
Mesenchymal Stem Cells and Induced Pluripotent Stem Cells as Therapies for Multiple Sclerosis

Abstract
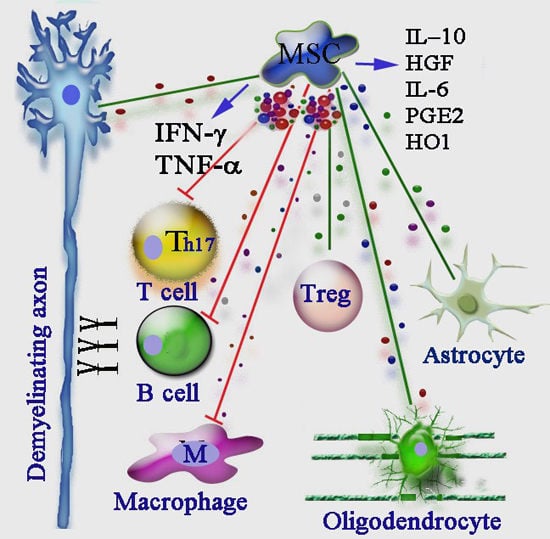
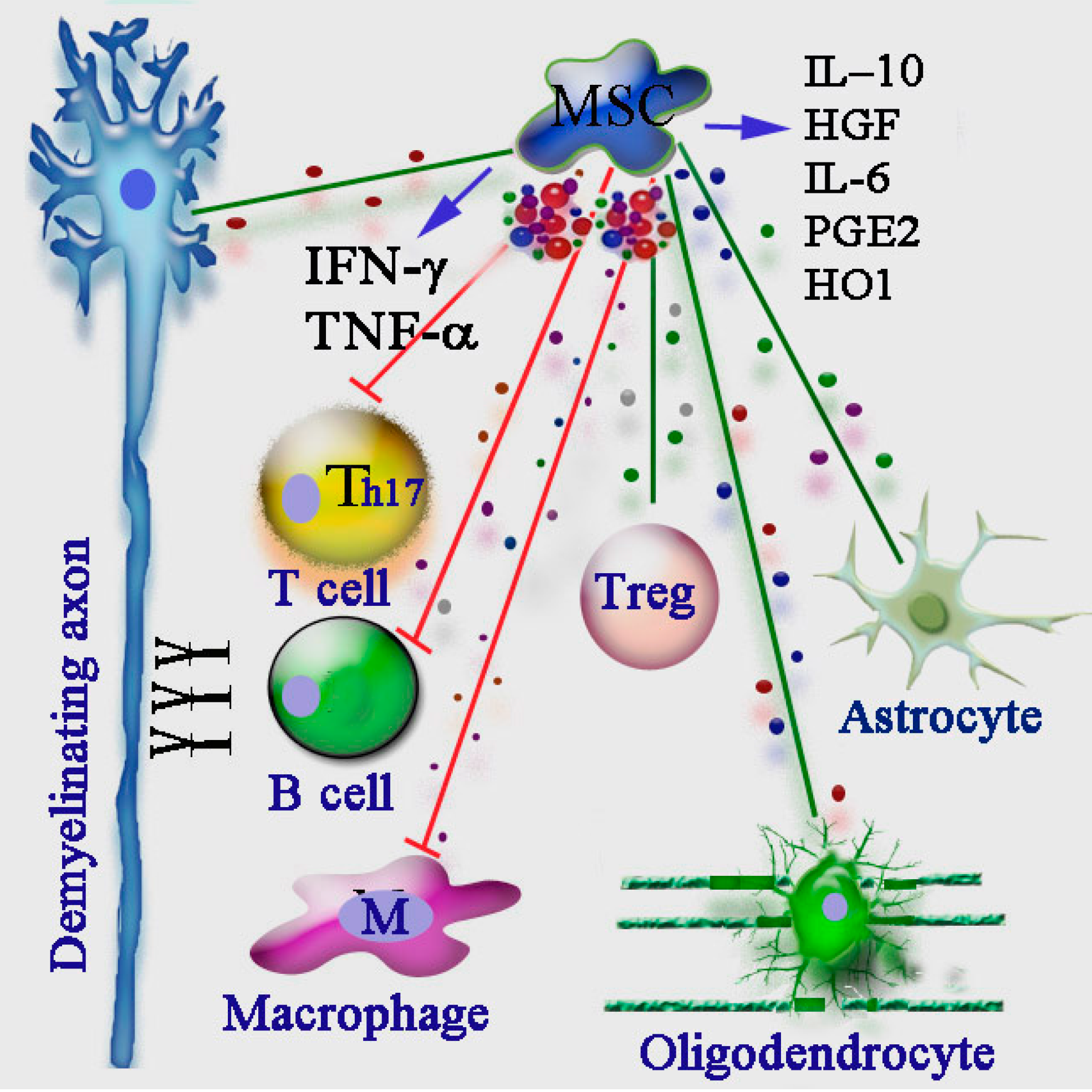
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Current Concepts in Multiple Sclerosis (MS) Pathology
3. Stem Cells as a “Biological Solution to a Biological Problem”
4. Mesenchymal Stem Cells (MSCs)
5. Efficacy of MSCs in Mouse Experimental Autoimmune Encephalomyelitis (EAE) Mouse: Current Evidence
6. Effect of the Inflammatory Environment of EAE on Endogenous MSCs
7. Efficacy of Genetically Engineered Human MSCs in Mouse EAE Models
8. Mechanisms of Action of MSCs
9. Immunosuppressive Effects of MSCs
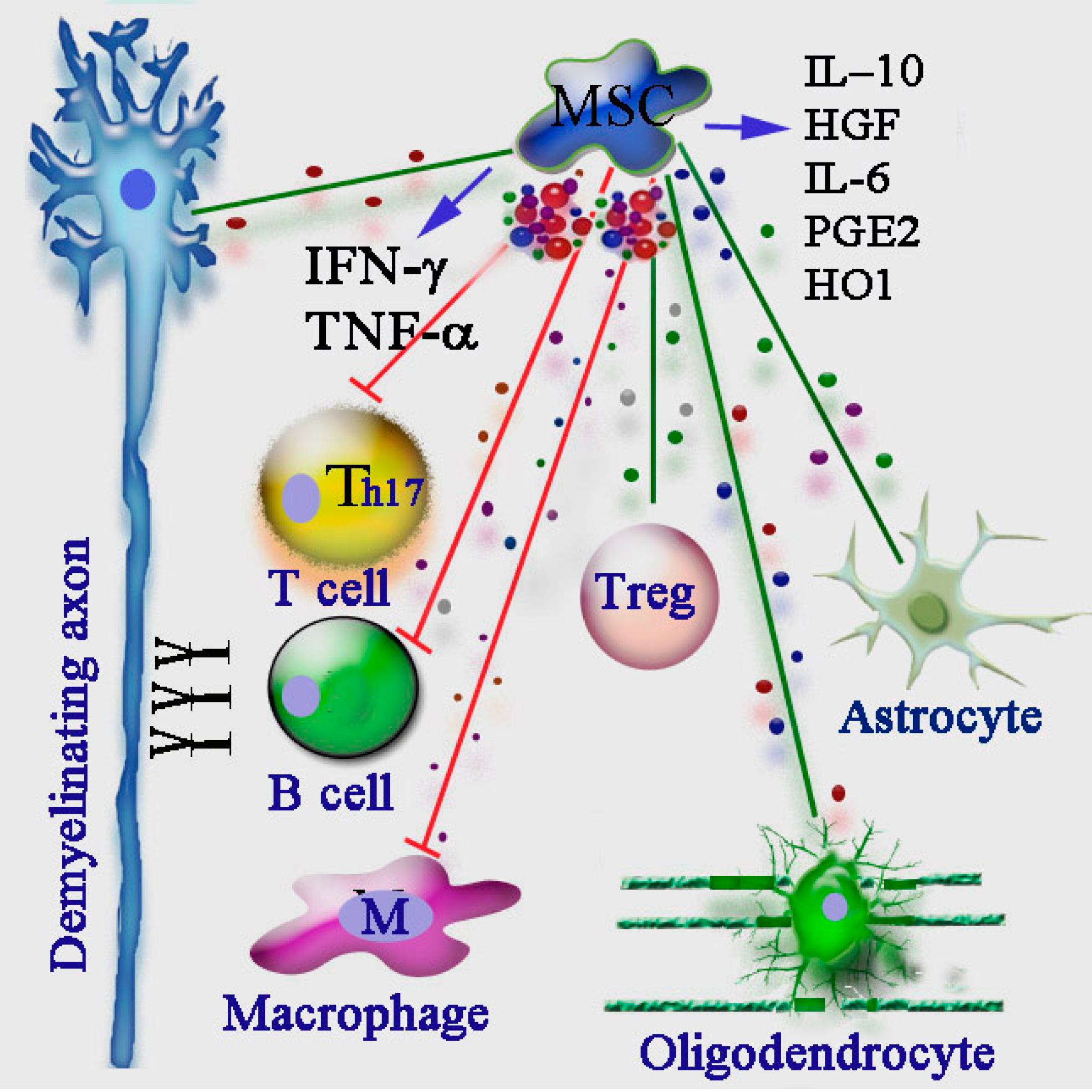
10. Immunomodulatory Action of MSCs in MS
11. Interaction of Endogenous MSCs and the Inflammatory Environment in EAE Determines the Outcome of Autoimmune Response
12. MSC-Derived Microvesicles (MVs) as Therapeutic Vehicle
13. Clinical Trials in MS Using MSCs
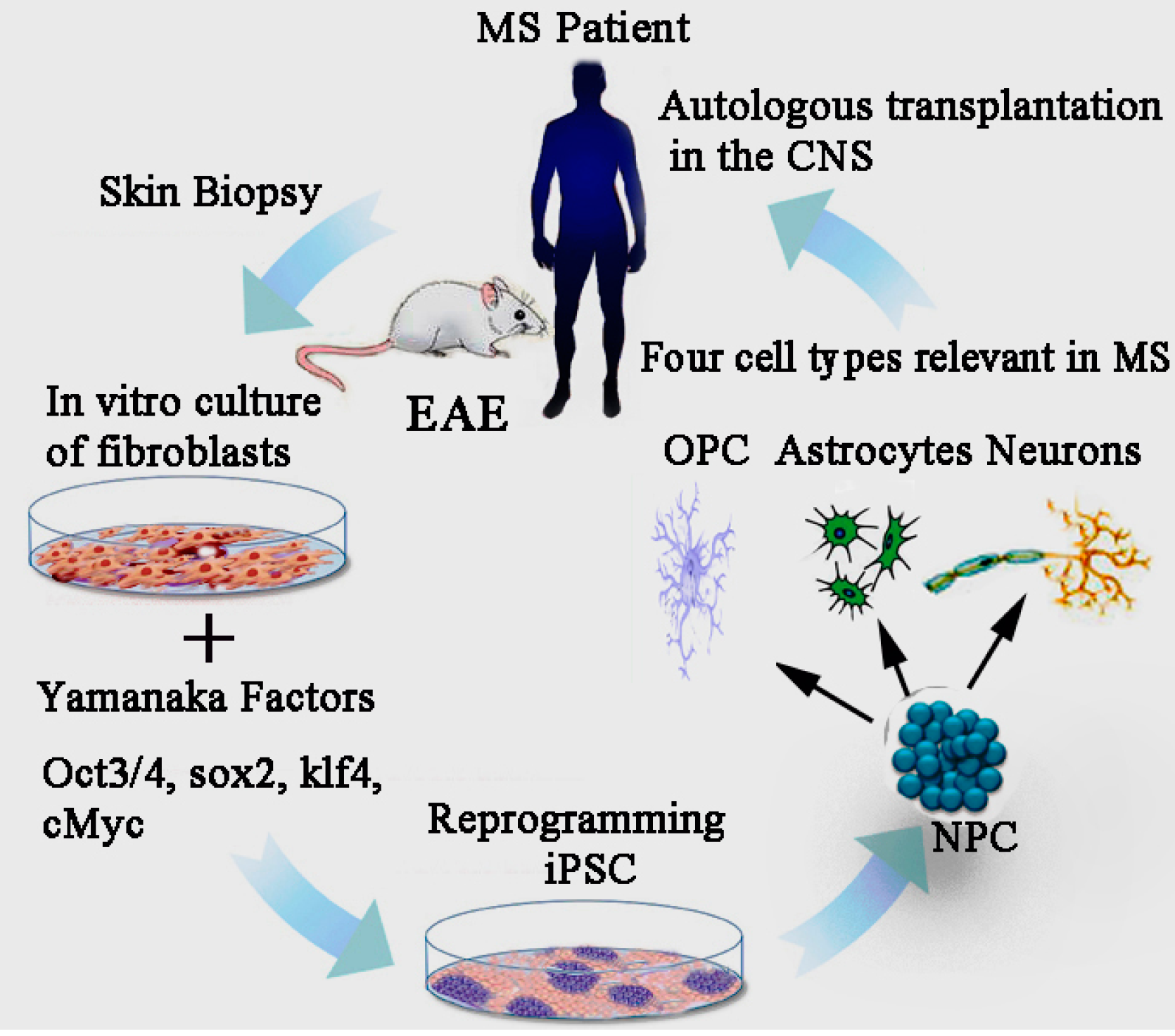
14. Induced Pluripotent Stem Cell (iPSCs)
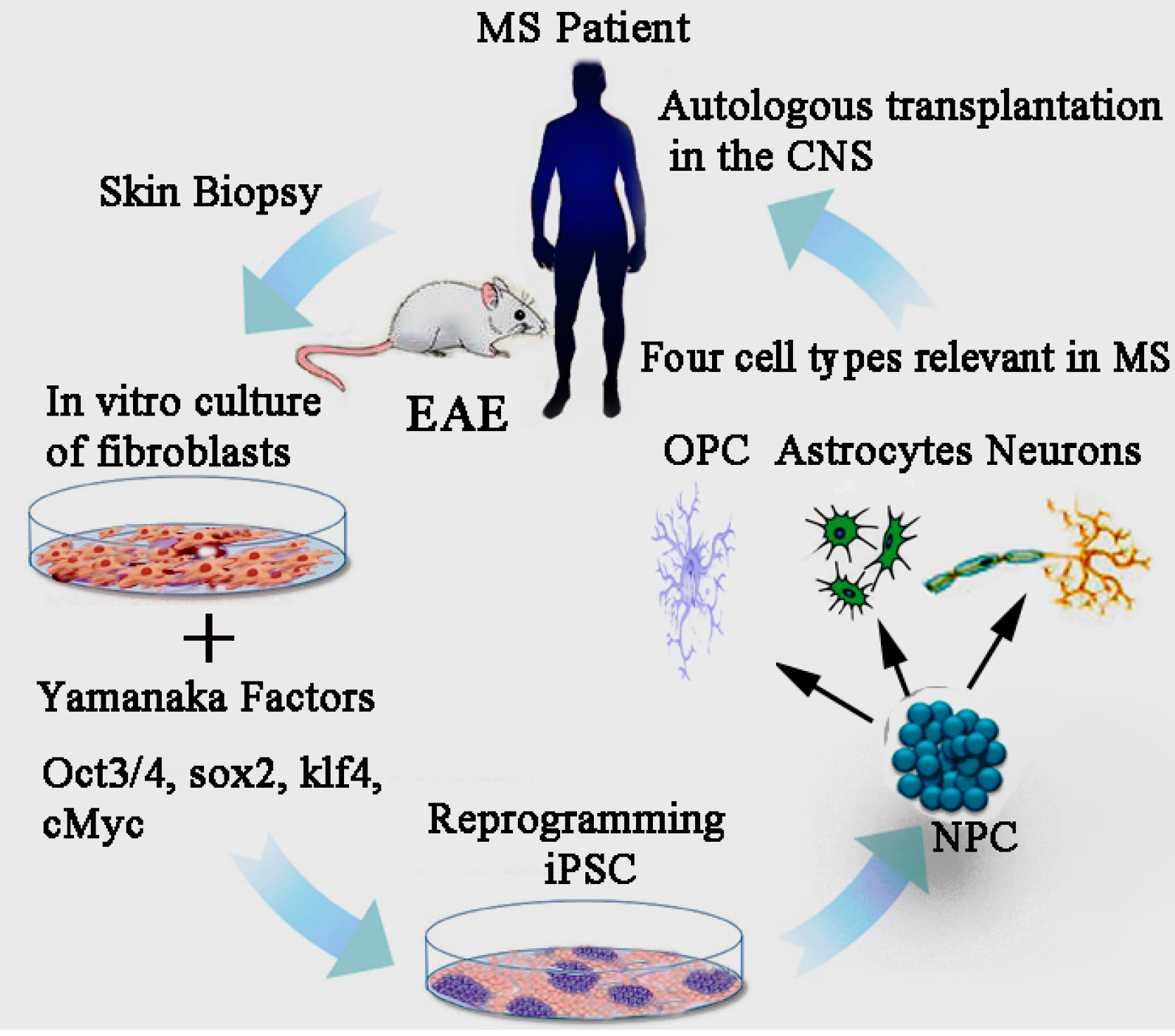
15. Effect of iPSCs Derived Neural Precursor Cells (NPCs) in EAE
16. Mechanisms of Action of iPSC–Neural Stem Cells (NSCs): Candidate Factors
17. iPSCs Derived Oligodendrocyte Precursor Cells (OPCs) as a Therapeutic Candidate for MS
18. Modelling MS Pathology though the iPSC Platform
19. Risks and Disadvantages with the Use of MSCs and iPSCs in Clinical Applications
20. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- McQueen, R.B.; Livingston, T.; Vollmer, T.; Corboy, J.; Buckley, B.; Allen, R.R.; Nair, K.; Campbell, J.D. Increased relapse activity for multiple sclerosis natalizumab users who become nonpersistent: A retrospective study. J. Manag. Care Spec. Pharm. 2015, 21, 210–218. [Google Scholar] [PubMed]
- Antel, J.; Antel, S.; Caramanos, Z.; Arnold, D.L.; Kuhlmann, T. Primary progressive multiple sclerosis: Part of the MS disease spectrum or separate disease entity? Acta Neuropathol. 2012, 123, 627–638. [Google Scholar] [CrossRef] [PubMed]
- Hedstrom, A.K.; Olsson, T.; Alfredsson, L. The role of environment and lifestyle in determining the risk of multiple sclerosis. Curr. Top. Behav. Neurosci. 2015. [Google Scholar] [CrossRef]
- Pantazou, V.; Schluep, M.; Du Pasquier, R. Environmental factors in multiple sclerosis. Presse Med. 2015, 44, e113–e120. [Google Scholar] [CrossRef] [PubMed]
- Kipp, M.; van der Valk, P.; Amor, S. Pathology of multiple sclerosis. CNS Neurol. Disord. 2012, 11, 506–517. [Google Scholar] [CrossRef]
- Hauser, S.L.; Josephson, S.A.; Johnston, S.C. Multiple sclerosis: Monotherapy rules. Ann. Neurol. 2013, 73, A5–A6. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M.; Hafler, D.A.; Lucchinetti, C.F. Multiple sclerosis-a quiet revolution. Nat. Rev. Neurol. 2015, 11, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Duddy, M. NICE approval of dimethyl fumarate could benefit thousands living with relapsing–remitting multiple sclerosis. Neurodegener. Dis. Manag. 2015, 5, 7–10. [Google Scholar] [CrossRef] [PubMed]
- Hauser, S.L.; Chan, J.R.; Oksenberg, J.R. Multiple sclerosis: Prospects and promise. Ann. Neurol. 2013, 74, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Rice, C.M.; Cottrell, D.; Wilkins, A.; Scolding, N.J. Primary progressive multiple sclerosis: Progress and challenges. J. Neurol. Neurosurg. Psychiatry 2013, 84, 1100–1106. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, J.M.; Robinson, A.P.; Miller, S.D. Strategies for protecting oligodendrocytes and enhancing remyelination in multiple sclerosis. Discov. Med. 2013, 16, 53–63. [Google Scholar] [PubMed]
- Robey, P.G. Stem cells near the century mark. J. Clin. Investig. 2000, 105, 1489–1491. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Ben-Hur, T.; Fainstein, N.; Nishri, Y. Cell-based reparative therapies for multiple sclerosis. Curr. Neurol. Neurosci. Rep. 2013, 13, 397. [Google Scholar] [CrossRef] [PubMed]
- Caplan, A.I. Mesenchymal stem cells. J. Orthop. Res. 1991, 9, 641–650. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Bowles, A.C.; Semon, J.A.; Scruggs, B.A.; Zhang, S.; Strong, A.L.; Gimble, J.M.; Bunnell, B.A. Transplantation of autologous adipose stem cells lacks therapeutic efficacy in the experimental autoimmune encephalomyelitis model. PLoS ONE 2014, 9, e85007. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Chai, J.; Shen, C.; Han, Y.; Sun, T. Human umbilical cord-derived mesenchymal stem cells differentiate into epidermal-like cells using a novel co-culture technique. Cytotechnology 2014, 66, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.; Keating, A.; Prockop, D.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Freedman, M.S.; Bar-Or, A.; Atkins, H.L.; Karussis, D.; Frassoni, F.; Lazarus, H.; Scolding, N.; Slavin, S.; Le Blanc, K.; Uccelli, A. The therapeutic potential of mesenchymal stem cell transplantation as a treatment for multiple sclerosis: Consensus report of the International MSCT Study Group. Mult. Scler. 2010, 16, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Zappia, E.; Casazza, S.; Pedemonte, E.; Benvenuto, F.; Bonanni, I.; Gerdoni, E.; Giunti, D.; Ceravolo, A.; Cazzanti, F.; Frassoni, F.; et al. Mesenchymal stem cells ameliorate experimental autoimmune encephalomyelitis inducing T-cell anergy. Blood 2005, 106, 1755–1761. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.A. Mesenchymal stem cell transplantation in multiple sclerosis. J. Neurol. Sci. 2013, 333, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Rivera, F.J.; Couillard-Despres, S.; Pedre, X.; Ploetz, S.; Caioni, M.; Lois, C.; Bogdahn, U.; Aigner, L. Mesenchymal stem cells instruct oligodendrogenic fate decision on adult neural stem cells. Stem Cells 2006, 24, 2209–2219. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Ren, G.; Huang, Y.; Su, J.; Han, Y.; Li, J.; Chen, X.; Cao, K.; Chen, Q.; Shou, P.; et al. Mesenchymal stem cells: A double-edged sword in regulating immune responses. Cell Death Differ. 2012, 19, 1505–1513. [Google Scholar] [CrossRef]
- Grigoriadis, N.; Lourbopoulos, A.; Lagoudaki, R.; Frischer, J.M.; Polyzoidou, E.; Touloumi, O.; Simeonidou, C.; Deretzi, G.; Kountouras, J.; Spandou, E.; et al. Variable behavior and complications of autologous bone marrow mesenchymal stem cells transplanted in experimental autoimmune encephalomyelitis. Exp. Neurol. 2011, 230, 78–89. [Google Scholar] [CrossRef]
- Glenn, J.D.; Smith, M.D.; Calabresi, P.A.; Whartenby, K.A. Mesenchymal stem cells differentially modulate effector CD8+ T cell subsets and exacerbate experimental autoimmune encephalomyelitis. Stem Cells 2014, 32, 2744–2755. [Google Scholar] [CrossRef] [PubMed]
- Rasini, V.; Dominici, M.; Kluba, T.; Siegel, G.; Lusenti, G.; Northoff, H.; Horwitz, E.M.; Schafer, R. Mesenchymal stromal/stem cells markers in the human bone marrow. Cytotherapy 2012, 15, 292–306. [Google Scholar] [CrossRef]
- Zacharaki, D.; Lagoudaki, R.; Touloumi, O.; Kotta, K.; Voultsiadou, A.; Poulatsidou, K.N.; Lourbopoulos, A.; Hadjigeorgiou, G.; Dardiotis, E.; Karacostas, D.; et al. Characterization of in vitro expanded bone marrow-derived mesenchymal stem cells isolated from experimental autoimmune encephalomyelitis mice. J. Mol. Neurosci. 2013, 51, 282–297. [Google Scholar] [CrossRef] [PubMed]
- Kassis, I.; Petrou, P.; Halimi, M.; Karussis, D. Mesenchymal stem cells (MSC) derived from mice with experimental autoimmune encephalomyelitis (EAE) suppress EAE and have similar biological properties with MSC from healthy donors. Immunol. Lett. 2013, 154, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Deshmane, S.L.; Kremlev, S.; Amini, S.; Sawaya, B.E. Monocyte chemoattractant protein-1 (MCP-1): An overview. J. Interferon Cytokine Res. 2009, 29, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Tsirka, S.E. Truncation of monocyte chemoattractant protein 1 by plasmin promotes blood-brain barrier disruption. J. Cell Sci. 2011, 124, 1486–1495. [Google Scholar] [CrossRef] [PubMed]
- Mallam, E.; Kemp, K.; Wilkins, A.; Rice, C.; Scolding, N. Characterization of in vitro expanded bone marrow-derived mesenchymal stem cells from patients with multiple sclerosis. Mult. Scler. 2010, 16, 909–918. [Google Scholar] [CrossRef] [PubMed]
- Ryu, C.H.; Park, K.Y.; Hou, Y.; Jeong, C.H.; Kim, S.M.; Jeun, S.S. Gene therapy of multiple sclerosis using interferon β-secreting human bone marrow mesenchymal stem cells. Biomed. Res. Int. 2013, 2013, 696738. [Google Scholar] [CrossRef] [PubMed]
- Payne, N.L.; Dantanarayana, A.; Sun, G.; Moussa, L.; Caine, S.; McDonald, C.; Herszfeld, D.; Bernard, C.C.; Siatskas, C. Early intervention with gene-modified mesenchymal stem cells over-expressing interleukin-4 enhances anti-inflammatory responses and functional recovery in experimental autoimmune demyelination. Cell Adhes. Migr. 2012, 6, 179–189. [Google Scholar] [CrossRef]
- Payne, N.L.; Sun, G.; McDonald, C.; Moussa, L.; Emerson-Webber, A.; Loisel-Meyer, S.; Medin, J.A.; Siatskas, C.; Bernard, C.C. Human adipose-derived mesenchymal stem cells engineered to secrete IL-10 inhibit APC function and limit CNS autoimmunity. Brain Behav. Immun. 2013, 30, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Maltman, D.J.; Hardy, S.A.; Przyborski, S.A. Role of mesenchymal stem cells in neurogenesis and nervous system repair. Neurochem. Int. 2011, 59, 347–356. [Google Scholar] [PubMed]
- Coles, A.J.; Compston, D.A.; Selmaj, K.W.; Lake, S.L.; Moran, S.; Margolin, D.H.; Norris, K.; Tandon, P.K. Alemtuzumab vs. interferon beta-1a in early multiple sclerosis. N. Engl. J. Med. 2008, 359, 1786–1801. [Google Scholar] [CrossRef] [PubMed]
- Krampera, M.; Cosmi, L.; Angeli, R.; Pasini, A.; Liotta, F.; Andreini, A.; Santarlasci, V.; Mazzinghi, B.; Pizzolo, G.; Vinante, F.; et al. Role for interferon-gamma in the immunomodulatory activity of human bone marrow mesenchymal stem cells. Stem Cells 2006, 24, 386–398. [Google Scholar] [CrossRef] [PubMed]
- De Miguel, M.P.; Fuentes-Julian, S.; Blazquez-Martinez, A.; Pascual, C.Y.; Aller, M.A.; Arias, J.; Arnalich-Montiel, F. Immunosuppressive properties of mesenchymal stem cells: Advances and applications. Curr. Mol. Med. 2012, 12, 574–591. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Ren, S.; Qu, X.; Ge, C.; Cheng, K.; Zhao, R.C. Mesenchymal stem cells inhibit Th17 cells differentiation via IFN-γ-mediated SOCS3 activation. Immunol. Res. 2015, 61, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Maccario, R.; Podesta, M.; Moretta, A.; Cometa, A.; Comoli, P.; Montagna, D.; Daudt, L.; Ibatici, A.; Piaggio, G.; Pozzi, S.; et al. Interaction of human mesenchymal stem cells with cells involved in alloantigen-specific immune response favors the differentiation of CD4+ T-cell subsets expressing a regulatory/suppressive phenotype. Haematologica 2005, 90, 516–525. [Google Scholar] [PubMed]
- Aggarwal, S.; Pittenger, M.F. Human mesenchymal stem cells modulate allogeneic immune cell responses. Blood 2005, 105, 1815–1822. [Google Scholar] [CrossRef] [PubMed]
- Czepiel, M.; Balasubramaniyan, V.; Schaafsma, W.; Stancic, M.; Mikkers, H.; Huisman, C.; Boddeke, E.; Copray, S. Differentiation of induced pluripotent stem cells into functional oligodendrocytes. Glia 2011, 59, 882–892. [Google Scholar] [CrossRef] [PubMed]
- Benkhoucha, M.; Santiago-Raber, M.L.; Schneiter, G.; Chofflon, M.; Funakoshi, H.; Nakamura, T.; Lalive, P.H. Hepatocyte growth factor inhibits CNS autoimmunity by inducing tolerogenic dendritic cells and CD25+Foxp3+ regulatory T cells. Proc. Natl. Acad. Sci. USA 2010, 107, 6424–6429. [Google Scholar] [CrossRef] [PubMed]
- Patanella, A.K.; Zinno, M.; Quaranta, D.; Nociti, V.; Frisullo, G.; Gainotti, G.; Tonali, P.A.; Batocchi, A.P.; Marra, C. Correlations between peripheral blood mononuclear cell production of BDNF, TNF-α, IL-6, IL-10 and cognitive performances in multiple sclerosis patients. J. Neurosci. Res. 2010, 88, 1106–1112. [Google Scholar] [PubMed]
- Douvaras, P.; Wang, J.; Zimmer, M.; Hanchuk, S.; O’Bara, M.A.; Sadiq, S.; Sim, F.J.; Goldman, J.; Fossati, V. Efficient generation of myelinating oligodendrocytes from primary progressive multiple sclerosis patients by induced pluripotent stem cells. Stem Cell Rep. 2014, 3, 250–259. [Google Scholar] [CrossRef]
- Trinschek, B.; Luessi, F.; Haas, J.; Wildemann, B.; Zipp, F.; Wiendl, H.; Becker, C.; Jonuleit, H. Kinetics of IL-6 production defines T effector cell responsiveness to regulatory T cells in multiple sclerosis. PLoS ONE 2013, 8, e77634. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.Z.; Su, W.R.; Shi, S.H.; Wilder-Smith, P.; Xiang, A.P.; Wong, A.; Nguyen, A.L.; Kwon, C.W.; Le, A.D. Human gingiva-derived mesenchymal stem cells elicit polarization of M2 macrophages and enhance cutaneous wound healing. Stem Cells 2010, 28, 1856–1868. [Google Scholar] [CrossRef] [PubMed]
- Dienz, O.; Rincon, M. The effects of IL-6 on CD4 T cell responses. Clin. Immunol. 2009, 130, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Kimbrel, E.A.; Ijichi, K.; Paul, D.; Lazorchak, A.S.; Chu, J.; Kouris, N.A.; Yavanian, G.J.; Lu, S.J.; Pachter, J.S.; et al. Human ESC-derived MSCs outperform bone marrow MSCs in the treatment of an EAE model of multiple sclerosis. Stem Cell Rep. 2014, 3, 115–130. [Google Scholar] [CrossRef]
- Bai, L.; Lennon, D.P.; Caplan, A.I.; DeChant, A.; Hecker, J.; Kranso, J.; Zaremba, A.; Miller, R.H. Hepatocyte growth factor mediates mesenchymal stem cell-induced recovery in multiple sclerosis models. Nat. Neurosci. 2012, 15, 862–870. [Google Scholar] [CrossRef] [PubMed]
- Copland, I.B.; Qayed, M.; Garcia, M.A.; Galipeau, J.; Waller, E.K. Bone marrow mesenchymal stromal cells from patients with acute and chronic graft-versus-host disease deploy normal phenotype, differentiation plasticity, and immune-suppressive activity. Biol. Blood Marrow Transplant. 2015. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, L.; Kikuiri, T.; Akiyama, K.; Chen, C.; Xu, X.; Yang, R.; Chen, W.; Wang, S.; Shi, S. Mesenchymal stem cell-based tissue regeneration is governed by recipient T lymphocytes via IFN-γ and TNF-α. Nat. Med. 2011, 17, 1594–1601. [Google Scholar] [CrossRef] [PubMed]
- Dang, S.; Xu, H.; Xu, C.; Cai, W.; Li, Q.; Cheng, Y.; Jin, M.; Wang, R.X.; Peng, Y.; Zhang, Y.; et al. Autophagy regulates the therapeutic potential of mesenchymal stem cells in experimental autoimmune encephalomyelitis. Autophagy 2014, 10, 1301–1315. [Google Scholar] [CrossRef] [PubMed]
- Sabin, K.; Kikyo, N. Microvesicles as mediators of tissue regeneration. Transl. Res. 2014, 163, 286–295. [Google Scholar] [CrossRef] [PubMed]
- Fierabracci, A.; del Fattore, A.; Luciano, R.; Muraca, M.; Teti, A. Recent advances in mesenchymal stem cell immunomodulation: The role of microvesicles. Cell Transplant. 2013, 24, 133–149. [Google Scholar] [CrossRef] [PubMed]
- Ju, G.Q.; Cheng, J.; Zhong, L.; Wu, S.; Zou, X.Y.; Zhang, G.Y.; Gu, D.; Miao, S.; Zhu, Y.J.; Sun, J.; et al. Microvesicles derived from human umbilical cord mesenchymal stem cells facilitate tubular epithelial cell dedifferentiation and growth via hepatocyte growth factor induction. PLoS ONE 2015, 10, e0121534. [Google Scholar] [CrossRef] [PubMed]
- Baglio, S.R.; Pegtel, D.M.; Baldini, N. Mesenchymal stem cell secreted vesicles provide novel opportunities in (stem) cell-free therapy. Front. Physiol. 2012, 3, 359. [Google Scholar] [CrossRef] [PubMed]
- Ardeshiry Lajimi, A.; Hagh, M.F.; Saki, N.; Mortaz, E.; Soleimani, M.; Rahim, F. Feasibility of cell therapy in multiple sclerosis: A systematic review of 83 studies. Int. J. Hematol. Oncol. Stem Cell Res. 2014, 7, 15–33. [Google Scholar]
- Llufriu, S.; Sepulveda, M.; Blanco, Y.; Marin, P.; Moreno, B.; Berenguer, J.; Gabilondo, I.; Martinez-Heras, E.; Sola-Valls, N.; Arnaiz, J.A.; et al. Randomized placebo-controlled phase II trial of autologous mesenchymal stem cells in multiple sclerosis. PLoS ONE 2014, 9, e113936. [Google Scholar] [CrossRef] [PubMed]
- Connick, P.; Kolappan, M.; Crawley, C.; Webber, D.J.; Patani, R.; Michell, A.W.; Du, M.Q.; Luan, S.L.; Altmann, D.R.; Thompson, A.J.; et al. Autologous mesenchymal stem cells for the treatment of secondary progressive multiple sclerosis: An open-label phase 2a proof-of-concept study. Lancet Neurol. 2012, 11, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Schadt, E.E.; Buchanan, S.; Brennand, K.J.; Merchant, K.M. Evolving toward a human-cell based and multiscale approach to drug discovery for CNS disorders. Front. Pharmacol. 2014, 5, 252. [Google Scholar] [CrossRef] [PubMed]
- Naegele, J.R.; Maisano, X.; Yang, J.; Royston, S.; Ribeiro, E. Recent advancements in stem cell and gene therapies for neurological disorders and intractable epilepsy. Neuropharmacology 2010, 58, 855–864. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Coleman, R.; Leang, R.; Tran, H.; Kopf, A.; Walsh, C.M.; Sears-Kraxberger, I.; Steward, O.; Macklin, W.B.; Loring, J.F.; et al. Human neural precursor cells promote neurologic recovery in a viral model of multiple sclerosis. Stem Cell Rep. 2014, 2, 825–837. [Google Scholar] [CrossRef]
- Laterza, C.; Merlini, A.; de Feo, D.; Ruffini, F.; Menon, R.; Onorati, M.; Fredrickx, E.; Muzio, L.; Lombardo, A.; Comi, G.; et al. iPSC-derived neural precursors exert a neuroprotective role in immune-mediated demyelination via the secretion of LIF. Nat. Commun. 2013, 4, 2597. [Google Scholar] [CrossRef] [PubMed]
- Marriott, M.P.; Emery, B.; Cate, H.S.; Binder, M.D.; Kemper, D.; Wu, Q.; Kolbe, S.; Gordon, I.R.; Wang, H.; Egan, G.; et al. Leukemia inhibitory factor signaling modulates both central nervous system demyelination and myelin repair. Glia 2008, 56, 686–698. [Google Scholar] [CrossRef] [PubMed]
- Butzkueven, H.; Emery, B.; Cipriani, T.; Marriott, M.P.; Kilpatrick, T.J. Endogenous leukemia inhibitory factor production limits autoimmune demyelination and oligodendrocyte loss. Glia 2006, 53, 696–703. [Google Scholar] [CrossRef] [PubMed]
- Levy, Y.A.; Mausner-Fainberg, K.; Vaknin-Dembinsky, A.; Amidror, T.; Regev, K.; Karni, A. Dysregulated production of leukemia inhibitory factor in immune cells of relapsing remitting multiple sclerosis patients. J. Neuroimmunol. 2015, 278, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Janssens, K.; van den Haute, C.; Baekelandt, V.; Lucas, S.; van Horssen, J.; Somers, V.; van Wijmeersch, B.; Stinissen, P.; Hendriks, J.J.; Slaets, H.; et al. Leukemia inhibitory factor tips the immune balance towards regulatory T cells in multiple sclerosis. Brain Behav. Immun. 2015, 45, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Bennett, J.; Basivireddy, J.; Kollar, A.; Biron, K.E.; Reickmann, P.; Jefferies, W.A.; McQuaid, S. Blood-brain barrier disruption and enhanced vascular permeability in the multiple sclerosis model EAE. J. Neuroimmunol. 2010, 229, 180–191. [Google Scholar] [CrossRef] [PubMed]
- Ben-Hur, T.; Goldman, S.A. Prospects of cell therapy for disorders of myelin. Ann. N. Y. Acad. Sci. 2008, 1142, 218–249. [Google Scholar] [CrossRef] [PubMed]
- Franklin, R.J.; Ffrench-Constant, C. Remyelination in the CNS: From biology to therapy. Nat. Rev. Neurosci. 2008, 9, 839–855. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, J.; Noble, M.; Mayer-Proschel, M. Characterization of A2B5+ glial precursor cells from cryopreserved human fetal brain progenitor cells. Glia 2002, 40, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Windrem, M.S.; Nunes, M.C.; Rashbaum, W.K.; Schwartz, T.H.; Goodman, R.A.; McKhann, G., 2nd; Roy, N.S.; Goldman, S.A. Fetal and adult human oligodendrocyte progenitor cell isolates myelinate the congenitally dysmyelinated brain. Nat. Med. 2004, 10, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Roy, N.S.; Wang, S.; Harrison-Restelli, C.; Benraiss, A.; Fraser, R.A.; Gravel, M.; Braun, P.E.; Goldman, S.A. Identification, isolation, and promoter-defined separation of mitotic oligodendrocyte progenitor cells from the adult human subcortical white matter. J. Neurosci. 1999, 19, 9986–9995. [Google Scholar] [PubMed]
- Hu, B.Y.; Du, Z.W.; Li, X.J.; Ayala, M.; Zhang, S.C. Human oligodendrocytes from embryonic stem cells: Conserved SHH signaling networks and divergent FGF effects. Development 2009, 136, 1443–1452. [Google Scholar] [CrossRef] [PubMed]
- Izrael, M.; Zhang, P.; Kaufman, R.; Shinder, V.; Ella, R.; Amit, M.; Itskovitz-Eldor, J.; Chebath, J.; Revel, M. Human oligodendrocytes derived from embryonic stem cells: Effect of noggin on phenotypic differentiation in vitro and on myelination in vivo. Mol. Cell. Neurosci. 2007, 34, 310–323. [Google Scholar] [CrossRef] [PubMed]
- Faulkner, J.; Keirstead, H.S. Human embryonic stem cell-derived oligodendrocyte progenitors for the treatment of spinal cord injury. Transpl. Immunol. 2005, 15, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Goldman, S.A.; Sim, F.J. Cell-based therapies for disorders of the brain and spinal cord. Neurotherapeutics 2011, 8, 537–538. [Google Scholar] [CrossRef] [PubMed]
- Windrem, M.S.; Schanz, S.J.; Guo, M.; Tian, G.F.; Washco, V.; Stanwood, N.; Rasband, M.; Roy, N.S.; Nedergaard, M.; Havton, L.A.; et al. Neonatal chimerization with human glial progenitor cells can both remyelinate and rescue the otherwise lethally hypomyelinated shiverer mouse. Cell Stem Cell 2008, 2, 553–565. [Google Scholar] [CrossRef] [PubMed]
- Windrem, M.S.; Roy, N.S.; Wang, J.; Nunes, M.; Benraiss, A.; Goodman, R.; McKhann, G.M., 2nd; Goldman, S.A. Progenitor cells derived from the adult human subcortical white matter disperse and differentiate as oligodendrocytes within demyelinated lesions of the rat brain. J. Neurosci. Res. 2002, 69, 966–975. [Google Scholar] [CrossRef] [PubMed]
- Keyoung, H.M.; Goldman, S.A. Glial progenitor-based repair of demyelinating neurological diseases. Neurosurg. Clin. N. Am. 2007, 18, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Bates, J.; Li, X.; Schanz, S.; Chandler-Militello, D.; Levine, C.; Maherali, N.; Studer, L.; Hochedlinger, K.; Windrem, M.; et al. Human iPSC-derived oligodendrocyte progenitor cells can myelinate and rescue a mouse model of congenital hypomyelination. Cell Stem Cell 2013, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Song, B.; Sun, G.; Herszfeld, D.; Sylvain, A.; Campanale, N.V.; Hirst, C.E.; Caine, S.; Parkington, H.C.; Tonta, M.A.; Coleman, H.A.; et al. Neural differentiation of patient specific iPS cells as a novel approach to study the pathophysiology of multiple sclerosis. Stem Cell Res. 2011, 8, 259–273. [Google Scholar] [CrossRef] [PubMed]
- Bar-Nur, O.; Russ, H.A.; Efrat, S.; Benvenisty, N. Epigenetic memory and preferential lineage-specific differentiation in induced pluripotent stem cells derived from human pancreatic islet beta cells. Cell Stem Cell 2011, 9, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Pelizzola, M.; Kida, Y.S.; Hawkins, R.D.; Nery, J.R.; Hon, G.; Antosiewicz-Bourget, J.; O’Malley, R.; Castanon, R.; Klugman, S.; et al. Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature 2011, 471, 68–73. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, J.; Yang, R.; Biswas, S.; Qin, X.; Zhang, M.; Deng, W. Mesenchymal Stem Cells and Induced Pluripotent Stem Cells as Therapies for Multiple Sclerosis. Int. J. Mol. Sci. 2015, 16, 9283-9302. https://doi.org/10.3390/ijms16059283
Xiao J, Yang R, Biswas S, Qin X, Zhang M, Deng W. Mesenchymal Stem Cells and Induced Pluripotent Stem Cells as Therapies for Multiple Sclerosis. International Journal of Molecular Sciences. 2015; 16(5):9283-9302. https://doi.org/10.3390/ijms16059283
Chicago/Turabian StyleXiao, Juan, Rongbing Yang, Sangita Biswas, Xin Qin, Min Zhang, and Wenbin Deng. 2015. "Mesenchymal Stem Cells and Induced Pluripotent Stem Cells as Therapies for Multiple Sclerosis" International Journal of Molecular Sciences 16, no. 5: 9283-9302. https://doi.org/10.3390/ijms16059283
APA StyleXiao, J., Yang, R., Biswas, S., Qin, X., Zhang, M., & Deng, W. (2015). Mesenchymal Stem Cells and Induced Pluripotent Stem Cells as Therapies for Multiple Sclerosis. International Journal of Molecular Sciences, 16(5), 9283-9302. https://doi.org/10.3390/ijms16059283