, H.; Elias, S.; Ciechanover, A. Components of ubiquitin-protein ligase system. Resolution, affinity purification, and role in protein breakdown. J. Biol. Chem. 1983, 258, 8206–8214. [Google Scholar] [PubMed]
- Eytan, E.; Ganoth, D.; Armon, T.; Hershko, A. ATP-dependent incorporation of 20S protease into the 26S complex that degrades proteins conjugated to ubiquitin. Proc. Natl. Acad. Sci. USA 1989, 86, 7751–7755. [Google Scholar] [CrossRef] [PubMed]
- Dahlmann, B.; Kuehn, L.; Reinauer, H. Studies on the activation by ATP of the 26 S proteasome complex from rat skeletal muscle. Biochem. J. 1995, 309, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Kleijnen, M.F.; Roelofs, J.; Park, S.; Hathaway, N.A.; Glickman, M.; King, R.W.; Finley, D. Stability of the proteasome can be regulated allosterically through engagement of its proteolytic active sites. Nat. Struct. Mol. Biol. 2007, 14, 1180–1188. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Zhang, X.; Li, S.; Liu, N.; Lian, W.; McDowell, E.; Zhou, P.; Zhao, C.; Guo, H.; Zhang, C.; et al. Physiological levels of ATP negatively regulate proteasome function. Cell Res. 2010, 20, 1372–1385. [Google Scholar] [CrossRef] [PubMed]
- Höglinger, G.U.; Carrard, G.; Michel, P.P.; Medja, F.; Lombès, A.; Ruberg, M.; Friguet, B.; Hirsch, E.C. Dysfunction of mitochondrial complex I and the proteasome: Interactions between two biochemical deficits in a cellular model of Parkinson’s disease. J. Neurochem. 2003, 86, 1297–1307. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Thorpe, J.; Ding, Q.; El-Amouri, I.S.; Keller, J.N. Proteasome synthesis and assembly are required for survival during stationary phase. Free Radic. Biol. Med. 2004, 37, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Vernace, V.A.; Arnaud, L.; Schmidt-Glenewinkel, T.; Figueiredo-Pereira, M.E. Aging perturbs 26S proteasome assembly in Drosophila melanogaster. FASEB J. 2007, 21, 2672–2682. [Google Scholar] [CrossRef] [PubMed]
- Keller, J.N.; Huang, F.F.; Markesbery, W.R. Decreased levels of proteasome activity and proteasome expression in aging spinal cord. Neuroscience 2000, 98, 149–156. [Google Scholar] [CrossRef]
- Dasuri, K.; Zhang, L.; Ebenezer, P.; Liu, Y.; Fernandez-Kim, S.O.; Keller, J.N. Aging and dietary restriction alter proteasome biogenesis and composition in the brain and liver. Mech. Ageing Dev. 2009, 130, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Ferrington, D.A.; Husom, A.D.; Thompson, L.V. Altered proteasome structure, function, and oxidation in aged muscle. FASEB J. 2005, 19, 644–646. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.S.; Hwang, J.S.; Chang, I.; Kim, S. Age-associated decrease in proteasome content and activities in human dermal fibroblasts: Restoration of normal level of proteasome subunits reduces aging markers in fibroblasts from elderly persons. J. Gerontol. A Biol. Sci. Med. Sci. 2007, 62, 490–499. [Google Scholar] [CrossRef] [PubMed]
- Chondrogianni, N.; Voutetakis, K.; Kapetanou, M.; Delitsikou, V.; Papaevgeniou, N.; Sakellari, M.; Lefaki, M.; Filippopoulou, K.; Gonos, E.S. Proteasome activation: An innovative promising approach for delaying aging and retarding age-related diseases. Ageing Res. Rev. 2015, 23, 37–55. [Google Scholar] [CrossRef] [PubMed]
- Vernace, V.A.; Schmidt-Glenewinkel, T.; Figueiredo-Pereira, M.E. Aging and regulated protein degradation: Who has the UPPer hand? Aging Cell. 2007, 6, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef] [PubMed]
- Schwarze, S.R.; Lee, C.M.; Chung, S.S.; Roecker, E.B.; Weindruch, R.; Aiken, J.M. High levels of mitochondrial DNA deletions in skeletal muscle of old rhesus monkeys. Mech. Ageing Dev. 1995, 83, 91–101. [Google Scholar] [CrossRef]
- Khaidakov, M.; Heflich, R.H.; Manjanatha, M.G.; Myers, M.B.; Aidoo, A. Accumulation of point mutations in mitochondrial DNA of aging mice. Mutat. Res. 2003, 526, 1–7. [Google Scholar] [CrossRef]
- Corral-Debrinski, M.; Horton, T.; Lott, M.T.; Shoffner, J.M.; Beal, M.F.; Wallace, D.C. Mitochondrial DNA deletions in human brain: Regional variability and increase with advanced age. Nat. Genet. 1992, 2, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Soong, N.W.; Hinton, D.R.; Cortopassi, G.; Arnheim, N. Mosaicism for a specific somatic mitochondrial DNA mutation in adult human brain. Nat. Genet. 1992, 2, 318–323. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.M.; Stewart, J.B.; Hagström, E.; Brené, S.; Mourier, A.; Coppotelli, G.; Freyer, C.; Lagouge, M.; Hoffer, B.J.; Olson, L.; et al. Germline mitochondrial DNA mutations aggravate ageing and can impair brain development. Nature 2013, 501, 412–415. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.M.; Öberg, J.; Brené, S.; Coppotelli, G.; Terzioglu, M.; Pernold, K.; Goiny, M.; Sitnikov, R.; Kehr, J.; Trifunovic, A.; et al. High brain lactate is a hallmark of aging and caused by a shift in the lactate dehydrogenase A/B ratio. Proc. Natl. Acad. Sci. USA 2010, 107, 20087–20092. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.M.; Coppotelli, G.; Hoffer, B.J.; Olson, L. Maternally transmitted mitochondrial DNA mutations can reduce lifespan. Sci. Rep. 2014. [Google Scholar] [CrossRef] [PubMed]
- Trifunovic, A.; Wredenberg, A.; Falkenberg, M.; Spelbrink, J.N.; Rovio, A.T.; Bruder, C.E.; Bohlooly, Y.M.; Gidlöf, S.; Oldfors, A.; Wibom, R.; et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 2004, 429, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Kujoth, G.C.; Hiona, A.; Pugh, T.D.; Someya, S.; Panzer, K.; Wohlgemuth, S.E.; Hofer, T.; Seo, A.Y.; Sullivan, R.; Jobling, W.A.; et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 2005, 309, 481–484. [Google Scholar] [CrossRef] [PubMed]
- Larsson, N.G.; Oldfors, A. Mitochondrial myopathies. Acta Physiol. Scand. 2001, 171, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Cottrell, D.A.; Turnbull, D.M. Mitochondria and ageing. Curr. Opin. Clin. Nutr. Metab. Care 2000, 3, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Cook, C.; Petrucelli, L. A critical evaluation of the ubiquitin-proteasome system in Parkinson’s disease. Biochim. Biophys. Acta 2009, 1792, 664–675. [Google Scholar] [CrossRef] [PubMed]
- Ekstrand, M.I.; Terzioglu, M.; Galter, D.; Zhu, S.; Hofstetter, C.; Lindqvist, E.; Thams, S.; Bergstrand, A.; Hansson, F.S.; Trifunovic, A.; et al. Progressive parkinsonism in mice with respiratory-chain-deficient dopamine neurons. Proc. Natl. Acad. Sci. USA 2007, 104, 1325–1330. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, N.; Ferger, B. Neurochemical findings in the MPTP model of Parkinson’s disease. J. Neural. Transm. 2001, 108, 1263–1282. [Google Scholar] [CrossRef] [PubMed]
- Harrington, C.R. The molecular pathology of Alzheimer’s disease. Neuroimaging Clin. N. Am. 2012, 22, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Riederer, B.M.; Leuba, G.; Vernay, A.; Riederer, I.M. The role of the ubiquitin proteasome system in Alzheimer’s disease. Exp. Biol. Med. 2011, 236, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Friedland-Leuner, K.; Stockburger, C.; Denzer, I.; Eckert, G.P.; Müller, W.E. Mitochondrial dysfunction: Cause and consequence of Alzheimer’s disease. Prog. Mol. Biol. Transl. Sci. 2014, 127, 183–210. [Google Scholar] [PubMed]
- Argyropoulou, A.; Aligiannis, N.; Trougakos, I.P.; Skaltsounis, A.L. Natural compounds with anti-ageing activity. Nat. Prod. Rep. 2013, 30, 1412–1437. [Google Scholar] [CrossRef] [PubMed]
- De Cabo, R.; Carmona-Gutierrez, D.; Bernier, M.; Hall, M.N.; Madeo, F. The search for antiaging interventions: From elixirs to fasting regimens. Cell 2014, 157, 1515–1526. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, S.; Moriguchi, E.H.; Ishikawa, P.; Hekman, P.; Nara, Y.; Mimura, G.; Moriguchi, Y.; Yamori, Y. Fish intake and cardiovascular risk among middle-aged Japanese in Japan and Brazil. J. Cardiovasc. Risk 1997, 4, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Huffman, K.M.; Slentz, C.A.; Bateman, L.A.; Thompson, D.; Muehlbauer, M.J.; Bain, J.R.; Stevens, R.D.; Wenner, B.R.; Kraus, V.B.; Newgard, C.B.; et al. Exercise-induced changes in metabolic intermediates, hormones, and inflammatory markers associated with improvements in insulin sensitivity. Diabetes Care 2011, 34, 174–176. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ross, J.M.; Olson, L.; Coppotelli, G. Mitochondrial and Ubiquitin Proteasome System Dysfunction in Ageing and Disease: Two Sides of the Same Coin? Int. J. Mol. Sci. 2015, 16, 19458-19476. https://doi.org/10.3390/ijms160819458
Ross JM, Olson L, Coppotelli G. Mitochondrial and Ubiquitin Proteasome System Dysfunction in Ageing and Disease: Two Sides of the Same Coin? International Journal of Molecular Sciences. 2015; 16(8):19458-19476. https://doi.org/10.3390/ijms160819458
Chicago/Turabian StyleRoss, Jaime M., Lars Olson, and Giuseppe Coppotelli. 2015. "Mitochondrial and Ubiquitin Proteasome System Dysfunction in Ageing and Disease: Two Sides of the Same Coin?" International Journal of Molecular Sciences 16, no. 8: 19458-19476. https://doi.org/10.3390/ijms160819458
APA StyleRoss, J. M., Olson, L., & Coppotelli, G. (2015). Mitochondrial and Ubiquitin Proteasome System Dysfunction in Ageing and Disease: Two Sides of the Same Coin? International Journal of Molecular Sciences, 16(8), 19458-19476. https://doi.org/10.3390/ijms160819458