
Deep Sequencing Analysis of miRNA Expression in Breast Muscle of Fast-Growing and Slow-Growing Broilers


 and
and
Abstract
:1. Introduction
2. Results
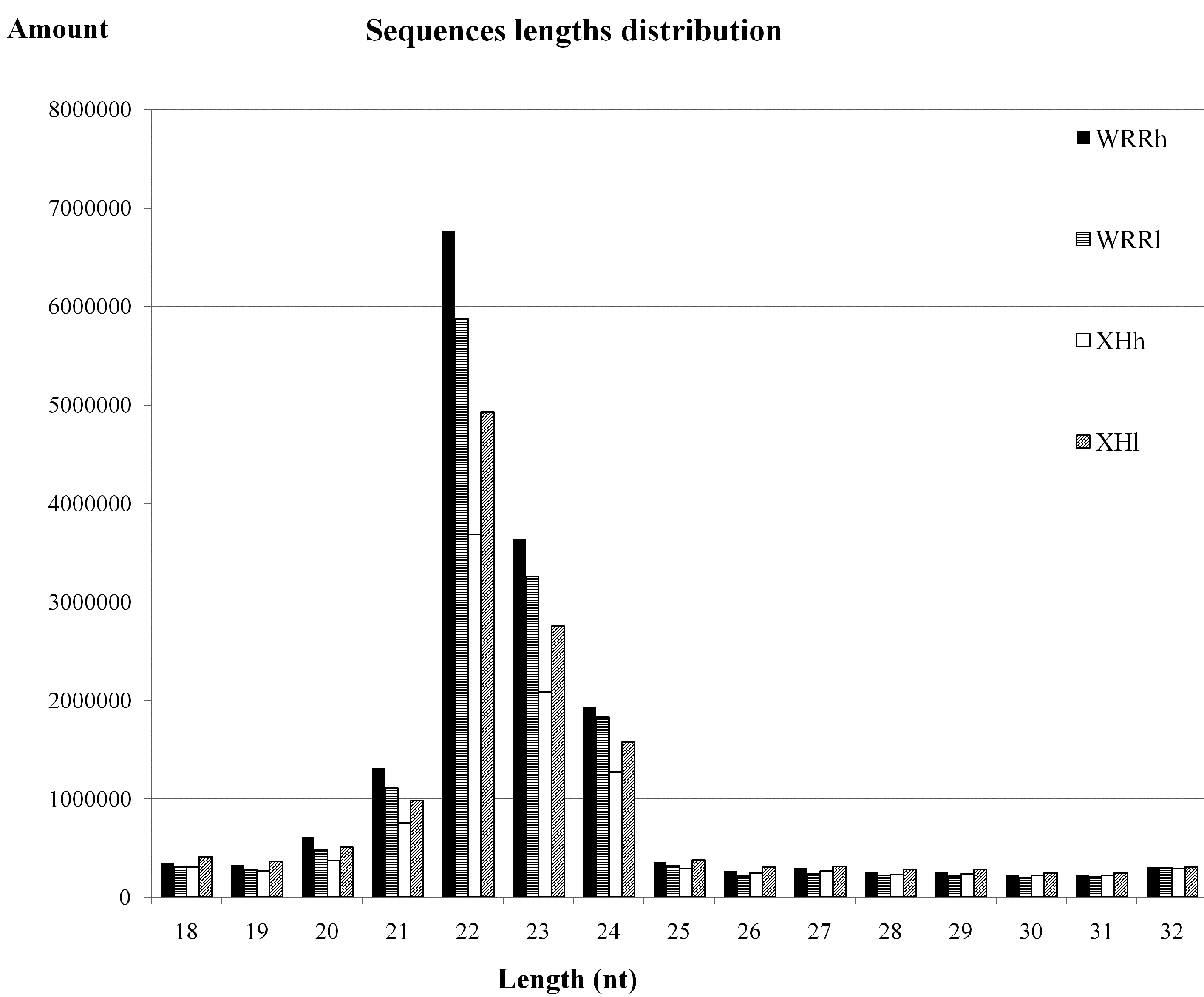
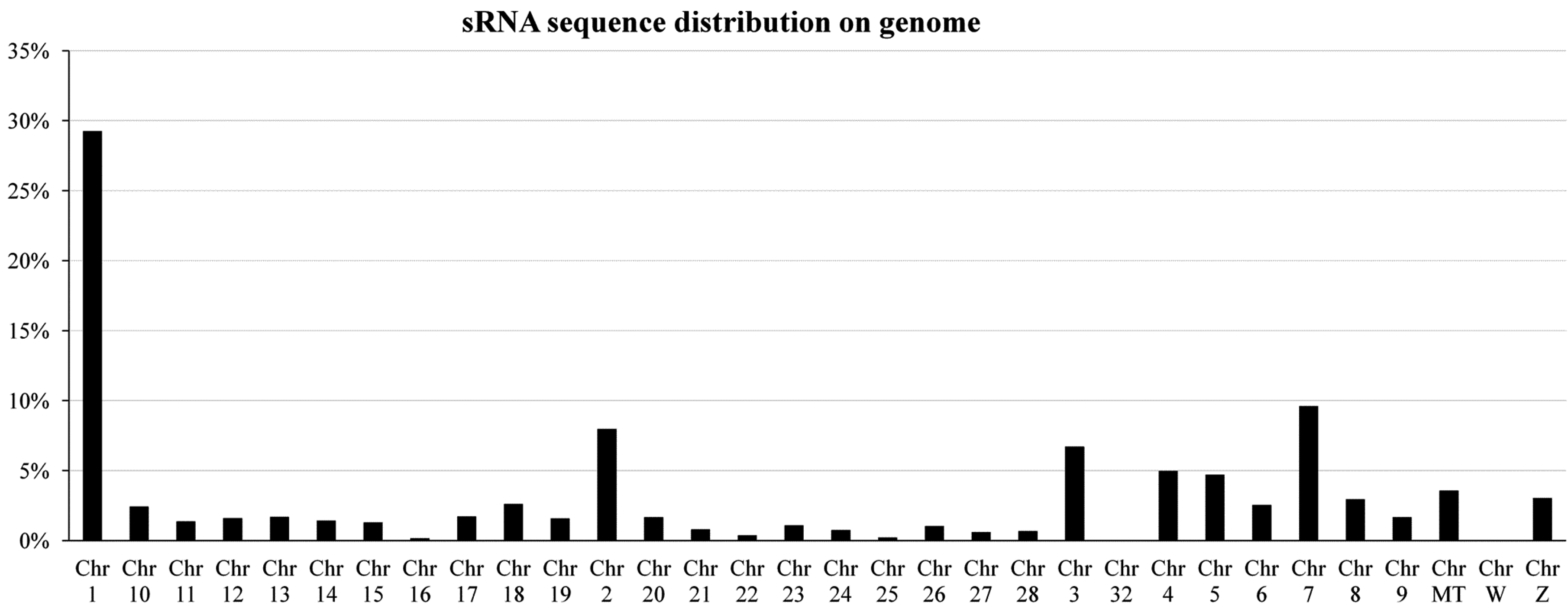
2.1. Overview of Small RNA Deep Sequencing Data

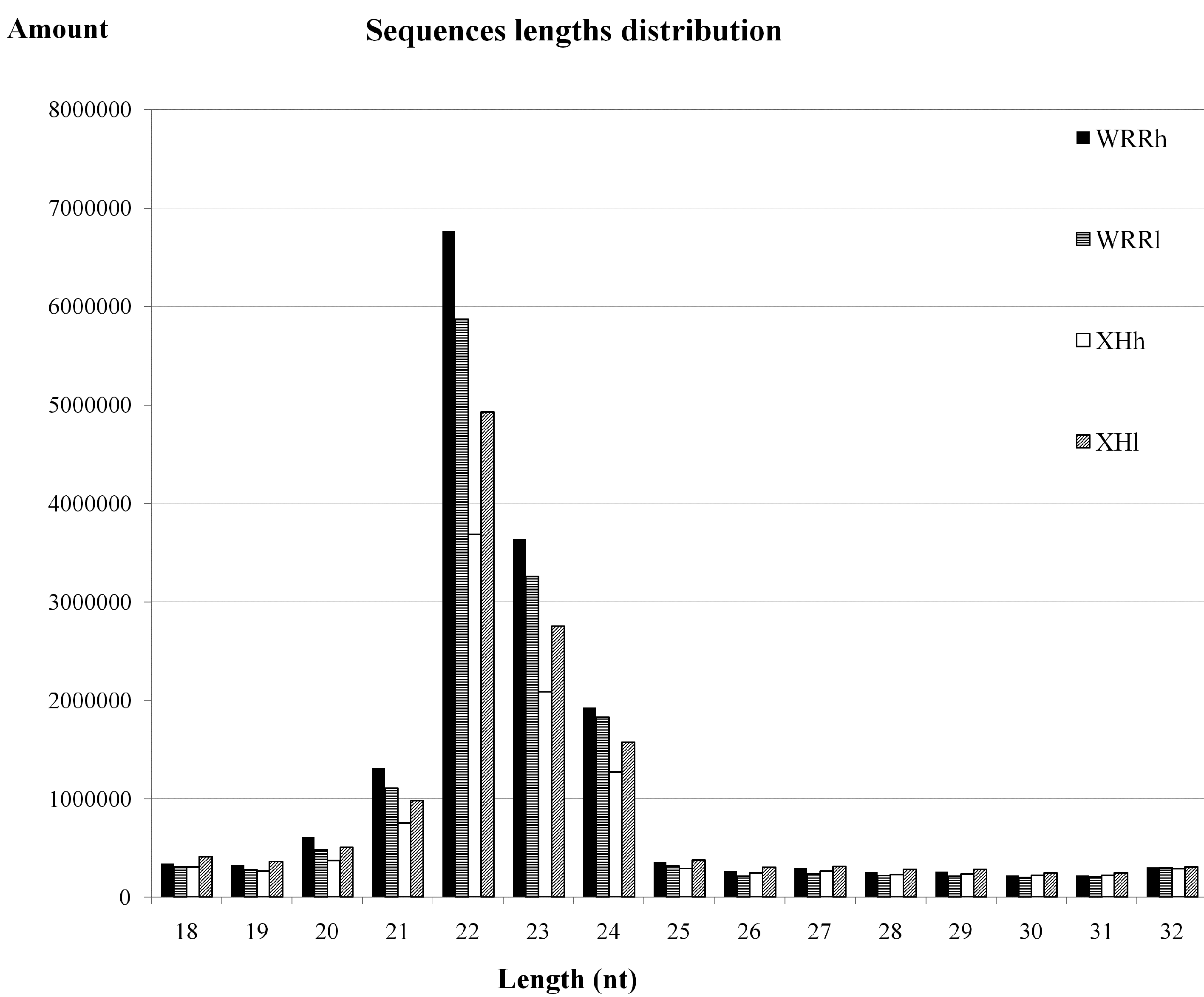{kind=link}
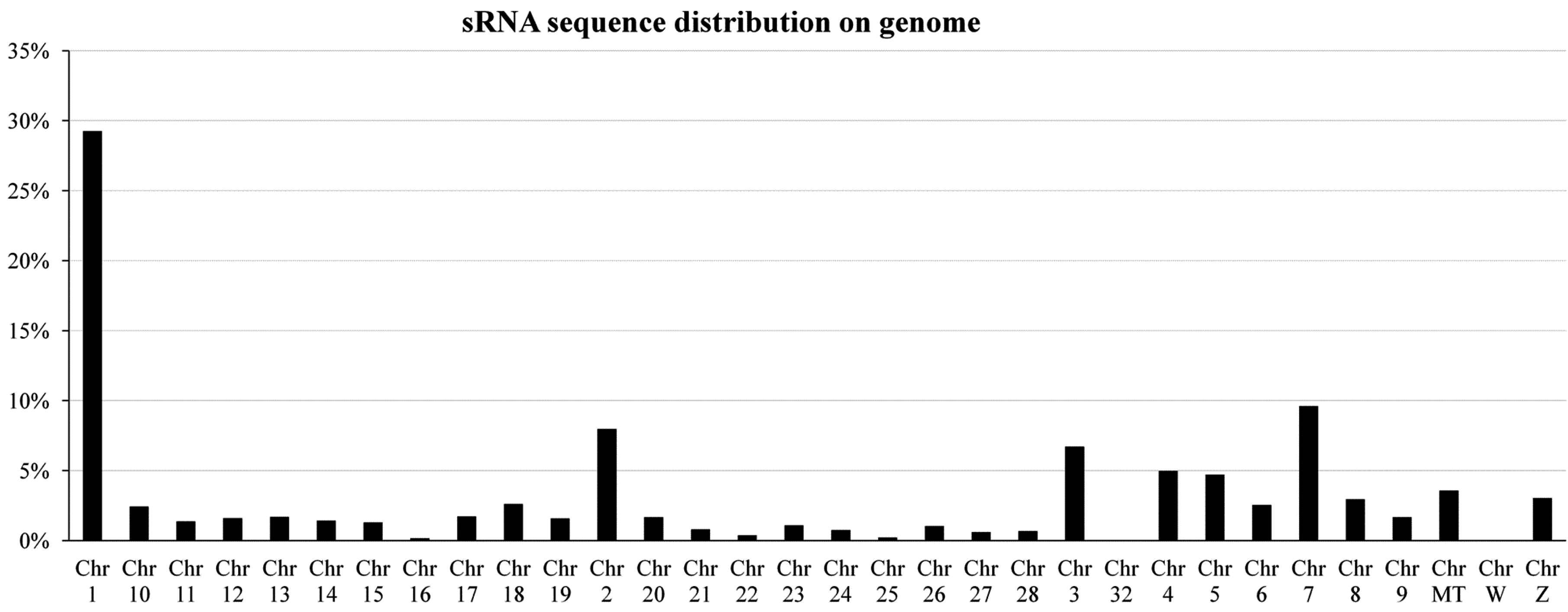{kind=link}
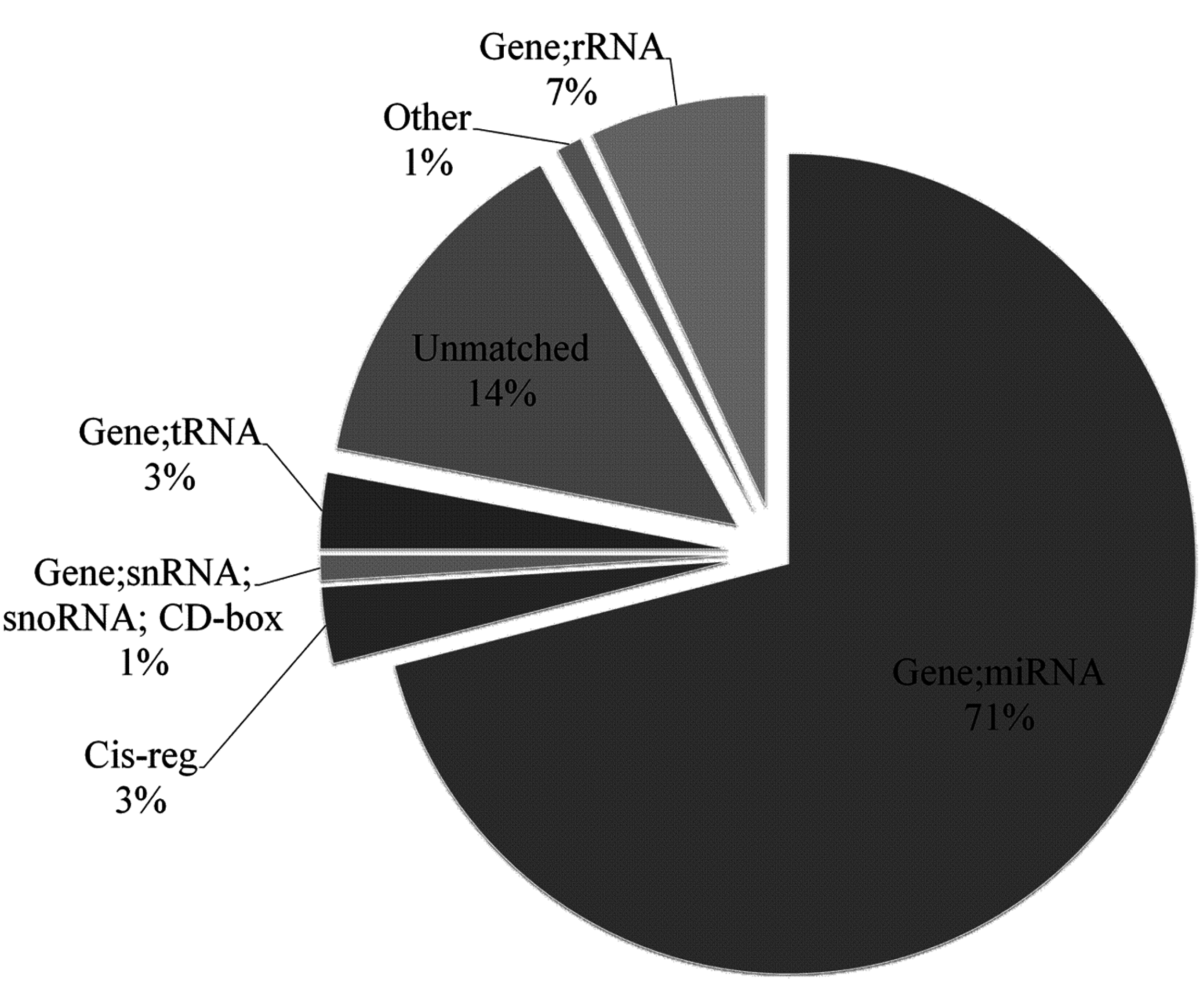{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Total Reads | Clean Reads | Unique Reads | Mapped Reads a | Percentage b |
|---|---|---|---|---|---|
| WRRh | 22,080,436 | 20,424,161 | 577,820 | 433,846 | 75.08% |
| WRRl | 19,917,025 | 18,160,137 | 513,726 | 392,323 | 76.37% |
| XHh | 16,410,221 | 14,806,039 | 494,539 | 366,898 | 74.19% |
| XHl | 19,473,651 | 17,295,270 | 625,063 | 482,989 | 77.27% |
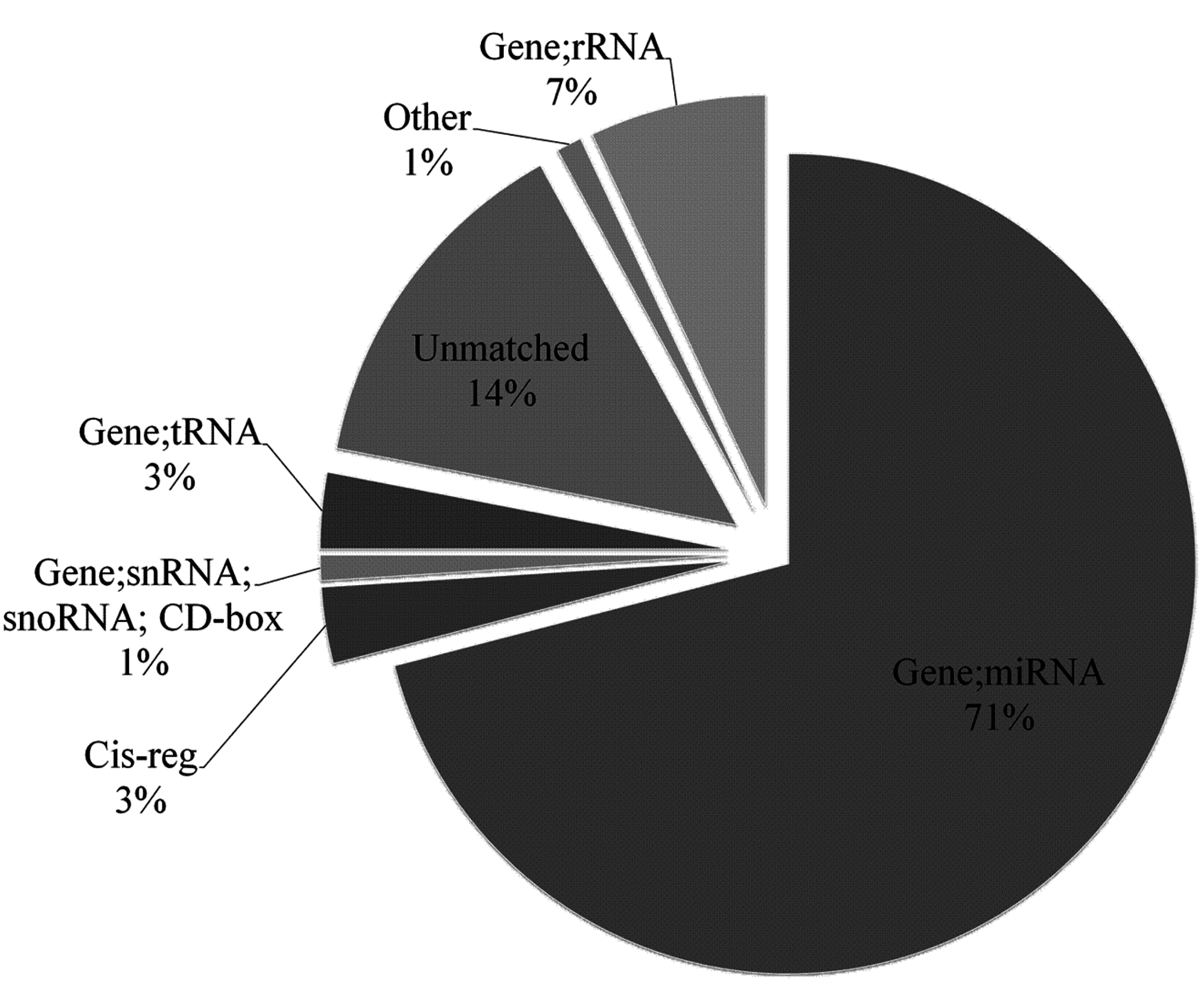
2.2. Characterization of Known miRNAs and Predicted Novel miRNAs
| miRNAs | Normalized Reads | Total Reads | |||
|---|---|---|---|---|---|
| WRRh | WRRl | XHh | XHl | ||
| gga-miR-133a | 3,558,683 | 3,069,071 | 1,997,286 | 2,607,787 | 11,232,827 |
| gga-miR-133c | 3,350,936 | 2,885,440 | 1,878,925 | 2,449,209 | 10,564,510 |
| gga-miR-133b | 3,326,848 | 2,864,578 | 1,864,721 | 2,431,274 | 10,487,421 |
| gga-let-7a | 1,699,621 | 1,513,865 | 857,210 | 1,133,532 | 5,204,228 |
| gga-miR-22-3p | 1,333,233 | 1,145,421 | 712,464 | 988,186 | 4,179,304 |
| gga-miR-30a-5p | 1,213,468 | 1,148,128 | 790,893 | 930,507 | 4,082,996 |
| gga-miR-26a | 1,212,635 | 1,054,689 | 691,456 | 1,006,522 | 3,965,302 |
| gga-miR-30d | 851,887 | 813,262 | 583,932 | 667,002 | 2,916,083 |
| gga-miR-181a-6p | 918,452 | 836,452 | 485,661 | 650,836 | 2,891,401 |
| gga-miR-10a-5p | 943,686 | 782,180 | 420,809 | 663,401 | 2,810,076 |
| gga-miR-10b | 911,725 | 757,564 | 398,852 | 633,567 | 2,701,708 |
| gga-miR-30e | 799,679 | 730,832 | 501,718 | 596,218 | 2,628,447 |
| gga-let-7j | 848,972 | 756,205 | 428,182 | 566,165 | 2,599,524 |
| gga-let-7f | 398,292 | 363,598 | 206,995 | 274,333 | 1,243,218 |
| gga-miR-148a | 288,585 | 300,432 | 144,015 | 180,973 | 914,005 |
| gga-miR-146c-5p | 224,147 | 207,782 | 171,443 | 132,712 | 736,084 |
| gga-let-7k | 211,853 | 206,518 | 118,297 | 155,412 | 692,080 |
| gga-let-7c | 242,661 | 189,820 | 111,118 | 139,257 | 682,856 |
| gga-miR-199-3p | 168,417 | 152,158 | 75,346 | 121,460 | 517,381 |
| gga-miR-126-5p | 139,914 | 109,805 | 89,607 | 86,681 | 426,007 |
2.3. Identification of Differentially Expressed miRNAs
| Sample 1 vs. Sample 2 | Up a | Down b | Total | Shared | |
|---|---|---|---|---|---|
| WRRh vs. WRRl | 116 (47) | 84 (24) | 200 (71) | 80 (22) | 26 (16) |
| XHh vs. XHl | 129 (44) | 150 (40) | 279 (84) | ||
| WRRh vs. XHh | 130 (31) | 127 (47) | 257 (78) | 110 (39) | |
| WRR vs. XHl | 137 (41) | 160 (60) | 297 (101) | ||
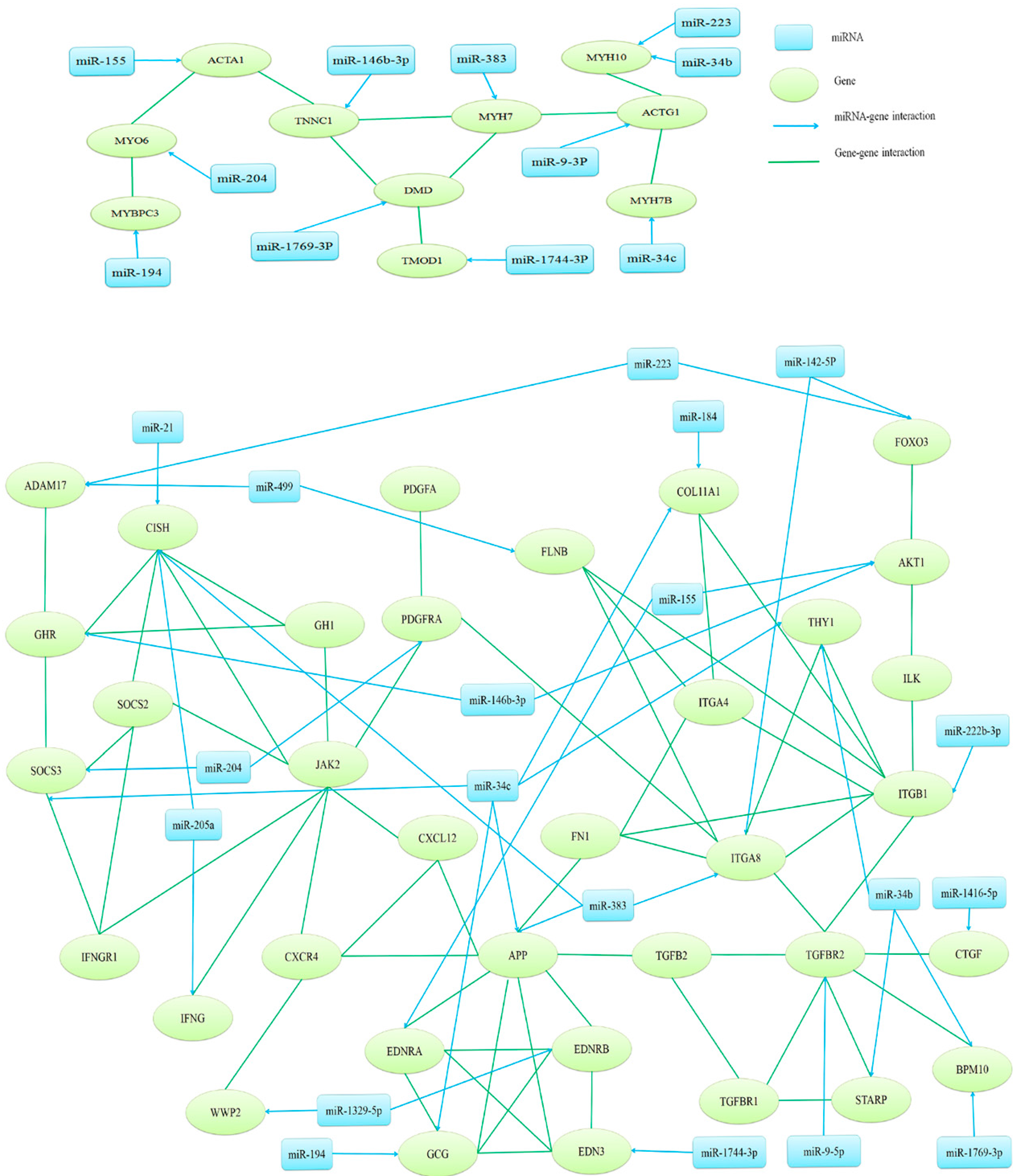
| miRNAs | Normalized Reads | Fold Change | Target Gene | |
|---|---|---|---|---|
| Sample 1 | Sample 2 | |||
| gga-miR-1329-5p | WRRh 459 | WRRl 1582 | 0.29 | EDNRB; WWP2 |
| WRRl 1582 | XHl 174 | 9.07 | ||
| gga-miR-1416-5p | XHh 5680 | XHl 2721 | 2.09 | CTGF; CXCL12 |
| WRRl 8061 | XHl 2721 | 2.96 | ||
| gga-miR-142-5p | WRRh 108,463 | XHh 54,059 | 2.01 | FOXO3; ITGA8 |
| WRRl 173,442 | XHl 71,860 | 2.41 | ||
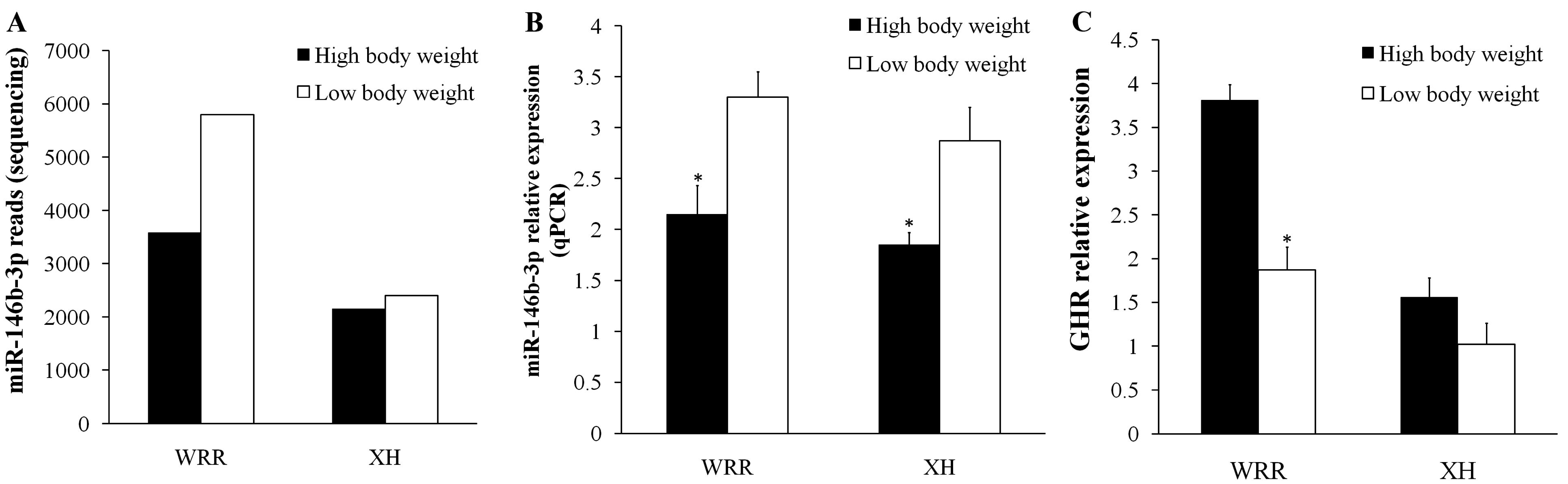
| gga-miR-146b-3p | WRRl 5800 | XHl 2404 | 2.41 | TNNC1; AKT1; GHR |
| gga-miR-146b-5p | WRRl 191,004 | XHl 71,169 | 2.68 | – |
| gga-miR-147 | WRRh 882 | WRRl 2824 | 0.31 | – |
| WRRl 2824 | XHl 928 | 3.04 | ||
| gga-miR-155 | WRRh 5205 | WRRl 14,058 | 0.37 | ACTA1; AKT1; EDNRA |
| WRRh 5205 | XHh 1759 | 2.96 | ||
| WRRl 14,058 | XHl 2018 | 6.97 | ||
| gga-miR-1744-3p | WRRh 2359 | WRRl 740 | 3.19 | TMOD1; EDN3 |
| gga-miR-1769-3p | WRRh 377 | WRRl 1120 | 0.34 | DMD; BMP10 |
| WRRl 1120 | XHl 498 | 2.25 | ||
| gga-miR-184 | WRRh 382 | XHh 1735 | 0.22 | COL11A1 |
| gga-miR-194 | WRRh 946 | WRRl 4669 | 0.20 | MYBPC3; GCG |
| WRRh 946 | XHh 1981 | 0.48 | ||
| WRRl 4669 | XHl 1470 | 3.18 | ||
| gga-miR-204 | XHh 2762 | XHl 5848 | 0.47 | MYO6; PDGFRA; SOCS3 |
| gga-miR-205a | WRRh 4900 | WRRl 2281 | 2.15 | CISH; IFNG |
| WRRh 4900 | XHh 2137 | 2.29 | ||
| WRRl 2281 | XHl 940 | 2.43 | ||
| gga-miR-21 | WRRh 489,673 | WRRl 1,060,669 | 0.46 | CISH |
| WRRl 1,060,669 | XHl 360,267 | 2.94 | ||
| gga-miR-222b-3p | WRRh 2477 | WRRl 6641 | 0.37 | ITGB1 |
| WRRh 2477 | XHh 658 | 3.77 | ||
| WRRl 6641 | XHl 791 | 8.40 | ||
| gga-miR-223 | WRRl 10,225 | XHl 4004 | 2.55 | MYH10; ADAM17; FOXO3 |
| gga-miR-34b | XHh 41 | XHl 1750 | 0.02 | MYH10; BMP10 |
| WRRl 218 | XHl 1750 | 0.12 | STRAP; THY1 | |
| gga-miR-34c | XHh 41 | XHl 1750 | 0.02 | MYH7B; APP |
| WRRl 218 | XHl 1750 | 0.12 | THY1; COL11A1; SOCS3; GCG | |
| gga-miR-383 | WRRh 373 | 1603 | 0.23 | MYH7; APP; ITGA8 |
| gga-miR-499 | WRRl 1754 | XHl 6084 | 0.29 | ADAM17; FLNB |
| gga-miR-9-3p | XHh 82 | XHl 1177 | 0.07 | ACTG1 |
| WRRl 81 | XHl 1177 | 0.07 | ||
| gga-miR-9-5p | XHh 1036 | XHl 3095 | 0.33 | TGFB2 |

2.4. Target Gene Prediction, Function Annotation and Network Analysis
| GO Accession | GO Terms | Gene Numbers | Fold Enrichment | p Value |
|---|---|---|---|---|
| GO:0040008 | regulation of growth | 29 | 1.5 | 0.016 |
| GO:0001558 | regulation of cell growth | 16 | 1.8 | 0.019 |
| GO:0045927 | positive regulation of growth | 11 | 2 | 0.029 |
| GO:0030307 | positive regulation of cell growth | 5 | 3.1 | 0.025 |
| GO:0017015 | regulation of transforming growth factor β receptor signaling pathway | 5 | 2.8 | 0.034 |
| GO:0055001 | muscle cell development | 11 | 2.4 | 0.034 |
| GO:0051146 | striated muscle cell differentiation | 13 | 1.8 | 0.040 |
| GO:0060415 | muscle tissue morphogenesis | 6 | 2.7 | 0.049 |
| GO:0003012 | muscle system process | 13 | 1.8 | 0.049 |
| GO:0060537 | muscle tissue development | 17 | 1.6 | 0.036 |
| GO:0055002 | striated muscle cell development | 8 | 2.1 | 0.035 |
| GO:0014706 | striated muscle tissue development | 16 | 1.5 | 0.027 |
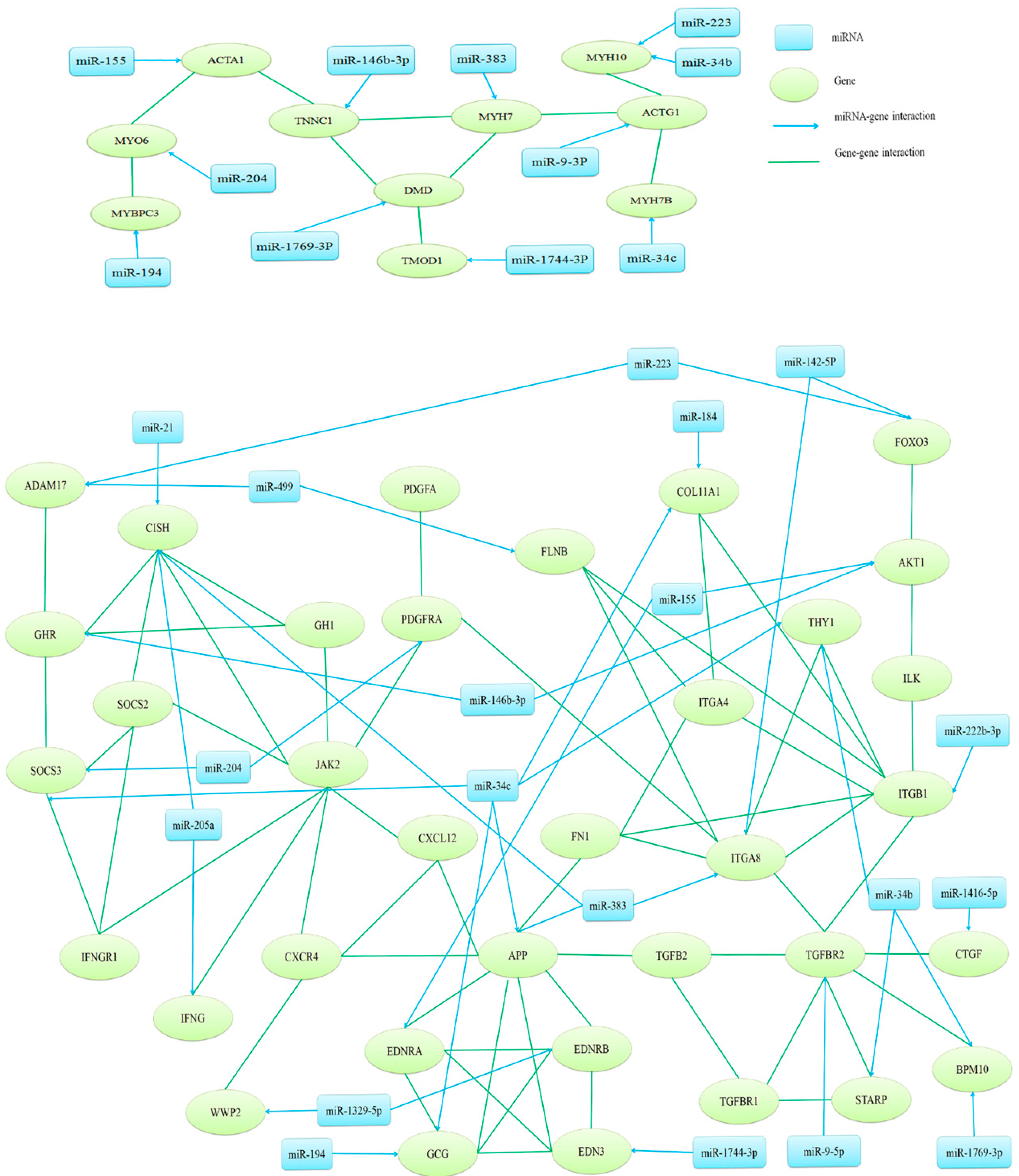
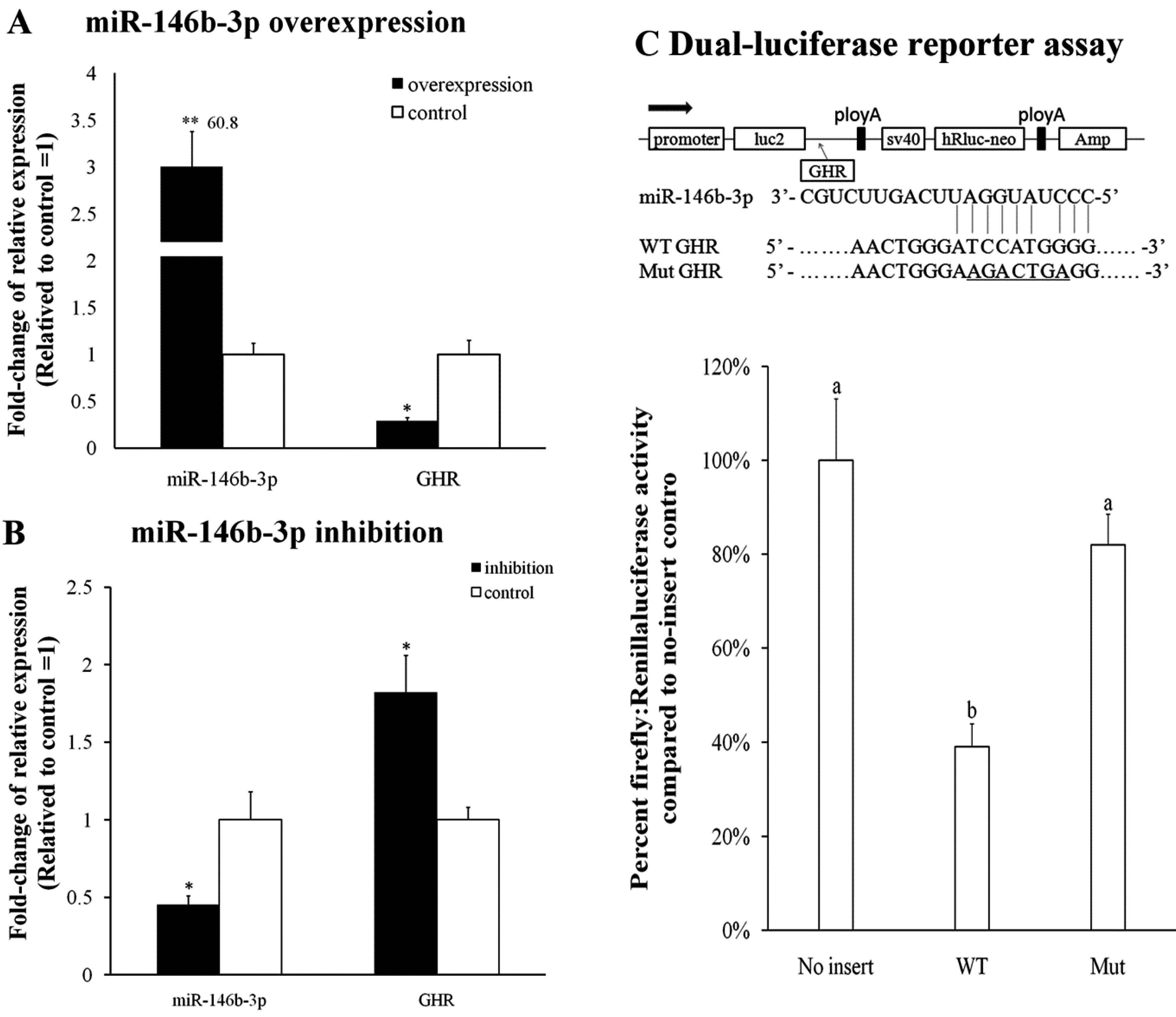
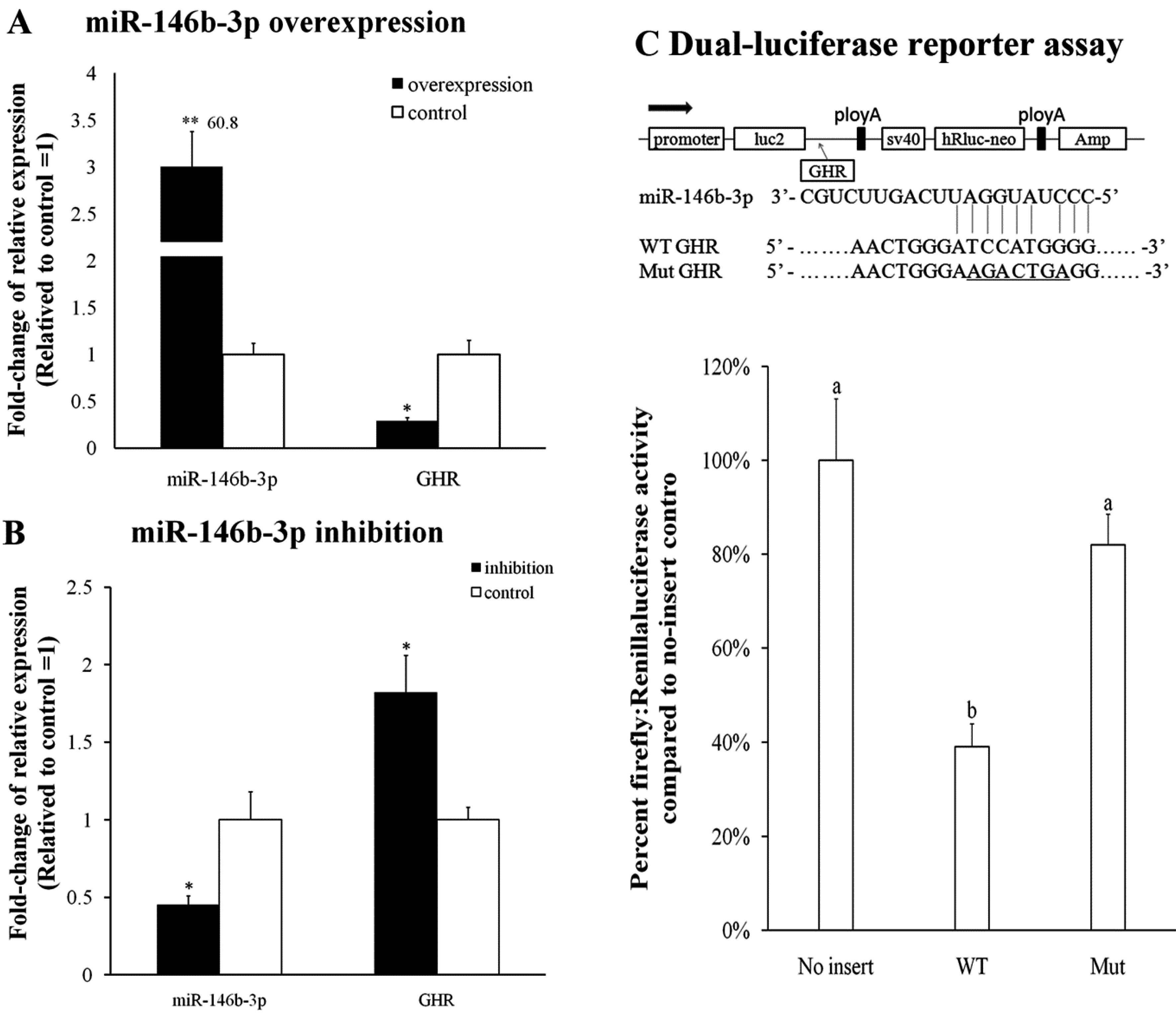
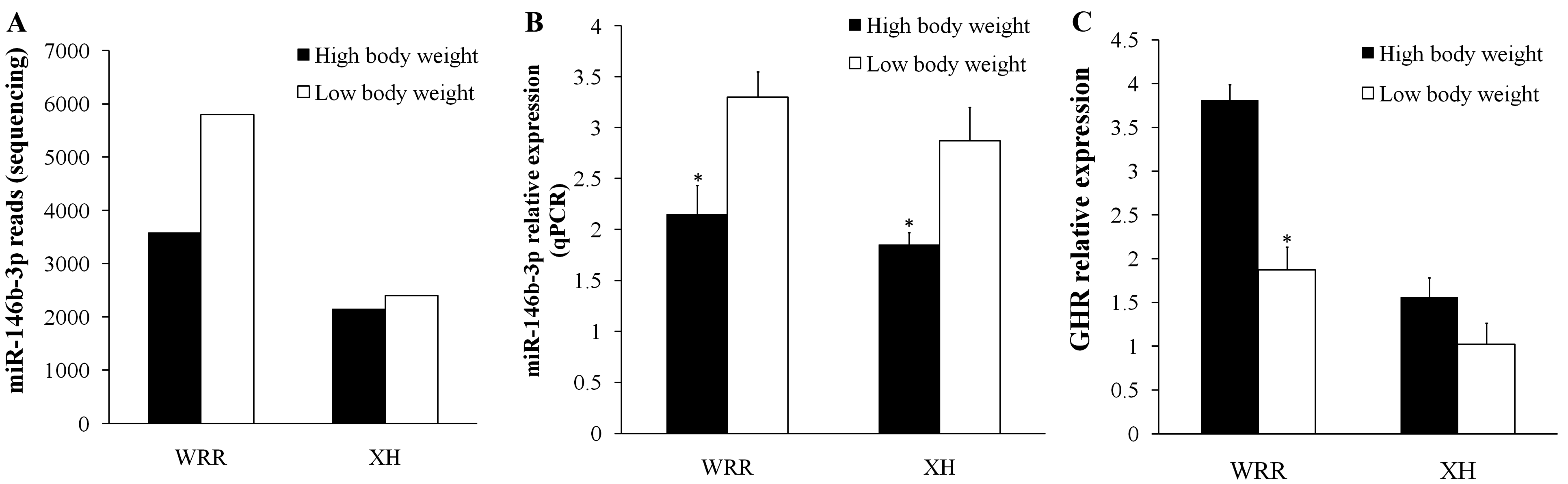
2.5. Validation of miR-146b-3p Targeted GHR Gene
3. Discussion
4. Experimental Section
4.1. Ethics Statement
4.2. Sample Preparation
4.3. Small RNA Library Construction and Solexa Sequencing
4.4. Sequence Analysis
4.5. Analysis of Differently Expressed miRNAs
4.6. Target Gene Prediction, Pathway and Network Analysis
4.7. RNA Oligonucleotides and Transfection
4.8. cDNA Synthesis and Quantitative Real Time PCR (qPCR)
4.9. pmirGLO Dual-Luciferase miRNA Target Expression Vector Construction and Dual-Luciferase Reporter Assay
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Scanes, C.G.; Harvey, S.; Marsh, J.A.; King, D.B. Hormones and growth in poultry. Poult. Sci. 1984, 63, 2062–2074. [Google Scholar] [CrossRef] [PubMed]
- Carlborg, O.; Kerje, S.; Schutz, K.; Jacobsson, L.; Jensen, P.; Andersson, L. A global search reveals epistatic interaction between QTL for early growth in the chicken. Genome Res. 2003, 13, 413. [Google Scholar] [CrossRef] [PubMed]
- Ankra-Badu, G.A.; Le Bihan-Duval, E.; Mignon-Grasteau, S.; Pitel, F.; Beaumont, C.; Duclos, M.J.; Simon, J.; Carré, W.; Porter, T.E.; Vignal, A.; et al. Mapping QTL for growth and shank traits in chickens divergently selected for high or low body weight. Anim. Genet. 2010, 41, 400–405. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Hu, X.X.; Du, Z.; Deng, X.M.; Huang, Y.H.; Fei, J.; Feng, J.D.; Liu, Z.L.; Da, Y.; Li, N. A genome scan for quantitative trait loci associated with body weight at different developmental stages in chickens. Anim. Genet. 2006, 37, 276–278. [Google Scholar] [CrossRef] [PubMed]
- Lei, M.; Peng, X.; Zhou, M.; Luo, C.; Nie, Q.; Zhang, X. Polymorphisms of the IGF1R gene and their genetic effects on chicken early growth and carcass traits. BMC Genet. 2008, 9, 70. [Google Scholar] [CrossRef] [PubMed]
- Darzi Niarami, M.; Masoudi, A.A.; Vaez Torshizi, R. Association of single nucleotide polymorphism of GHSR and TGFB2 genes with growth and body composition traits in sire and dam lines of a broiler chicken. Anim. Biotechnol. 2014, 25, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Nie, Q.H.; Fang, M.X.; Xie, L.; Shen, X.; Liu, J.; Luo, Z.P.; Shi, J.J.; Zhang, X.Q. Associations of ATGL gene polymorphisms with chicken growth and fat traits. J. Appl. Genet. 2003, 51, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Feng, C.; Ma, L.; Song, C.; Wang, Y.; Da, Y.; Li, H.; Chen, K.; Ye, S.; Ge, C.; et al. Genome-wide association study of body weight in chicken F2 resource population. PLoS ONE 2011, 6, e21872. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Luo, C.; Zhang, C.; Zhang, R.; Tang, J.; Nie, Q.; Ma, L.; Hu, X.; Li, N.; Da, Y.; et al. Genome-wide association study identified a narrow chromosome 1 region associated with chicken growth traits. PLoS ONE 2012, 7, e30910. [Google Scholar] [CrossRef] [PubMed]
- Ahsan, M.; Li, X.; Lundberg, A.E.; Kierczak, M.; Siegel, P.B.; Carlborg, O.; Marklund, S. Identification of candidate genes and mutations in QTL regions for chicken growth using bioinformatic analysis of NGS and SNP-chip data. Front. Genet. 2013, 4, 226. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Z.; Pettersson, M.E.; Hu, X.; Luo, C.; Qu, H.; Shu, D.; Shen, X.; Carlborg, O.; Li, N. Genetic dissection of growth traits in a Chinese indigenous × commercial broiler chicken cross. BMC Genomics 2013, 14, 151. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Misztal, I.; Aguilar, I.; Legarra, A.; Fernando, R.L.; Vitezica, Z.; Okimoto, R.; Wing, T.; Hawken, R.; Muir, W.M. Genome-wide association mapping including phenotypes from relatives without genotypes in a single-step (ssGWAS) for 6-week body weight in broiler chickens. Front. Genet. 2014, 5, 134. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.H.; Wang, Q.L.; Pan, X.P. MicroRNAs and their regulatory roles in animals and plants. J. Cell. Physiol. 2007, 210, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Cullen, B.R. MicroRNAs as mediators of viral evasion of the immune system. Nat. Immunol. 2013, 14, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Muers, M. Small RNA: Long-lived microRNA complexes. Nat. Rev. Genet. 2013, 14, 78. [Google Scholar] [CrossRef] [PubMed]
- Hitachi, K.; Tsuchida, K. Role of microRNAs in skeletal muscle hypertrophy. Front. Physiol. 2014, 4, 408. [Google Scholar] [CrossRef] [PubMed]
- Duan, R.; Pak, C.; Jin, P. Single nucleotide polymorphism associated with mature miR-125a alters the processing of pri-miRNA. Hum. Mol. Genet. 2007, 16, 1124–1131. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Li, Y.; Zhou, R.M.; Wang, G.Y.; Wang, C.M.; Chen, Z.F.; Liu, W. Hsa-miR-196a2 functional SNP is associated with the risk of ESCC in individuals under 60 years old. Biomarkers 2014, 19, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Cordes, K.R.; Srivastava, D.; Ivey, K.N. MicroRNAs in cardiac development. Pediatr. Cardiol. 2010, 31, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Glazov, E.A.; Cottee, P.A.; Barris, W.C.; Moore, R.J.; Dalrymple, B.P.; Tizard, M.L. A microRNA catalog of the developing chicken embryo identified by a deep sequencing approach. Genome Res. 2008, 18, 957–964. [Google Scholar] [CrossRef] [PubMed]
- Hicks, J.A.; Tembhurne, P.; Liu, H.C. MicroRNA expression in chicken embryos. Poult. Sci. 2008, 87, 2335–2343. [Google Scholar] [CrossRef] [PubMed]
- Hicks, J.A.; Trakooljul, N.; Liu, H.C. Discovery of chicken microRNAs associated with lipogenesis and cell proliferation. Physiol. Genomics 2010, 41, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Hicks, J.A.; Tembhurne, P.A.; Liu, H.C. Identification of microRNA in the developing chick immune organs. Immunogenetics 2009, 61, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Lian, L.; Qu, L.; Chen, Y.; Lamont, S.J.; Yang, N. A systematic analysis of miRNA transcriptome in Marek’s disease virus-induced lymphoma reveals novel and differentially expressed miRNAs. PLoS ONE 2012, 7, e51003. [Google Scholar] [CrossRef] [PubMed]
- Tian, F.; Luo, J.; Zhang, H.; Chang, S.; Song, J. MiRNAs expression signatures induced by Marek’s disease virus infection in chickens. Genomics 2012, 99, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Li, H.; Mu, H.; Luo, W.; Li, Y.; Jia, X.; Wang, S.; Nie, Q.; Li, Y.; Zhang, X. Let-7b regulates the expression of the growth hormone receptor gene in deletion-type dwarf chickens. BMC Genomics 2012, 13, 306. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information, Gene Expression Omnibus. Available online: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE62971 (accessed on 5 November 2014).
- Friedländer, M.R.; Chen, W.; Adamidi, C.; Maaskola, J.; Einspanier, R.; Knespel, S.; Rajewsky, N. Discovering microRNAs from deep sequencing data using miRDeep. Nat. Biotechnol. 2008, 26, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Kuchenbauer, F.; Morin, R.D.; Argiropoulos, B.; Petriv, O.I.; Griffith, M.; Heuser, M.; Yung, E.; Piper, J.; Delaney, A.; Prabhu, A.L.; et al. Humphries RK: In-depth characterization of the microRNA transcriptome in a leukemia progression model. Genome Res. 2009, 18, 1787–1797. [Google Scholar] [CrossRef] [PubMed]
- Betel, D.; Wilson, M.; Gabow, A.; Marks, D.S.; Sander, C. The microRNA.org resource: Targets and expression. Nucleic Acids Res. 2008, 36, D149–D153. [Google Scholar] [CrossRef] [PubMed]
- Rehmsmeier, M.; Steffen, P.; Hoechsmann, M.; Giegerich, R. Fast and effective prediction of microRNA/target duplexes RNA. RNA 2004, 10, 1507–1517. [Google Scholar] [CrossRef] [PubMed]
- Franceschini, A.; Szklarczyk, D.; Frankild, S.; Kuhn, M.; Simonovic, M.; Roth, A.; Lin, J.; Minguez, P.; Bork, P.; Jensen, L.J. STRING v9.1: Protein–protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013, 41, D808–D815. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Nie, Q.; Zhang, X. Overview of genomic insights into chicken growth traits based on genome-wide association study and microRNA regulation. Curr. Genomics 2013, 14, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Gu, Z.; Jiang, H. MicroRNAs in farm animals. Animal 2013, 7, 1567–1575. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Zhang, J.; Hou, X.; Zan, L.; Wang, N.; Tang, Z.; Li, K. OLFML3 expression is decreased during prenatal muscle development and regulated by microRNA-155 in pigs. Int. J. Biol. Sci. 2012, 8, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Wu, R.; Zhang, Y.; Zhu, D. A systematic analysis of the skeletal muscle miRNA transcriptome of chicken varieties with divergent skeletal muscle growth identifies novel miRNAs and differentially expressed miRNAs. BMC Genomics 2011, 12, 186. [Google Scholar] [CrossRef] [PubMed]
- Kang, L.; Cui, X.; Zhang, Y.; Yang, C.; Jiang, Y. Identification of miRNAs associated with sexual maturity in chicken ovary by Illumina small RNA deep sequencing. BMC Genomics 2013, 14, 352. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.F.; Mandel, E.M.; Thomson, J.M.; Wu, Q.; Callis, T.E.; Hammond, S.M.; Conlon, F.L.; Wang, D.Z. The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat. Genet. 2006, 38, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Xu, H.; Xie, S.; Zhou, H.; Qu, L. Insulin-like growth factor-1 receptor is regulated by microRNA-133 during skeletal myogenesis. PLoS ONE 2011, 6, e29173. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yu, J.; Zhang, Y.; Gong, D.; Gu, Z. Identification and characterization of microRNA from chicken adipose tissue and skeletal muscle. Poult. Sci. 2012, 91, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Rathjen, T.; Pais, H.; Sweetman, D.; Moulton, V.; Munsterberg, A.; Dalmay, T. High throughput sequencing of microRNAs in chicken somites. FEBS Lett. 2009, 583, 1422–1426. [Google Scholar] [CrossRef] [PubMed]
- Darnell, D.K.; Kaur, S.; Stanislaw, S.; Konieczka, J.K.; Yatskievych, T.A.; Antin, P.B. MicroRNA expression during chick embryo development. Dev. Dyn. 2006, 235, 3156–3165. [Google Scholar] [CrossRef] [PubMed]
- Goljanek-Whysall, K.; Sweetman, D.; Abu-Elmagd, M.; Chapnik, E.; Dalmay, T.; Hornstein, E.; Münsterberg, A. MicroRNA regulation of the paired-box transcription factor Pax3 confers robustness to developmental timing of myogenesis. Proc. Natl. Acad. Sci. USA 2011, 19, 11936–11941. [Google Scholar] [CrossRef] [PubMed]
- Thum, T.; Gross, C.; Fiedler, J.; Fischer, T.; Kissler, S.; Bussen, M.; Galuppo, P.; Just, S.; Rottbauer, W.; Frantz, S.; et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature 2008, 456, 980–984. [Google Scholar] [CrossRef] [PubMed]
- Papagiannakopoulos, T.; Shapiro, A.; Kosik, K.S. MicroRNA-21 targets a network of key tumor-suppressive pathways in glioblastoma cells. Cancer Res. 2008, 68, 8164–8172. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Gan, H.; Zhang, H.; Tang, W.; Sun, Y.; Tang, X.; Kong, D.; Zhou, J.; Wang, Y.; Zhu, Y. MicroRNA-21 inhibits SMAD7 expression through a target sequence in the 3′ untranslated region and inhibits proliferation of renal tubular epithelial cells. Mol. Med. Rep. 2014, 10, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, E.R.; Geelissen, S.M.; van der, G.S.; Darras, V.M. The release of growth hormone (GH): Relation to the thyrotropic and corticotropic axis in the chicken. Domest. Anim. Endocrinol. 2006, 29, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Nie, Q.; Sun, B.; Zhang, D.; Luo, C.; Ishag, N.A.; Lei, M.; Yang, G.; Zhang, X. High diversity of the chicken growth hormone gene and effects on growth and carcass traits. J. Hered. 2005, 96, 698–703. [Google Scholar] [CrossRef] [PubMed]
- Greco, S.; Perfetti, A.; Fasanaro, P.; Cardani, R.; Capogrossi, M.C.; Meola, G.; Martelli, F. Deregulated microRNAs in myotonic dystrophy type 2. PLoS ONE 2012, 7, e39732.46. [Google Scholar] [CrossRef] [PubMed]
- Khanna, N.; Ge, Y.; Chen, J. MicroRNA-146b promotes myogenic differentiation and modulates multiple gene targets in muscle cells. PLoS ONE 2014, 9, e100657. [Google Scholar] [CrossRef] [PubMed]
- Comer, B.S.; Camoretti-Mercado, B.; Kogut, P.C.; Halayko, A.J.; Solway, J.; Gerthoffer, W.T. MicroRNA-146a and microRNA-146b expression and anti-inflammatory function in human airway smooth muscle. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L727-34.48. [Google Scholar] [CrossRef] [PubMed]
- Tamiya, T.; Kashiwagi, I.; Takahashi, R.; Yasukawa, H.; Yoshimura, A. Suppressors of cytokine signaling (SOCS) proteins and JAK/STAT pathways: Regulation of T-cell inflammation by SOCS1 and SOCS3. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 980–985. [Google Scholar] [CrossRef] [PubMed]
- Rui, L.; Yuan, M.; Frantz, D.; Shoelson, S.; White, M. SOCS-1 and SOCS-3 block insulin signaling by ubiquitin-mediated degradation of IRS1 and IRS2. J. Biol. Chem. 2002, 277, 42394–42398. [Google Scholar] [CrossRef] [PubMed]
- Ram, P.A.; Waxman, D.J. Role of the cytokine-inducible SH2 protein CIS in desensitization of STAT5b signaling by continuous growth hormone. J. Biol. Chem. 2000, 275, 39487–39496. [Google Scholar] [CrossRef] [PubMed]
- Elia, L.; Contu, R.; Quintavalle, M.; Varrone, F.; Chimenti, C.; Russo, M.A.; Cimino, V.; de Marinis, L.; Frustaci, A.; Condorelli, G. Reciprocal regulation of microRNA-1 and insulin-like growth factor-1 signal transduction cascade in cardiac and skeletal muscle in physiological and pathological conditions. Circulation 2009, 120, 2377–2385. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, C.M.; Emanuelli, B.; Kahn, C.R. Critical nodes in signalling pathways: Insights into insulin action. Nat. Rev. Mol. Cell Biol. 2006, 7, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Goldberg, A.L. SIRT1 protein; by blocking the activities of transcription factors FoxO1 and FoxO3; inhibits muscle atrophy and promotes muscle growth. J. Biol. Chem. 2013, 288, 30515–30526. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Geles, K.G.; Paik, J.H.; DePinho, R.A.; Tjian, R. Codependent activators direct myoblast-specific MyoD transcription. Dev. Cell 2008, 15, 534–546. [Google Scholar] [CrossRef] [PubMed]
- Haneklaus, M.; Gerlic, M.; OʼNeill, L.A.; Masters, S.L. miR-223: Infection, inflammation and cancer. J. Intern. Med. 2013, 274, 215–226. [Google Scholar] [CrossRef] [PubMed]
- McGirt, L.Y.; Adams, C.M.; Baerenwald, D.A.; Zwerner, J.P.; Zic, J.A.; Eischen, C.M. miR-223 regulates cell growth and targets proto-oncogenes in mycosis fungoides/cutaneous T-cell lymphoma. J. Investig. Dermatol. 2013, 134, 1101–1107. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Fisslthaler, B.; Zippel, N.; Frömel, T.; Hu, J.; Elgheznawy, A.; Heide, H.; Popp, R.; Fleming, I. MicroRNA-223 antagonizes angiogenesis by targeting β1 integrin and preventing growth factor signaling in endothelial cells. Circ. Res. 2013, 113, 1320–1330. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.H.; Cai, Q.Q.; Dong, Y.W.; Wang, R.; He, B.M.; Qi, B.; Xu, C.J.; Wu, X.Z. Decoy oligonucleotide rescues IGF1R expression from microRNA-223 suppression. PLoS ONE 2013, 8, e82167. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Li, H.; Jia, C.Y.; Cheng, W.; Yu, M.; Peng, M.; Zhu, Y.; Zhao, Q.; Dong, Y.W.; Shao, K.; et al. MicroRNA-223 regulates FOXO1 expression and cell proliferation. FEBS Lett. 2012, 586, 1038–1043. [Google Scholar] [CrossRef] [PubMed]
- Kee, H.J.; Park, S.; Kwon, J.S.; Choe, N.; Ahn, Y.; Kook, H.; Jeong, M.H. B cell translocation gene; a direct target of miR-142-5p; inhibits vascular smooth muscle cell proliferation by down-regulating cell cycle progression. FEBS Lett. 2013, 587, 2385–2392. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.H.; Zhou, Z.; Yang, K.P.; Wang, X.G.; Zhu, Y.; Fa, X.E. miR-142-5p and miR-9 may be involved in squamous lung cancer by regulating cell cycle related genes. Eur. Rev. Med. Pharmacol. Sci. 2013, 17, 3213–3220. [Google Scholar] [PubMed]
- Wrana, J.L.; Attisano, L.; Carcamo, J. TGF-β signals through a heteromeric protein kinase receptor complex. Cell 1992, 71, 1003–1014. [Google Scholar] [CrossRef]
- Massague, J.; Chen, Y.G. Controlling TGF-β signaling. Genes Dev. 2000, 14, 627–644. [Google Scholar] [PubMed]
- Hu, Y.; Xu, H.; Li, Z.; Zheng, X.; Jia, X.; Nie, Q.; Zhang, X. Comparison of the genome-wide DNA methylation profiles between fast-growing and slow-growing broilers. PLoS ONE 2013, 8, e56411. [Google Scholar] [CrossRef] [PubMed]
- FASTX-Toolkit. Available online: http://hannonlab.cshl.edu/fastx_toolkit (accessed on 2 February 2010).
- Bowtie. Available online: http://bowtie-bio.sourceforge.net/index.shtml (accessed on 9 April 2013).
- Wang, L.; Feng, Z.; Wang, X.; Zhang, X. DEGSeq: An R package for identifying differentially expressed genes from RNA-Seq data. Bioinformatics 2010, 26, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Zheng, X.; Yang, J.; Imamichi, T. Extracting biological meaning from large gene lists with DAVID. Curr. Protoc. Bioinform. 2009. [Google Scholar] [CrossRef]
- Jansen, R.; Greenbaum, D.; Gerstein, M. Relating whole-genome expression data with protein–protein interactions. Genome Res. 2002, 12, 37–46. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ouyang, H.; He, X.; Li, G.; Xu, H.; Jia, X.; Nie, Q.; Zhang, X. Deep Sequencing Analysis of miRNA Expression in Breast Muscle of Fast-Growing and Slow-Growing Broilers. Int. J. Mol. Sci. 2015, 16, 16242-16262. https://doi.org/10.3390/ijms160716242
Ouyang H, He X, Li G, Xu H, Jia X, Nie Q, Zhang X. Deep Sequencing Analysis of miRNA Expression in Breast Muscle of Fast-Growing and Slow-Growing Broilers. International Journal of Molecular Sciences. 2015; 16(7):16242-16262. https://doi.org/10.3390/ijms160716242
Chicago/Turabian StyleOuyang, Hongjia, Xiaomei He, Guihuan Li, Haiping Xu, Xinzheng Jia, Qinghua Nie, and Xiquan Zhang. 2015. "Deep Sequencing Analysis of miRNA Expression in Breast Muscle of Fast-Growing and Slow-Growing Broilers" International Journal of Molecular Sciences 16, no. 7: 16242-16262. https://doi.org/10.3390/ijms160716242
APA StyleOuyang, H., He, X., Li, G., Xu, H., Jia, X., Nie, Q., & Zhang, X. (2015). Deep Sequencing Analysis of miRNA Expression in Breast Muscle of Fast-Growing and Slow-Growing Broilers. International Journal of Molecular Sciences, 16(7), 16242-16262. https://doi.org/10.3390/ijms160716242