
Direct Injection of CRISPR/Cas9-Related mRNA into Cytoplasm of Parthenogenetically Activated Porcine Oocytes Causes Frequent Mosaicism for Indel Mutations

 , , and
, , and
Abstract
:
1. Introduction
2. Results and Discussion
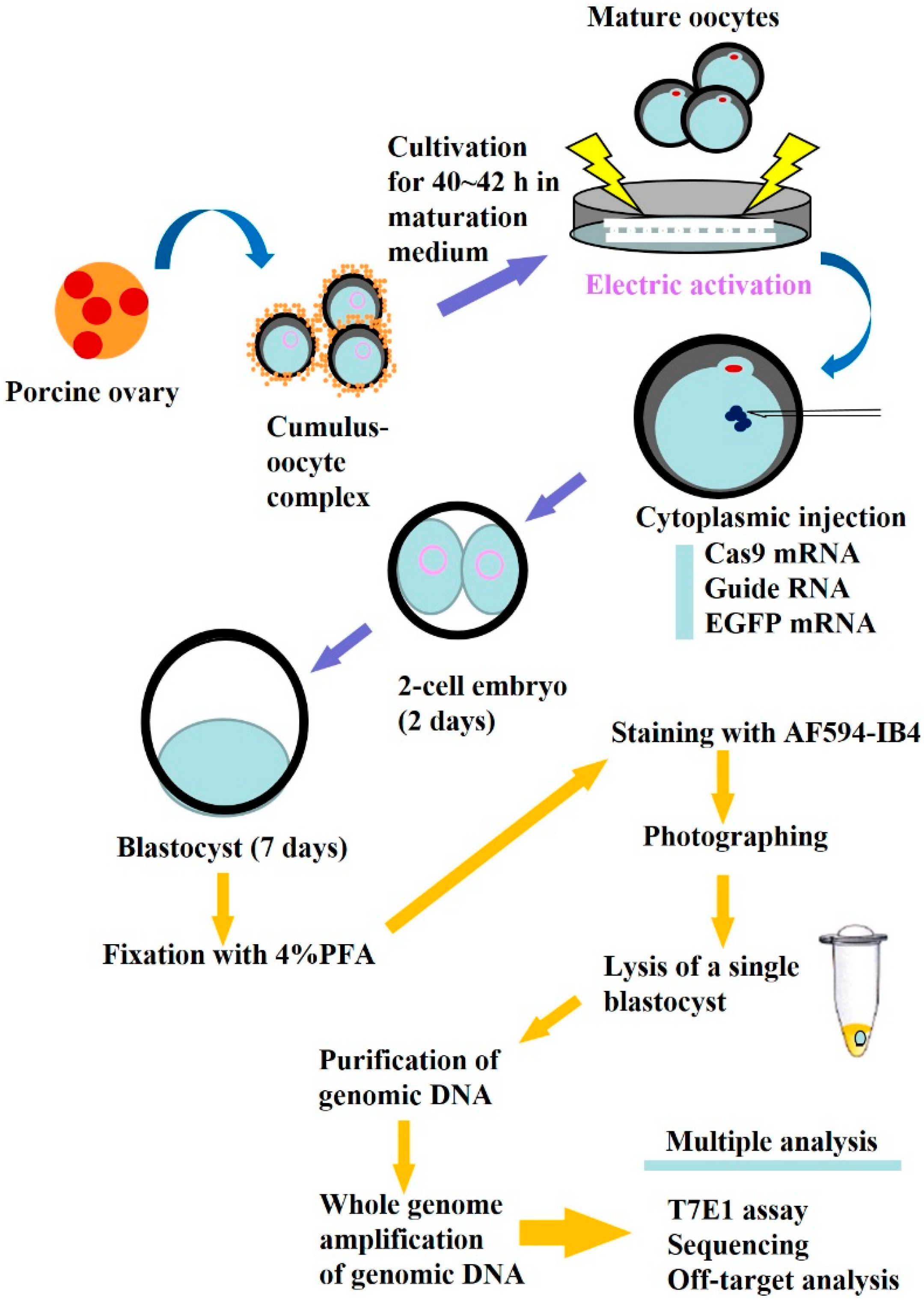
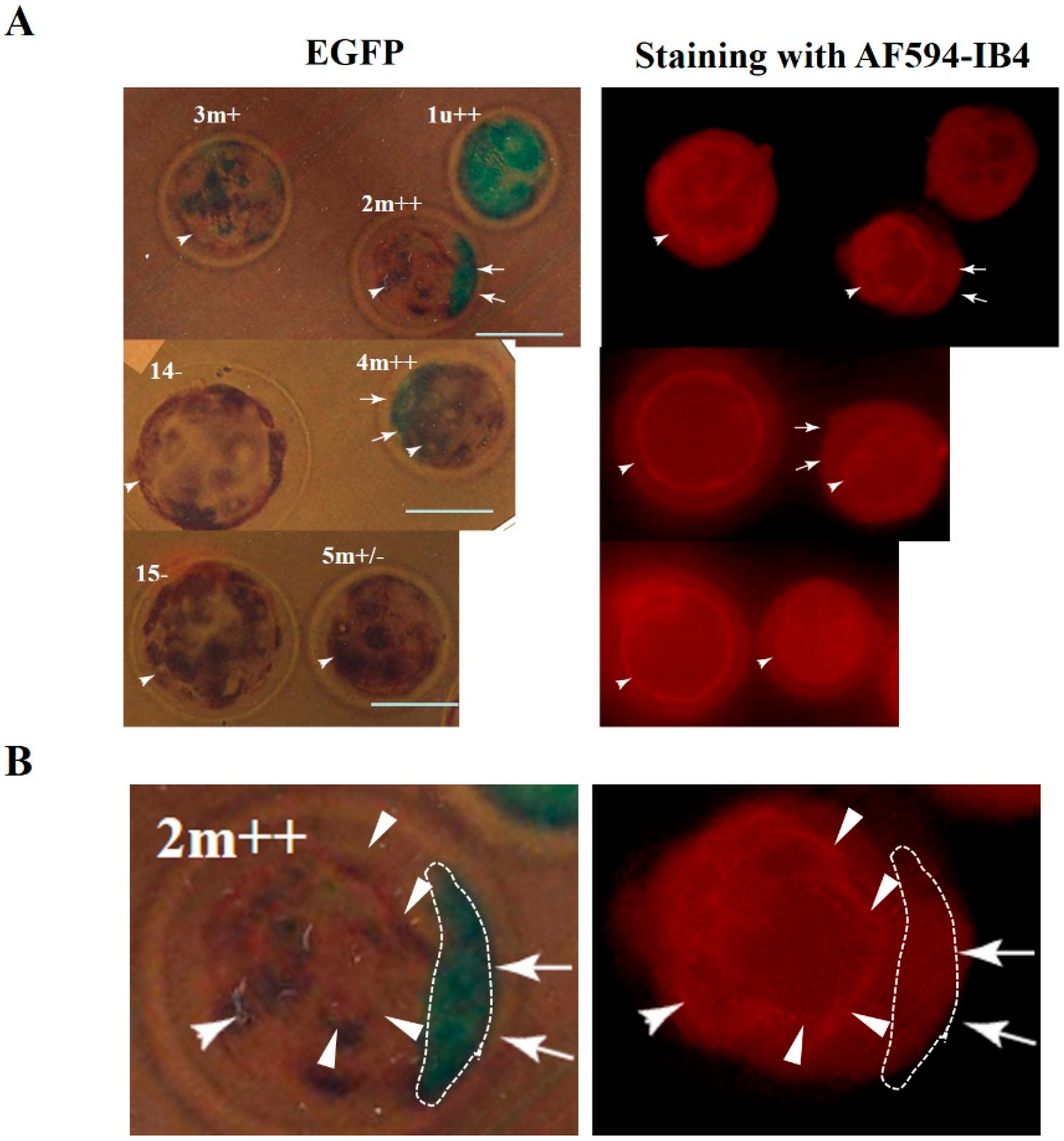
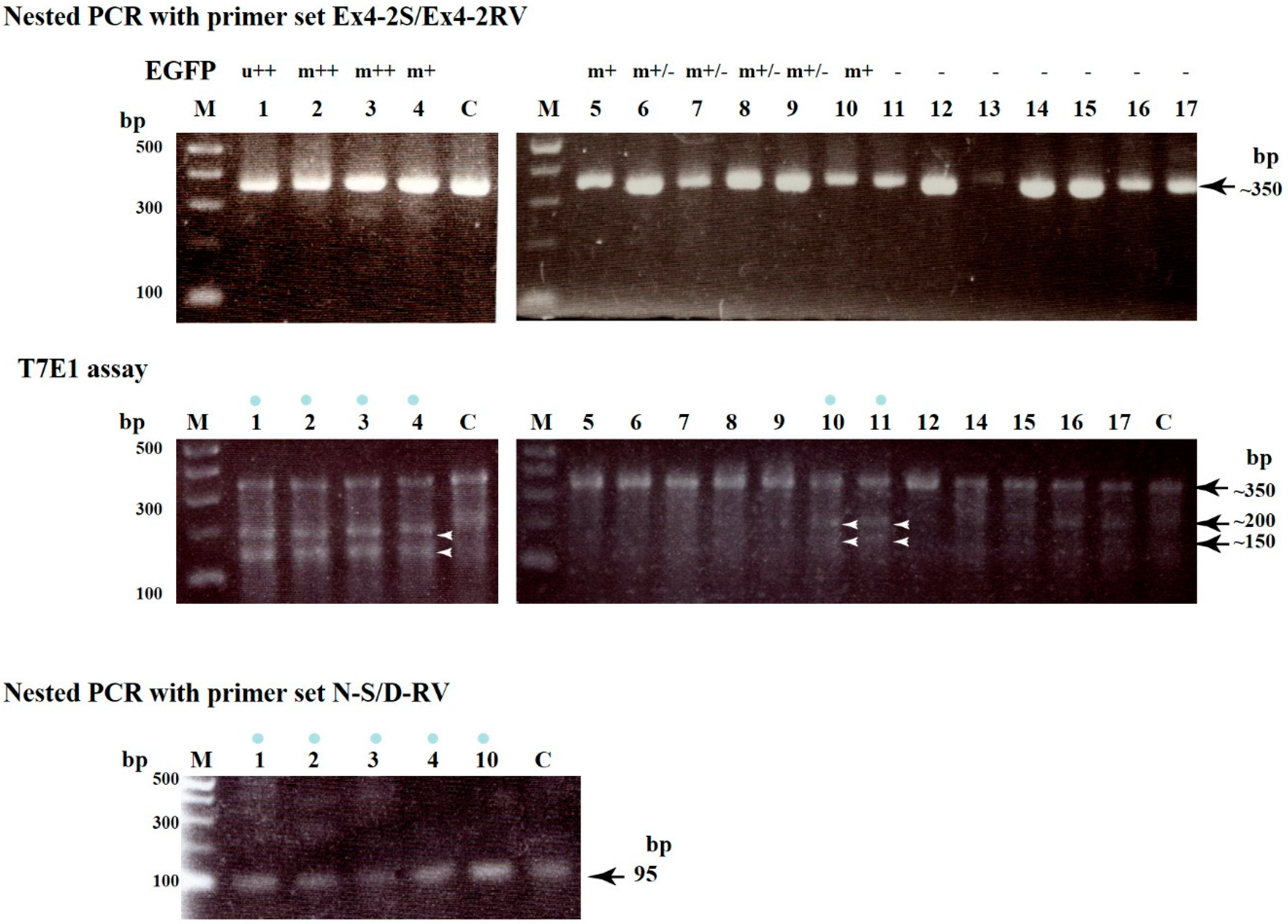
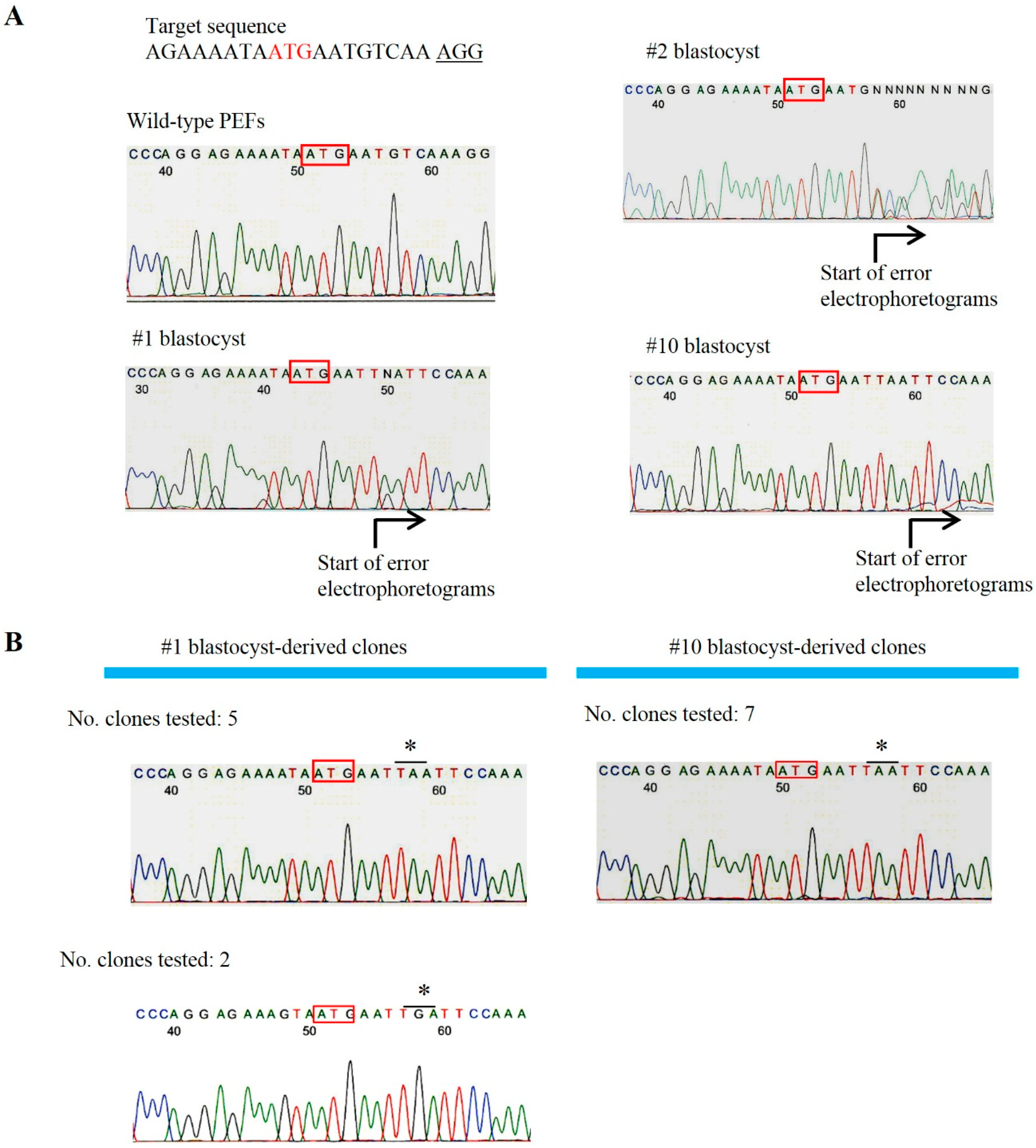
2.1. Evaluation of CRISPR/Cas9-Based Genome Editing Ability in Porcine Embryos

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
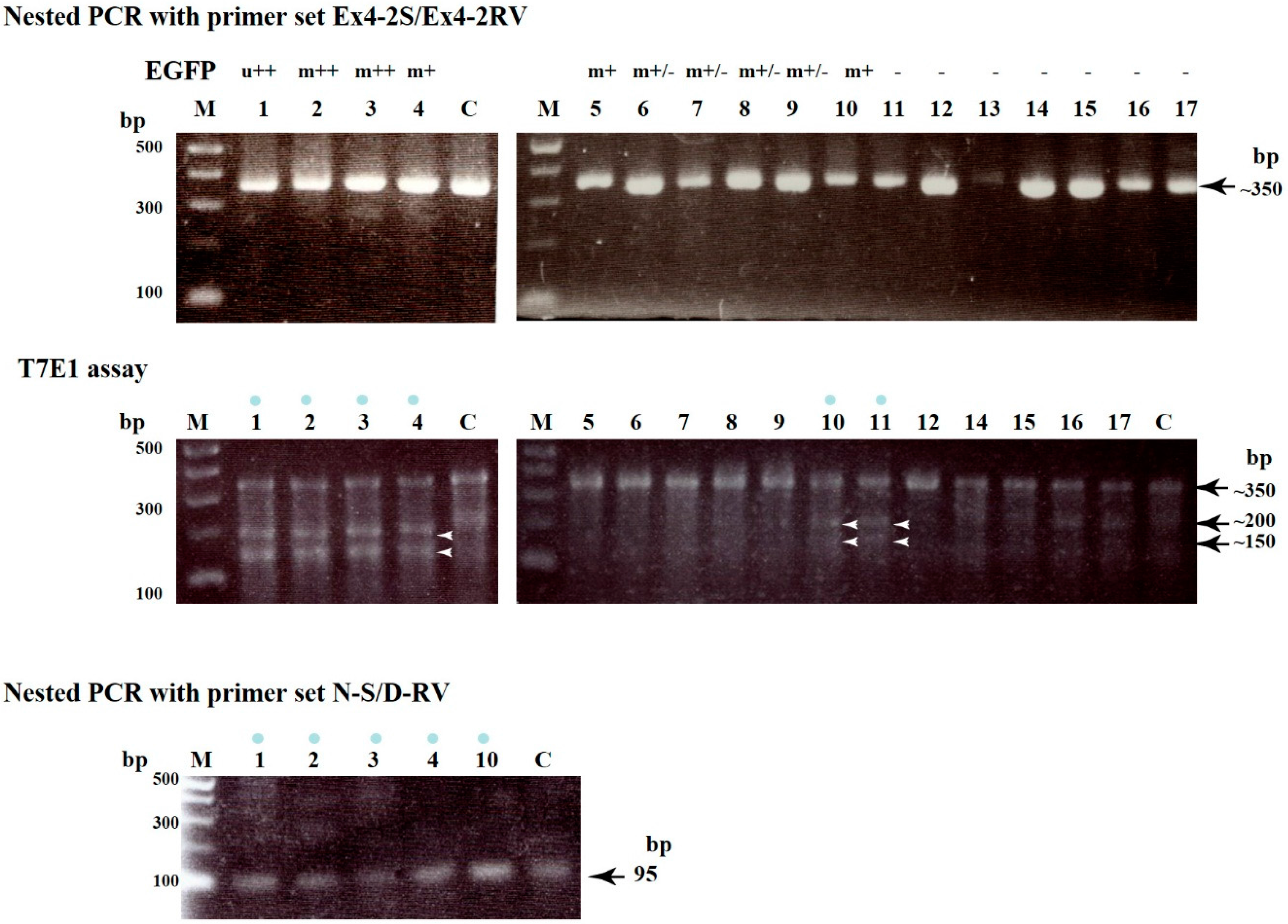
{kind=link}
| Name of Blastocysts | Fluorescence Observation after Staining with AF594-IB4 EGFP-Derived | Production of ~350 bp Fragment d AF594-Derived | Indel Mutations after T7E1 Assay e | |
|---|---|---|---|---|
| Fluorescence b | Fluorescence c | |||
| 1 | u+ | Totally decrease | OK | Yes |
| 2 | m++ | Mosaic | OK | Yes |
| 3 | m+ | Mosaic | OK | Yes |
| 4 | m++ | Mosaic | OK | Yes |
| 5 | m+/− | Uniform | OK | No |
| 6 | m+/− | Uniform | OK | No |
| 7 | m+/− | Uniform | OK | No |
| 8 | m+/− | Uniform | OK | No |
| 9 | m+/− | Uniform | OK | No |
| 10 | m+ | Mosaic | OK | Yes |
| 11 | − | Uniform | OK | Yes |
| 12 | − | Uniform | OK | No |
| 13 | − | Uniform | Insufficient | ND |
| 14 | − | Uniform | OK | No |
| 15 | − | Uniform | OK | No |
| 16 | − | Uniform | OK | No |
| 17 | − | Uniform | OK | No |
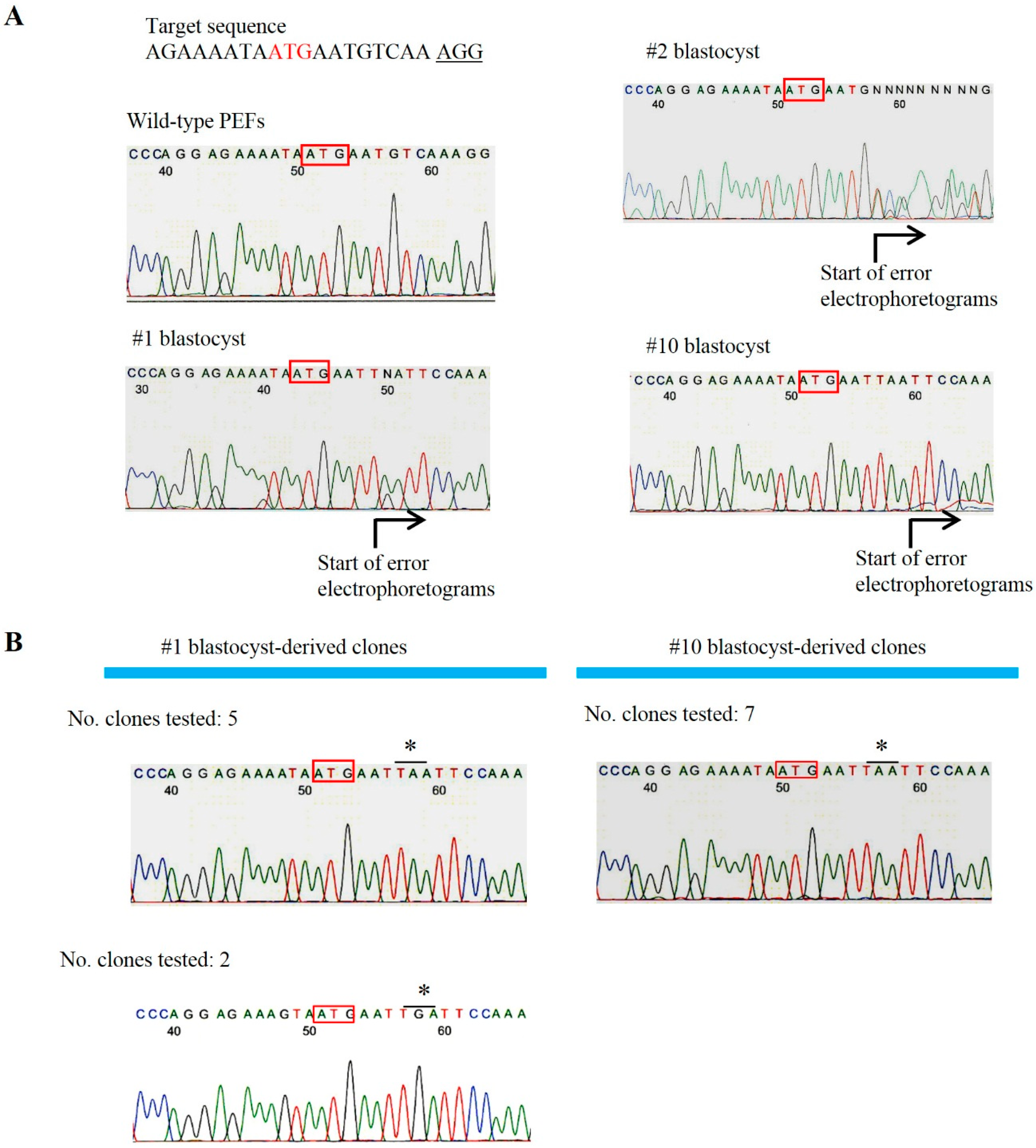
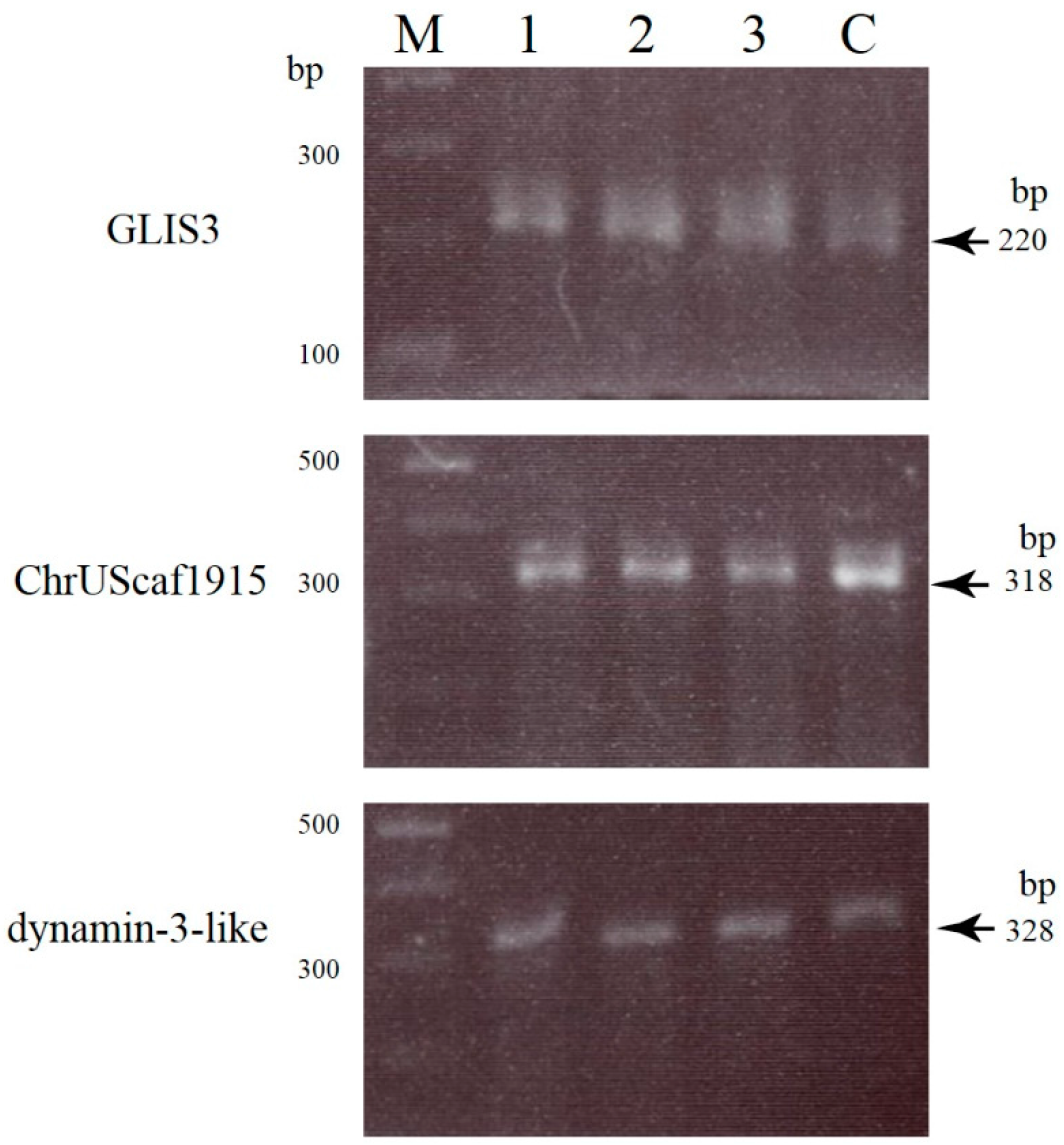
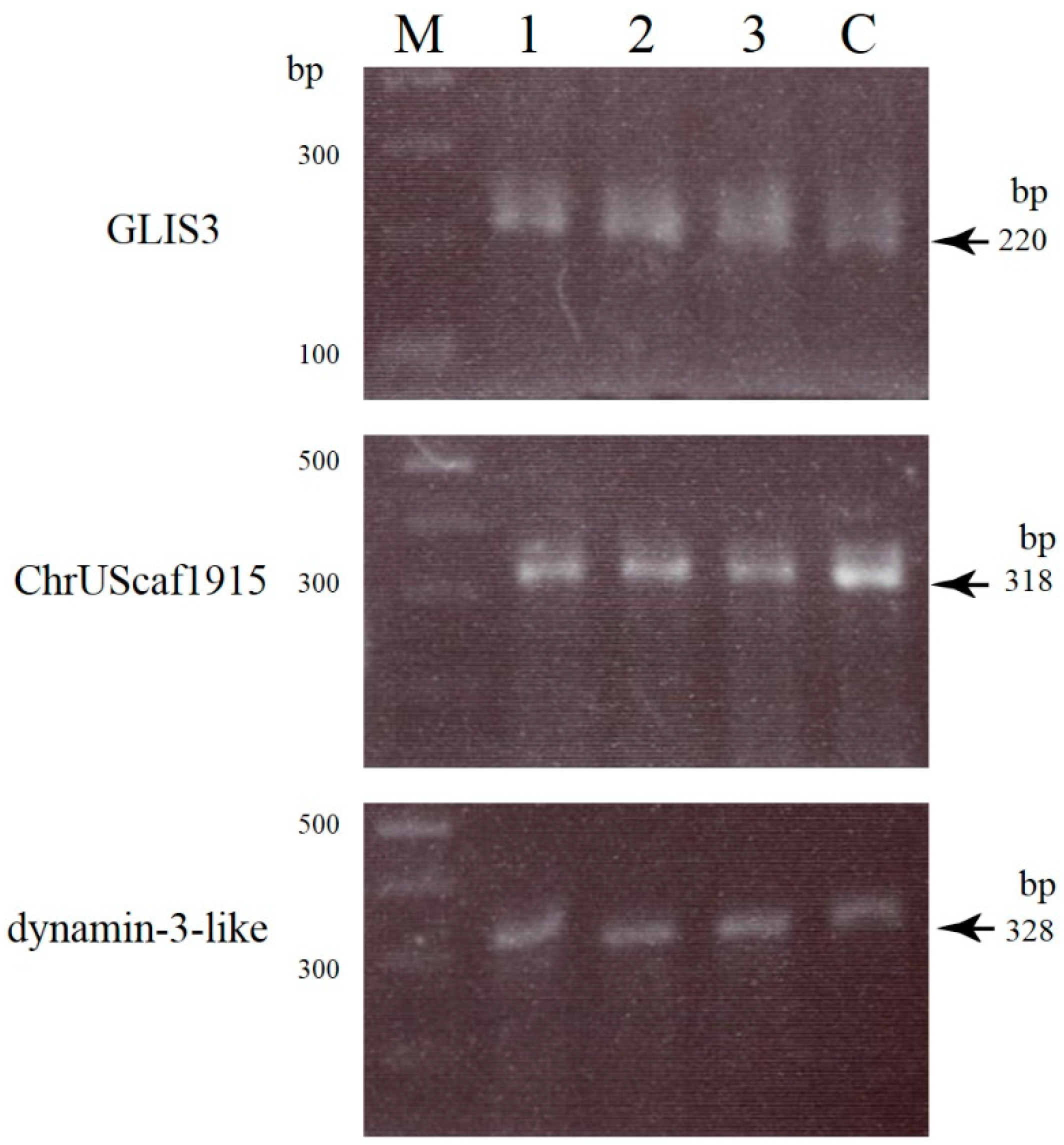
2.2. Uncovering Possible Off-Target Mutations in the CRISPR/Cas9-Related mRNA-Injected Porcine Embryos
3. Experimental Section
3.1. In Vitro Synthesis of Cas9 mRNA, gRNA and EGFP mRNA
3.2. Production of PA Embryos and Cytoplasmic Microinjection of RNAs
3.3. T7E1-Based Assay and Sanger Sequencing of Mutated Sites
3.4. Screening and Detection of Off-Target Sites
3.5. Staining with AF594-IB4 and Fluorescence Detection
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Vajta, G.; Zhang, Y.; Macháty, Z. Somatic cell nuclear transfer in pigs: Recent achievements and future possibilities. Reprod. Fertil. Dev. 2007, 19, 403–423. [Google Scholar]
- Lee, K.; Prather, R.S. Advancements in somatic cell nuclear transfer and future perspectives. Anim. Front. 2013, 3, 56–61. [Google Scholar]
- Harrison, M.M.; Jenkins, B.V.; O’Connor-Giles, K.M.; Wildonger, J.A. CRISPR view of development. Genes Dev. 2014, 28, 1859–1872. [Google Scholar]
- Hsu, P.D.; Lander, E.S.; Zhang, F. Development and applications of CRISPR-Cas9 for genome engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef]
- Horvath, P.; Barrangou, R. CRISPR/Cas, the immune system of bacteria and archaea. Science 2010, 327, 167–170. [Google Scholar] [CrossRef] [PubMed]
- Bhaya, D.; Davison, M.; Barrangou, R. CRISPR-Cas systems in bacteria and archaea: Versatile small RNAs for adaptive defense and regulation. Annu. Rev. Genet. 2011, 45, 273–297. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Gaj, T.; Gersbach, C.A.; Barbas, C.F. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013, 31, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Gratz, S.J.; Cummings, A.M.; Nguyen, J.N.; Hamm, D.C.; Donohue, L.K.; Harrison, M.M.; Wildonger, J.; O’Connor-Giles, K.M. Genome engineering of Drosophila with the CRISPR RNA-guided Cas9 nuclease. Genetics 2013, 194, 1029–1035. [Google Scholar] [CrossRef] [PubMed]
- Bassett, A.R.; Tibbit, C.; Ponting, C.P.; Liu, J.L. Highly efficient targeted mutagenesis of Drosophila with the CRISPR/Cas9 system. Cell Rep. 2013, 4, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Ren, M.; Wang, Z.; Zhang, B.; Rong, Y.S.; Jiao, R.; Gao, G. Highly efficient genome modifications mediated by CRISPR/Cas9 in Drosophila. Genetics 2013, 195, 289–291. [Google Scholar] [CrossRef] [PubMed]
- Friedland, A.E.; Tzur, Y.B.; Esvelt, K.M.; Colaiacovo, M.P.; Church, G.M.; Calarco, J.A. Heritable genome editing in C. elegans via a CRISPR-Cas9 system. Nat. Methods 2013, 10, 741–743. [Google Scholar] [CrossRef] [PubMed]
- Chang, N.; Sun, C.; Gao, L.; Zhu, D.; Xu, X.; Zhu, X.; Xiong, J.-W.; Xi, J.J. Genome editing with RNA-guided Cas9 nuclease in zebrafish embryos. Cell Res. 2013, 23, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Hwang, W.Y.; Fu, Y.; Reyon, D.; Maeder, M.L.; Tsai, S.Q.; Sander, J.D.; Peterson, R.T.; Yeh, J.-R.J.; Joung, J.K. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat. Biotechnol. 2013, 31, 227–229. [Google Scholar] [CrossRef] [PubMed]
- Auer, T.O.; Duroure, K.; de Cian, A.; Concordet, J.P.; del Bene, F. Highly efficient CRISPR/Cas9-mediated knock-in in zebrafish by homology-independent DNA repair. Genome Res. 2014, 24, 142–153. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Zhang, J.; Wu, H.; Wang, J.; Ma, K.; Li, Z.; Zhang, X.; Zhang, P.; Huang, X. Generation of gene-modified mice via Cas9/RNA-mediated gene targeting. Cell Res. 2013, 23, 720–723. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yang, H.; Shivalila, C.S.; Dawlaty, M.M.; Cheng, A.W.; Zhang, F.; Jaenisch, R. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 2013, 153, 910–918. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, H.; Shivalila, C.S.; Cheng, A.W.; Shi, L.; Jaenisch, R. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell 2013, 154, 1370–1379. [Google Scholar] [CrossRef] [PubMed]
- Fujii, W.; Kawasaki, K.; Sugiura, K.; Naito, K. Efficient generation of large-scale genome-modified mice using gRNA and CAS9 endonuclease. Nucleic Acids Res. 2013, 41, e187. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Xue, W.; Chen, S.; Bogorad, R.L.; Benedetti, E.; Grompe, M.; Koteliansky, V.; Sharp, P.A.; Jacks, T.; Anderson, D.G. Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nat. Biotechnol. 2014, 32, 551–553. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Xu, Y.; Yu, S.; Lu, L.; Ding, M.; Cheng, J.; Song, G.; Gao, X.; Yao, L.; Fan, D.; et al. An efficient genotyping method for genome-modified animals and human cells generated with CRISPR/Cas9 system. Sci. Rep. 2014, 4, 6420. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Chang, N.; Wang, X.; Zhou, F.; Zhou, X.; Zhu, X.; Xiong, J.-W. Heritable gene-targeting with gRNA/Cas9 in rats. Cell Res. 2013, 23, 1322–1325. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Zhang, X.; Shen, B.; Lu, Y.; Chan, W.; Ma, J.; Bai, L.; Huang, X.; Zhang, L. Generating rats with conditional alleles using CRISPR/Cas9. Cell Res. 2014, 24, 122–125. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Ma, J.; Zhang, X.; Chen, W.; Yu, L.; Lu, Y.; Bai, L.; Shen, B.; Huang, X.; Zhang, L. Generation of eGFP and Cre knockin rats by CRISPR/Cas9. FEBS J. 2014, 281, 3779–3790. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Teng, F.; Li, T.; Zhou, Q. Simultaneous generation and germline transmission of multiple gene mutations in rat using CRISPR-Cas systems. Nat. Biotechnol. 2013, 31, 684–686. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Xu, J.; Zhu, T.; Fan, J.; Lai, L.; Zhang, J.; Chen, Y.E. Effective gene targeting in rabbits using RNA-guided Cas9 nucleases. J. Mol. Cell Biol. 2014, 6, 97–99. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Miyoshi, K.; Nagao, Y.; Nishi, Y.; Ohtsuka, M.; Nakamura, S.; Sakurai, T.; Watanabe, S. The combinational use of CRISPR/Cas9-based gene editing and targeted toxin technology enables efficient biallelic knockout of the α-1,3-galactosyltransferase gene in porcine embryonic fibroblasts. Xenotransplantation 2014, 21, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Hai, T.; Teng, F.; Guo, R.; Li, W.; Zhou, Q. One-step generation of knockout pigs by zygote injection of CRISPR/Cas system. Cell Res. 2014, 24, 372–375. [Google Scholar] [CrossRef] [PubMed]
- Whitworth, K.M.; Lee, K.; Benne, J.A.; Beaton, B.P.; Spate, L.D.; Murphy, S.L.; Samuel, M.S.; Mao, J.; O’Gorman, C.; Walters, E.M.; et al. Use of the CRISPR/Cas9 system to produce genetically engineered pigs from in vitro-derived oocytes and embryos. Biol. Reprod. 2014, 91, 78. [Google Scholar] [CrossRef] [PubMed]
- Heo, Y.T.; Quan, X.; Xu, Y.N.; Baek, S.; Choi, H.; Kim, N.-H.; Kim, J. CRISPR/Cas9 nuclease-mediated gene knock-in in bovine-induced pluripotent cells. Stem Cells Dev. 2015, 24, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Shen, B.; Cui, Y.; Chen, Y.; Wang, J.; Wang, L.; Kang, Y.; Zhao, X.; Si, W.; Li, W.; et al. Generation of gene-modified cynomolgus monkey via Cas9/RNA-mediated gene targeting in one-cell embryos. Cell 2014, 156, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.W.; Kim, S.; Kim, J.M.; Kim, J.S. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat. Biotechnol. 2013, 31, 230–232. [Google Scholar] [CrossRef] [PubMed]
- Park, A.; Won, S.T.; Pentecost, M.; Bartkowski, W.; Lee, B. CRISPR/Cas9 allows efficient and complete knock-in of a destabilization domain-tagged essential protein in a human cell line, allowing rapid knockdown of protein function. PLoS ONE 2014, 9, e95101. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Wei, J.J.; Sabatini, D.M.; Lander, E.S. Genetic screens in human cells using the CRISPR-Cas9 system. Science 2014, 343, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Carlson, D.F.; Fahrenkrug, S.C.; Hackett, P.B. Targeting DNA with fingers and TALENs. Mol. Ther. Nucleic Acids 2012, 1, e3. [Google Scholar] [CrossRef] [PubMed]
- Chi, H.; Shinohara, M.; Yokomine, T.; Sato, M.; Takao, S.; Yoshida, M.; Miyoshi, K. Successful suppression of endogenous α-1,3-galactosyltransferase expression by RNA interference in pig embryos generated in vitro. J. Reprod. Dev. 2012, 58, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, T.; Watanabe, S.; Kamiyoshi, A.; Sato, M.; Shindo, T. A single blastocyst assay optimized for detecting CRISPR/Cas9 system-induced indel mutations in mice. BMC Biotechnol. 2014, 14, 69. [Google Scholar] [CrossRef] [PubMed]
- Chi, H.; Sato, M.; Yoshida, M.; Miyoshi, K. Expression analysis of a α-1,3-galactosyltransferase, an enzyme that creates xenotransplantation-related α-Gal epitope, in pig preimplantation embryos. Anim. Sci. J. 2012, 83, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, H.A.; Loveland, B.E.; Sandrin, M.S. Gal(α1–3)Gal is the major xenoepitope expressed on pig endothelial cells recognized by naturally occurring cytotoxic human antibodies. Transplantation 1994, 58, 879–882. [Google Scholar] [CrossRef] [PubMed]
- Akasaka, E.; Ozawa, A.; Mori, H.; Mizobe, Y.; Yoshida, M.; Miyoshi, K.; Sato, M. Whole-genome amplification-based GenomiPhi for multiple genomic analysis of individual early porcine embryos. Theriogenology 2011, 75, 1543–1549. [Google Scholar] [CrossRef] [PubMed]
- Kure-bayashi, S.; Miyake, M.; Katayama, M.; Miyano, T.; Kato, S. Development of porcine blastocysts from in vitro-matured and activated haploid and diploid oocytes. Theriogenology 1996, 46, 1027–1036. [Google Scholar] [CrossRef]
- Himaki, T.; Mizobe, Y.; Tsuda, K.; Suetomo, M.; Yamakuchi, H.; Miyoshi, K.; Takao, S.; Yoshida, M. Effect of postactivation treatment with latrunculin A on in vitro and in vivo development of cloned embryos derived from kidney fibroblasts of an aged Clawn miniature boar. J. Reprod. Dev. 2012, 58, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Foden, J.A.; Khayter, C.; Maeder, M.L.; Reyon, D.; Joung, J.K.; Sander, J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013, 31, 822–826. [Google Scholar] [CrossRef] [PubMed]
- Pattanayak, V.; Lin, S.; Guilinger, J.P.; Ma, E.; Doudna, J.A.; Liu, D.R. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat. Biotechnol. 2013, 31, 839–843. [Google Scholar] [CrossRef] [PubMed]
- Galili, U. The α-gal epitope and the anti-Gal antibody in xenotransplantation and in cancer immunotherapy. Immunol. Cell Biol. 2005, 83, 674–686. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Akasaka, E.; Saitoh, I.; Ohtsuka, M.; Nakamura, S.; Sakurai, T.; Watanabe, S. Targeted toxin-based selectable drug-free enrichment of mammalian cells with high transgene expression. Biology 2013, 2, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Hosono, S.; Faruqi, A.F.; Dean, F.B.; Du, Y.; Sun, Z.; Wu, X.; Du, J.; Kingsmore, S.F.; Egholm, M.; Lasken, R.S. Unbiased whole-genome amplification directly from clinical samples. Genome Res. 2003, 13, 954–964. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Yoshida, M.; Miyoshi, K. Utility of ultrasound stimulation for activation of pig oocytes matured in vitro. Mol. Reprod. Dev. 2005, 72, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, K.; Suzuki, C.; Tanaka, A.; Anas, I.M.K.; Iwamura, S. Birth of piglets derived from porcine zygotes cultured in a chemically defined medium. Biol. Reprod. 2002, 66, 112–119. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sato, M.; Koriyama, M.; Watanabe, S.; Ohtsuka, M.; Sakurai, T.; Inada, E.; Saitoh, I.; Nakamura, S.; Miyoshi, K. Direct Injection of CRISPR/Cas9-Related mRNA into Cytoplasm of Parthenogenetically Activated Porcine Oocytes Causes Frequent Mosaicism for Indel Mutations. Int. J. Mol. Sci. 2015, 16, 17838-17856. https://doi.org/10.3390/ijms160817838
Sato M, Koriyama M, Watanabe S, Ohtsuka M, Sakurai T, Inada E, Saitoh I, Nakamura S, Miyoshi K. Direct Injection of CRISPR/Cas9-Related mRNA into Cytoplasm of Parthenogenetically Activated Porcine Oocytes Causes Frequent Mosaicism for Indel Mutations. International Journal of Molecular Sciences. 2015; 16(8):17838-17856. https://doi.org/10.3390/ijms160817838
Chicago/Turabian StyleSato, Masahiro, Miyu Koriyama, Satoshi Watanabe, Masato Ohtsuka, Takayuki Sakurai, Emi Inada, Issei Saitoh, Shingo Nakamura, and Kazuchika Miyoshi. 2015. "Direct Injection of CRISPR/Cas9-Related mRNA into Cytoplasm of Parthenogenetically Activated Porcine Oocytes Causes Frequent Mosaicism for Indel Mutations" International Journal of Molecular Sciences 16, no. 8: 17838-17856. https://doi.org/10.3390/ijms160817838
APA StyleSato, M., Koriyama, M., Watanabe, S., Ohtsuka, M., Sakurai, T., Inada, E., Saitoh, I., Nakamura, S., & Miyoshi, K. (2015). Direct Injection of CRISPR/Cas9-Related mRNA into Cytoplasm of Parthenogenetically Activated Porcine Oocytes Causes Frequent Mosaicism for Indel Mutations. International Journal of Molecular Sciences, 16(8), 17838-17856. https://doi.org/10.3390/ijms160817838