
Visfatin Mediates SCLC Cells Migration across Brain Endothelial Cells through Upregulation of CCL2

Abstract
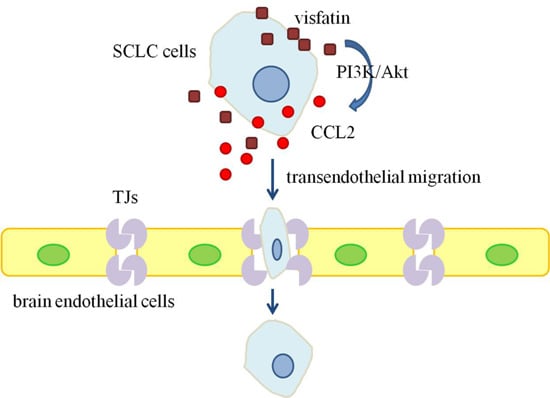
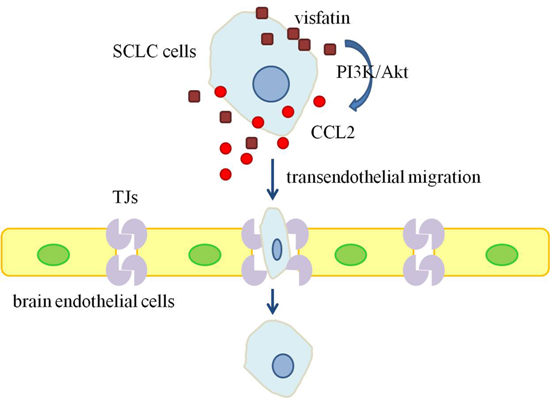
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
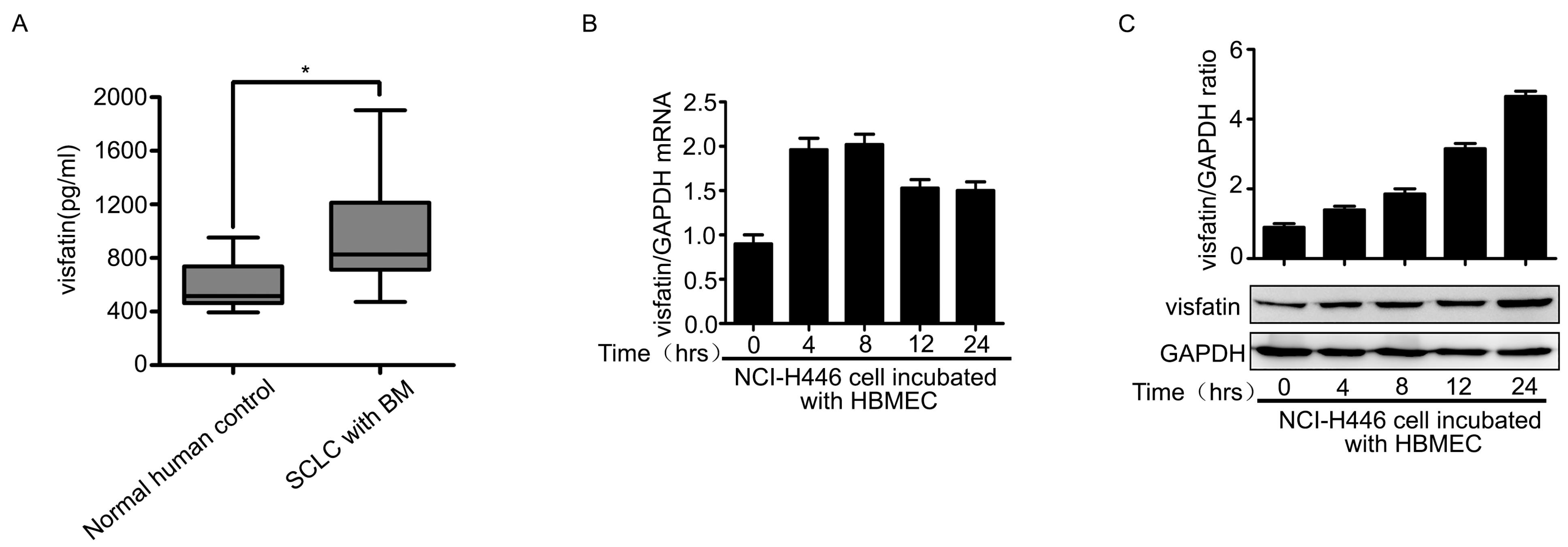
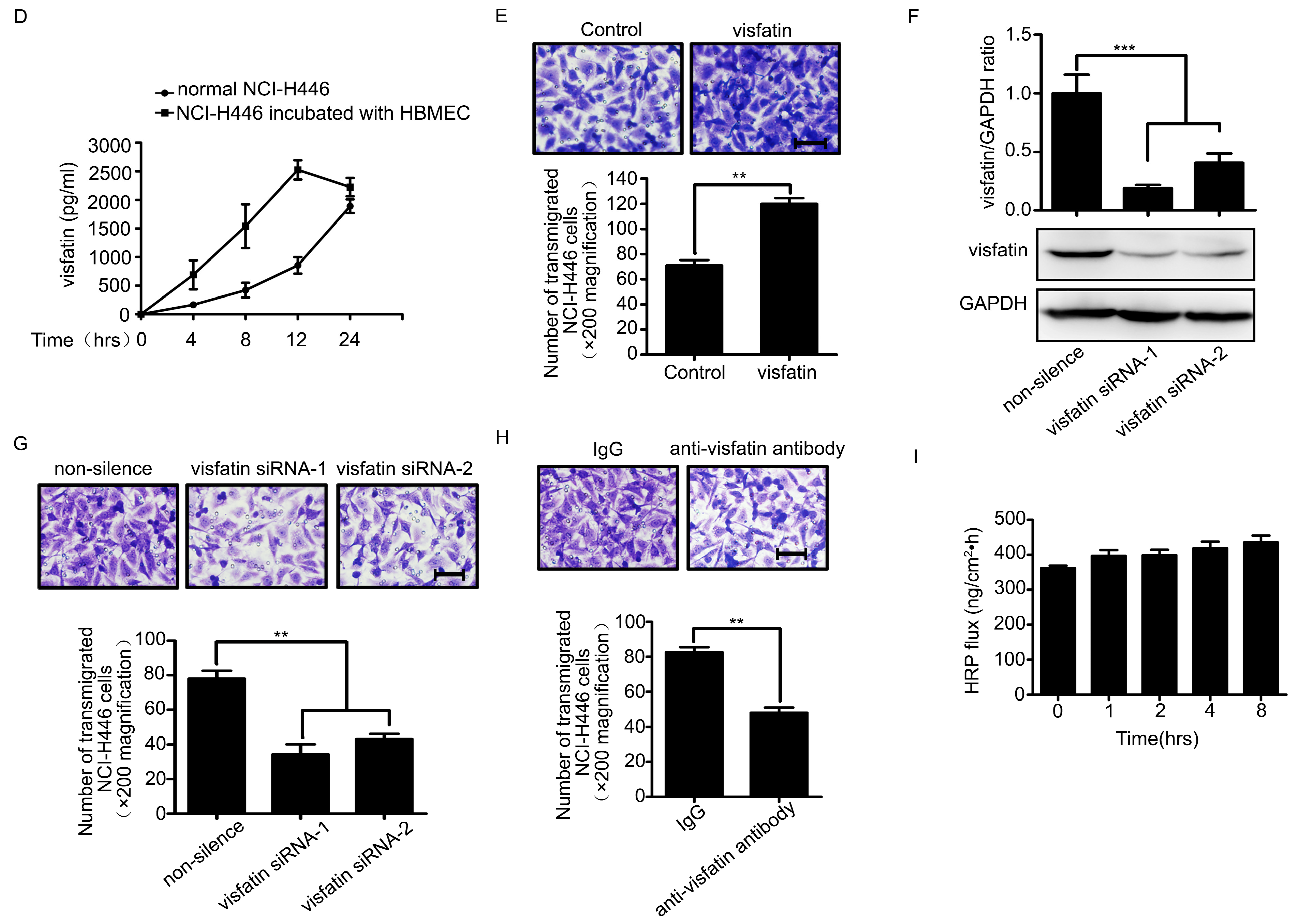
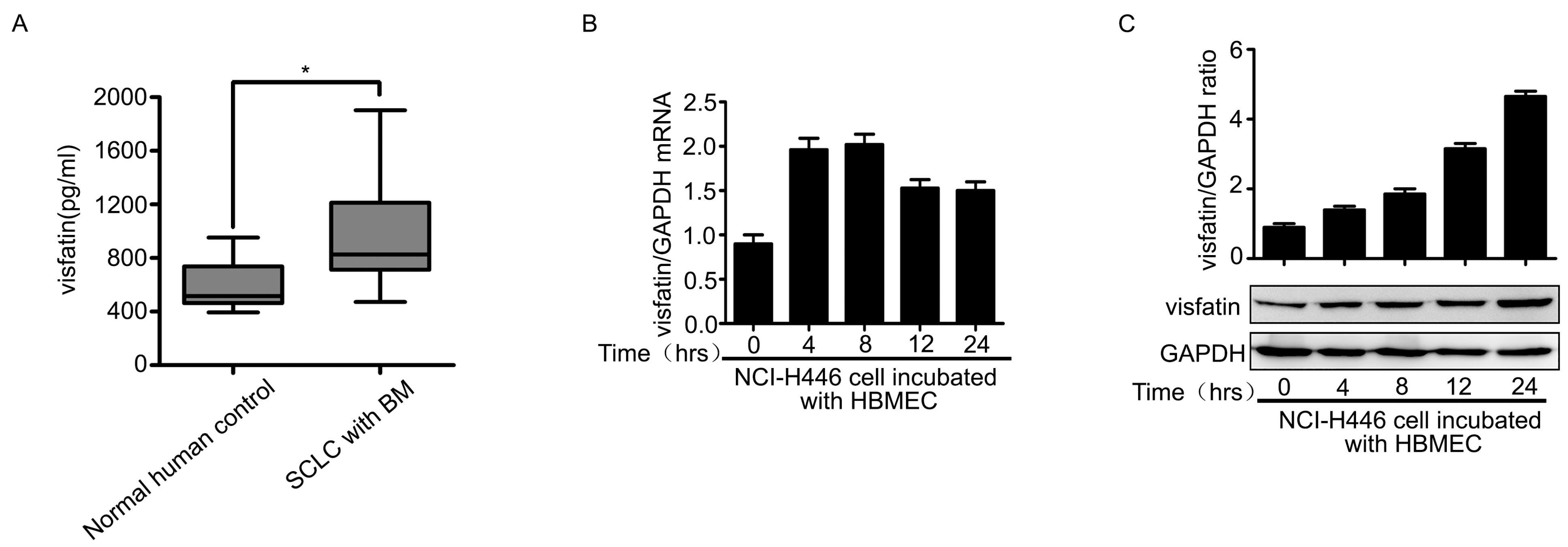
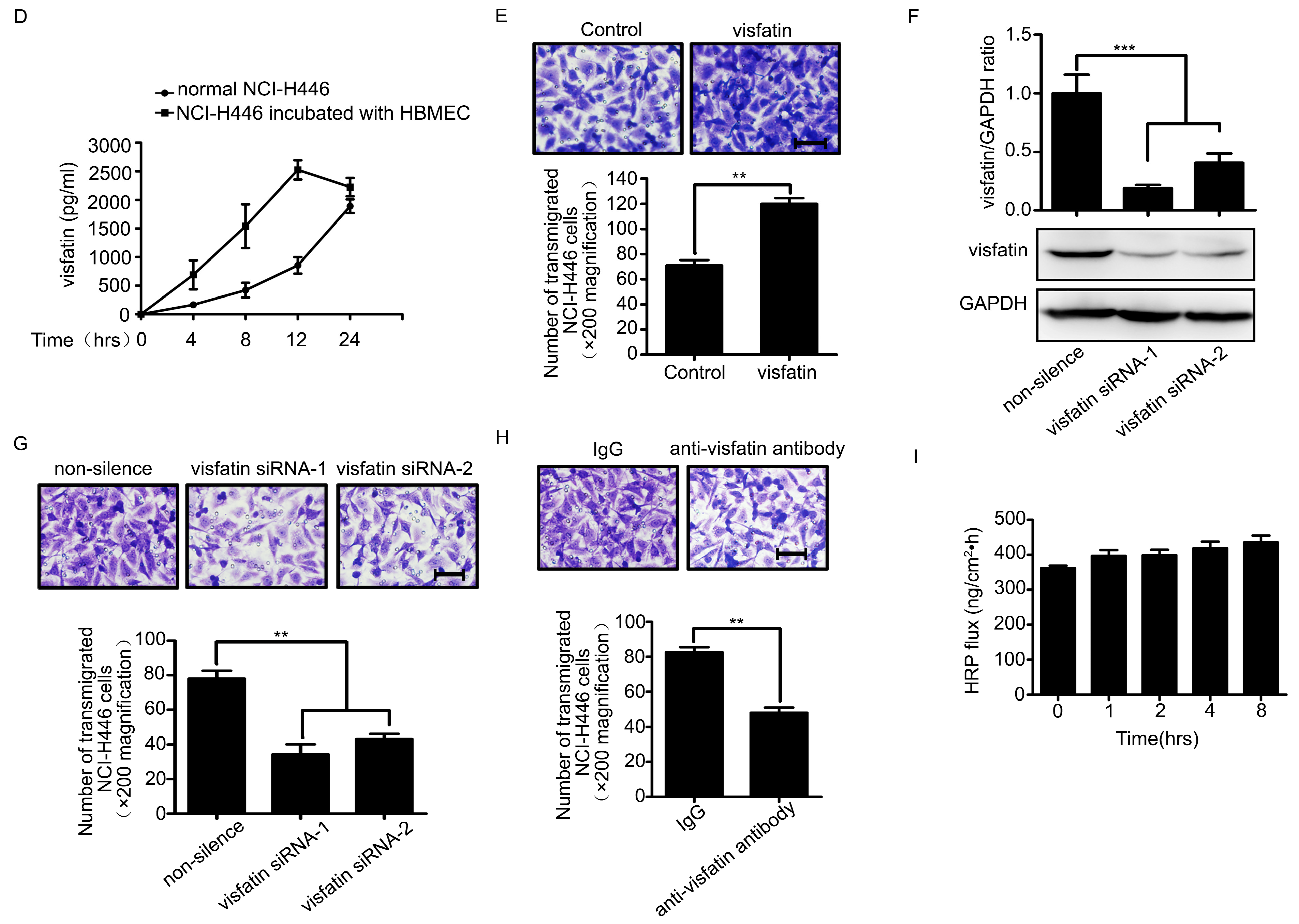
2.1. Visfatin Contributed to NCI-H446 Cells Migration across HBMEC Monolayer
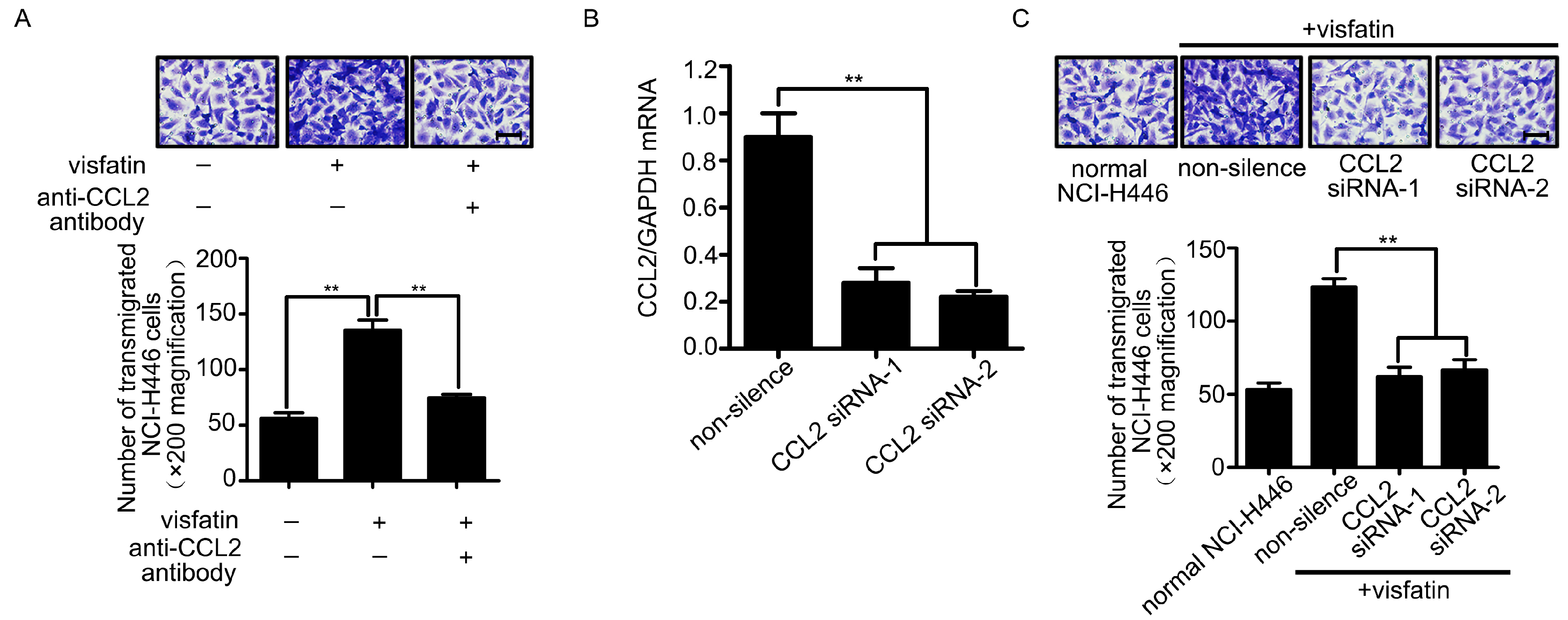
2.2. CCL2 Was Involved in Visfatin-Mediated NCI-H446 Cells Transendothelial Migration
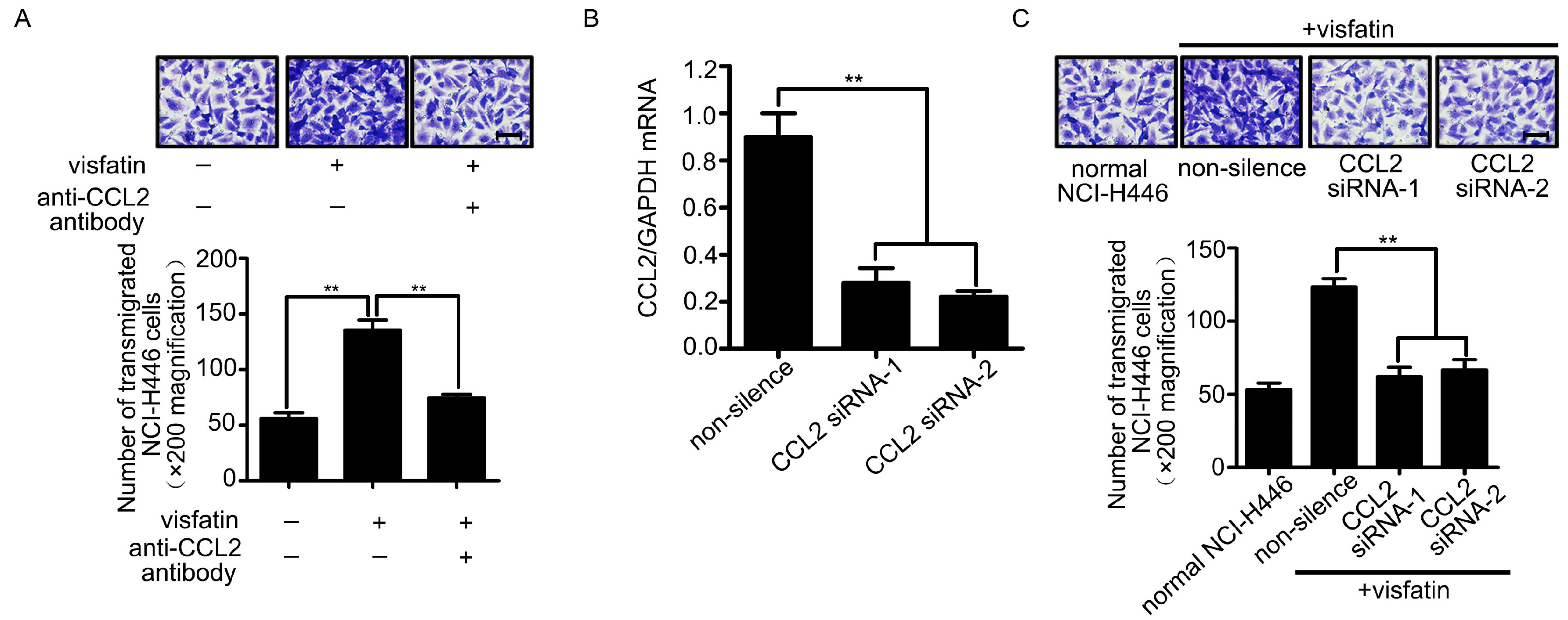
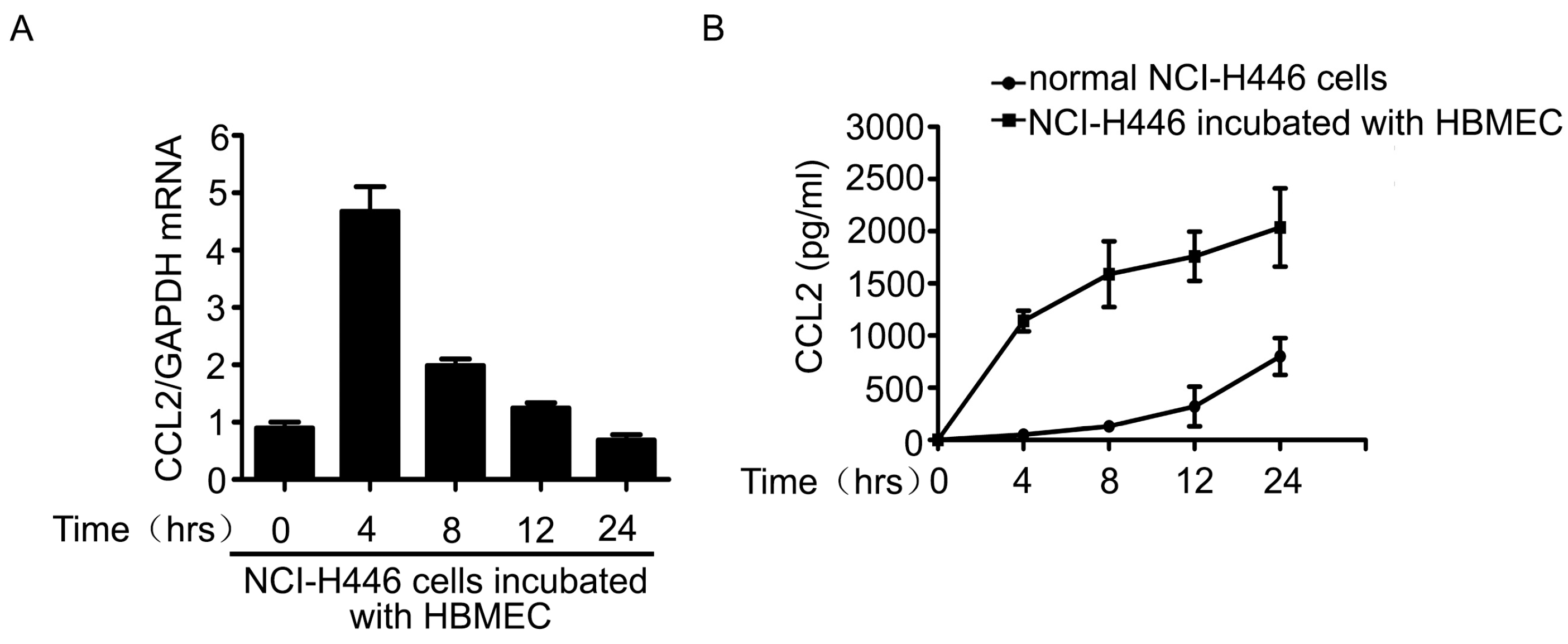
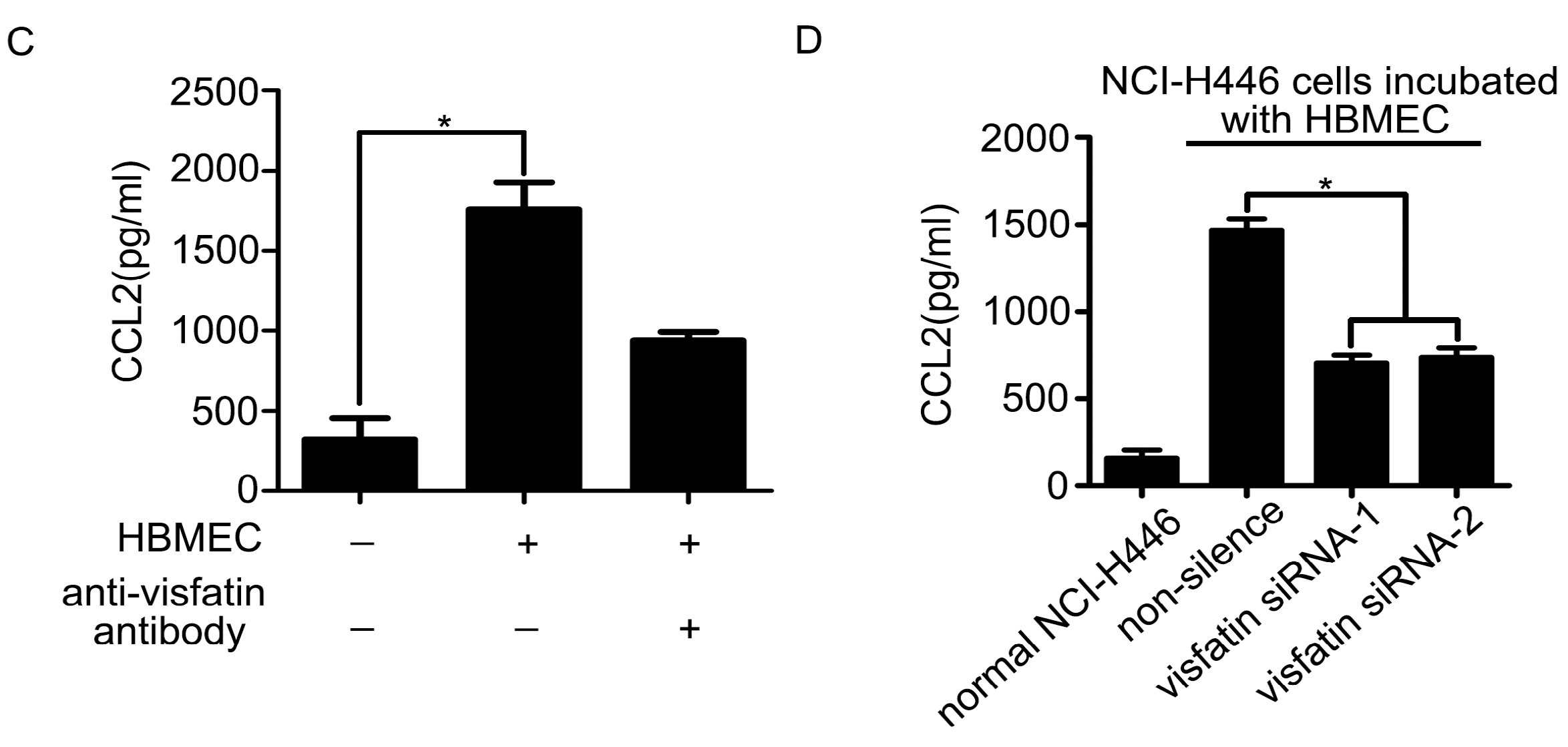
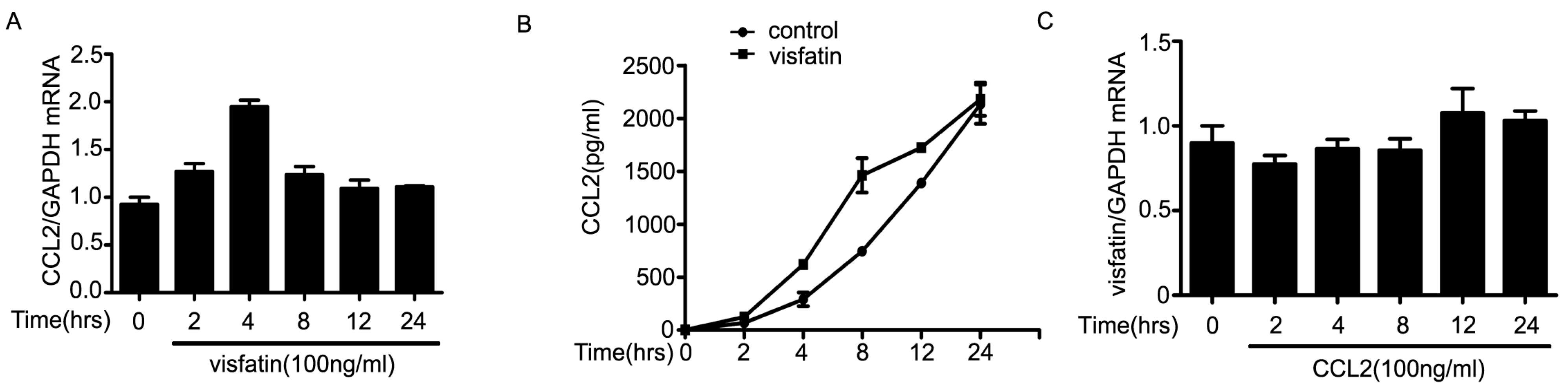
2.3. The Upregulation of CCL2 Was Induced by Visfatin in the Co-Culture System of NCI-H446 Cells and HBMEC
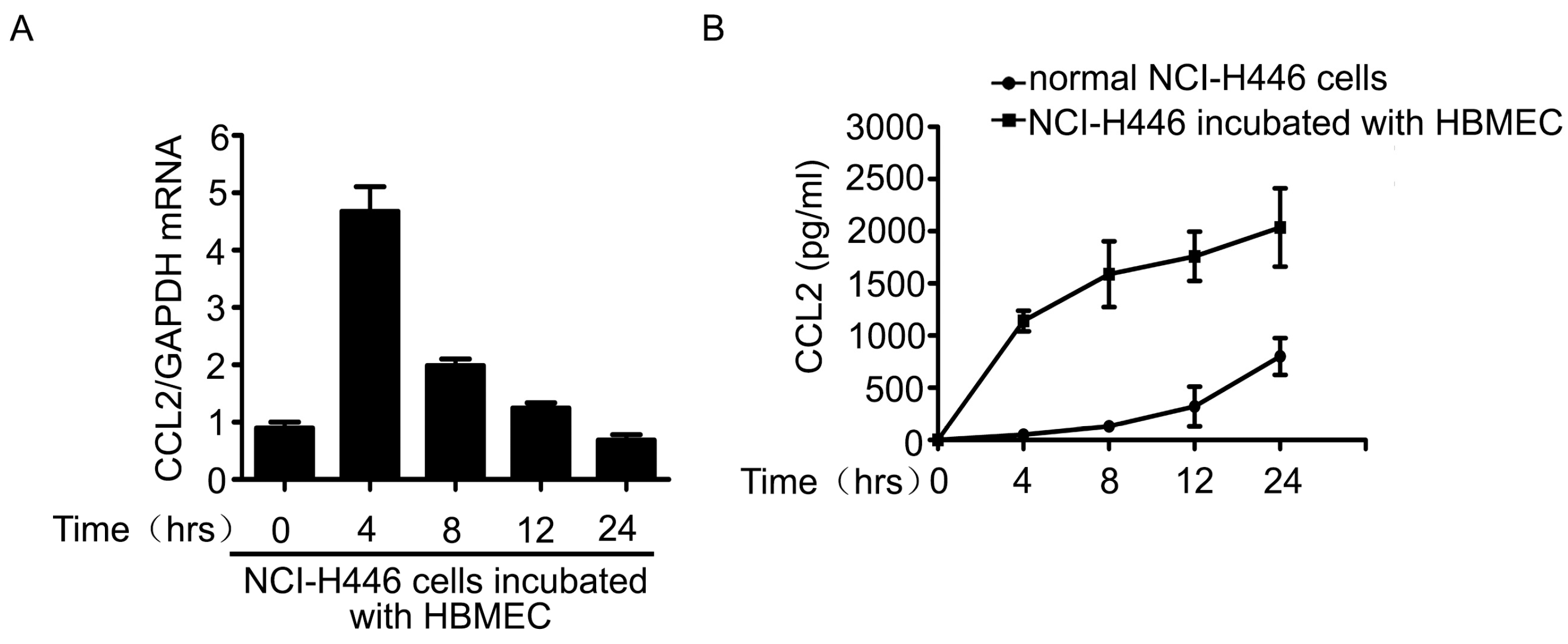
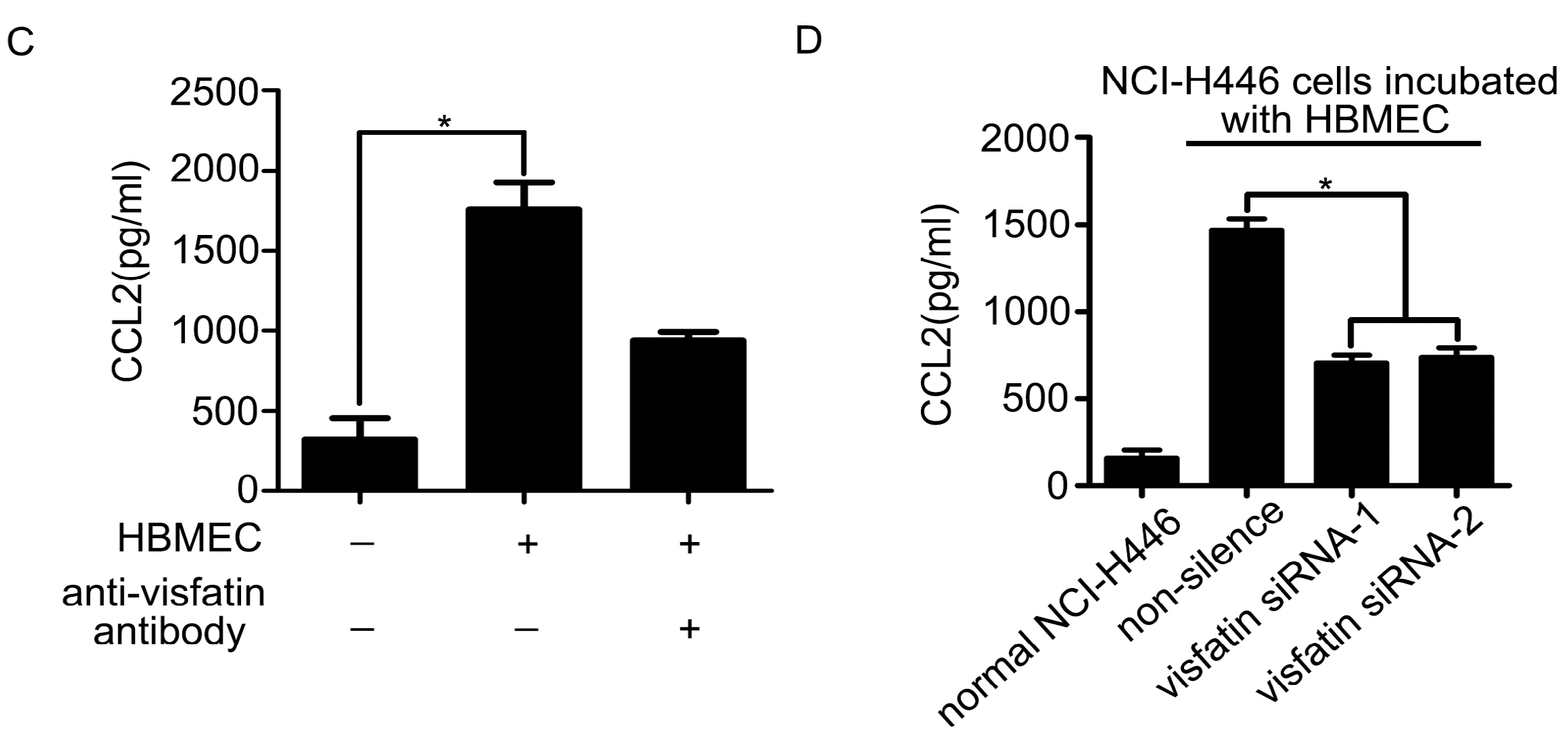
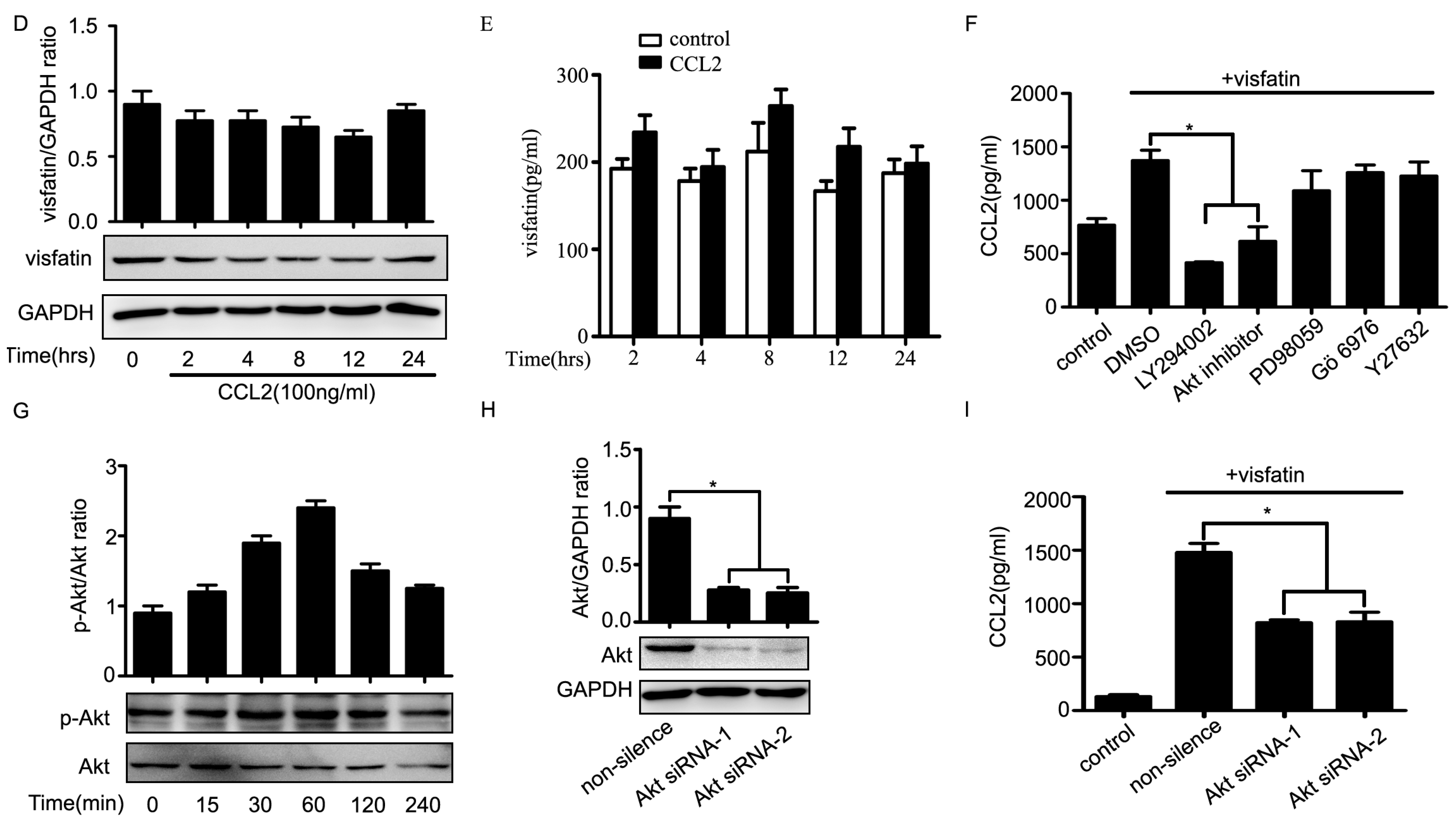
2.4. PI3K/Akt Signaling Was Involved in Visfatin-Induced the Upregulation of CCL2
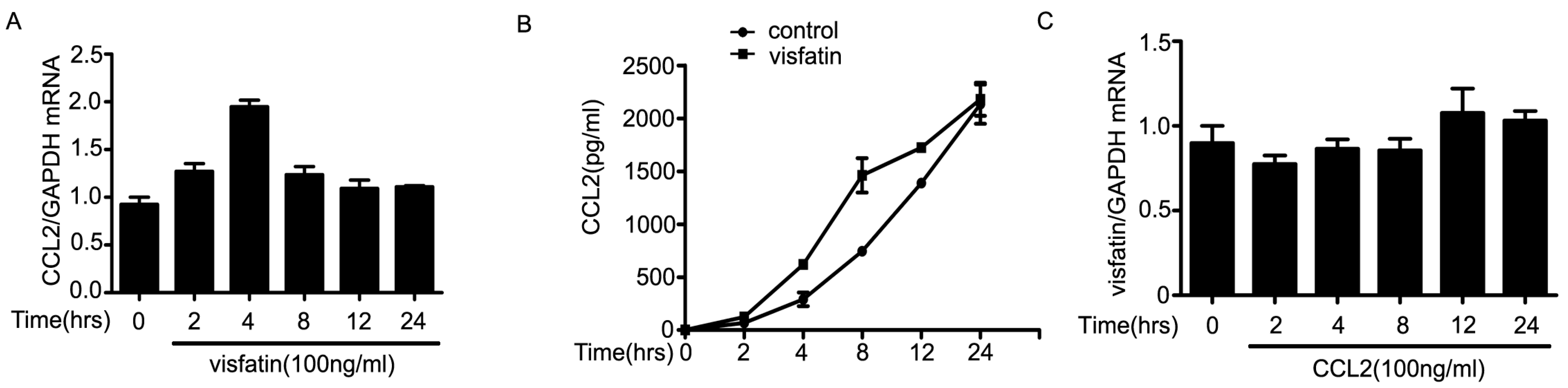
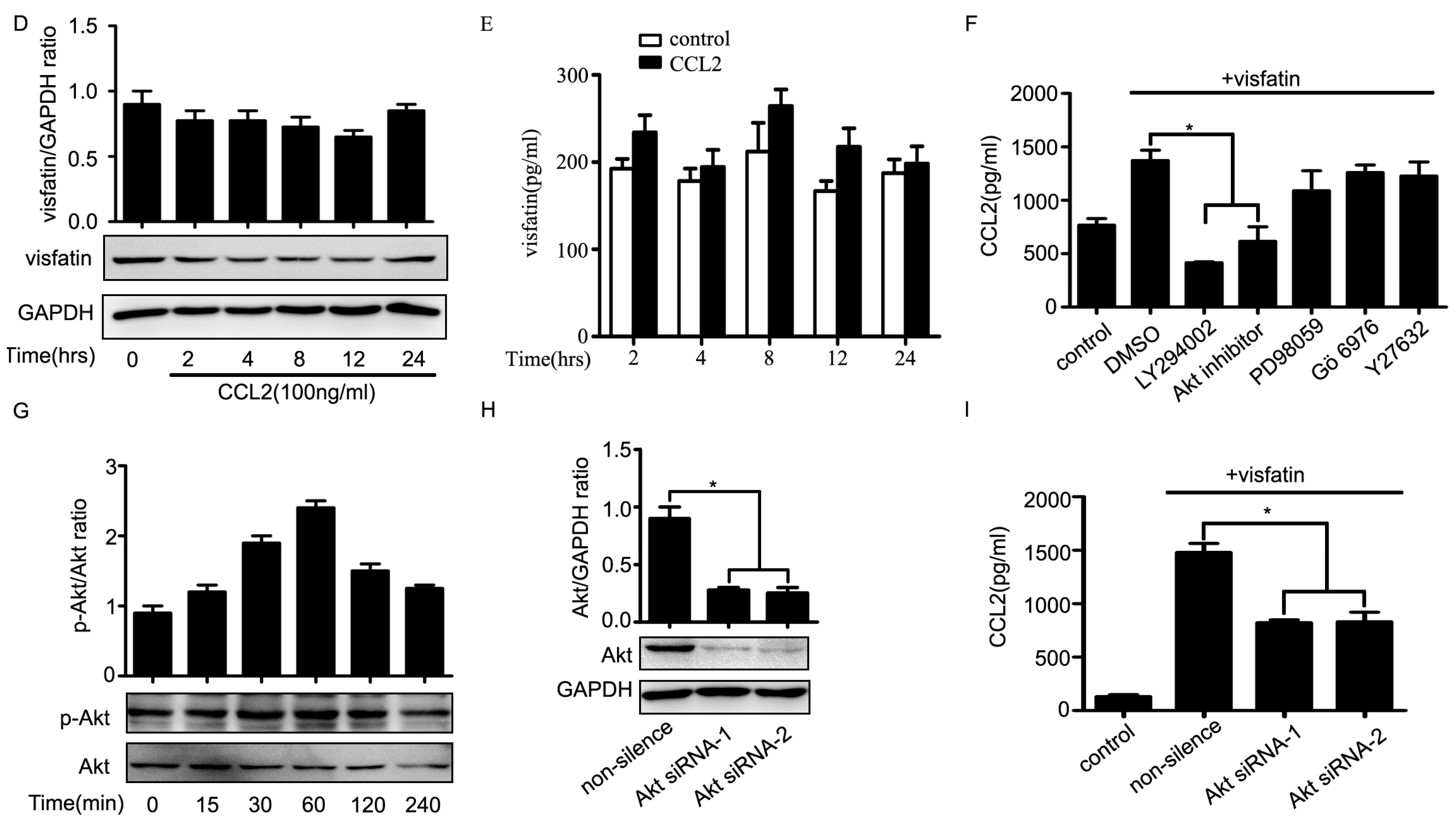
3. Discussion
4. Experimental Section
4.1. Patients and Specimens
4.2. Cell Lines and Cell Culture
4.3. Real-Time PCR
4.4. Cell Fractionation and Western Blot
4.5. Enzyme-Linked Immunosorbent Assay
4.6. Transendothelial Migration Assay
4.7. siRNA Transfection
4.8. HRP Flux Measurement
4.9. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Langer, C.J.; Mehta, M.P. Current management of brain metastases, with a focus on systemic options. J. Clin. Oncol. 2005, 23, 6207–6219. [Google Scholar] [CrossRef] [PubMed]
- Orr, F.W.; Wang, H.H.; Lafrenie, R.M.; Scherbarth, S.; Nance, D.M. Interactions between cancer cells and the endothelium in metastasis. J. Pathol. 2000, 190, 310–329. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Patabendige, A.A.K.; Dolman, D.E.M.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Langley, R.R.; Fidler, I.J. The seed and soil hypothesis revisited-the role of tumor-stroma interactions in metastasis to different organs. Int. J. Cancer 2011, 128, 2527–2535. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Zhao, W.D.; Tan, Z.M.; Fang, W.G.; Zhu, L.; Chen, Y.H. Involvement of Rho/ROCK signalling in small cell lung cancer migration through human brain microvascular endothelial cells. FEBS Lett. 2006, 580, 4252–4260. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Wang, C.; Zhang, Y.; Zhao, X.Y.; Huang, B.; Wu, P.F.; Li, Q.; Li, H.; Liu, Y.S.; Cao, L.Y.; et al. Elevated PLGF contributes to small-cell lung cancer brain metastasis. Oncogene 2013, 32, 2952–2962. [Google Scholar] [CrossRef] [PubMed]
- Curat, C.A.; Wegner, V.; Sengenes, C.; Miranville, A.; Tonus, C.; Busse, R.; Bouloumie, A. Macrophages in human visceral adipose tissue: Increased accumulation in obesity and a source of resistin and visfatin. Diabetologia 2006, 49, 744–747. [Google Scholar] [CrossRef] [PubMed]
- Fukuhara, A.; Matsuda, M.; Nishizawa, M.; Segawa, K.; Tanaka, M.; Kishimoto, K.; Matsuki, Y.; Murakami, M.; Ichisaka, T.; Murakami, H.; et al. Visfatin: A protein secreted by visceral fat that mimics the effects of insulin. Science 2005, 307, 426–430. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.P.; Oka, S.; Shao, D.; Hariharan, N.; Sadoshima, J. Nicotinamide phosphoribosyltransferase regulates cell survival through NAD+ synthesis in cardiac myocytes. Circ. Res. 2009, 105, 481–491. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.S.; Song, H.K.; Lee, M.H.; Ko, G.J.; Han, J.Y.; Han, S.Y.; Han, K.H.; Kim, H.K.; Cha, D.R. Visfatin is upregulated in type-2 diabetic rats and targets renal cells. Kidney Int. 2010, 78, 170–181. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.G.; Kim, E.O.; Jeong, B.R.; Min, Y.J.; Park, J.W.; Kim, E.S.; Namgoong, I.S.; Kim, Y.I.; Lee, B.J. Visfatin stimulates proliferation of MCF-7 human breast cancer cells. Mol. Cells 2010, 30, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Moschen, A.R.; Gerner, R.R.; Tilg, H. Pre-B cell colony enhancing factor/nampt/visfatin in inflammation and obesity-related disorders. Curr. Pharm. Des. 2010, 16, 1913–1920. [Google Scholar] [CrossRef] [PubMed]
- Preusser, M.; Capper, D.; Ilhan-Mutlu, A.; Berghoff, A.S.; Birner, P.; Bartsch, R.; Marosi, C.; Zielinski, C.; Mehta, M.P.; Winkler, F.; et al. Brain metastases: Pathobiology and emerging targeted therapies. Acta Neuropathol. 2012, 123, 205–222. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, I.; Fazakas, C.; Krizbai, I.A. In vitro models of the blood-brain barrier. Acta Neurobiol. Exp. 2011, 71, 113–128. [Google Scholar]
- Liu, Y.; Kosaka, A.; Ikeura, M.; Kohanbash, G.; Fellows-Mayle, W.; Snyder, L.A.; Okada, H. Premetastatic soil and prevention of breast cancer brain metastasis. Neuro-Oncology 2013, 15, 891–903. [Google Scholar] [CrossRef] [PubMed]
- Olarescu, N.C.; Ueland, T.; Lekva, T.; Dahl, T.B.; Halvorsen, B.; Aukrust, P.; Bollerslev, J. Adipocytes as a source of increased circulating levels of nicotinamide phosphoribosyltransferase/visfatin in active acromegaly. J. Clin. Endocr. Metab. 2012, 97, 1355–1362. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, D.; Denkins, Y.; Reiland, J.; Greiter-Wilke, A.; Galjour, J.; Murry, B.; Blust, J.; Roy, M. Brain-metastatic melanoma: A neurotrophic perspective. Pathol. Oncol. Res. 2003, 9, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.X.; Ngo, J.A.; Wetzel, M.D.; Marchetti, D. Heparanase mediates a novel mechanism in lapatinib-resistant brain metastatic breast cancer. Neoplasia 2015, 17, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Kafka, A.; Tomas, D.; Beros, V.; Pecina, H.I.; Zeljko, M.; Pecina-Slaus, N. Brain metastases from lung cancer show increased expression of DVL1, DVL3 and β-catenin and downregulation of E-cadherin. Int. J. Mol. Sci. 2014, 15, 10635–10651. [Google Scholar] [CrossRef] [PubMed]
- Lim, E.; Lin, N.U. Updates on the management of breast cancer brain metastases. Oncology 2014, 28, 572–578. [Google Scholar] [PubMed]
- Castrucci, W.A.; Knisely, J.P.S. An update on the treatment of cns metastases in small cell lung cancer. Cancer J. 2008, 14, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.C.; Yang, Y.H.; Su, J.H.; Chang, H.L.; Hou, M.F.; Yuan, S.S.F. High visfatin expression in breast cancer tissue is associated with poor survival. Cancer Epidemiol. Biomark. 2011, 20, 1892–1901. [Google Scholar] [CrossRef]
- Ntikoudi, E.; Kiagia, M.; Boura, P.; Syrigos, K.N. Hormones of adipose tissue and their biologic role in lung cancer. Cancer Treat. Rev. 2014, 40, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Okumura, S.; Sasaki, T.; Minami, Y.; Ohsaki, Y. Nicotinamide phosphoribosyltransferase: A potent therapeutic target in non-small cell lung cancer with epidermal growth factor receptor-gene mutation. J. Thorac. Oncol. 2012, 7, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Santidrian, A.F.; LeBoeuf, S.E.; Wold, E.D.; Ritland, M.; Forsyth, J.S.; Felding, B.H. Nicotinamide phosphoribosyltransferase can affect metastatic activity and cell adhesive functions by regulating integrins in breast cancer. DNA Repair 2014, 23, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.J.; Wu, C.S.; Lin, H.; Lee, I.T.; Wu, C.M.; Tseng, J.J.; Chou, M.M.; Sheu, W.H.H. Visfatin-induced expression of inflammatory mediators in human endothelial cells through the NF-κB pathway. Int. J. Obes. 2009, 33, 465–472. [Google Scholar] [CrossRef]
- Brentano, F.; Schorr, O.; Ospelt, C.; Stanczyk, J.; Gay, R.E.; Gay, S.; Kyburz, D. Pre-B cell colony-enhancing factor/visfatin, a new marker of inflammation in rheumatoid arthritis with proinflammatory and matrix-degrading activities. Arthritis Rheum. 2007, 56, 2829–2839. [Google Scholar] [CrossRef] [PubMed]
- Keklikoglou, I.; de Palma, M. Cancer metastasis risk after anti-macrophage therapy. Nature 2014, 515, 46–47. [Google Scholar] [CrossRef] [PubMed]
- Lebel-Haziv, Y.; Meshel, T.; Soria, G.; Yeheskel, A.; Mamon, E.; Ben-Baruch, A. Breast cancer: Coordinated regulation of CCL2 secretion by intracellular glycosaminoglycans and chemokine motifs. Neoplasia 2014, 16, 723–740. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Patel, L.; Pienta, K.J. Cc chemokine ligand 2 (CCL2) promotes prostate cancer tumorigenesis and metastasis. Cytokine Growth Factor Rev. 2010, 21, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Stamatovic, S.M.; Dimitrijevic, O.B.; Keep, R.F.; Andjelkovic, A.V. Protein kinase c α-RhoA cross-talk in CCL2-induced alterations in brain endothelial permeability. J. Biol. Chem. 2006, 281, 8379–8388. [Google Scholar] [CrossRef] [PubMed]
- Stamatovic, S.M.; Shakui, P.; Keep, R.F.; Moore, B.B.; Kunkel, S.L.; van Rooijen, N.; Andjelkovic, A.V. Monocyte chemoattractant protein-1 regulation of blood-brain barrier permeability. J. Cerebr. Blood Flow Metab. 2005, 25, 593–606. [Google Scholar] [CrossRef]
- Adya, R.; Tan, B.K.; Chen, J.; Randeva, H.S. Pre-B cell colony enhancing factor (PBEF)/visfatin induces secretion of MCP-1 in human endothelial cells: Role in visfatin-induced angiogenesis. Atherosclerosis 2009, 205, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.W.; Qiao, S.B.; Yuan, J.S.; Liu, D.Q. Visfatin stimulates production of monocyte chemotactic protein-1 and interleukin-6 in human vein umbilical endothelial cells. Horm. Metab. Res. 2009, 41, 281–286. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, T.; Miao, Z.; Jiang, J.; Yuan, S.; Fang, W.; Li, B.; Chen, Y. Visfatin Mediates SCLC Cells Migration across Brain Endothelial Cells through Upregulation of CCL2. Int. J. Mol. Sci. 2015, 16, 11439-11451. https://doi.org/10.3390/ijms160511439
Liu T, Miao Z, Jiang J, Yuan S, Fang W, Li B, Chen Y. Visfatin Mediates SCLC Cells Migration across Brain Endothelial Cells through Upregulation of CCL2. International Journal of Molecular Sciences. 2015; 16(5):11439-11451. https://doi.org/10.3390/ijms160511439
Chicago/Turabian StyleLiu, Tingting, Ziwei Miao, Jiusheng Jiang, Shuai Yuan, Wengang Fang, Bo Li, and Yuhua Chen. 2015. "Visfatin Mediates SCLC Cells Migration across Brain Endothelial Cells through Upregulation of CCL2" International Journal of Molecular Sciences 16, no. 5: 11439-11451. https://doi.org/10.3390/ijms160511439
APA StyleLiu, T., Miao, Z., Jiang, J., Yuan, S., Fang, W., Li, B., & Chen, Y. (2015). Visfatin Mediates SCLC Cells Migration across Brain Endothelial Cells through Upregulation of CCL2. International Journal of Molecular Sciences, 16(5), 11439-11451. https://doi.org/10.3390/ijms160511439