
Identification and Expression Analysis of Candidate Genes Associated with Defense Responses to Phytophthora capsici in Pepper Line “PI 201234”

Abstract
:1. Introduction
2. Results
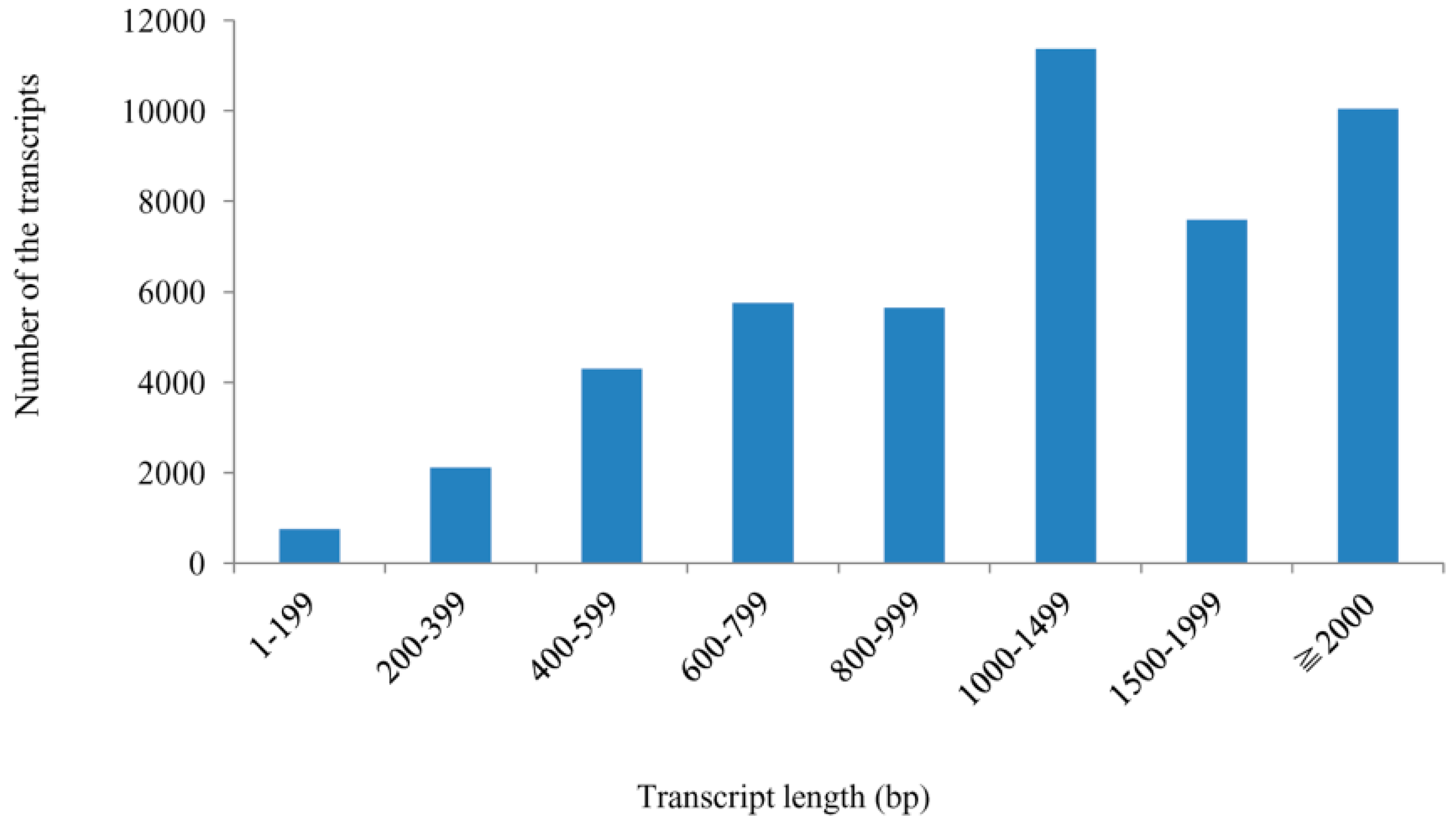
2.1. Sequencing Output and Mapping Reads to the Genome

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Class | Library A | Library CK | ||||
|---|---|---|---|---|---|---|
| Number | Total Length (bp) | Percentage (%) | Number | Total Length (bp) | Percentage (%) | |
| Raw reads | 79,250,598 | 7,925,059,800 | 75,339,602 | 7,533,960,200 | ||
| Clean reads | 76,015,888 | 7,152,478,994 | 95.92 | 72,130,988 | 6,782,519,810 | 95.74 |
| Mapping to genome | 71,904,454 | 94.60 | 67,803,241 | 94.00 | ||
| Total mapping position | 86,610,410 | 86,962,176 | ||||
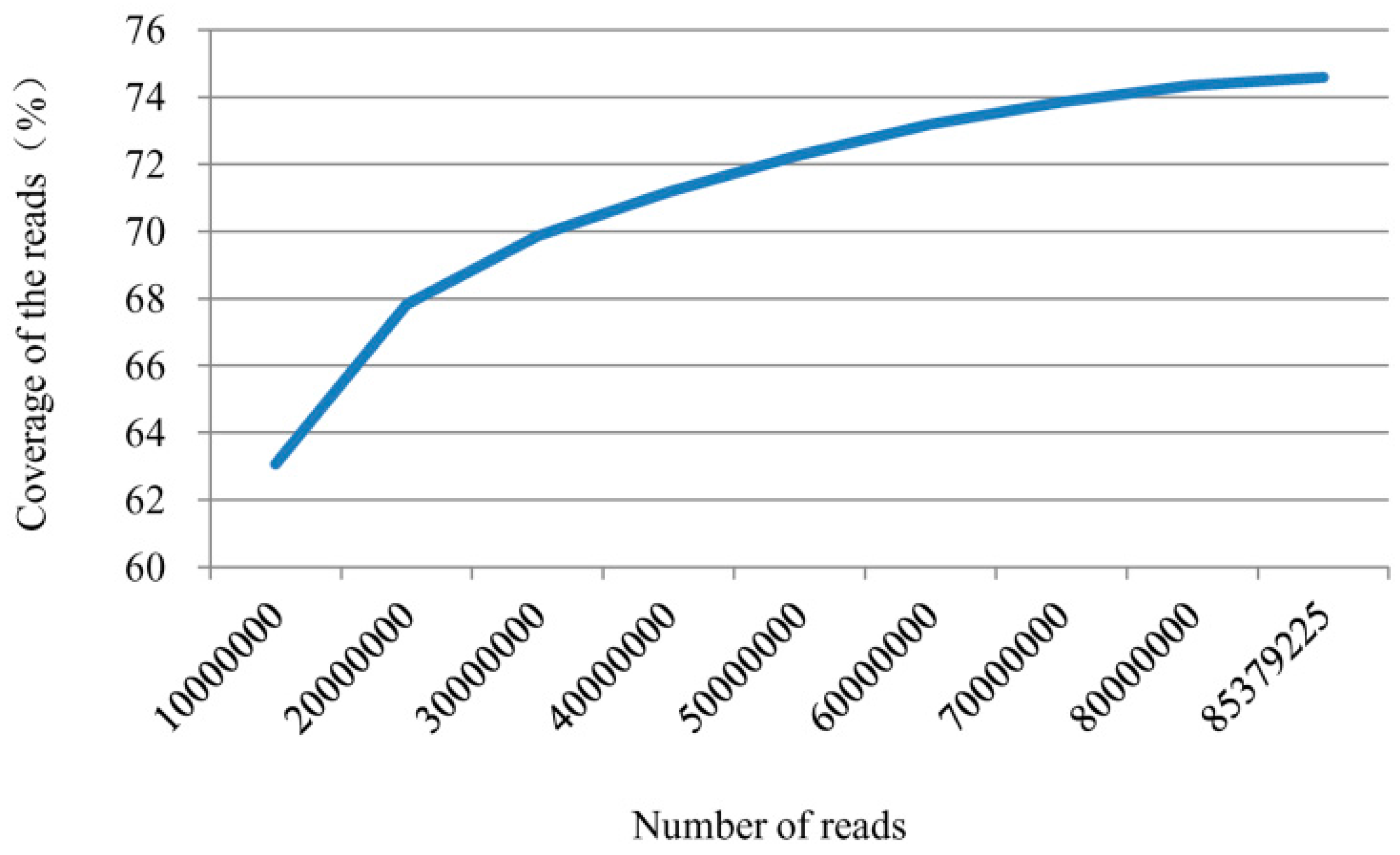
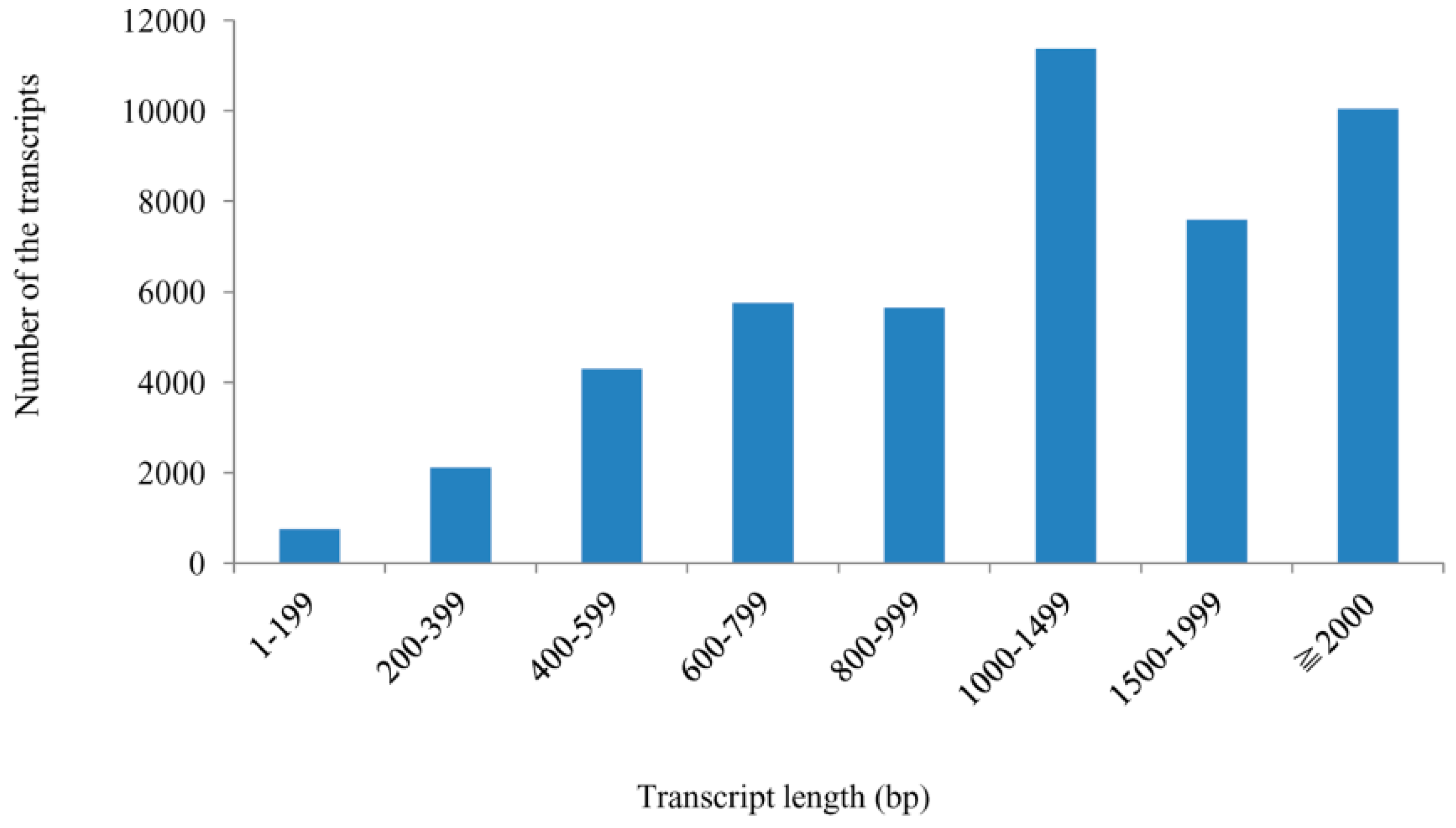
| Class | Number | Percentage (%) |
|---|---|---|
| Total genes | 30,106 | 100 |
| Expressed genes | 30,090 | 99.95 |
| Expressed in library A | 29,988 | 99.66 |
| Expressed in library CK | 29,972 | 99.61 |
| Expressed both | 29,870 | 99.27 |
| Expressed only in library A | 118 | 0.39 |
| Expressed only in library CK | 102 | 0.34 |
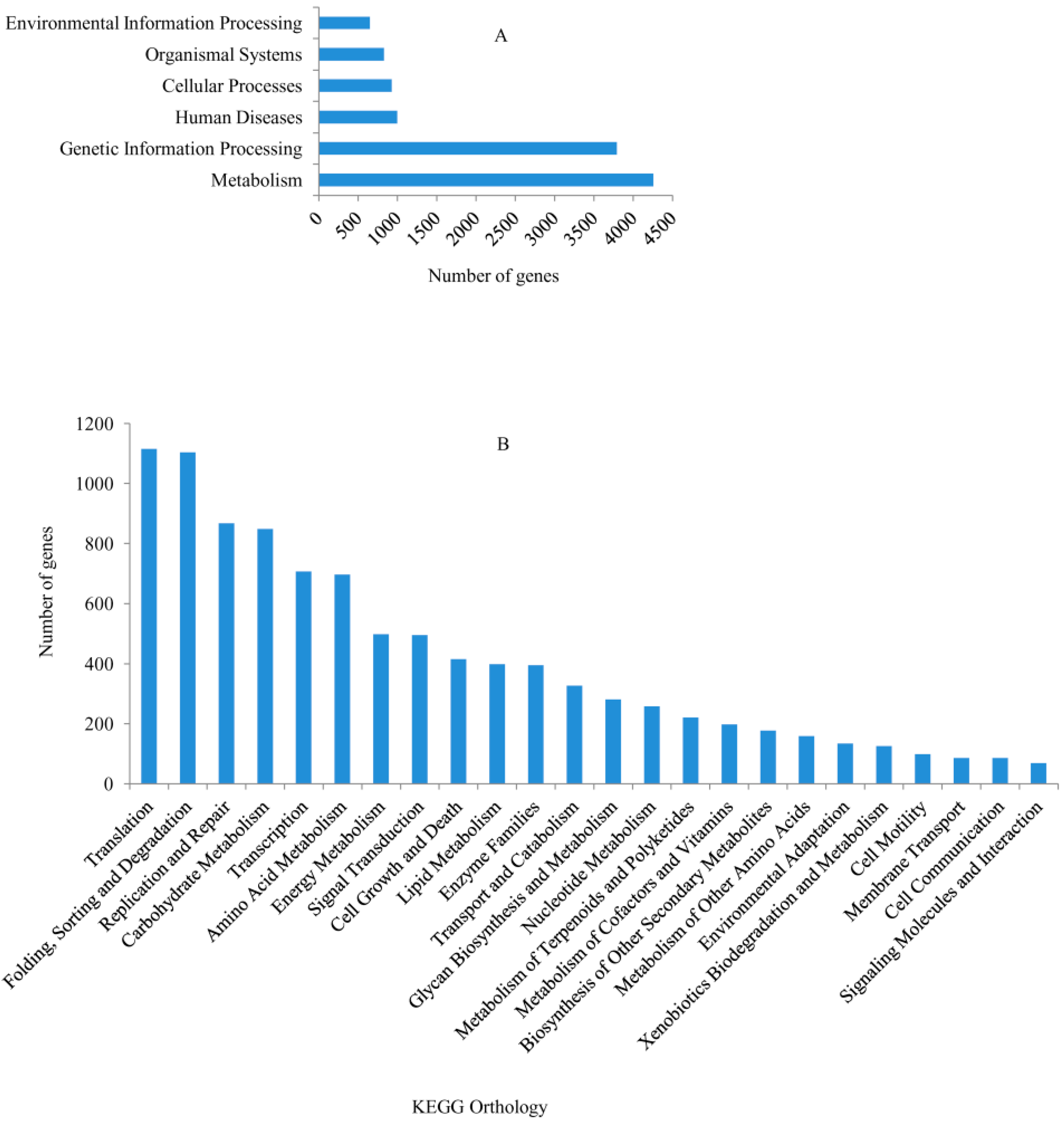
2.2. Kyoto Encyclopedia of Genes and Genomes (KEGG) Functional Classifications

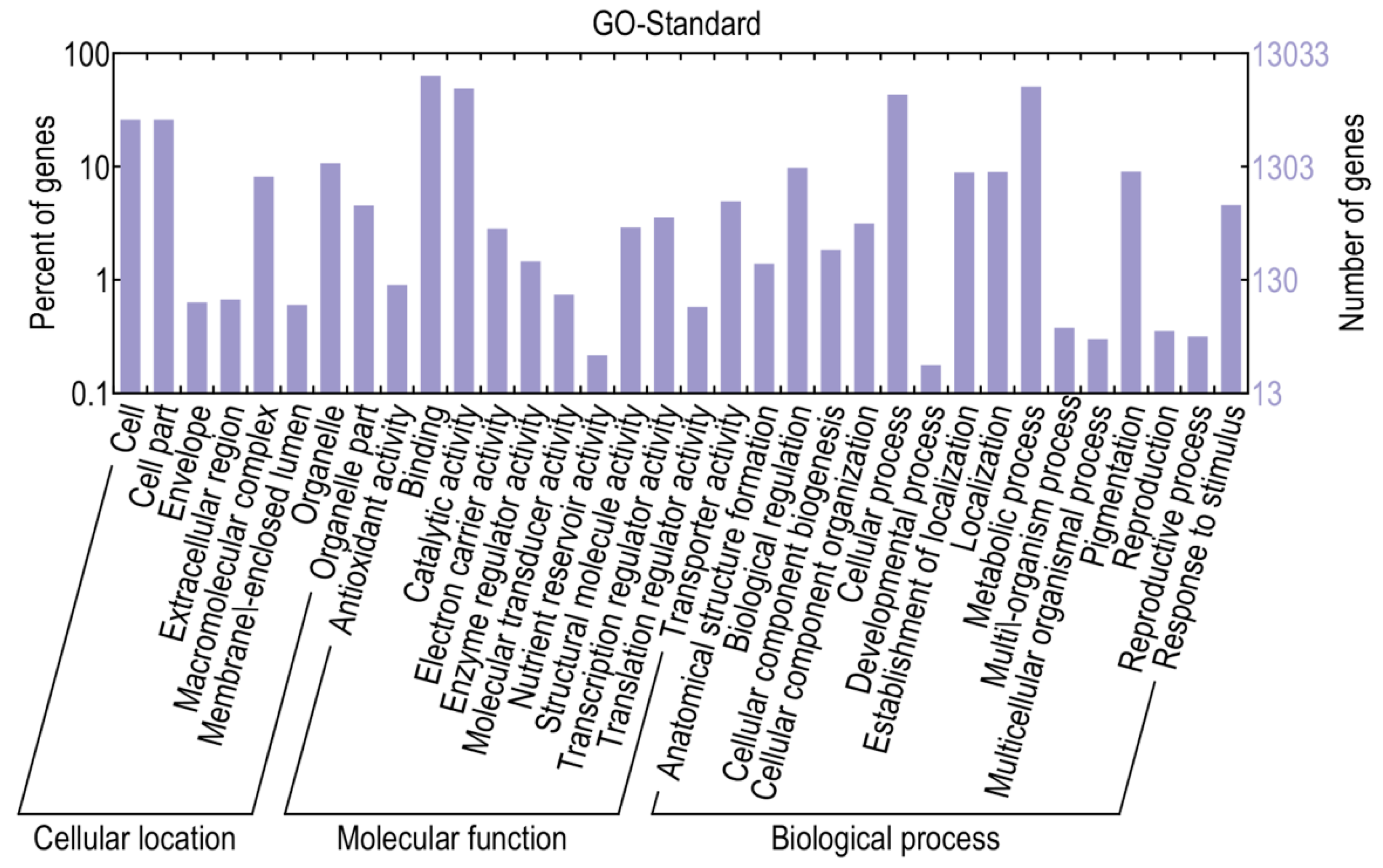
2.3. Functional Classifications via Interpro and Gene Ontology (GO)
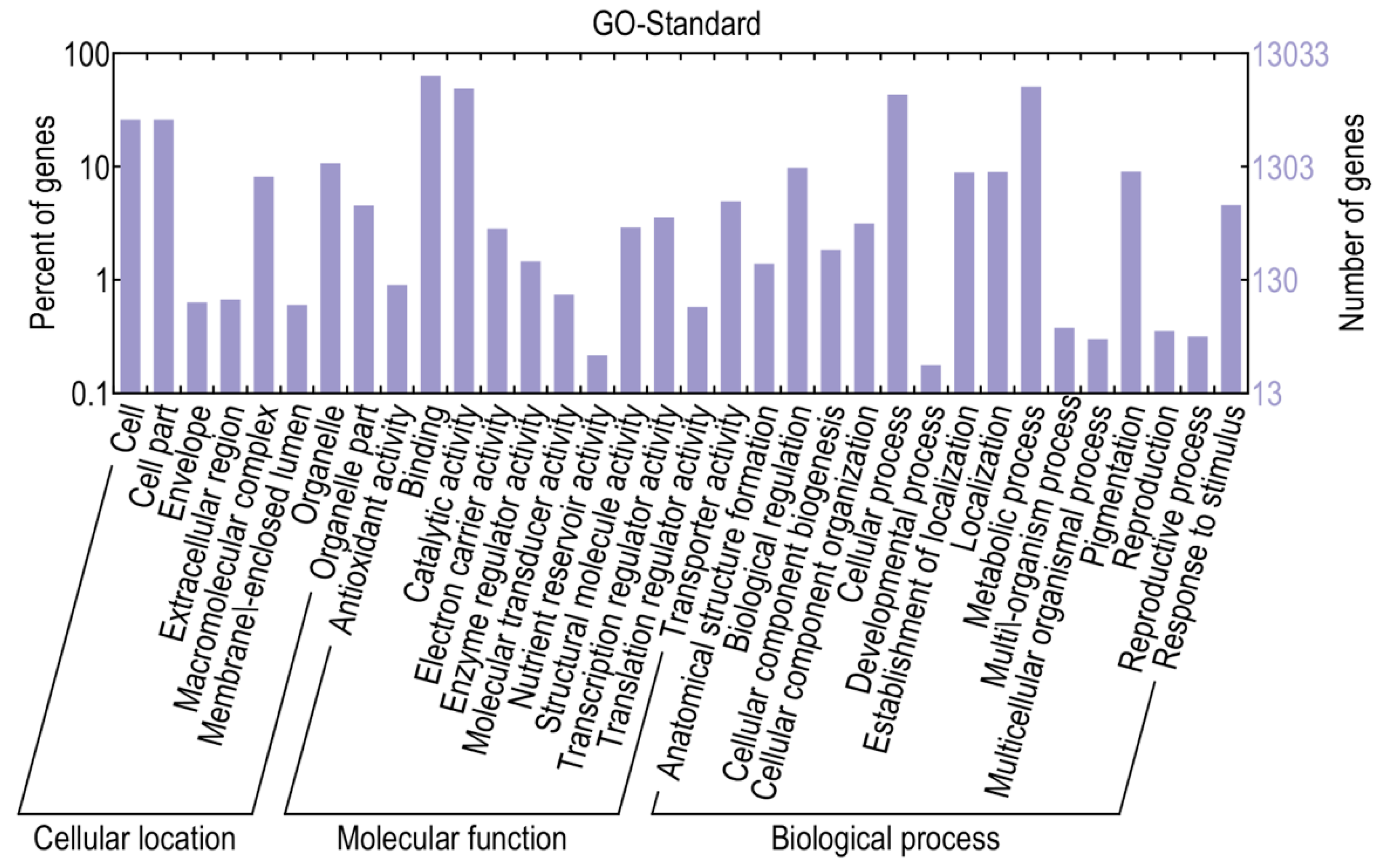
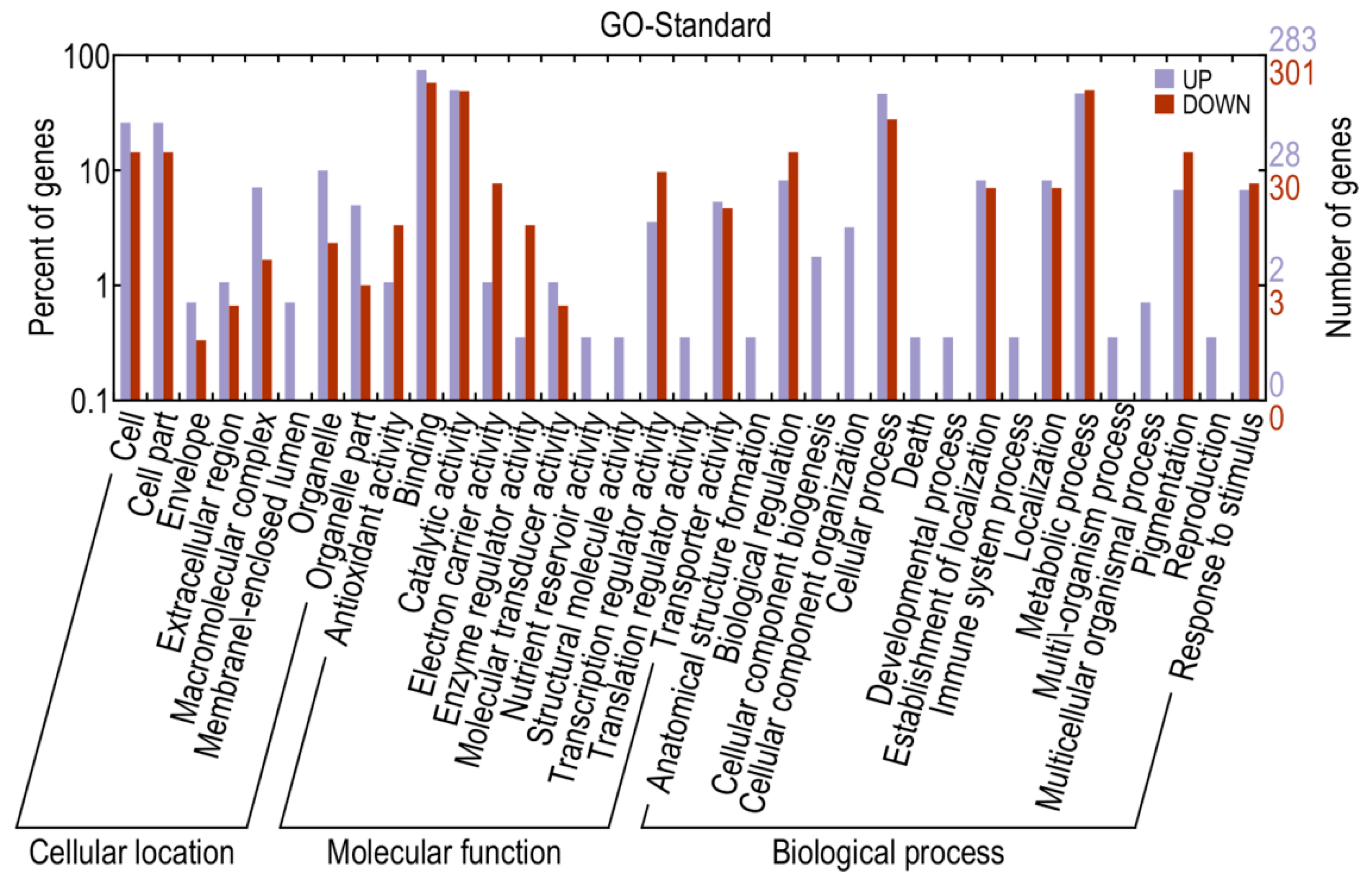
2.4. Identification and Annotation of Potential Differentially Expressed Genes
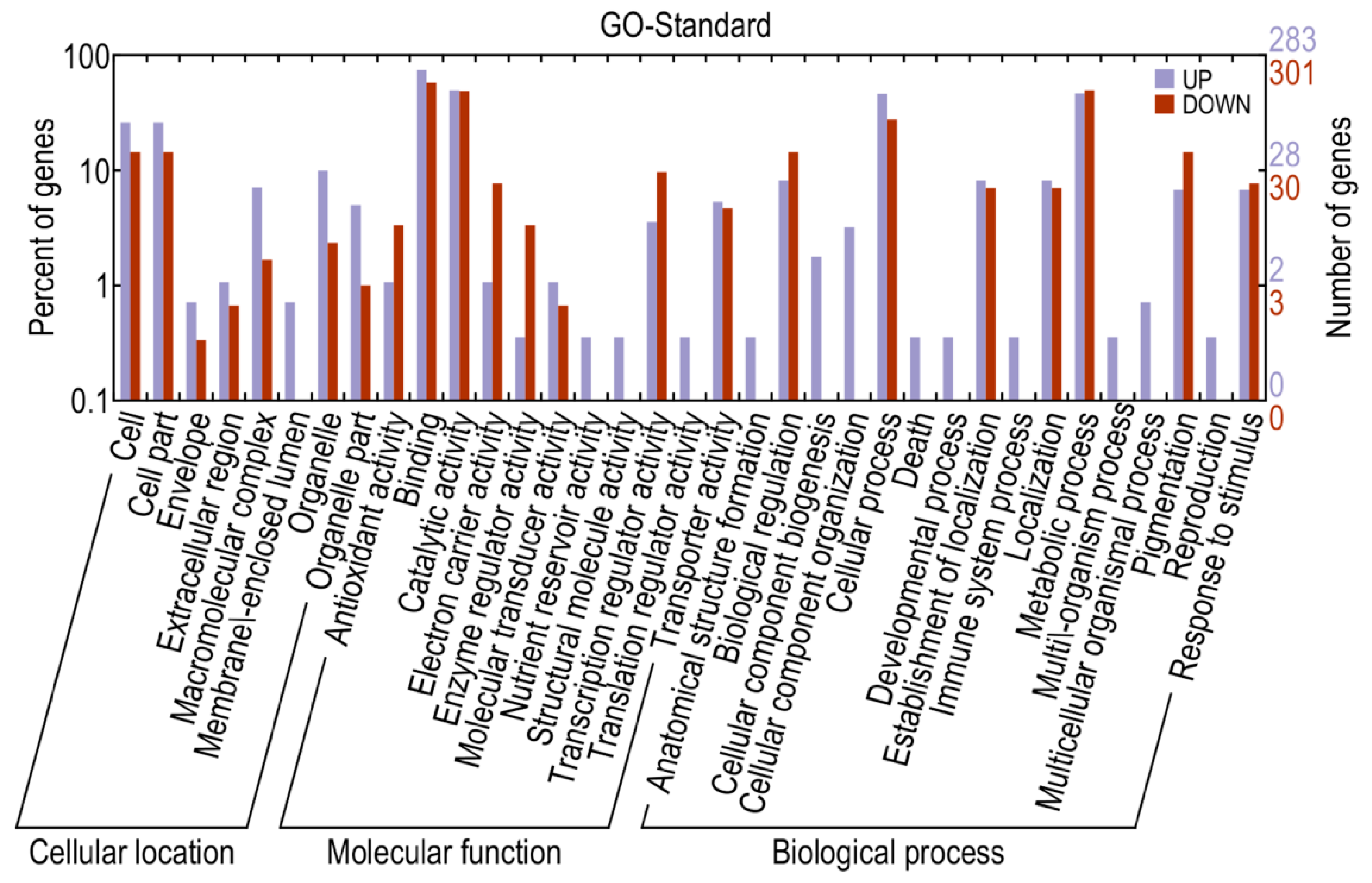
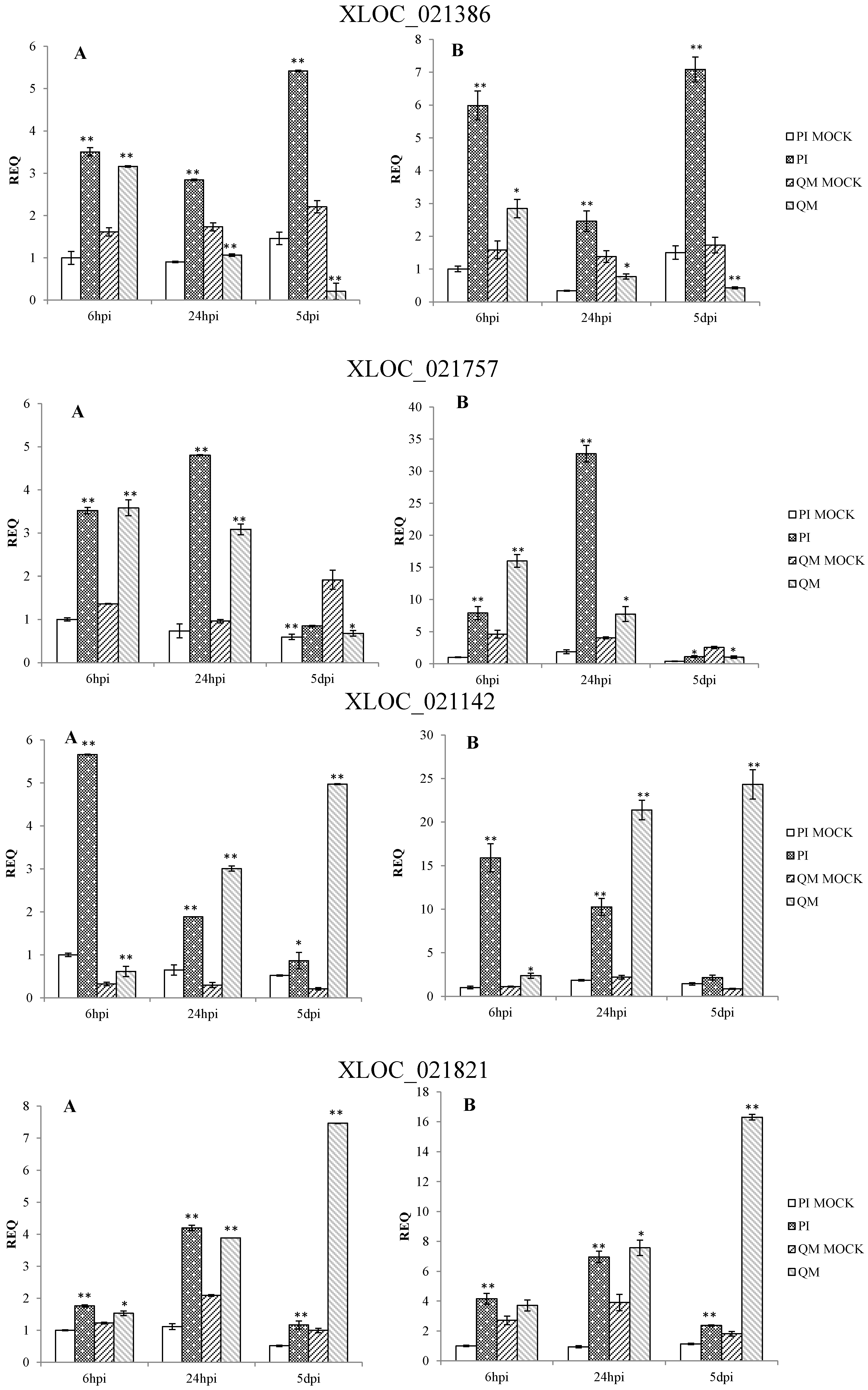
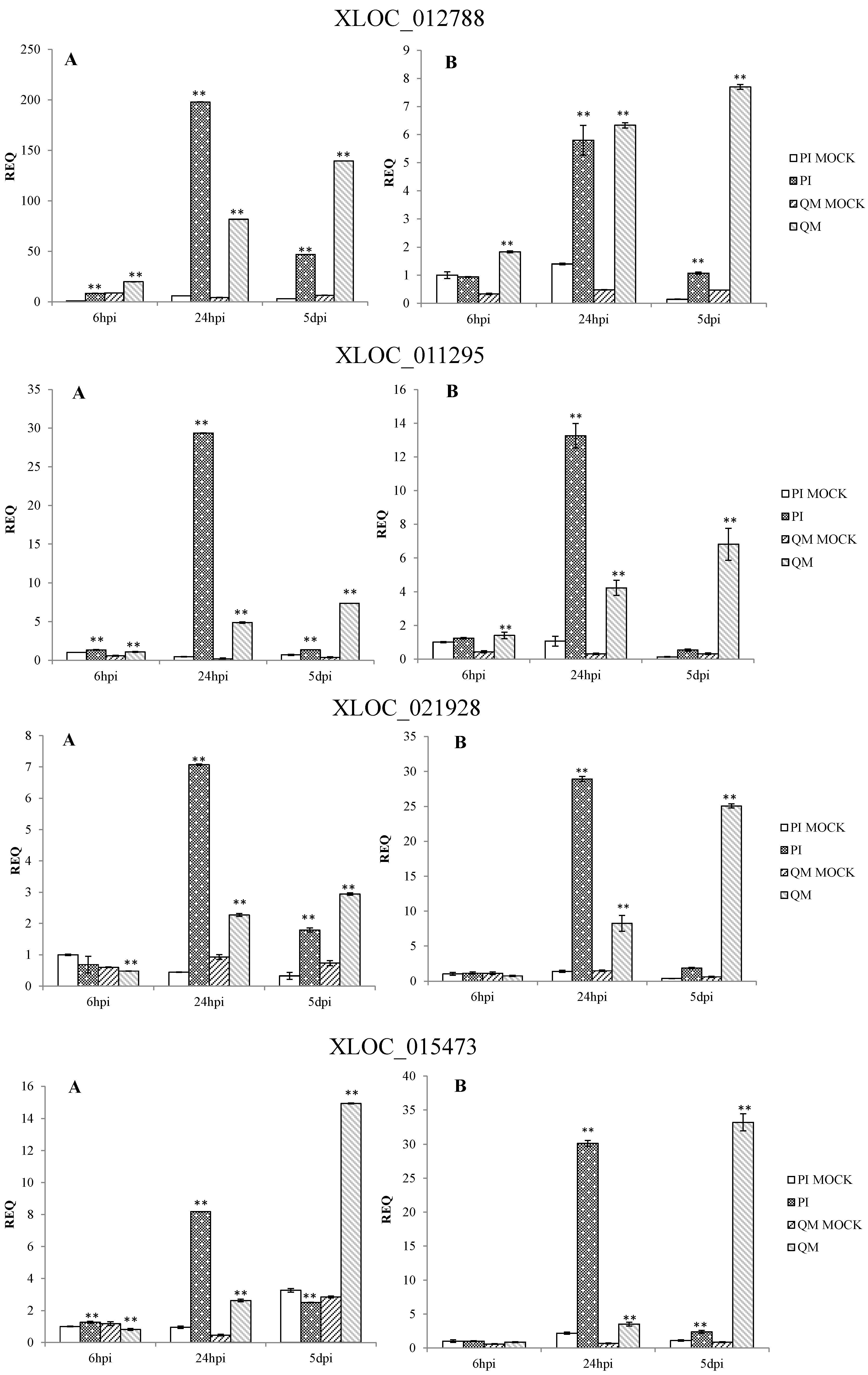
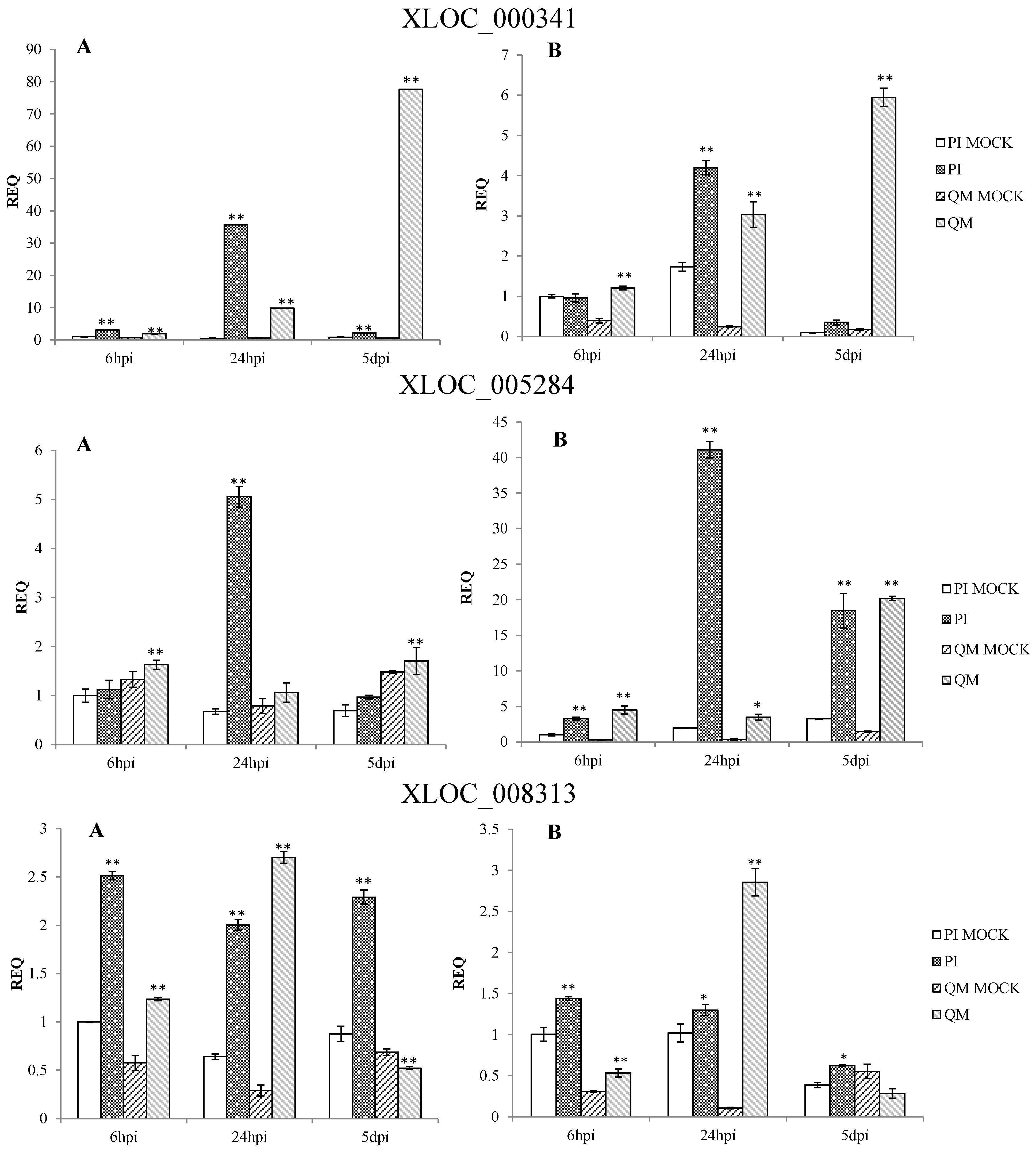
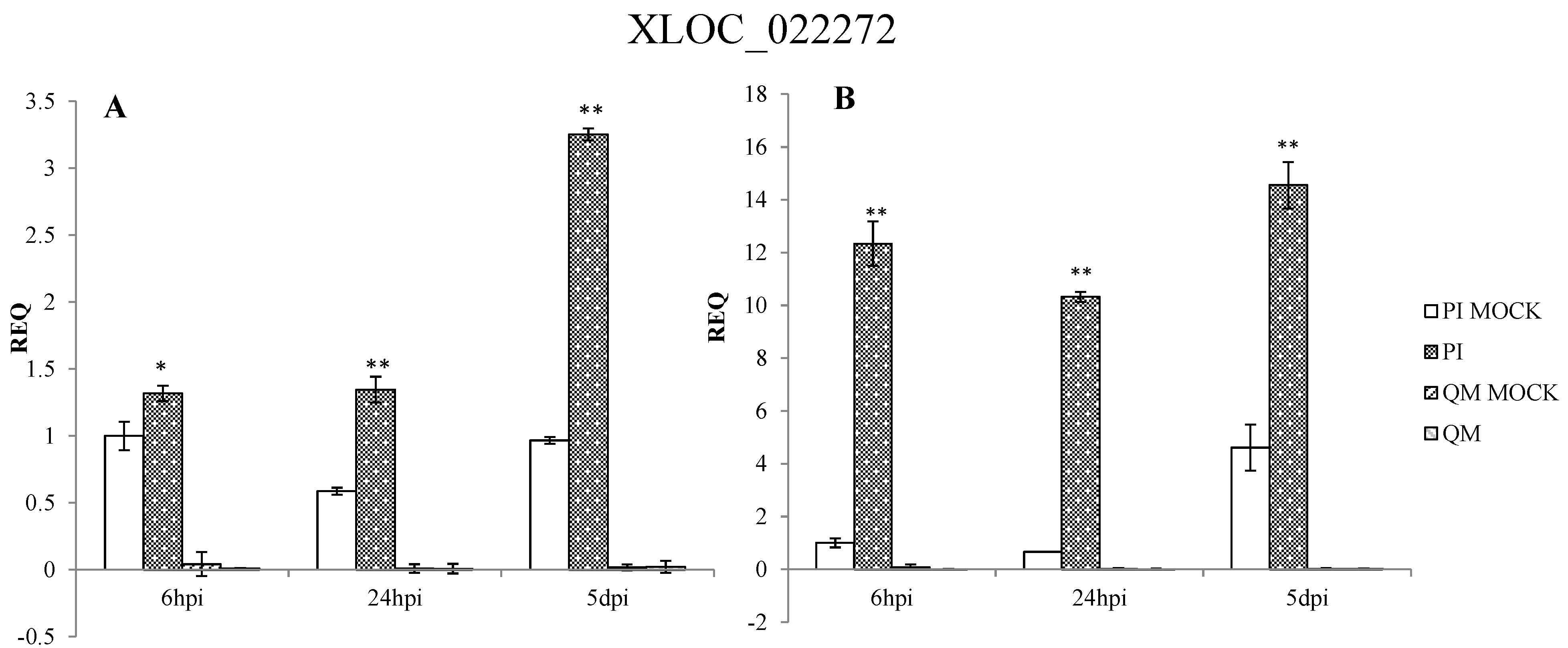
2.5. RNA-Seq Validation and Selection of Potential Defense-Related Genes
| Gene Name | Reference Gene | Annotation | Primer (5'–3') |
|---|---|---|---|
| XLOC_023615 | Capana06g000792 | SWEET sugar transporter | F: ATTGCTCCAAAGCCACCACC |
| R: TGGCAGCATCGTCTCGTTCA | |||
| XLOC_004633 | Capana01g001100 | Major intrinsic protein, conserved site | F: TTGTGGCTGTTTCAGTGTCA |
| R: GGTAGCAATCTTGAGGAGGA | |||
| XLOC_021386 | Capana05g000172 | Proteinase inhibitor I25, cystatin, conserved region | F: AGGCGAAGACAAATCTGGAAT |
| R: TGCTAAATAGTTATGTGGCGAGTC | |||
| XLOC_021757 | Capana05g001613 | Plant peroxidase | F: GTATTACTCGGCAGAAGGGACTC |
| R: GTGGTTGGGCTTGTGGTGT | |||
| XLOC_021200 | Capana05g002178 | Plant peroxidase | F: CTTTTCCACGATTGTTTTGTTAGG |
| R: CGACCTGCTGGCACTGAAT | |||
| XLOC_021142 | Capana05g001951 | AP2/ERF domain | F: TCCTCATACCTAAACGAACCCA |
| R: AGTTGTTGTCGTGTGTTGGATTG | |||
| XLOC_021821 | Capana05g001948 | AP2/ERF domain | F: TTGAAAGAATCTCGGACACCC |
| R: GAAATTGAACGGCGACCAG |
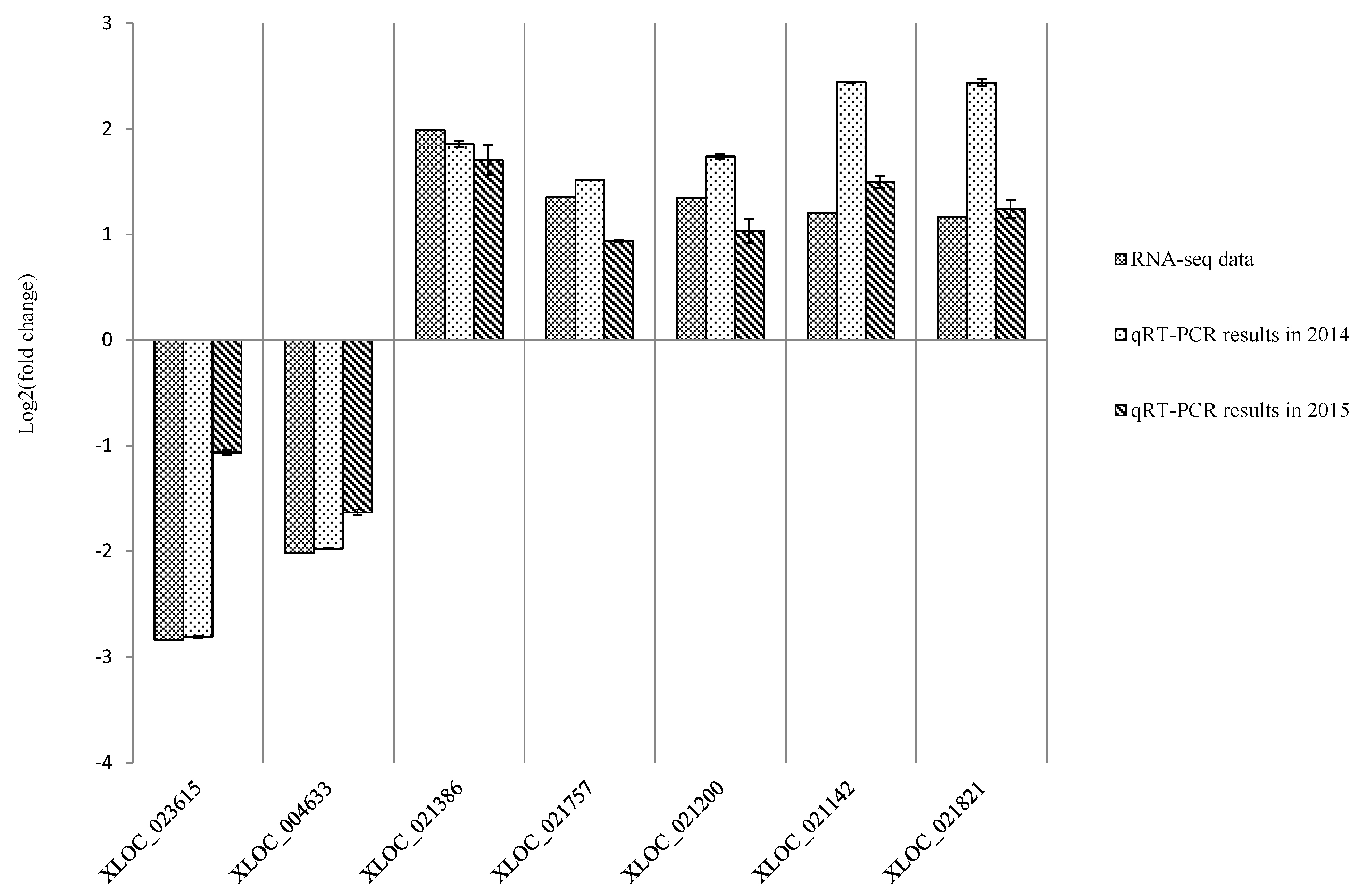
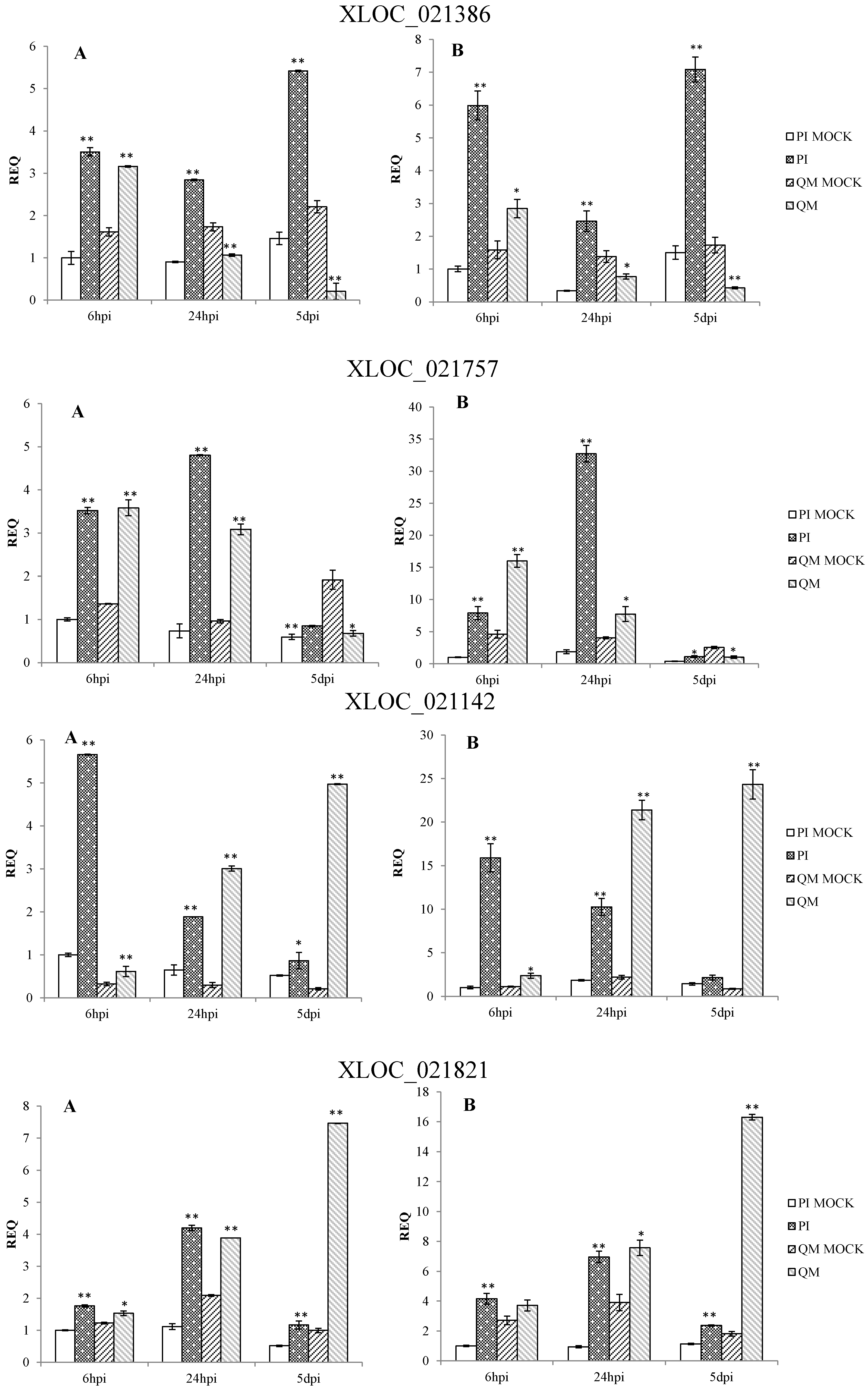

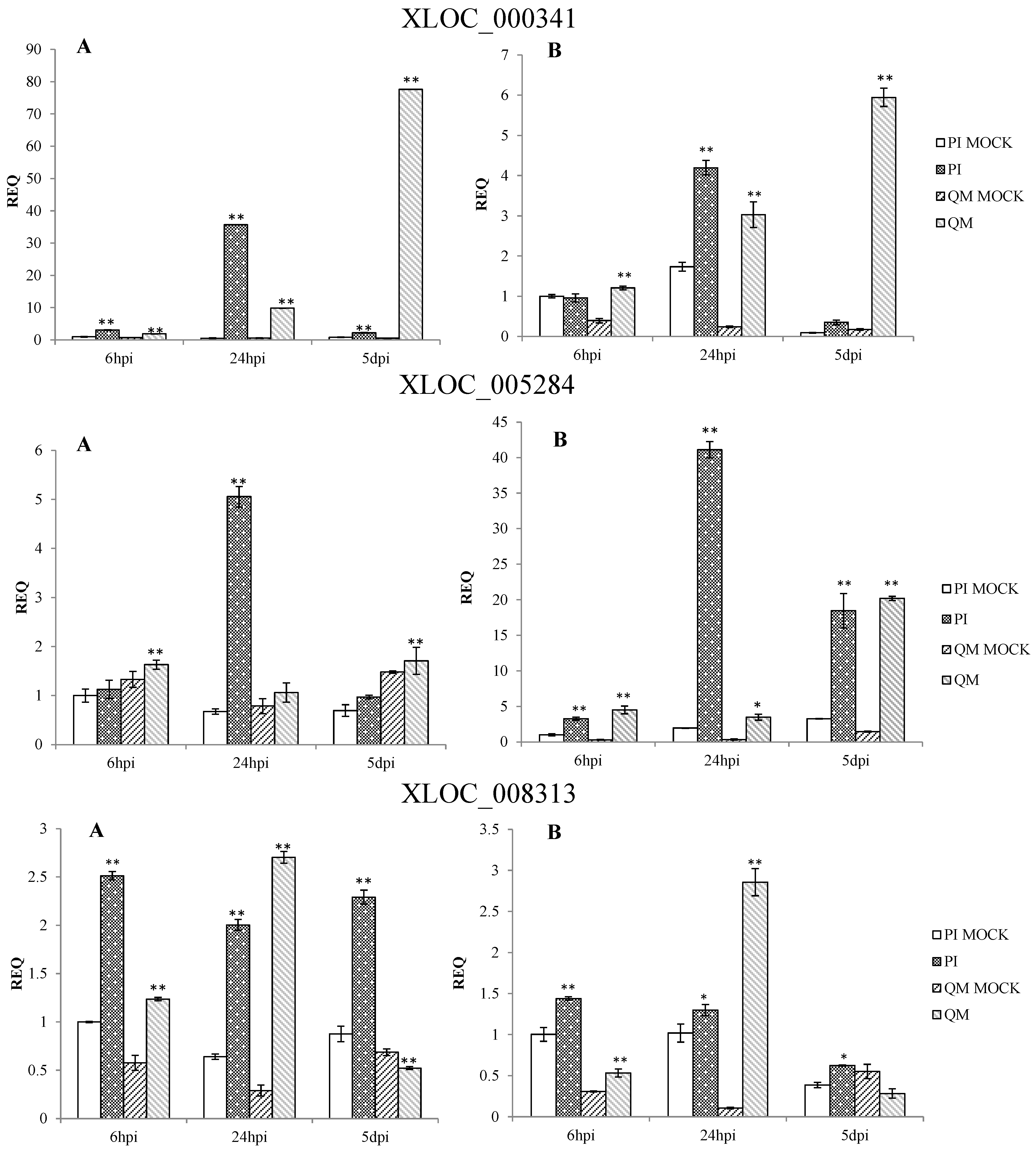
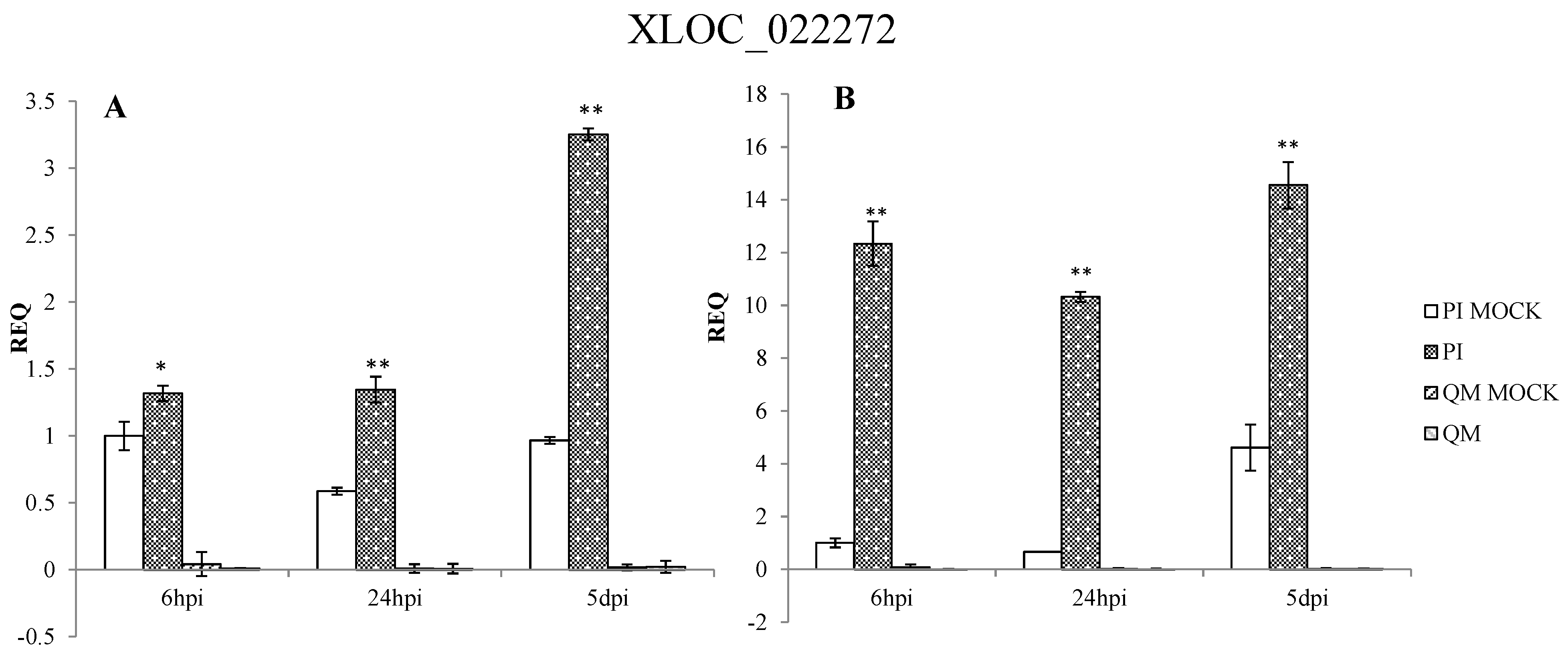
3. Discussion
3.1. RNA-Seq Dataset Analysis
3.2. Potential Defense Related Genes against P. capsici
3.2.1. Cell Wall Modification
3.2.2. Phytoalexins
3.2.3. Phytohormones
3.2.4. Analysis of a Novel Gene on Chromosome 5 in the Zunla-1 Genome
4. Experimental Section
4.1. Biological Material for RNA-Seq
4.2. Biological Material for Selection of Candidate Genes
4.3. Inoculation Procedure and Sample Collection
4.4. RNA Extraction and Library Preparation for Transcriptome Sequencing
4.5. Transcriptome Data Processing and Assembly
4.6. Functional Annotation and Classification
4.7. Detection of Differentially Expressed Genes
4.8. RNA-Seq Validation via qPCR
4.9. Selection of Potential Defense-Related Genes
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Walker, S.J.; Bosland, P.W. Inheritance of Phytophthora root rot and foliar blight resistance in pepper. J. Am. Soc. Hortic. Sci. 1999, 124, 14–18. [Google Scholar]
- Ristaino, J.B.; Johnston, S.A. Ecologically based approaches to management of Phytophthora blight on bell pepper. Plant Dis. 1999, 83, 1080–1089. [Google Scholar] [CrossRef]
- Parra, G.; Ristaino, J.B. Resistance to mefenoxam and metalaxyl among field isolates of Phytophthora capsici causing Phytophthora blight of bell pepper. Plant Dis. 2001, 85, 1069–1075. [Google Scholar] [CrossRef]
- Lamour, K.; Hausbeck, M. The dynamics of mefenoxam insensitivity in a recombining population of Phytophthora capsici characterized with amplified fragment length polymorphism markers. Phytopathology 2001, 91, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Boedo, C.; le Clerc, V.; Briard, M.; Simoneau, P.; Chevalier, M.; Georgeault, S.; Poupard, P. Impact of carrot resistance on development of the Alternaria leaf blight pathogen (Alternaria dauci). Eur. J. Plant Pathol. 2008, 121, 55–66. [Google Scholar] [CrossRef]
- Thabuis, A.; Palloix, A.; Pflieger, S.; Daubeze, A.M.; Caranta, C.; Lefebvre, V. Comparative mapping of Phytophthora resistance loci in pepper germplasm: Evidence for conserved resistance loci across Solanaceae and for a large genetic diversity. Theor. Appl. Genet. 2003, 106, 1473–1485. [Google Scholar] [PubMed]
- Saini, S.; Sharma, P. Inheritance of resistance to fruit rot (Phytophthora capsici Leon.) and induction of resistance in bell pepper (Capsicum annuum L.). Euphytica 1978, 27, 721–723. [Google Scholar] [CrossRef]
- Kim, B.; Hur, J. Inheritance of resistance to bacterial spot and to Phytophthora blight in peppers. J. Korean Soc. Hortic. Sci. 1990, 31, 350–357. [Google Scholar]
- Barksdale, T.H.; Papavizas, G.C.; Johnston, S.A. Resistance to foliar blight and crown rot of pepper caused by Phytophthora capsici. Plant Dis. 1984, 68, 506–509. [Google Scholar] [CrossRef]
- Guerrero-Moreno, A.; Laborde, J. Current status of pepper breeding for resistance to Phytophthora capsici in Mexico. In Proceedings of the Synopses 4th Meeting Capsicum Working Group Eucarpia, IVT, Wageningen, The Netherlands, 14–16 October 1980; pp. 52–56.
- Reifschneider, F.J.B.; Boiteux, L.S.; Dellavecchia, P.T.; Poulos, J.M.; Kuroda, N. Inheritance of adult-plant resistance to Phytophthora capsici in pepper. Euphytica 1992, 62, 45–49. [Google Scholar] [CrossRef]
- Ortega, R.G.; PalazónEspañol, C.; Zueco, J.C. Genetic relationships among four pepper genotypes resistant to Phytophthora capsici. Plant Breed. 1992, 108, 118–125. [Google Scholar] [CrossRef]
- Lefebvre, V.; Palloix, A. Both epistatic and additive effects of QTLs are involved in polygenic induced resistance to disease: A case study, the interaction pepper–Phytophthora capsici Leonian. Theor. Appl. Genet. 1996, 93, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, J.; Danan, S.; Boudet, C.; Barchi, L.; Sage-Palloix, A.M.; Caromel, B.; Palloix, A.; Lefebvre, V. Are the polygenic architectures of resistance to Phytophthora capsici and P. parasitica independent in pepper? Theor. Appl. Genet. 2007, 115, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Nahm, S.H.; Lee, H.R.; Yoon, G.B.; Kim, K.T.; Kang, B.C.; Choi, D.; Kweon, O.Y.; Cho, M.C.; Kwon, J.K.; et al. BAC-derived markers converted from RFLP linked to Phytophthora capsici resistance in pepper (Capsicum annuum L.). Theor. Appl. Genet. 2008, 118, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Minamiyama, Y.; Tsuro, M.; Kubo, T.; Hirai, M. QTL analysis for resistance to Phytophthora capsici in pepper using a high density SSR-based map. Breed. Sci. 2007, 57, 129–134. [Google Scholar] [CrossRef]
- Ogundiwin, E.A.; Berke, T.F.; Massoudi, M.; Black, L.L.; Huestis, G.; Choi, D.; Lee, S.; Prince, J.P. Construction of 2 intraspecific linkage maps and identification of resistance QTLs for Phytophthora capsici root-rot and foliar-blight diseases of pepper (Capsicum annuum L.). Genome 2005, 48, 698–711. [Google Scholar] [CrossRef] [PubMed]
- Sugita, T.; Yamaguchi, K.; Kinoshita, T.; Yuji, K.; Sugimura, Y.; Nagata, R.; Kawasaki, S.; Todoroki, A. QTL analysis for resistance to Phytophthora blight (Phytophthora capsici Leon.) using an intraspecific doubled-haploid population of Capsicum annuum. Breed. Sci. 2006, 56, 137–145. [Google Scholar] [CrossRef]
- Thabuis, A.; Palloix, A.; Servin, B.; Daubeze, A.M.; Signoret, P.; Hospital, F.; Lefebvre, V. Marker-assisted introgression of 4 Phytophthora capsici resistance QTL alleles into a bell pepper line: Validation of additive and epistatic effects. Mol. Breed. 2004, 14, 9–20. [Google Scholar] [CrossRef]
- Hai, T.H.T.; Kim, J.H.; Cho, M.C.; Chae, S.Y.; Lee, H.E. Identification and development of molecular markers linked to Phytophthora root rot resistance in pepper (Capsicum annuum L.). Eur. J. Plant Pathol. 2013, 135, 289–297. [Google Scholar] [CrossRef]
- Mallard, S.; Cantet, M.; Massire, A.; Bachellez, A.; Ewert, S.; Lefebvre, V. A key QTL cluster is conserved among accessions and exhibits broad-spectrum resistance to Phytophthora capsici: A valuable locus for pepper breeding. Mol. Breed. 2013, 32, 349–364. [Google Scholar] [CrossRef]
- Rehrig, W.Z.; Ashrafi, H.; Hill, T.; Prince, J.; van Deynze, A. CaDMR1 Cosegregates with QTL Pc5.1 for Resistance to Phytophthora capsici in Pepper (Capsicum annuum). Plant Genome-Us 2014, 7. [Google Scholar] [CrossRef]
- Liu, W.Y.; Kang, J.H.; Jeong, H.S.; Choi, H.J.; Yang, H.B.; Kim, K.T.; Choi, D.; Choi, G.J.; Jahn, M.; Kang, B.C. Combined use of bulked segregant analysis and microarrays reveals SNP markers pinpointing a major QTL for resistance to Phytophthora capsici in pepper. Theor. Appl. Genet. 2014, 127, 2503–2513. [Google Scholar] [CrossRef] [PubMed]
- Venturini, L.; Ferrarini, A.; Zenoni, S.; Tornielli, G.B.; Fasoli, M.; dal Santo, S.; Minio, A.; Buson, G.; Tononi, P.; Zago, E.D.; et al. De novo transcriptome characterization of Vitis vinifera cv. Corvina unveils varietal diversity. BMC Genomics 2013, 14, 41. [Google Scholar] [CrossRef]
- Bottino, M.C.; Rosario, S.; Grativol, C.; Thiebaut, F.; Rojas, C.A.; Farrineli, L.; Hemerly, A.S.; Ferreira, P.C.G. High-throughput sequencing of small RNA transcriptome reveals salt stress regulated microRNAs in sugarcane. PLoS ONE 2013, 8, e59423. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Wang, Y.; Wang, S.L.; Wu, X.; Yang, K.; Niu, Y.J.; Dai, S.L. Transcriptome-wide survey and expression analysis of stress-responsive NAC genes in Chrysanthemum lavandulifolium. Plant Sci. 2012, 193, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Pan, Y.; Liu, Z.; Zhu, X.W.; Zhai, L.L.; Xu, L.; Yu, R.G.; Gong, Y.Q.; Liu, L.W. De novo transcriptome sequencing of radish (Raphanus sativus L.) and analysis of major genes involved in glucosinolate metabolism. BMC Genomics 2013, 14, 836. [Google Scholar] [CrossRef] [PubMed]
- Socquet-Juglard, D.; Kamber, T.; Pothier, J.F.; Christen, D.; Gessler, C.; Duffy, B.; Patocchi, A. Comparative RNA-seq analysis of early-infected peach leaves by the invasive phytopathogen Xanthomonas arboricola pv. pruni. PLoS ONE 2013, 8, e54196. [Google Scholar] [CrossRef]
- Haas, B.J.; Zody, M.C. Advancing RNA-seq analysis. Nat. Biotechnol. 2010, 28, 421–423. [Google Scholar] [CrossRef] [PubMed]
- Kitajima, S.; Sato, F. Plant pathogenesis-related proteins: Molecular mechanisms of gene expression and protein function. J. Biochem. 1999, 125, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Kader, J.C. Lipid-transfer proteins in plants. Annu. Rev. Plant Phys. 1996, 47, 627–654. [Google Scholar] [CrossRef]
- Jung, H.W.; Kim, W.; Hwang, B.K. Three pathogen-inducible genes encoding lipid transfer protein from pepper are differentially activated by pathogens, abiotic, and environmental stresses. Plant Cell Environ. 2003, 26, 915–928. [Google Scholar] [CrossRef] [PubMed]
- Diao, G.P.; Wang, Y.C.; Wang, C.; Yang, C.P. Cloning and functional characterization of a novel glutathione S-transferase gene from Limonium bicolor. Plant Mol. Biol. Rep. 2011, 29, 77–87. [Google Scholar] [CrossRef]
- Liao, W.H.; Ji, L.X.; Wang, J.; Chen, Z.; Ye, M.X.; Ma, H.D.; An, X.M. Identification of glutathione S-transferase genes responding to pathogen infestation in Populus tomentosa. Funct. Integr. Genomic 2014, 14, 517–529. [Google Scholar] [CrossRef]
- Pirselova, B.; Matusikova, I. Callose: The plant cell wall polysaccharide with multiple biological functions. Acta Physiol. Plant 2013, 35, 635–644. [Google Scholar] [CrossRef]
- Jacobs, A.K.; Lipka, V.; Burton, R.A.; Panstruga, R.; Strizhov, N.; Schulze-Lefert, P.; Fincher, G.B. An Arabidopsis callose synthase, GSL5, is required for wound and papillary callose formation. Plant Cell 2003, 15, 2503–2513. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, M.T.; Stein, M.; Hou, B.H.; Vogel, J.P.; Edwards, H.; Somerville, S.C. Loss of a callose synthase results in salicylic acid-dependent disease resistance. Science 2003, 301, 969–972. [Google Scholar] [CrossRef] [PubMed]
- Kudlicka, K.; Brown, R.M. Cellulose and callose biosynthesis in higher plants. I. Solubilization and separation of (1−>3)- and (1−>4)-β-glucan synthase activities from mung bean. Plant Physiol. 1997, 115, 643–656. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.Y.; Hong, Z.L.; Chatterjee, J.; Kim, S.H.; Verma, D.P.S. Expression of callose synthase genes and its connection with Npr1 signaling pathway during pathogen infection. Planta 2008, 229, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Espelie, K.E.; Franceschi, V.R.; Kolattukudy, P.E. Immunocytochemical localization and time course of appearance of an anionic peroxidase associated with suberization in wound-healing potato-tuber tissue. Plant Physiol. 1986, 81, 487–492. [Google Scholar] [CrossRef] [PubMed]
- Young, S.A.; Guo, A.; Guikema, J.A.; White, F.F.; Leach, J.E. Rice cationic peroxidase accumulates in xylem vessels during incompatible interactions with Xanthomonas oryzae pv oryzae. Plant Physiol. 1995, 107, 1333–1341. [Google Scholar] [CrossRef] [PubMed]
- Lagrimini, L.M.; Burkhart, W.; Moyer, M.; Rothstein, S. Molecular-cloning of complementary-DNA encoding the lignin-forming peroxidase from tobacco: Molecular analysis and tissue-specific expression. Proc. Natl. Acad. Sci. USA 1987, 84, 7542–7546. [Google Scholar] [CrossRef] [PubMed]
- Ahuja, I.; Kissen, R.; Bones, A.M. Phytoalexins in defense against pathogens. Trends Plant Sci. 2012, 17, 73–90. [Google Scholar] [CrossRef] [PubMed]
- Holland, K.W.; O’Keefe, S.F. Recent applications of peanut phytoalexins. Recent. Pat. Food Nutr. Agric. 2010, 2, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Tzi, B.N.; Ye, X.J.; Wong, J.H.; Fang, E.F.; Chan, Y.S.; Pan, W.L.; Ye, X.Y.; Sze, S.C.W.; Zhang, K.Y.B.; Liu, F.; et al. Glyceollin, a soybean phytoalexin with medicinal properties. Appl. Microbiol. Biotechnol. 2011, 90, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.Y.; Browning, J.D.; Awika, J.M. Sorghum 3-deoxyanthocyanins possess strong phase II enzyme inducer activity and cancer cell growth inhibition properties. J. Agric. Food Chem. 2009, 57, 1797–1804. [Google Scholar] [CrossRef] [PubMed]
- Smoliga, J.M.; Baur, J.A.; Hausenblas, H.A. Resveratrol and health—A comprehensive review of human clinical trials. Mol. Nutr. Food Res. 2011, 55, 1129–1141. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Jikumaru, Y.; Okada, A.; Okada, K.; Koga, J.; Umemura, K.; Minami, E.; Shibuya, N.; Hasegawa, M.; Kodama, O.; et al. Effects of a bile acid elicitor, cholic acid, on the biosynthesis of diterpenoid phytoalexins in suspension-cultured rice cells. Phytochemistry 2008, 69, 973–981. [Google Scholar] [CrossRef] [PubMed]
- Huffaker, A.; Kaplan, F.; Vaughan, M.M.; Dafoe, N.J.; Ni, X.Z.; Rocca, J.R.; Alborn, H.T.; Teal, P.E.A.; Schmelz, E.A. Novel acidic sesquiterpenoids constitute a dominant class of pathogen-induced phytoalexins in maize. Plant Physiol. 2011, 156, 2082–2097. [Google Scholar] [CrossRef] [PubMed]
- Araceli, A.C.; Elda, C.M.; Edmundo, L.G.; Ernesto, G.P. Capsidiol production in pepper fruits (Capsicum annuum L.) induced by arachidonic acid is dependent of an oxidative burst. Physiol. Mol. Plant Pathol. 2007, 70, 69–76. [Google Scholar] [CrossRef]
- Broekaert, W.F.; Delaure, S.L.; de Bolle, M.F.C.; Cammue, B.P.A. The role of ethylene in host–pathoven interactions. Annu. Rev. Phytopathol. 2006, 44, 393–416. [Google Scholar] [CrossRef] [PubMed]
- Van Loon, L.C.; Geraats, B.P.J.; Linthorst, H.J.M. Ethylene as a modulator of disease resistance in plants. Trends Plant Sci. 2006, 11, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Núñez-Pastrana, R.; Arcos-Ortega, G.F.; Souza-Perera, R.A.; Sánchez-Borges, C.A.; Nakazawa-Ueji, Y.E.; García-Villalobos, F.J.; Guzmán-Antonio, A.A.; Zúñiga-Aguilar, J.J. Ethylene, but not salicylic acid or methyl jasmonate, induces a resistance response against Phytophthora capsici in Habanero pepper. Eur. J. Plant Pathol. 2011, 131, 669–683. [Google Scholar] [CrossRef]
- Francia, D.; Demaria, D.; Calderini, O.; Ferraris, L.; Valentino, D.; Arcioni, S.; Tamietti, G.; Cardinale, F. Wounding induces resistance to pathogens with different lifestyles in tomato: Role of ethylene in cross-protection. Plant Cell Environ. 2007, 30, 1357–1365. [Google Scholar] [CrossRef] [PubMed]
- Balaji, V.; Mayrose, M.; Sherf, O.; Jacob-Hirsch, J.; Eichenlaub, R.; Iraki, N.; Manulis-Sasson, S.; Rechavi, G.; Barash, I.; Sessa, G. Tomato transcriptional changes in response to Clavibacter michiganensis subsp michiganensis reveal a role for ethylene in disease development. Plant Physiol. 2008, 146, 1797–1809. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.Q.; Yang, C.; Thara, V.K.; Zhou, J.; Martin, G.B. Pti4 is induced by ethylene and salicylic acid, and its product is phosphorylated by the Pto kinase. Plant Cell 2000, 12, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.Q.; Wildermuth, M.C.; Chakravarthy, S.; Loh, Y.T.; Yang, C.M.; He, X.H.; Han, Y.; Martin, G.B. Tomato transcription factors Pti4, Pti5, and Pti6 activate defense responses when expressed in Arabidopsis. Plant Cell 2002, 14, 817–831. [Google Scholar] [CrossRef] [PubMed]
- Ballare, C.L. Jasmonate-induced defenses: A tale of intelligence, collaborators and rascals. Trends Plant Sci. 2011, 16, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Chini, A.; Fonseca, S.; Fernandez, G.; Adie, B.; Chico, J.M.; Lorenzo, O.; Garcia-Casado, G.; Lopez-Vidriero, I.; Lozano, F.M.; Ponce, M.R.; et al. The JAZ family of repressors is the missing link in jasmonate signalling. Nature 2007, 448, 666–671. [Google Scholar] [CrossRef] [PubMed]
- Demianski, A.J.; Chung, K.M.; Kunkel, B.N. Analysis of Arabidopsis JAZ gene expression during Pseudomonas syringae pathogenesis. Mol. Plant Pathol. 2012, 13, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Van Damme, E.J.M.; Barre, A.; Rouge, P.; Peumans, W.J. Cytoplasmic/nuclear plant lectins: A new story. Trends Plant Sci. 2004, 9, 484–489. [Google Scholar] [CrossRef] [PubMed]
- Hwang, I.S.; Hwang, B.K. The pepper mannose-binding lectin gene CaMBL1 is required to regulate cell death and defense responses to microbial pathogens. Plant Physiol. 2011, 155, 447–463. [Google Scholar] [CrossRef] [PubMed]
- Pang, Z.L.; Shao, J.P.; Chen, L.; Lu, X.H.; Hu, J.; Qin, Z.H.; Liu, X.L. Resistance to the novel fungicide pyrimorph in Phytophthora capsici: Risk assessment and detection of point mutations in CesA3 that confer resistance. PLoS ONE 2013, 8, e56513. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Qin, C.; Yu, C.; Shen, Y.; Fang, X.; Chen, L.; Min, J.; Cheng, J.; Zhao, S.; Xu, M.; Luo, Y.; et al. Whole-genome sequencing of cultivated and wild peppers provides insights into Capsicum domestication and specialization. Proc. Natl. Acad. Sci. USA 2014, 111, 5135–5140. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Hattori, M.; Aoki-Kinoshita, K.F.; Itoh, M.; Kawashima, S.; Katayama, T.; Araki, M.; Hirakawa, M. From genomics to chemical genomics: New developments in KEGG. Nucleic Acids Res. 2006, 34, D354–D357. [Google Scholar] [CrossRef] [PubMed]
- Zdobnov, E.M.; Apweiler, R. InterProScan—An integration platform for the signature-recognition methods in InterPro. Bioinformatics 2001, 17, 847–848. [Google Scholar] [CrossRef] [PubMed]
- Harris, M.A.; Clark, J.; Ireland, A.; Lomax, J.; Ashburner, M.; Foulger, R.; Eilbeck, K.; Lewis, S.; Marshall, B.; Mungall, C.; et al. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 2004, 32, D258–D261. [Google Scholar] [CrossRef] [PubMed]
- Vencio, R.Z.N.; Brentani, H.; Pereira, C.A.B. Using credibility intervals instead of hypothesis tests in SAGE analysis. Bioinformatics 2003, 19, 2461–2464. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, P.; Liu, X.; Guo, J.; Liu, C.; Fu, N.; Shen, H. Identification and Expression Analysis of Candidate Genes Associated with Defense Responses to Phytophthora capsici in Pepper Line “PI 201234”. Int. J. Mol. Sci. 2015, 16, 11417-11438. https://doi.org/10.3390/ijms160511417
Wang P, Liu X, Guo J, Liu C, Fu N, Shen H. Identification and Expression Analysis of Candidate Genes Associated with Defense Responses to Phytophthora capsici in Pepper Line “PI 201234”. International Journal of Molecular Sciences. 2015; 16(5):11417-11438. https://doi.org/10.3390/ijms160511417
Chicago/Turabian StyleWang, Pingyong, Xiaodan Liu, Jinju Guo, Chen Liu, Nan Fu, and Huolin Shen. 2015. "Identification and Expression Analysis of Candidate Genes Associated with Defense Responses to Phytophthora capsici in Pepper Line “PI 201234”" International Journal of Molecular Sciences 16, no. 5: 11417-11438. https://doi.org/10.3390/ijms160511417
APA StyleWang, P., Liu, X., Guo, J., Liu, C., Fu, N., & Shen, H. (2015). Identification and Expression Analysis of Candidate Genes Associated with Defense Responses to Phytophthora capsici in Pepper Line “PI 201234”. International Journal of Molecular Sciences, 16(5), 11417-11438. https://doi.org/10.3390/ijms160511417