
Small-Nucleic-Acid-Based Therapeutic Strategy Targeting the Transcription Factors Regulating the Vascular Inflammation, Remodeling and Fibrosis in Atherosclerosis

Abstract
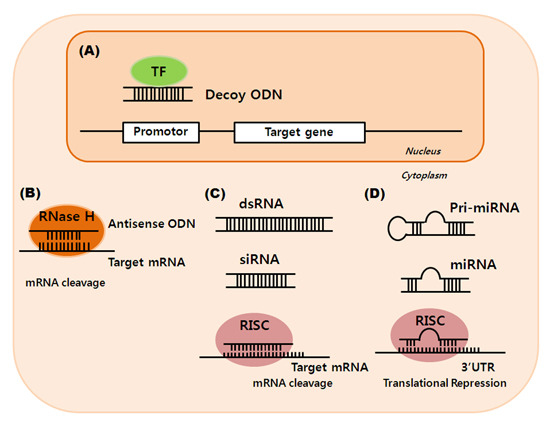
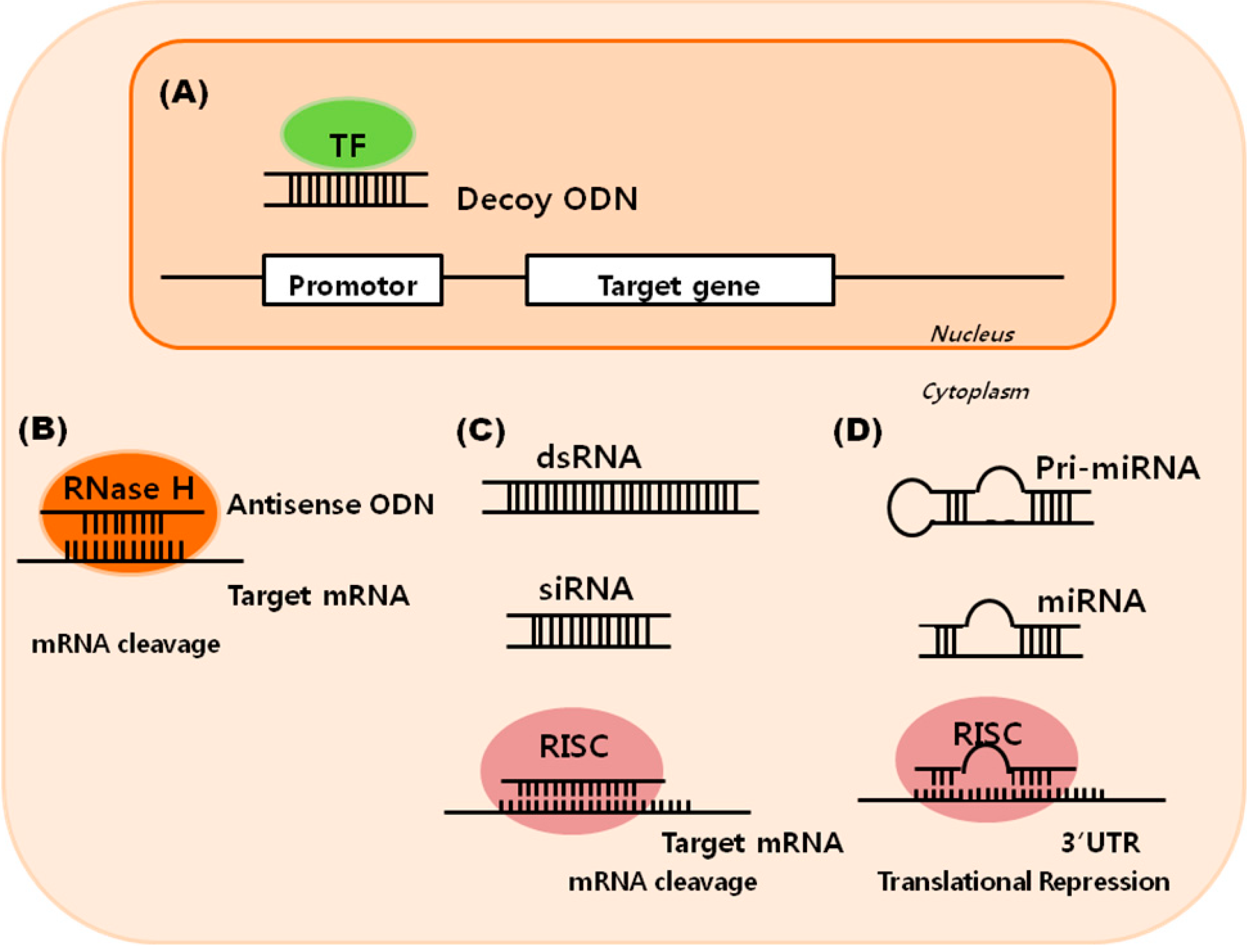
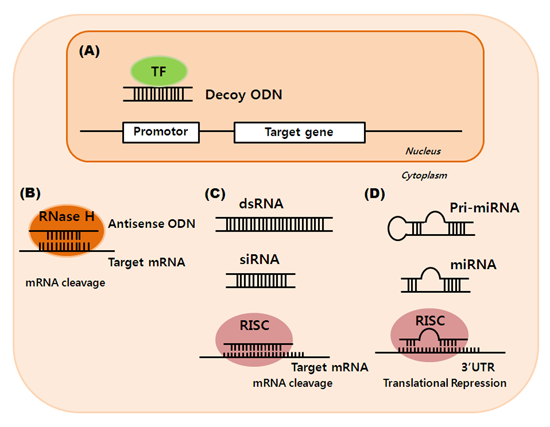
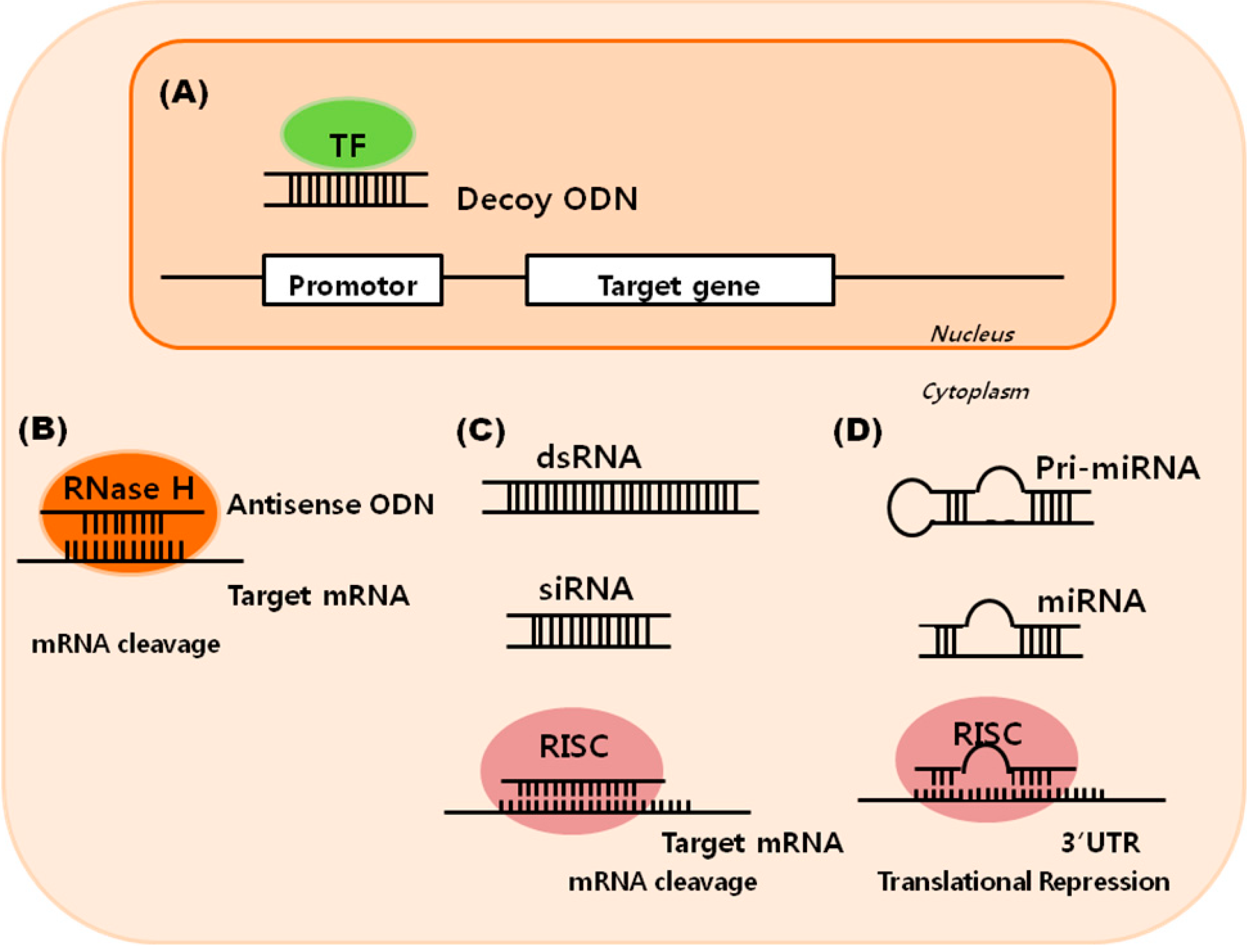
:
1. Introduction

{kind=link}
{kind=link}
| Strategy and Target Gene | Biological Effects | References |
|---|---|---|
| Antisense ODN | ||
| ACE | The inhibition of ACE by antisense ODN attenuates neointimal formation | [9] |
| TGF | TβR-targeted antisense ODN inhibits the expression of TGF-β1, and local delivery of an antisense TGF-β1 construct inhibits intimal hyperplasia in autogenous vein grafts in rats | [10,11] |
| STAT | Antisense ODN targeting SOCS3 exacerbates the atherosclerotic process in apolipoprotein E(−/−) mice by increasing the size, leukocyte content, and chemokine expression in the lesions | [12] |
| siRNA | ||
| TGF | TGF-β1 siRNA effectively reduces high-glucose-induced TGF-β1, PAI-1, and collagen type I mRNA, and protein expression in kidney and liver fibrosis | [13,14,15] |
| Ang II | Ang II induces ECM turnover and fibrosis via both TGF-β-dependent and -independent Smad3 signaling pathways. Ang II is involved in several intracellular signaling systems common to TGF-β, including activation of the Smad pathway, protein kinases (MAPK and ρ-kinase), and production of reactive oxygen species | [16,17,18,19] |
| Matrix metalloproteinase | RNA silencing targeted to matrix metalloproteinase-2 and -9 suppresses VSMC migration and neointimal formation in mice | [20,21] |
| VCAM-1 | Transfection of mice aorta VSMCs with siRNAs targeting VCAM-1 results in a reduction in the number of migrated SMCs | [22] |
| JAK2/STAT3 | IL-6, an inducer of the acute-phase response, induces the phosphorylation of STAT3, the proliferation of VSMCs, and the release of MCP-1. A specific JAK2 inhibitor AG490 partially inhibits STAT3 activation and MCP-1 production | [23] |
| STAT1 | STAT1 is a point of convergence for proatherogenic IFN-γ and LPS-mediated TLR4-dependent signal transduction. A specific STAT1 inhibitor, fludarabine, inhibits IFN- and LPS-dependent adhesion of monocytes to ECs | [24,25,26] |
| c-Jun | The effect of IL-6 on P4Hα1 expression is mediated by c-Jun through the Raf-MEK1/2-ERK1/2 MAPK pathway, and silencing c-Jun with siRNA significantly reduces IL-6-induced plaque instability | [27] |
| NF-κB | siRNA knockdown of NF-κB inhibits the IL-17-mediated up-regulation of VCAM-1 expression | [28] |
| ERK1/2 | ERK1/2 silencing attenuates IFN-induced ox-LDL uptake in macrophages and reverses high-glucose-induced CTGF-mediated proliferation and ECM production in VSMCs | [29,30] |
| CD74 | CD74 siRNA decreases IFN-γ-induced NF-κB activation and MCP-1 production in VSMCs | [31] |
| TLR4 | TLR4 silencing decreases ox-LDL-associated NF-κB activity, MCP-1, and IL-8 | [32,33] |
| miRNA | ||
| miR-21 | miR-21 is increased in ligated or balloon-injured rat carotid arteries and in human atherosclerotic lesions | [34,35] |
| miR-143/145 | miR-145 is the most abundant miRNA in VSMCs of normal rat carotid arteries | [36] |
| miR-146a | miR-146a targeting the KLF4 3ʹ UTR promotes VSMC proliferation in vitro and vascular neointimal hyperplasia in vivo | [37] |
| miR-126 | miR-126 plays an antiatherogenic role by enhancing endothelial repair and repressing VCAM-1 | [38,39] |
| miR-181 | Overexpression of miR-181 inhibits importin-α3 expression and repression of NF-κB nuclear translocation, and subsequent repression of NF-κB-responsive genes such as those encoding the adhesion molecules VCAM-1 and E-selectin | [40,41] |
| miR-155 | Expression of miR-155 is induced by TLR ligands such as LPS, mildly ox-LDL, and IFN-γ. miR-155 suppresses SHIP1, SOCS1, TGF-activated kinase 1/MAP3K7-binding protein 2, and Bcl-6. miR-155 also modulates TGF signaling in macrophages via the targeting of Smad2 | [42,43] |
| Decoy ODN | ||
| E2F | Double-stranded decoy ODNs corresponding to E2F binding sites block the activation of genes mediating cell-cycle progression, and inhibit VSMC proliferation and intimal hyperplasia in injured vessels | [44,45,46] |
| Sp1 | Inhibition of the Sp1-binding promoter region was found to suppress Ang I receptor expression and decrease the blood pressure in rats | [47] |
| NF-κB | Treatment with a NF-κB decoy ODN reduces the activities of inflammatory cytokines such as TNF-α and IL-1β, and the expression of adhesion molecules was found to be reduced in an animal model | [48,49,50] |
| NF-κB + Sp1 | Chimeric decoy ODN alleviates atherosclerotic changes and reduces serum cholesterol, inflammatory cytokines, and the expressions of atherosclerotic markers | [51] |
2. Antisense Oligodeoxynucleotide (ODN) Strategy for Vascular Disease
2.1. Antisense ODN Targeted for Ang II
2.2. Antisense ODN Targeted for TGF-β1
2.3. Antisense ODN Targeted for JAK/STAT
2.4. Clinical Trial of Antisense ODN Strategy
3. siRNA Strategy for Vascular Disease
3.1. siRNA Targeted for TGF-β/Smad Signaling Pathways
3.2. siRNA Targeted for Ang-II-Induced Signaling Pathways
3.3. siRNA Targeted to Matrix Metalloproteinases (MMPs)
3.4. siRNA Targeted to Adhesion Molecules
3.5. siRNA Targeted for JAK/STAT
3.6. siRNA Targeted to Nuclear Factor-κB and Its Associated Noncanonical Transcription Factors
3.7. Clinical Trial of siRNA Strategy
4. miRNA Strategy for Vascular Disease
4.1. miR-21
4.2. miR-143/145 and miR-146a
4.3. miR-10
4.4. miR-24
4.5. miR-181b
4.6. miR-126
4.7. miR-221/222
4.8. miR-155
4.9. Clinical Trial of miRNA Strategy
5. Decoy ODN Strategy for Vascular Disease
5.1. Decoy ODN Targeting E2F
5.2. Decoy ODN Targeting Sp1
5.3. Decoy ODN Targeting NF-κB
5.4. Decoy ODN Targeting Two or More Transcription Factors
5.5. Ring-Type Decoy ODN
5.6. Clinical Trial of Decoy ODN Strategy
6. Conclusions
Acknowledgments
Author Contributions
Abbreviations
| ABCA | ATP-binding cassette transporter |
| ACE | Angiotensin-converting enzyme |
| Ang | Angiotensin |
| AP-1 | Activator protein-1 |
| AT1 | Angiotensin II type 1 receptor |
| ASO | Antisense oligonucleotide |
| Bcl-2 | B-cell leukemia/lymphoma 2 |
| Co-Smad | Common mediator Smad |
| CSE | Cystathionine lyase |
| CTGF | Connective-tissue growth factor |
| dsRNA | Double-stranded RNA |
| EC | Endothelial cell |
| ECM | Extracellular matrix |
| EndMT | Endothelial-to-mesenchymal transition |
| ERK | Extracellular-signal-regulated kinase |
| Ets | E26 transformation-specific |
| GC | Guanine-cytosine |
| ICAM | Intercellular adhesion molecule |
| IFN | Interferon |
| IκB | Nuclear factor of κ light polypeptide gene enhancer in B-cell inhibitor |
| IL | Interleukin |
| JAK | Janus kinase |
| KLF | Krüppel-like factor |
| LDL | Low-density lipoprotein |
| LPS | Lipopolysaccharide |
| LTBP | Latent transforming-growth-factor-β-binding protein |
| LXRα | Liver X receptor α |
| MAPK | Mitogen-activated protein kinase |
| MCP | Monocyte chemotactic protein |
| MEK | Mitogen-activated protein kinase kinase |
| miRNA | MicroRNA |
| MMP | Matrix metalloproteinase |
| mRNA | Messenger RNA |
| NF-κB | Nuclear factor κB |
| ODN | Oligodeoxynucleotide |
| RISC | RNA-induced silencing complex |
| RNAi | RNA interference |
| ox-LDL | Oxidized low-density lipoprotein |
| P4Hα1 | Prolyl-4-hydroxylase α1 |
| PAI-1 | Plasminogen activator inhibitor-1 |
| PDGF | Platelet-derived growth factor |
| PPAR | Peroxisome proliferator-activated receptor |
| Pre-miRNA | Precursor microRNA |
| Pri-miRNA | Primary RNA precursor of microRNA |
| PS | Phosphorothioate |
| PTEN | Phosphatase and tensin homolog |
| RNase H | Ribonuclease H |
| R-Smad | Receptor-regulated Smad |
| SHIP1 | Polyphosphate-5-phosphatase |
| siRNA | Small interfering RNA |
| SMC | Smooth-muscle cell |
| SOCS | Suppressors of cytokine signaling |
| Sp1 | Specificity protein 1 |
| STAT | Signal transducer and activator of transcription |
| TβRI | TGF-β1 serine/threonine kinase receptor I |
| TβRII | TGF-β1 serine/threonine kinase receptor II |
| TGF | Transforming growth factor |
| TLR | Toll-like receptor |
| TNF | Tumor necrosis factor |
| UTR | Untranslated region |
| VCAM | Vascular cellular adhesion molecule |
| VSMC | Vascular smooth-muscle cell |
Conflicts of Interest
References
- Pasternak, R.C.; Criqui, M.H.; Benjamin, E.J.; Fowkes, F.G.; Isselbacher, E.M.; McCullough, P.A.; Wolf, P.A.; Zheng, Z.J. Atherosclerotic vascular disease conference: Writing group I: Epidemiology. Circulation 2004, 109, 2605–2612. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.C., Jr.; Milani, R.V.; Arnett, D.K.; Crouse, J.R., 3rd; McDermott, M.M.; Ridker, P.M.; Rosenson, R.S.; Taubert, K.A.; Wilson, P.W. Atherosclerotic vascular disease conference: Writing group II: Risk factors. Circulation 2004, 109, 2613–2616. [Google Scholar] [CrossRef] [PubMed]
- Joosten, M.M.; Pai, J.K.; Bertoia, M.L.; Rimm, E.B.; Spiegelman, D.; Mittleman, M.A.; Mukamal, K.J. Associations between conventional cardiovascular risk factors and risk of peripheral artery disease in men. JAMA 2012, 308, 1660–1667. [Google Scholar] [CrossRef] [PubMed]
- Grainger, D.J. TGF-β and atherosclerosis in man. Cardiovasc. Res. 2007, 74, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Ross, R. Atherosclerosis—An inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef]
- Tedgui, A.; Mallat, Z. Cytokines in atherosclerosis: Pathogenic and regulatory pathways. Physiol. Rev. 2006, 86, 515–581. [Google Scholar] [CrossRef] [PubMed]
- Ikedo, H.; Tamaki, K.; Ueda, S.; Kato, S.; Fujii, M.; Ten Dijke, P.; Okuda, S. Smad protein and TGF-β signaling in vascular smooth muscle cells. Int. J. Mol. Med. 2003, 11, 645–650. [Google Scholar] [CrossRef] [PubMed]
- Samarakoon, R.; Overstreet, J.M.; Higgins, P.J. TGF-β signaling in tissue fibrosis: Redox controls, target genes and therapeutic opportunities. Cell Signal. 2013, 25, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Morishita, R.; Gibbons, G.H.; Tomita, N.; Zhang, L.; Kaneda, Y.; Ogihara, T.; Dzau, V.J. Antisense oligodeoxynucleotide inhibition of vascular angiotensin-converting enzyme expression attenuates neointimal formation: Evidence for tissue angiotensin-converting enzyme function. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 915–922. [Google Scholar] [CrossRef] [PubMed]
- Kemaladewi, D.U.; Pasteuning, S.; van der Meulen, J.W.; van Heiningen, S.H.; van Ommen, G.J.; Ten Dijke, P.; Aartsma-Rus, A.; ’t Hoen, P.A.; Hoogaars, W.M. Targeting TGF-β signaling by antisense oligonucleotide-mediated knockdown of TGF-β type I receptor. Mol. Ther. Nucleic Acids 2014, 3, e156. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.X.; Liu, Z.; Tan, X.D.; Cui, D.X.; Wang, B.S.; Dai, X.W. Nanoparticle-mediated local delivery of an antisense TGF-β1 construct inhibits intimal hyperplasia in autogenous vein grafts in rats. PLoS ONE 2012, 7, e41857. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Muñoz, G.; Martin-Ventura, J.L.; Hernandez-Vargas, P.; Mallavia, B.; Lopez-Parra, V.; Lopez-Franco, O.; Muñoz-Garcia, B.; Fernandez-Vizarra, P.; Ortega, L.; Egido, J.; et al. Suppressors of cytokine signaling modulate JAK/STAT-mediated cell responses during atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Hwang, M.; Kim, H.J.; Noh, H.J.; Chang, Y.C.; Chae, Y.M.; Kim, K.H.; Jeon, J.P.; Lee, T.S.; Oh, H.K.; Lee, Y.S.; et al. TGF-β1 siRNA suppresses the tubulointerstitial fibrosis in the kidney of ureteral obstruction. Exp. Mol. Pathol. 2006, 81, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Noh, H.J.; Kim, H.C.; Lee, S.S.; Kang, Y.N.; Chae, Y.M.; Park, K.K. The inhibitory effect of siRNAs on the high glucose-induced over-expression of TGF-β1 in mesangial cells. J. Korean Med. Sci. 2006, 21, 430–435. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Kim, H.C.; Hwang, M.Y.; Oh, H.K.; Lee, T.S.; Chang, Y.C.; Song, H.J.; Won, N.H.; Park, K.K. The antifibrotic effect of TGF-β1 siRNAs in murine model of liver cirrhosis. Biochem. Biophys. Res. Commun. 2006, 343, 1072–1078. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Chung, A.C.; Huang, X.R.; Lan, H.Y. Angiotensin II induces connective tissue growth factor and collagen I expression via transforming growth factor-β-dependent and -independent Smad pathways: The role of Smad3. Hypertension 2009, 54, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Vita, J.; Sánchez-López, E.; Esteban, V.; Rupérez, M.; Egido, J.; Ruiz-Ortega, M. Angiotensin II activates the Smad pathway in vascular smooth muscle cells by a transforming growth factor-β-independent mechanism. Circulation 2005, 111, 2509–2517. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P.K.; Griendling, K.K. Angiotensin II cell signaling: Physiological and pathological effects in the cardiovascular system. Am. J. Physiol. Cell Physiol. 2007, 292, C82–C97. [Google Scholar] [CrossRef] [PubMed]
- Carvajal, G.; Rodríguez-Vita, J.; Rodrigues-Díez, R.; Sánchez-López, E.; Rupérez, M.; Cartier, C.; Esteban, V.; Ortiz, A.; Egido, J.; Mezzano, S.A.; et al. Angiotensin II activates the Smad pathway during epithelial mesenchymal transdifferentiation. Kidney Int. 2008, 74, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.L.; Dwivedi, A.; Somerville, M.; George, S.J.; Newby, A.C. Matrix metalloproteinase (MMP)-3 activates MMP-9 mediated vascular smooth muscle cell migration and neointima formation in mice. Arterioscler. Thromb. Vasc. Biol. 2011, 31, e35–e44. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Lee, D.; Jang, Y.L.; Chae, S.Y.; Choi, D.; Jeong, J.H.; Kim, S.H. Facial amphipathic deoxycholic acid-modified polyethyleneimine for efficient MMP-2 siRNA delivery in vascular smooth muscle cells. Eur. J. Pharm. Biopharm. 2012, 81, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Petersen, E.J.; Miyoshi, T.; Yuan, Z.; Hirohata, S.; Li, J.Z.; Shi, W.; Angle, J.F. siRNA silencing reveals role of vascular cell adhesion molecule-1 in vascular smooth muscle cell migration. Atherosclerosis 2008, 198, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Mu, W.; Kahn, A.; Jing, N.; Li, J.H.; Lan, H.Y.; Nakagawa, T.; Ohashi, R.; Johnson, R.J. Role of JAK/STAT pathway in IL-6-induced activation of vascular smooth muscle cells. Am. J. Nephrol. 2004, 24, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Sikorski, K.; Chmielewski, S.; Przybyl, L.; Heemann, U.; Wesoly, J.; Baumann, M.; Bluyssen, H.A. STAT1-mediated signal integration between IFNγ and LPS leads to increased EC and SMC activation and monocyte adhesion. Am. J. Physiol. Cell Physiol. 2011, 300, C1337–C1344. [Google Scholar] [CrossRef] [PubMed]
- Sikorski, K.; Czerwoniec, A.; Bujnicki, J.M.; Wesoly, J.; Bluyssen, H.A. STAT1 as a novel therapeutical target in pro-atherogenic signal integration of IFNγ, TLR4 and IL-6 in vascular disease. Cytokine Growth Factor Rev. 2011, 22, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Sikorski, K.; Chmielewski, S.; Olejnik, A.; Wesoly, J.Z.; Heemann, U.; Baumann, M.; Bluyssen, H. STAT1 as a central mediator of IFNγ and TLR4 signal integration in vascular dysfunction. JAKSTAT 2012, 1, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Huang, X.Z.; Li, X.N.; Feng, M.; Li, L.; Cai, X.J.; Zhang, C.; Liu, X.L.; Zhang, M.X.; Zhang, Y.; et al. Interleukin 6 destabilizes atherosclerotic plaques by down-regulating prolyl-4-hydroxylase α1 via a mitogen-activated protein kinase and c-Jun pathway. Arch. Biochem. Biophys. 2012, 528, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chen, J.; Liu, X.; Awar, L.; McMickle, A.; Bai, F.; Nagarajan, S.; Yu, S. IL-17 induces expression of vascular cell adhesion molecule through signalling pathway of NF-κB, but not Akt1 and TAK1 in vascular smooth muscle cells. Scand. J. Immunol. 2013, 77, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Mclaren, J.E.; Michael, D.R.; Clement, M.; Fielding, C.A.; Ramji, D.P. ERK is integral to the IFN-γ-mediated activation of STAT1, the expression of key genes implicated in atherosclerosis, and the uptake of modified lipoproteins by human macrophages. J. Immunol. 2010, 185, 3041–3048. [Google Scholar] [CrossRef] [PubMed]
- Ha, Y.M.; Lee, D.H.; Kim, M.; Kang, Y.J. High glucose induces connective tissue growth factor expression and extracellular matrix accumulation in rat aorta vascular smooth muscle cells via extracellular signal-regulated kinase 1/2. Korean J. Physiol. Pharmacol. 2013, 17, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Martín-Ventura, J.L.; Madrigal-Matute, J.; Muñoz-Garcia, B.; Blanco-Colio, L.M.; van Oostrom, M.; Zalba, G.; Fortuño, A.; Gomez-Guerrero, C.; Ortega, L.; Ortiz, A.; et al. Increased CD74 expression in human atherosclerotic plaques: Contribution to inflammatory responses in vascular cells. Cardiovasc. Res. 2009, 83, 586–594. [Google Scholar] [CrossRef] [PubMed]
- Geng, H.; Wang, A.; Rong, G.; Zhu, B.; Deng, Y.; Chen, J.; Zhong, R. The effects of ox-LDL in human atherosclerosis may be mediated in part via the toll-like receptor 4 pathway. Mol. Cell. Biochem. 2010, 342, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Meng, Z.; Yan, C.; Deng, Q.; Dong, X.; Duan, Z.M.; Gao, D.F.; Niu, X.L. Oxidized low-density lipoprotein induces inflammatory responses in cultured human mast cells via Toll-like receptor 4. Cell Physiol. Biochem. 2013, 31, 842–853. [Google Scholar] [CrossRef] [PubMed]
- Ji, R.; Cheng, Y.; Yue, J.; Yang, J.; Liu, X.; Chen, H.; Dean, D.B.; Zhang, C. MicroRNA expression signature and antisense-mediated depletion reveal an essential role of MicroRNA in vascular neointimal lesion formation. Circ. Res. 2007, 100, 1579–1588. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Pei, Y.; Cao, Q.; Wang, R. MicroRNA-21 represses human cystathionine γ-lyase expression by targeting at specificity protein-1 in smooth muscle cells. J. Cell. Physiol. 2012, 227, 3192–3200. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Liu, X.; Yang, J.; Lin, Y.; Xu, D.Z.; Lu, Q.; Deitch, E.A.; Huo, Y.; Delphin, E.S.; Zhang, C. MicroRNA-145, a novel smooth muscle cell phenotypic marker and modulator, controls vascular neointimal lesion formation. Circ. Res. 2009, 105, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.G.; Zheng, B.; Han, M.; Fang, X.M.; Li, H.X.; Miao, S.B.; Su, M.; Han, Y.; Shi, H.J.; Wen, J.K. miR-146a and Krüppel-like factor 4 form a feedback loop to participate in vascular smooth muscle cell proliferation. EMBO Rep. 2011, 12, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Harris, T.A.; Yamakuchi, M.; Ferlito, M.; Mendell, J.T.; Lowenstein, C.J. MicroRNA-126 regulates endothelial expression of vascular cell adhesion molecule 1. Proc. Natl. Acad. Sci. USA 2008, 105, 1516–1521. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Aurora, A.B.; Johnson, B.A.; Qi, X.; McAnally, J.; Hill, J.A.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. The endothelial-specific microRNA miR-126 governs vascular integrity and angiogenesis. Dev. Cell 2008, 15, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Icli, B.; Wara, A.K.; Belkin, N.; He, S.; Kobzik, L.; Hunninghake, G.M.; Vera, M.P.; MICU Registry; Blackwell, T.S.; et al. MicroRNA-181b regulates NF-κB-mediated vascular inflammation. J. Clin. Investig. 2012, 122, 1973–1990. [Google Scholar] [PubMed]
- Sun, X.; He, S.; Wara, A.K.; Icli, B.; Shvartz, E.; Tesmenitsky, Y.; Belkin, N.; Li, D.; Blackwell, T.S.; Sukhova, G.K.; et al. Systemic delivery of microRNA-181b inhibits nuclear factor-κB activation, vascular inflammation, and atherosclerosis in apolipoprotein E-deficient mice. Circ. Res. 2014, 3, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Chen, T.; Yang, L.; Li, Z.; Wong, M.M.; Zheng, X.; Pan, X.; Zhang, L.; Yan, H. Regulation of microRNA-155 in atherosclerotic inflammatory responses by targeting MAP3K10. PLoS ONE 2012, 7, e46551. [Google Scholar] [CrossRef] [PubMed]
- Louafi, F.; Martinez-Nunez, R.T.; Sanchez-Elsner, T. MicroRNA-155 targets SMAD2 and modulates the response of macrophages to transforming growth factor-β. J. Biol. Chem. 2010, 285, 41328–41336. [Google Scholar] [CrossRef] [PubMed]
- Morishita, R.; Gibbons, G.H.; Horiuchi, M.; Ellison, K.E.; Nakama, M.; Zhang, L.; Kaneda, Y.; Ogihara, T.; Dzau, V.J. A gene therapy strategy using a transcription factor decoy of the E2F binding site inhibits smooth muscle proliferation in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 5855–5859. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.D.; Morishita, R.; Kaneda, Y.; Kim, H.S.; Chang, Y.C.; Lee, K.U.; Park, J.Y.; Lee, H.W.; Kim, Y.H.; Lee, I.K. Novel E2F decoy oligodeoxynucleotides inhibit in vitro vascular smooth muscle cell proliferation and in vivo neointimal hyperplasia. Gene Ther. 2002, 9, 1682–1692. [Google Scholar] [CrossRef] [PubMed]
- Hashiya, N.; Aoki, M.; Tachibana, K.; Taniyama, Y.; Yamasaki, K.; Hiraoka, K.; Makino, H.; Yasufumi, K.; Ogihara, T.; Morishita, R. Local delivery of E2F decoy oligodeoxynucleotides using ultrasound with microbubble agent (Optison) inhibits intimal hyperplasia after balloon injury in rat carotid artery model. Biochem. Biophys. Res. Commun. 2004, 317, 508–514. [Google Scholar] [CrossRef] [PubMed]
- Kubo, T.; Kinjyo, N.; Ikezawa, A.; Kambe, T.; Fukumori, R. Sp1 decoy oligodeoxynucleotide decreases angiotensin receptor expression and blood pressure in spontaneously hypertensive rats. Brain Res. 2003, 992, 1–8. [Google Scholar] [CrossRef]
- Yoshimura, S.; Morishita, R.; Hayashi, K.; Yamamoto, K.; Nakagami, H.; Kaneda, Y.; Sakai, N.; Ogihara, T. Inhibition of intimal hyperplasia after balloon injury in rat carotid artery model using cis-element “decoy” of nuclear factor-κB binding site as a novel molecular strategy. Gene Ther. 2001, 8, 1635–1642. [Google Scholar]
- Yamasaki, K.; Asai, T.; Shimizu, M.; Aoki, M.; Hashiya, N.; Sakonjo, H.; Makino, H.; Kaneda, Y.; Ogihara, T.; Morishita, R. Inhibition of NF-κB activation using cis-element “decoy” of NFκB binding site reduces neointimal formation in porcine balloon-injured coronary artery model. Gene Ther. 2003, 10, 356–364. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Park, J.H.; Kim, K.H.; Lee, W.R.; Lee, S.; Kwon, O.C.; Kim, K.S.; Park, K.K. Effect of NF-κB decoy oligodeoxynucleotide on LPS/high-fat diet-induced atherosclerosis in an animal model. Basic Clin. Pharmacol. Toxicol. 2010, 107, 925–930. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.R.; Kim, K.H.; An, H.J.; Park, Y.Y.; Kim, K.S.; Lee, C.K.; Min, B.K.; Park, K.K. Effects of chimeric decoy oligodeoxynucleotide in the regulation of transcription factors NF-κB and Sp1 in an animal model of atherosclerosis. Basic Clin. Pharmacol. Toxicol. 2013, 112, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.H.; Lim, S.; Wong, W.S. Antisense oligonucleotides: From design to therapeutic application. Clin. Exp. Pharmacol. Physiol. 2006, 33, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Isaka, Y.; Imai, E.; Takahara, S.; Rakugi, H. Oligonucleotidic therapeutics. Expert Opin. Drug Discov. 2008, 3, 991–996. [Google Scholar] [CrossRef] [PubMed]
- Kole, R.; Krainer, A.R.; Altman, S. RNA therapeutics: Beyond RNA interference and antisense oligonucleotides. Nat. Rev. Drug Discov. 2012, 11, 125–140. [Google Scholar] [CrossRef] [PubMed]
- Heeneman, S.; Sluimer, J.C.; Daemen, M.J. Angiotensin-converting enzyme and vascular remodeling. Circ. Res. 2007, 101, 441–454. [Google Scholar] [CrossRef] [PubMed]
- Bobik, A. Transforming growth factor-βs and vascular disorders. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1712–1720. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Ortega, M.; Rodríguez-Vita, J.; Sanchez-Lopez, E.; Carvajal, G.; Egido, J. TGF-β signaling in vascular fibrosis. Cardiovasc. Res. 2007, 74, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Tamiya, T.; Kashiwagi, I.; Takahashi, R.; Yasukawa, H.; Yoshimura, A. Suppressors of cytokine signaling (SOCS) proteins and JAK/STAT pathways: Regulation of T-cell inflammation by SOCS1 and SOCS3. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 980–985. [Google Scholar] [CrossRef] [PubMed]
- Recio, C.; Oguiza, A.; Lazaro, I.; Mallavia, B.; Egido, J.; Gomez-Guerrero, C. Suppressor of cytokine signaling 1-derived peptide inhibits Janus kinase/signal transducers and activators of transcription pathway and improves inflammation and atherosclerosis in diabetic mice. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1953–1960. [Google Scholar] [CrossRef] [PubMed]
- Jabs, D.A.; Griffiths, P.D. Fomivirsen for the treatment of cytomegalovirus retinitis. Am. J. Ophthalmol. 2002, 133, 552–556. [Google Scholar] [CrossRef]
- Raal, F.J.; Santos, R.D.; Blom, D.J.; Marais, A.D.; Charng, M.J.; Cromwell, W.C.; Lachmann, R.H.; Gaudet, D.; Tan, J.L.; Chasan-Taber, S.; et al. Mipomersen, an apolipoprotein B synthesis inhibitor, for lowering of LDL cholesterol concentrations in patients with homozygous familial hypercholesterolaemia: A randomised, double-blind, placebo-controlled trial. Lancet 2010, 375, 998–1006. [Google Scholar] [CrossRef]
- Visser, M.E.; Wagener, G.; Baker, B.F.; Geary, R.S.; Donovan, J.M.; Beuers, U.H.; Nederveen, A.J.; Verheij, J.; Trip, M.D.; Basart, D.C.; et al. Mipomersen, an apolipoprotein B synthesis inhibitor, lowers low-density lipoprotein cholesterol in high-risk statin-intolerant patients: A randomized, double-blind, placebo-controlled trial. Eur. Heart J. 2012, 33, 1142–1149. [Google Scholar] [CrossRef] [PubMed]
- Stein, E.A.; Dufour, R.; Gagne, C.; Gaudet, D.; East, C.; Donovan, J.M.; Chin, W.; Tribble, D.L.; McGowan, M. Apolipoprotein B synthesis inhibition with mipomersen in heterozygous familial hypercholesterolemia: Results of a randomized, double-blind, placebo-controlled trial to assess efficacy and safety as add-on therapy in patients with coronary artery disease. Circulation 2012, 126, 2283–2292. [Google Scholar] [CrossRef] [PubMed]
- Thomas, G.S.; Cromwell, W.C.; Ali, S.; Chin, W.; Flaim, J.D.; Davidson, M. Mipomersen, an apolipoprotein B synthesis inhibitor, reduces atherogenic lipoproteins in patients with severe hypercholesterolemia at high cardiovascular risk: A randomized, double-blind, placebo-controlled trial. J. Am. Coll. Cardiol. 2013, 62, 2178–2184. [Google Scholar] [CrossRef] [PubMed]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Dorsett, Y.; Tuschl, T. siRNAs: Applications in functional genomics and potential as therapeutics. Nat. Rev. Drug Discov. 2004, 3, 318–329. [Google Scholar] [CrossRef] [PubMed]
- Kowalski, P.S.; Leus, N.G.; Scherphof, G.L.; Ruiters, M.H.; Kamps, J.A.; Molema, G. Targeted siRNA delivery to diseased microvascular endothelial cells: Cellular and molecular concepts. IUBMB Life 2011, 63, 648–658. [Google Scholar] [CrossRef] [PubMed]
- Kowalski, P.S.; Lintermans, L.L.; Morselt, H.W.; Leus, N.G.; Ruiters, M.H.; Molema, G.; Kamps, J.A. Anti-VCAM-1 and anti-E-selectin SAINT-O-Somes for selective delivery of siRNA into inflammation-activated primary endothelial cells. Mol. Pharm. 2013, 10, 3033–3044. [Google Scholar] [CrossRef] [PubMed]
- Leus, N.G.; Talman, E.G.; Ramana, P.; Kowalski, P.S.; Woudenberg-Vrenken, T.E.; Ruiters, M.H.; Molema, G.; Kamps, J.A. Effective siRNA delivery to inflamed primary vascular endothelial cells by anti-E-selectin and anti-VCAM-1 PEGylated SAINT-based lipoplexes. Int. J. Pharm. 2014, 459, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Kalinina, N.; Agrotis, A.; Antropova, Y.; Ilyinskaya, O.; Smirnov, V.; Tararak, E.; Bobik, A. Smad expression in human atherosclerotic lesions: Evidence for impaired TGF-β/Smad signaling in smooth muscle cells of fibrofatty lesions. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1391–1396. [Google Scholar] [CrossRef] [PubMed]
- Rupérez, M.; Lorenzo, O.; Blanco-Colio, L.M.; Esteban, V.; Egido, J.; Ruiz-Ortega, M. Connective tissue growth factor is a mediator of angiotensin II-induced fibrosis. Circulation 2003, 108, 1499–1505. [Google Scholar] [CrossRef] [PubMed]
- Leask, A.; Holmes, A.; Black, C.M.; Abraham, D.J. Connective tissue growth factor gene regulation. Requirements for its induction by transforming growth factor-β2 in fibroblasts. J. Biol. Chem. 2003, 278, 13008–13015. [Google Scholar] [CrossRef] [PubMed]
- Naito, T.; Masaki, T.; Nikolic-Paterson, D.J.; Tanji, C.; Yorioka, N.; Kohno, N. Angiotensin II induces thrombospondin-1 production in human mesangial cells via p38 MAPK and JNK: A mechanism for activation of latent TGF-β1. Am. J. Physiol. Renal Physiol. 2004, 286, F278–F287. [Google Scholar] [CrossRef] [PubMed]
- Mallawaarachchi, C.M.; Weissberg, P.L.; Siow, R.C. Smad7 gene transfer attenuates adventitial cell migration and vascular remodeling after balloon injury. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1383–1387. [Google Scholar] [CrossRef] [PubMed]
- Suwanabol, P.A.; Kent, K.C.; Liu, B. TGF-β and restenosis revisited: A Smad link. J. Surg. Res. 2011, 167, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Hayashida, T.; Decaestecker, M.; Schnaper, H.W. Cross-talk between ERK MAP kinase and Smad signaling pathways enhances TGF-β-dependent responses in human mesangial cells. FASEB J. 2003, 17, 1576–1578. [Google Scholar] [PubMed]
- Samarakoon, R.; Higgins, C.E.; Higgins, S.P.; Kutz, S.M.; Higgins, P.J. Plasminogen activator inhibitor type-1 gene expression and induced migration in TGF-β1-stimulated smooth muscle cells is pp 60(c-Src)/MEK-dependent. J. Cell. Physiol. 2005, 204, 236–246. [Google Scholar] [CrossRef] [PubMed]
- Seay, U.; Sedding, D.; Krick, S.; Hecker, M.; Seeger, W.; Eickelberg, O. Transforming growth factor-β-dependent growth inhibition in primary vascular smooth muscle cells is p38-dependent. J. Pharmacol. Exp. Ther. 2005, 315, 1005–1012. [Google Scholar] [CrossRef] [PubMed]
- Rupérez, M.; Rodrigues-Díez, R.; Blanco-Colio, L.M.; Sánchez-López, E.; Rodríguez-Vita, J.; Esteban, V.; Carvajal, G.; Plaza, J.J.; Egido, J.; Ruiz-Ortega, M. HMG-CoA reductase inhibitors decrease angiotensin II-induced vascular fibrosis: Role of RhoA/ROCK and MAPK pathways. Hypertension 2007, 50, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues Díez, R.; Rodrigues-Díez, R.; Lavoz, C.; Rayego-Mateos, S.; Civantos, E.; Rodríguez-Vita, J.; Mezzano, S.; Ortiz, A.; Egido, J.; Ruiz-Ortega, M. Statins inhibit angiotensin II/Smad pathway and related vascular fibrosis, by a TGF-β-independent process. PLoS ONE 2010, 5, e14145. [Google Scholar] [CrossRef] [PubMed]
- Samarakoon, R.; Higgins, P.J. Integration of non-SMAD and SMAD signaling in TGF-β1-induced plasminogen activator inhibitor type-1 gene expression in vascular smooth muscle cells. Thromb. Haemost. 2008, 100, 976–983. [Google Scholar] [CrossRef] [PubMed]
- Mei, S.; Gu, H.; Ward, A.; Yang, X.; Guo, H.; He, K.; Liu, Z.; Cao, W. p38 mitogen-activated protein kinase (MAPK) promotes cholesterol ester accumulation in macrophages through inhibition of macroautophagy. J. Biol. Chem. 2012, 287, 11761–11768. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, V.; Golledge, J.; Heywood, E.B.; Bruemmer, D.; Daugherty, A. Regulation of peroxisome proliferator-activated receptor-γ by angiotensin II via transforming growth factor-β1-activated p38 mitogen-activated protein kinase in aortic smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.H.; Karnovsky, M.J. Increased MMP-2 expression in connective tissue growth factor over-expression vascular smooth muscle cells. J. Biol. Chem. 2002, 277, 9800–9805. [Google Scholar] [CrossRef] [PubMed]
- Newby, A.C. Dual role of matrix metalloproteinases (matrixins) in intimal thickening and atherosclerotic plaque rupture. Physiol. Rev. 2005, 85, 1–31. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhao, D.; Spinetti, G.; Zhang, J.; Jiang, L.Q.; Pintus, G.; Monticone, R.; Lakatta, E.G. Matrix metalloproteinase 2 activation of transforming growth factor-β1 (TGF-β1) and TGF-β1-type II receptor signaling within the aged arterial wall. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1503–1509. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Jin, M.; Yang, F.; Zhu, J.; Xiao, Q.; Zhang, L. Matrix metalloproteinases: Inflammatory regulators of cell behaviors in vascular formation and remodeling. Mediators Inflamm. 2013, 2013, 928315. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Mohammad, G.; Dos Santos, J.M.; Zhong, Q. Abrogation of MMP-9 gene protects against the development of retinopathy in diabetic mice by preventing mitochondrial damage. Diabetes 2011, 60, 3023–3033. [Google Scholar] [CrossRef] [PubMed]
- Walker, T.; Wendel, H.P.; Raabe, C.; Wiechnik, P.; Spranger, L.; Heidenreich, O.; Scheule, A.M.; Nordheim, A.; Ziemer, G. Graft protection in bypass surgery: siRNA-mediated silencing of adhesion molecules. Oligonucleotides 2009, 19, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Walker, T.; Saup, E.; Nolte, A.; Simon, P.; Kornberger, A.; Steger, V.; Adolph, O.; Wendel, H.P. Transfection of short-interfering RNA silences adhesion molecule expression on cardiac microvascular cells. Thorac. Cardiovasc. Surg. 2011, 59, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.W.; de Waal Malefyt, R.; Coffman, R.L.; O’Garra, A. Interleukin-10 and the interleukin-10 receptor. Annu. Rev. Immunol. 2001, 19, 683–765. [Google Scholar] [CrossRef] [PubMed]
- Aaronson, D.S.; Horvath, C.M. A road map for those who don’t know JAK-STAT. Science 2002, 296, 1653–1655. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, J.J.; Pesu, M.; Borie, D.C.; Changelian, P.S. A new modality for immunosuppression: Targeting the JAK/STAT pathway. Nat. Rev. Drug Discov. 2004, 3, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Hao, X.R.; Cao, D.L.; Hu, Y.W.; Li, X.X.; Liu, X.H.; Xiao, J.; Liao, D.F.; Xiang, J.; Tang, C.K. IFN-γ down-regulates ABCA1 expression by inhibiting LXRα in a JAK/STAT signaling pathway-dependent manner. Atherosclerosis 2009, 203, 417–428. [Google Scholar] [CrossRef] [PubMed]
- Brand, K.; Page, S.; Rogler, G.; Bartsch, A.; Brandl, R.; Knuechel, R.; Page, M.; Kaltschmidt, C.; Baeuerle, P.A.; Neumeier, D. Activated transcription factor nuclear factor-κB is present in the atherosclerotic lesion. J. Clin. Investig. 1996, 97, 1715–1722. [Google Scholar] [CrossRef] [PubMed]
- Bourcier, T.; Sukhova, G.; Libby, P. The nuclear factor-κB signaling pathway participates in dysregulation of vascular smooth muscle cells in vitro and in human atherosclerosis. J. Biol. Chem. 1997, 272, 15817–15824. [Google Scholar] [CrossRef] [PubMed]
- Hajra, L.; Evans, A.I.; Chen, M.; Hyduk, S.J.; Collins, T.; Cybulsky, M.I. The NF-κB signal transduction pathway in aortic endothelial cells is primed for activation in regions predisposed to atherosclerotic lesion formation. Proc. Natl. Acad. Sci. USA 2000, 97, 9052–9057. [Google Scholar] [CrossRef] [PubMed]
- De Martin, R.; Hoeth, M.; Hofer-Warbinek, R.; Schmid, J.A. The transcription factor NF-κB and the regulation of vascular cell function. Arterioscler. Thromb. Vasc. Biol. 2000, 20, E83–E88. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.H.; Best, P.J.; Edwards, W.D.; Holmes, D.R., Jr.; Carlson, P.J.; Celermajer, D.S.; Lerman, A. Nuclear factor-κB immunoreactivity is present in human coronary plaque and enhanced in patients with unstable angina pectoris. Atherosclerosis 2002, 160, 147–153. [Google Scholar] [CrossRef]
- Caamaño, J.; Hunter, C.A. NF-κB family of transcription factors: Central regulators of innate and adaptive immune functions. Clin. Microbiol. Rev. 2002, 15, 414–429. [Google Scholar] [CrossRef] [PubMed]
- Brasier, A.R. The nuclear factor-κB-interleukin-6 signalling pathway mediating vascular inflammation. Cardiovasc. Res. 2010, 86, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Moon, S.K.; Cha, B.Y.; Kim, C.H. ERK1/2 mediates TNF-α-induced matrix metalloproteinase-9 expression in human vascular smooth muscle cells via the regulation of NF-κB and AP-1: Involvement of the ras dependent pathway. J. Cell. Physiol. 2004, 198, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Coelho, T.; Adams, D.; Silva, A.; Lozeron, P.; Hawkins, P.N.; Mant, T.; Perez, J.; Chiesa, J.; Warrington, S.; Tranter, E.; et al. Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N. Engl. J. Med. 2013, 369, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Urbich, C.; Kuehbacher, A.; Dimmeler, S. Role of microRNAs in vascular diseases, inflammation, and angiogenesis. Cardiovasc. Res. 2008, 79, 581–588. [Google Scholar] [CrossRef] [PubMed]
- Hobert, O. Gene regulation by transcription factors and microRNAs. Science 2008, 319, 1785–1786. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Krol, J.; Loedige, I.; Filipowicz, W. The widespread regulation of microRNA biogenesis, function and decay. Nat. Rev. Genet. 2010, 11, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Raitoharju, E.; Lyytikäinen, L.P.; Levula, M.; Oksala, N.; Mennander, A.; Tarkka, M.; Klopp, N.; Illig, T.; Kähönen, M.; Karhunen, P.J.; et al. miR-21, miR-210, miR-34a, and miR-146a/b are up-regulated in human atherosclerotic plaques in the Tampere Vascular Study. Atherosclerosis 2011, 219, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Raitoharju, E.; Oksala, N.; Lehtimäki, T. MicroRNAs in the atherosclerotic plaque. Clin. Chem. 2013, 59, 1708–1721. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.N.; Hilyard, A.C.; Lagna, G.; Hata, A. SMAD proteins control DROSHA-mediated microRNA maturation. Nature 2008, 454, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Tsoyi, K.; Jang, H.J.; Nizamutdinova, I.T.; Park, K.; Kim, Y.M.; Kim, H.J.; Seo, H.G.; Lee, J.H.; Chang, K.C. PTEN differentially regulates expressions of ICAM-1 and VCAM-1 through PI3K/Akt/GSK-3β/GATA-6 signaling pathways in TNF-α-activated human endothelial cells. Atherosclerosis 2010, 213, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.; Baker, M.B.; Moore, J.P.; Searles, C.D. MiR-21 is induced in endothelial cells by shear stress and modulates apoptosis and eNOS activity. Biochem. Biophys. Res. Commun. 2010, 393, 643–648. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wang, K.C.; Wu, W.; Subramaniam, S.; Shyy, J.Y.; Chiu, J.J.; Li, J.Y.; Chien, S. MicroRNA-21 targets peroxisome proliferators-activated receptor-α in an autoregulatory loop to modulate flow-induced endothelial inflammation. Proc. Natl. Acad. Sci. USA 2011, 108, 10355–10360. [Google Scholar] [CrossRef] [PubMed]
- Kumarswamy, R.; Volkmann, I.; Jazbutyte, V.; Dangwal, S.; Park, D.H.; Thum, T. Transforming growth factor-β-induced endothelial-to-mesenchymal transition is partly mediated by microRNA-21. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Cordes, K.R.; Sheehy, N.T.; White, M.P.; Berry, E.C.; Morton, S.U.; Muth, A.N.; Lee, T.H.; Miano, J.M.; Ivey, K.N.; Srivastava, D. miR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature 2009, 460, 705–710. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Becker Buscaglia, L.E.; Barker, J.R.; Li, Y. MicroRNAs in NF-κB signaling. J. Mol. Cell Biol. 2011, 3, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Shi, C.; Manduchi, E.; Civelek, C.; Davies, P.F. MicroRNA-10a regulation of proinflammatory phenotype in athero-susceptible endothelium in vivo and in vitro. Proc. Natl. Acad. Sci. USA 2010, 107, 13450–13455. [Google Scholar] [CrossRef] [PubMed]
- Suárez, Y.; Wang, C.; Manes, T.D.; Pober, J.S. Cutting edge: TNF-induced microRNAs regulate TNF-induced expression of E-selectin and intercellular adhesion molecule-1 on human endothelial cells: Feedback control of inflammation. J. Immunol. 2010, 184, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Jovanovic, I.; Zivkovic, M.; Jovanovic, J.; Djuric, T.; Stankovic, A. The co-inertia approach in identification of specific microRNA in early and advanced atherosclerosis plaque. Med. Hypotheses 2014, 83, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Di Gregoli, K.; Jenkins, N.; Salter, R.; White, S.; Newby, A.C.; Johnson, J.L. MicroRNA-24 regulates macrophage behavior and retards atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1990–2000. [Google Scholar] [CrossRef] [PubMed]
- Maegdefessel, L.; Spin, J.M.; Raaz, U.; Eken, S.M.; Toh, R.; Azuma, J.; Adam, M.; Nagakami, F.; Heymann, H.M.; Chernugobova, E.; et al. miR-24 limits aortic vascular inflammation and murine abdominal aneurysm development. Nat. Commun. 2014, 31, 5214. [Google Scholar] [CrossRef] [PubMed]
- Chan, M.C.; Hilyard, A.C.; Wu, C.; Davis, B.N.; Hill, N.S.; Lal, A.; Lieberman, J.; Lagna, G.; Hata, A. Molecular basis for antagonism between PDGF and the TGFβ family of signalling pathways by control of miR-24 expression. EMBO J. 2010, 29, 559–573. [Google Scholar] [CrossRef] [PubMed]
- Pober, J.S.; Sessa, W.C. Evolving functions of endothelial cells in inflammation. Nat. Rev. Immunol. 2007, 7, 803–815. [Google Scholar] [CrossRef] [PubMed]
- Gareus, R.; Kotsaki, E.; Xanthoulea, S.; van der Made, I.; Gijbels, M.J.; Kardakaris, R.; Polykratis, A.; Kollias, G.; de Winther, M.P.; Pasparakis, M. Endothelial cell-specific NF-κB inhibition protects mice from atherosclerosis. Cell Metable 2008, 8, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Pierce, J.W.; Schoenleber, R.; Jesmok, G.; Best, J.; Moore, S.A.; Collins, T.; Gerritsen, M.E. Novel inhibitors of cytokine-induced IκBα phosphorylation and endothelial cell adhesion molecule expression show anti-inflammatory effects in vivo. J. Biol. Chem. 1997, 272, 21096–21103. [Google Scholar] [CrossRef] [PubMed]
- Asgeirsdóttir, S.A.; van Solingen, C.; Kurniati, N.F.; Zwiers, P.J.; Heeringa, P.; van Meurs, M.; Satchell, S.C.; Saleem, M.A.; Mathieson, P.W.; Banas, B.; et al. MicroRNA-126 contributes to renal microvascular heterogeneity of VCAM-1 protein expression in acute inflammation. Am. J. Physiol. Renal. Physiol. 2012, 302, F1630–F1639. [Google Scholar] [CrossRef] [PubMed]
- Harris, T.A.; Yamakuchi, M.; Kondo, M.; Oettgen, P.; Lowenstein, C.J. Ets-1 and Ets-2 regulate the expression of microRNA-126 in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1990–1997. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Cheng, Y.; Yang, J.; Xu, L.; Zhang, C. Cell-specific effects of miR-221/222 in vessels: Molecular mechanism and therapeutic application. J. Mol. Cell. Cardiol. 2012, 52, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Dentelli, P.; Rosso, A.; Orso, F.; Olgasi, C.; Taverna, D.; Brizzi, M.F. MicroRNA-222 controls neovascularization by regulating signal transducer and activator of transcription 5A expression. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1562–1568. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, R.M.; Taganov, K.D.; Boldin, M.P.; Cheng, G.; Baltimore, D. MicroRNA-155 is induced during the macrophage inflammatory response. Proc. Natl. Acad. Sci. USA 2007, 104, 1604–1609. [Google Scholar] [CrossRef] [PubMed]
- McCoy, C.E.; Sheedy, F.J.; Qualls, J.E.; Doyle, S.L.; Quinn, S.R.; Murray, P.J.; O’Neill, L.A. IL-10 inhibits miR-155 induction by toll-like receptors. J. Biol. Chem. 2010, 285, 20492–20498. [Google Scholar] [CrossRef] [PubMed]
- Janssen, H.L.; Reesink, H.W.; Lawitz, E.J.; Zeuzem, S.; Rodriguez-Torres, M.; Patel, K.; van der Meer, A.J.; Patick, A.K.; Chen, A.; Zhou, Y.; et al. Treatment of HCV infection by targeting microRNA. N. Engl. J. Med. 2013, 368, 1685–1694. [Google Scholar] [CrossRef] [PubMed]
- Mann, M.J.; Dzau, V.J. Therapeutic applications of transcription factor decoy oligonucleotides. J. Clin. Investig. 2000, 106, 1071–1075. [Google Scholar] [CrossRef] [PubMed]
- Dzau, V.J. Transcription factor decoy. Circ. Res. 2002, 90, 1234–1236. [Google Scholar] [CrossRef] [PubMed]
- Park, K.K.; Deok Ahn, J.; Lee, I.K.; Magae, J.; Heintz, N.H.; Kwak, J.Y.; Lee, Y.C.; Cho, Y.S.; Kim, H.C.; Chae, Y.M.; et al. Inhibitory effects of novel E2F decoy oligodeoxynucleotides on mesangial cell proliferation by coexpression of E2F/DP. Biochem. Biophys. Res. Commun. 2003, 308, 689–697. [Google Scholar] [CrossRef]
- Geiser, A.G.; Busam, K.J.; Kim, S.J.; Lafyatis, R.; O’Reilly, M.A.; Webbink, R.; Roberts, A.B.; Sporn, M.B. Regulation of the transforming growth factor-β1 and -β3 promoters by transcription factor Sp1. Gene 1993, 129, 223–228. [Google Scholar] [CrossRef]
- Kambe, T.; Kinjyo, N.; Hiruki, H.; Kubo, T. Basal transcriptional regulation of rat AT1 angiotensin II receptor gene expression. Clin. Exp. Pharmacol. Physiol. 2004, 31, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Chae, Y.M.; Park, K.K.; Magae, J.; Lee, I.S.; Kim, C.H.; Kim, H.C.; Hong, S.; Lee, J.G.; Choi, I.J.; Kim, H.S.; et al. Sp1-decoy oligodeoxynucleotide inhibits high glucose-induced mesangial cell proliferation. Biochem. Biophys. Res. Commun. 2004, 319, 550–555. [Google Scholar] [CrossRef] [PubMed]
- Goto, T.; Matsui, Y.; Fernandes, R.J.; Hanson, D.A.; Kubo, T.; Yukata, K.; Michigami, T.; Komori, T.; Fujita, T.; Yang, L.; et al. Sp1 family of transcription factors regulates the human α2 (XI) collagen gene (COL11A2) in Saos-2 osteoblastic cells. J. Bone Miner. Res. 2006, 21, 661–673. [Google Scholar] [CrossRef] [PubMed]
- Kum, Y.S.; Kim, K.H.; Park, T.I.; Suh, I.S.; Oh, H.K.; Cho, C.H.; Park, J.B.; Chang, Y.C.; Park, J.H.; Lee, K.G.; et al. Antifibrotic effect via the regulation of transcription factor Sp1 in lung fibrosis. Biochem. Biophys. Res. Commun. 2007, 363, 368–374. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.H.; Chae, Y.M.; Park, K.K.; Kim, C.H.; Lee, I.S.; Chang, Y.C. Suppression of mesangial cell proliferation and extracellular matrix production in streptozotocin-induced diabetic rats by Sp1 decoy oligodeoxynucleotide in vitro and in vivo. J. Cell. Biochem. 2008, 103, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Jo, J.H.; Kim, K.H.; Kim, S.J.; Lee, W.R.; Park, K.K.; Park, J.B. Antifibrotic effect through the regulation of transcription factor using ring type-Sp1 decoy oligodeoxynucleotide in carbon tetrachloride-induced liver fibrosis. J. Gene Med. 2009, 11, 824–833. [Google Scholar] [CrossRef] [PubMed]
- Chae, Y.M.; Park, K.K.; Lee, I.K.; Kim, J.K.; Kim, C.H.; Chang, Y.C. Ring-Sp1 decoy oligonucleotide effectively suppresses extracellular matrix gene expression and fibrosis of rat kidney induced by unilateral ureteral obstruction. Gene Ther. 2006, 13, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zhou, Y.; Chen, K.Q.; An, G.; Ji, S.Y.; Chen, Q.K. Anti-fibrotic effects via regulation of transcription factor Sp1 on hepatic stellate cells. Cell Physiol. Biochem. 2012, 29, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Morishita, R.; Sugimoto, T.; Aoki, M.; Kida, I.; Tomita, N.; Moriguchi, A.; Maeda, K.; Sawa, Y.; Kaneda, Y.; Higaki, J.; et al. In vivo transfection of cis element “decoy” against nuclear factor-κB binding site prevents myocardial infarction. Nat. Med. 1997, 3, 894–899. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Lee, E.S.; Cha, S.H.; Park, J.H.; Park, J.S.; Chang, Y.C.; Park, K.K. Transcriptional regulation of NF-κB by ring type decoy oligodeoxynucleotide in an animal model of nephropathy. Exp. Mol. Pathol. 2009, 86, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Lee, W.R.; Kang, Y.N.; Chang, Y.C.; Park, K.K. Inhibitory effect of nuclear factor-κB decoy oligodeoxynucleotide on liver fibrosis through regulation of the epithelial-mesenchymal transition. Hum. Gene Ther. 2014, 25, 721–729. [Google Scholar] [CrossRef] [PubMed]
- Miyake, T.; Aoki, M.; Morishita, R. Inhibition of anastomotic intimal hyperplasia using a chimeric decoy strategy against NF-κB and E2F in a rabbit model. Cardiovasc. Res. 2008, 79, 706–714. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, H.; Aoki, M.; Miyake, T.; Kawasaki, T.; Iwai, M.; Jo, N.; Oishi, M.; Kataoka, K.; Ohgi, S.; Ogihara, T.; et al. Inhibition of experimental abdominal aortic aneurysm in the rat by use of decoy oligodeoxynucleotides suppressing activity of nuclear factor-κB and ets transcription factors. Circulation 2004, 109, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Miyake, T.; Aoki, M.; Osako, M.K.; Shimamura, M.; Nakagami, H.; Morishita, R. Systemic administration of ribbon-type decoy oligodeoxynucleotide against nuclear factor-κB and ets prevents abdominal aortic aneurysm in rat model. Mol. Ther. 2011, 19, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.F.; Huang, H.; Li, X.Y.; Guo, W.; Xing, W.; Sun, Z.Y.; Liang, H.P.; Yu, J.; Chen, D.F.; Wang, Z.G.; et al. A dual AP-1 and SMAD decoy ODN suppresses tissue fibrosis and scarring in mice. J. Investig. Dermatol. 2013, 133, 1080–1087. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Park, J.H.; Lee, W.R.; Park, J.S.; Kim, H.C.; Park, K.K. The inhibitory effect of chimeric decoy oligodeoxynucleotide against NF-κB and Sp1 in renal interstitial fibrosis. J. Mol. Med. 2013, 91, 573–586. [Google Scholar] [CrossRef] [PubMed]
- Sung, W.J.; Kim, K.H.; Kim, YJ.; Chang, Y.C.; Lee, I.H.; Park, K.K. Antifibrotic effect of synthetic Smad/Sp1 chimeric decoy oligodeoxynucleotide through the regulation of epithelial mesenchymal transition in unilateral ureteral obstruction model of mice. Exp. Mol. Pathol. 2013, 95, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Jun-Ichi, S.; Hiroshi, I.; Ryo, G.; Ryuichi, M.; Kensuke, E.; Mitsuaki, I. Initial clinical cases of the use of a NF-κB decoy at the site of coronary stenting for the prevention of restenosis. Circ. J. 2004, 68, 270–271. [Google Scholar] [CrossRef] [PubMed]
- Egashira, K.; Suzuki, J.; Ito, H.; Aoki, M.; Isobe, M.; Morishita, R. Long-term follow up of initial clinical cases with NF-κB decoy oligodeoxynucleotide transfection at the site of coronary stenting. J. Gene Med. 2008, 10, 805–809. [Google Scholar] [CrossRef] [PubMed]
- Mann, M.J.; Whittemore, A.D.; Donaldson, M.C.; Belkin, M.; Conte, M.S.; Polak, J.F.; Orav, E.J.; Ehsan, A.; Dell’Acqua, G.; Dzau, V.J. Ex-vivo gene therapy of human vascular bypass grafts with E2F decoy: The PREVENT single-centre, randomised, controlled trial. Lancet 1999, 354, 1493–1498. [Google Scholar] [CrossRef]
- Conte, M.S.; Bandyk, D.F.; Clowes, A.W.; Moneta, G.L.; Seely, L.; Lorenz, T.J.; Namini, H.; Hamdan, A.D.; Roddy, S.P.; Belkin, M.; et al. PREVENT III Investigators. Results of PREVENT III: A multicenter, randomized trial of edifoligide for the prevention of vein graft failure in lower extremity bypass surgery. J. Vasc. Surg. 2006, 43, 742–751. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Youn, S.W.; Park, K.-K. Small-Nucleic-Acid-Based Therapeutic Strategy Targeting the Transcription Factors Regulating the Vascular Inflammation, Remodeling and Fibrosis in Atherosclerosis. Int. J. Mol. Sci. 2015, 16, 11804-11833. https://doi.org/10.3390/ijms160511804
Youn SW, Park K-K. Small-Nucleic-Acid-Based Therapeutic Strategy Targeting the Transcription Factors Regulating the Vascular Inflammation, Remodeling and Fibrosis in Atherosclerosis. International Journal of Molecular Sciences. 2015; 16(5):11804-11833. https://doi.org/10.3390/ijms160511804
Chicago/Turabian StyleYoun, Sung Won, and Kwan-Kyu Park. 2015. "Small-Nucleic-Acid-Based Therapeutic Strategy Targeting the Transcription Factors Regulating the Vascular Inflammation, Remodeling and Fibrosis in Atherosclerosis" International Journal of Molecular Sciences 16, no. 5: 11804-11833. https://doi.org/10.3390/ijms160511804
APA StyleYoun, S. W., & Park, K.-K. (2015). Small-Nucleic-Acid-Based Therapeutic Strategy Targeting the Transcription Factors Regulating the Vascular Inflammation, Remodeling and Fibrosis in Atherosclerosis. International Journal of Molecular Sciences, 16(5), 11804-11833. https://doi.org/10.3390/ijms160511804