
De Novo Assembly and Characterization of the Transcriptome of the Chinese Medicinal Herb, Gentiana rigescens



Abstract
:1. Introduction
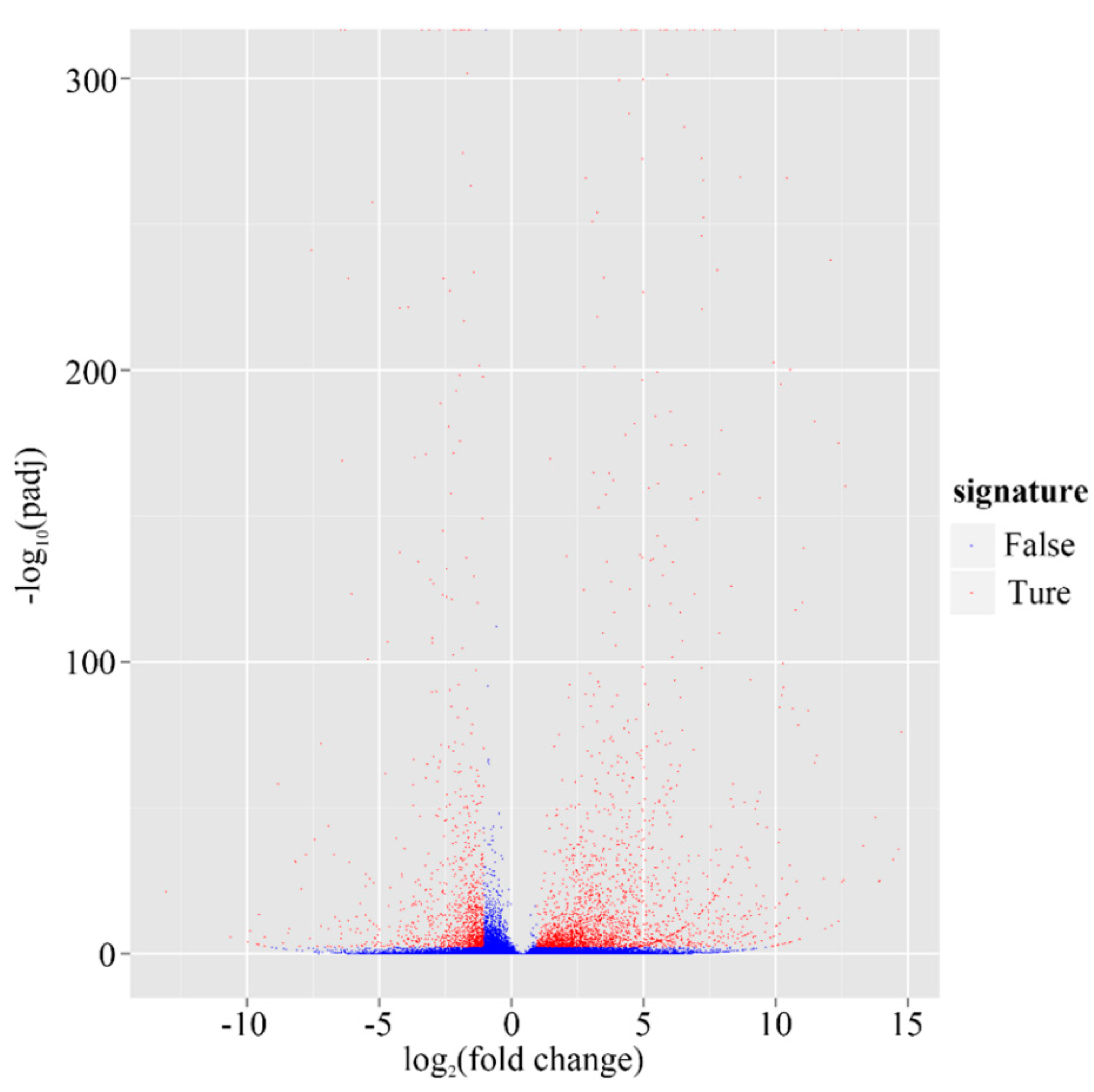
2. Results and Discussion
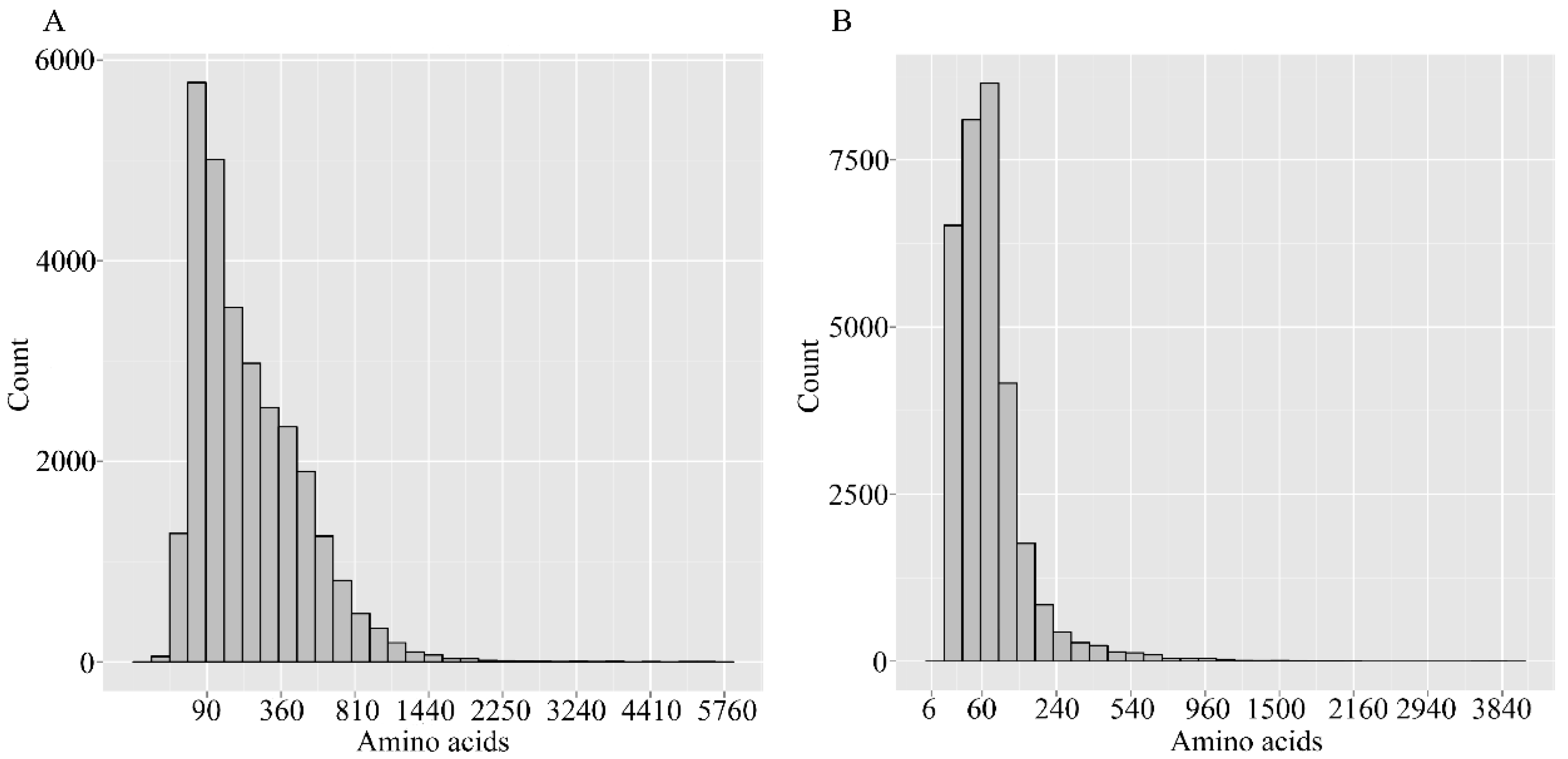
2.1. Sequencing and Assembly

{kind=link}
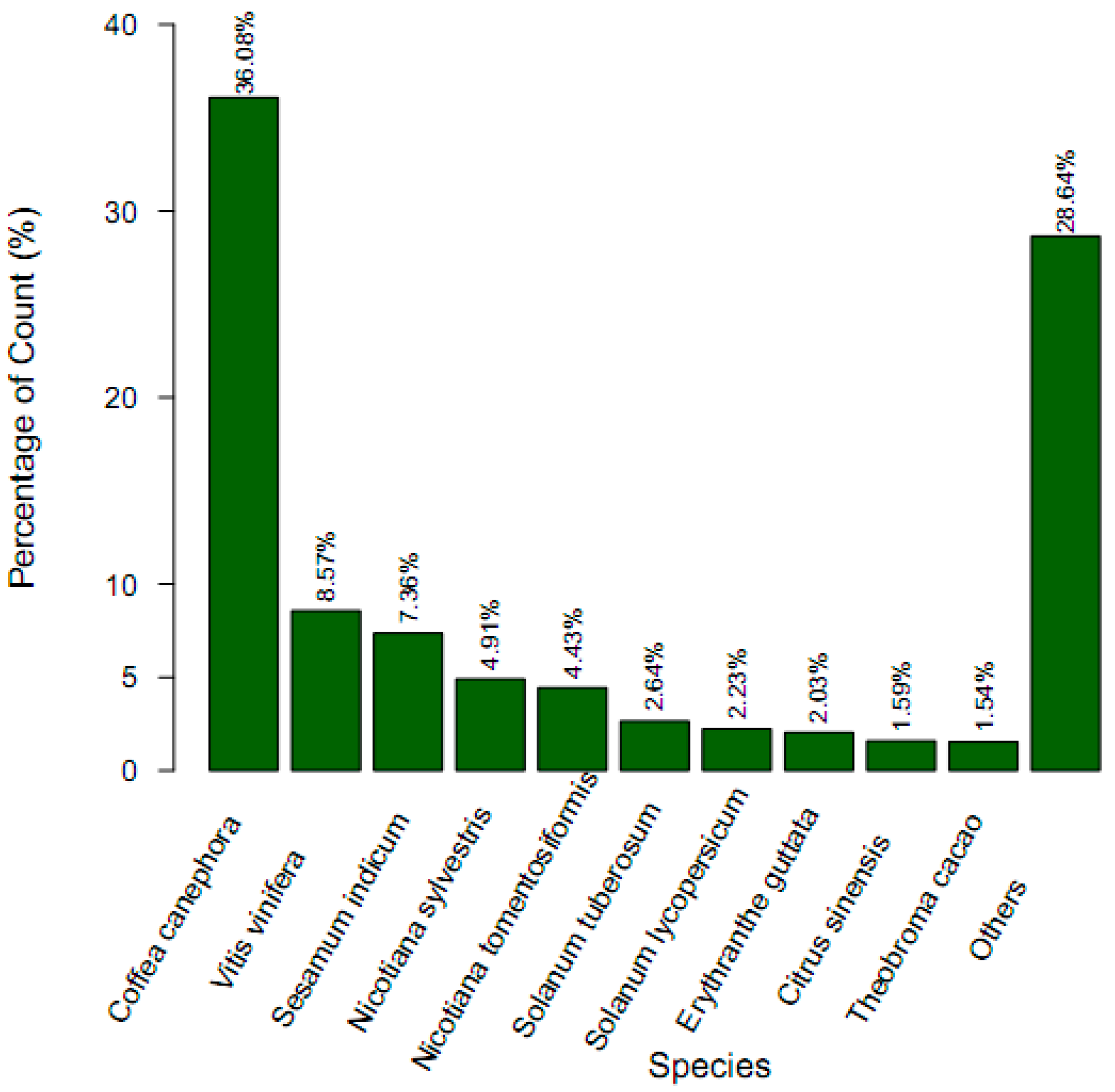{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Item | Sample | Raw Reads | Clean Reads | Clean Bases (G) | Error (%) | Q20 (%) | Q30 (%) | GC (%) |
|---|---|---|---|---|---|---|---|---|
| Leaf | Leaf_1 | 57,802,913 | 56,289,486 | 5.63 | 0.03 | 97.97 | 93.09 | 43.00 |
| Leaf_2 | 57,802,913 | 56,289,486 | 5.63 | 0.03 | 97.48 | 92.61 | 43.04 | |
| Root | Root_1 | 53,933,882 | 50,596,096 | 5.06 | 0.03 | 97.02 | 90.79 | 43.23 |
| Root_2 | 53,933,882 | 50,596,096 | 5.06 | 0.03 | 96.59 | 90.34 | 43.31 |
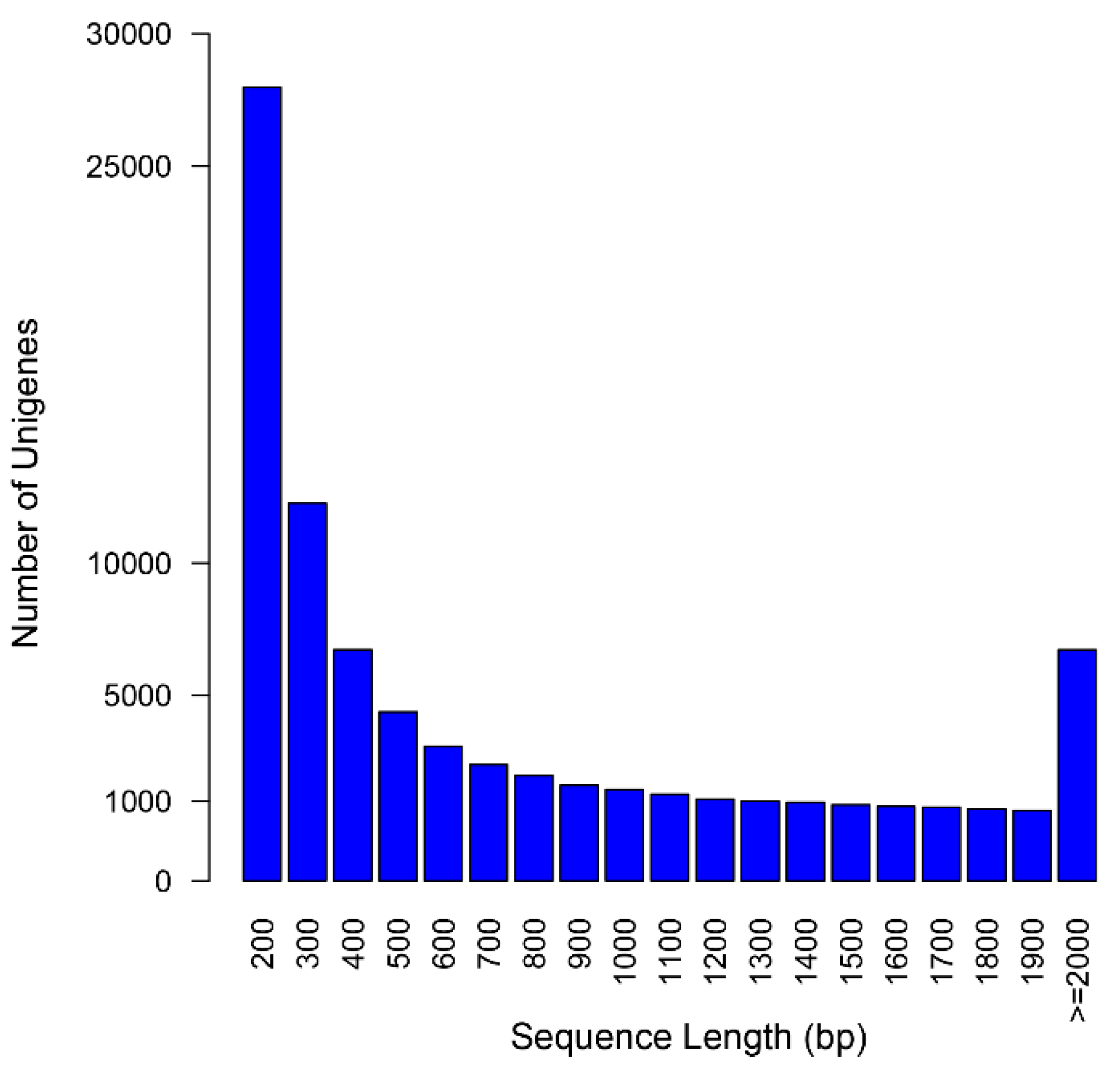
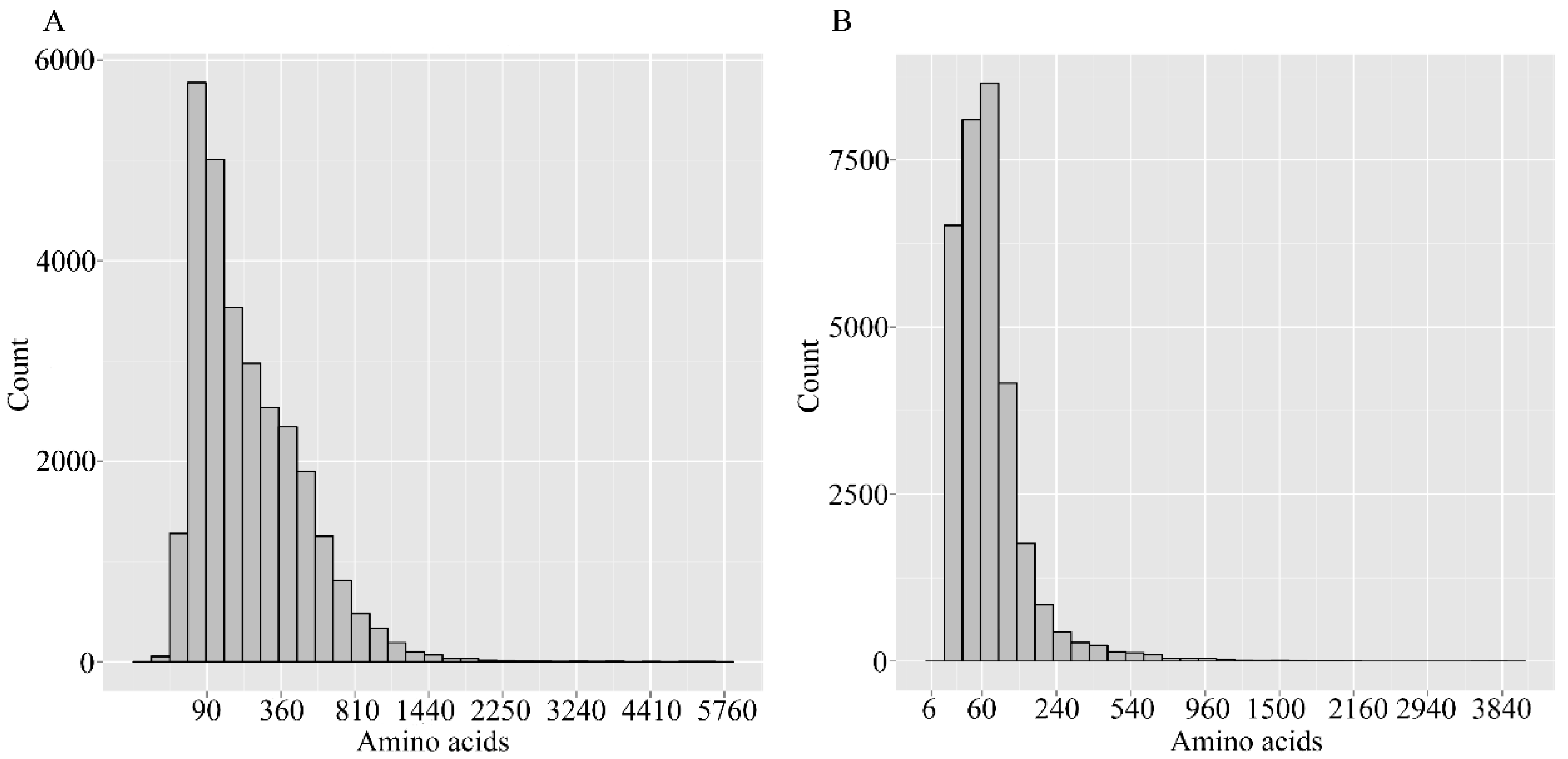
2.2. Assembly Assessment
| Item | Contigs | Unigenes |
|---|---|---|
| Total number | 189,576 | 76,717 |
| Total length (bp) | 228,624,912 | 57,734,637 |
| Mean length (bp) | 1206 | 753 |
| N50 (bp) | 1996 | 1384 |
| GC content (%) | 39.7 | 39.5 |
| Number of length ≥ 500 bp | 120,525 | 29,795 |
| Number of length ≥ 1000 bp | 81,919 | 16,332 |
| Reads mapping rate (%) | 92.72 | - |
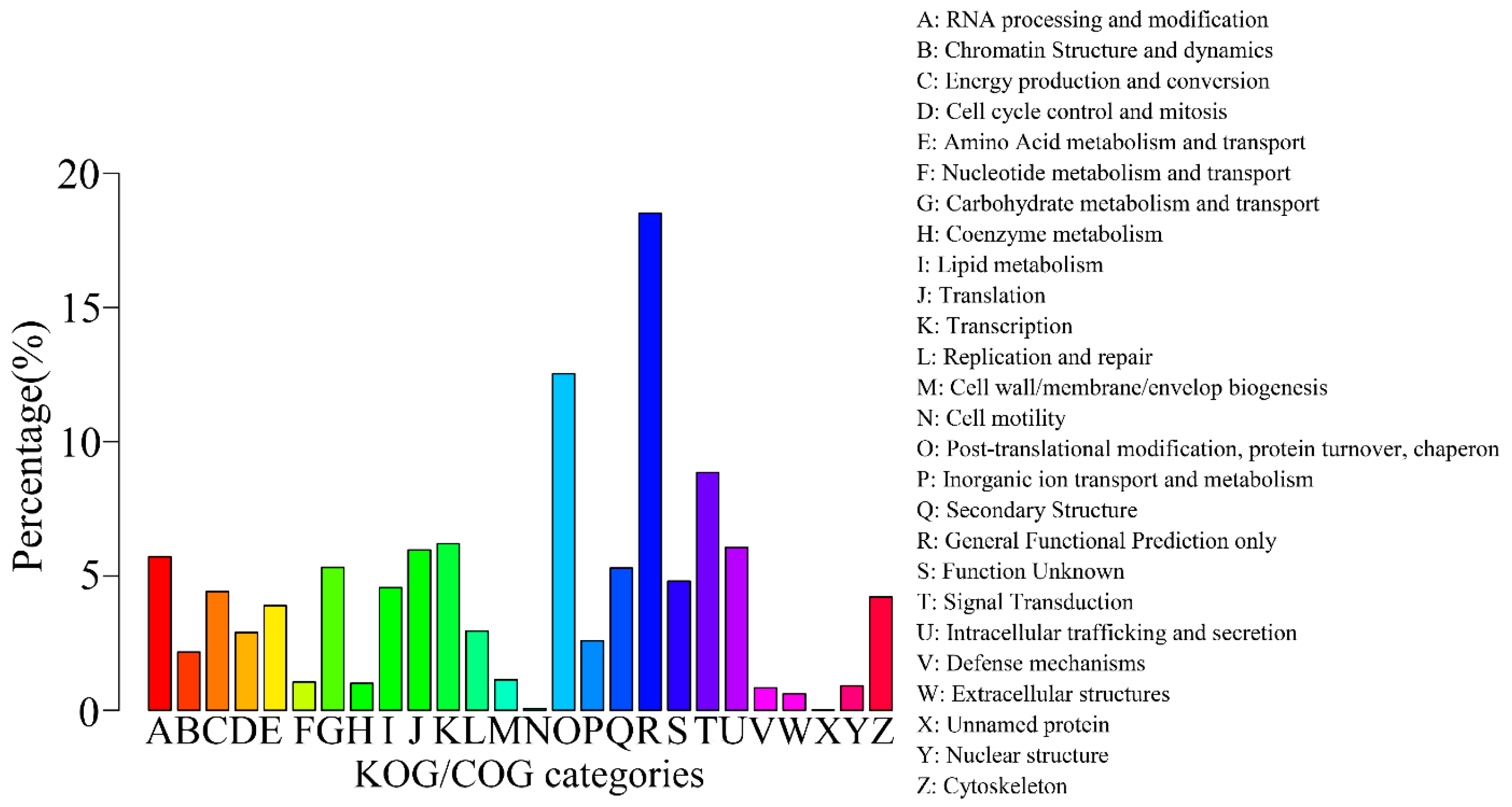
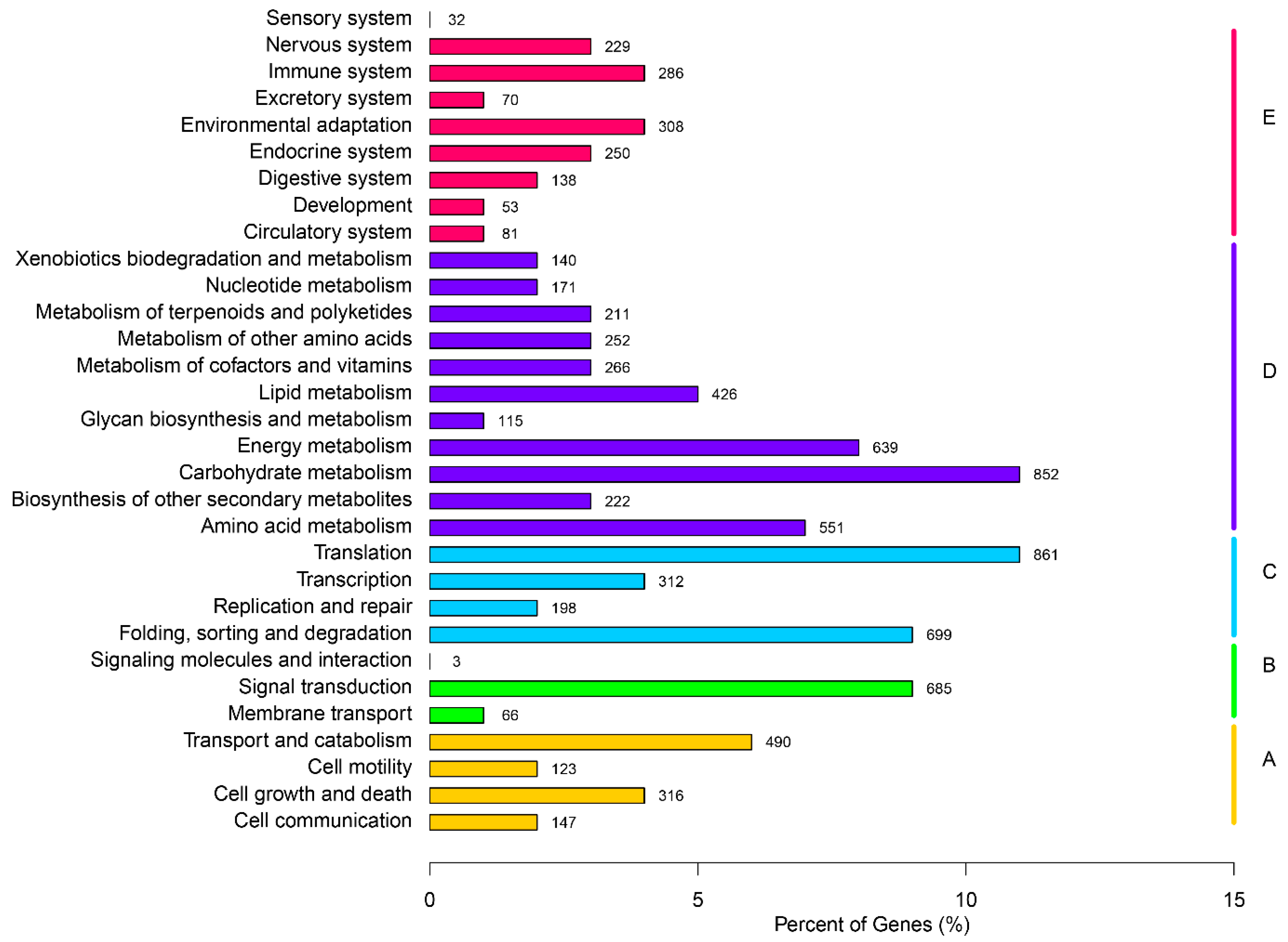
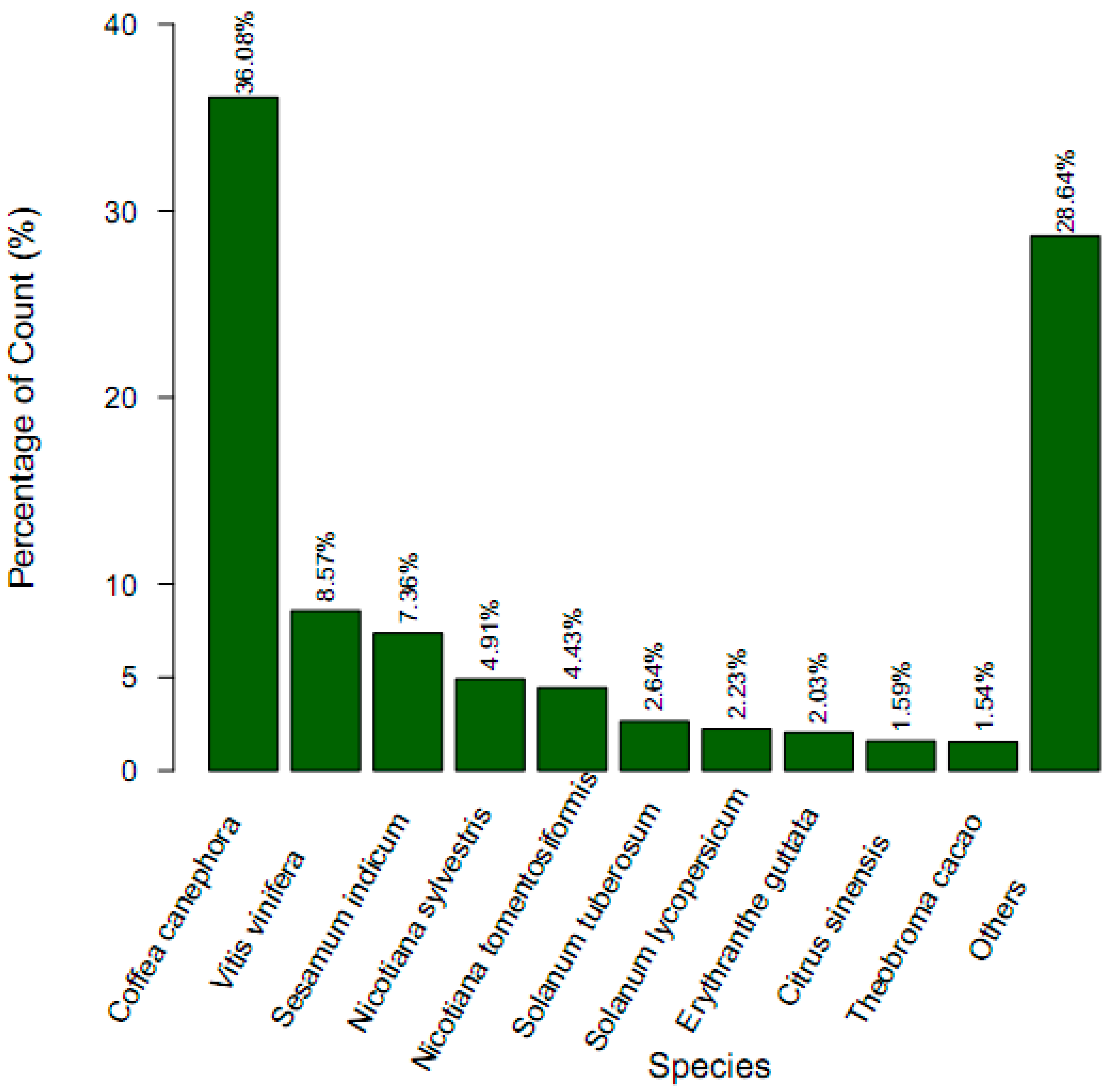
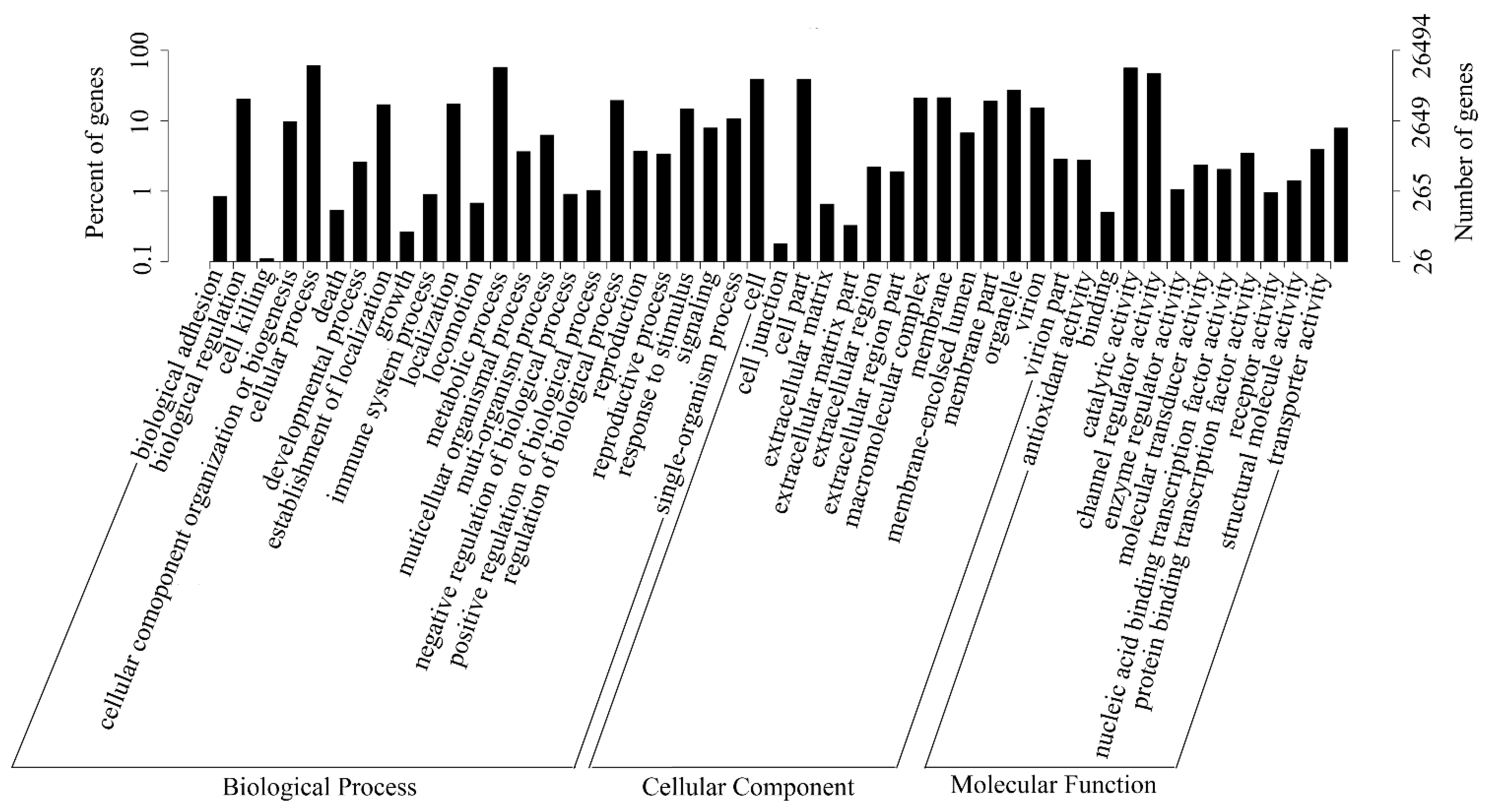
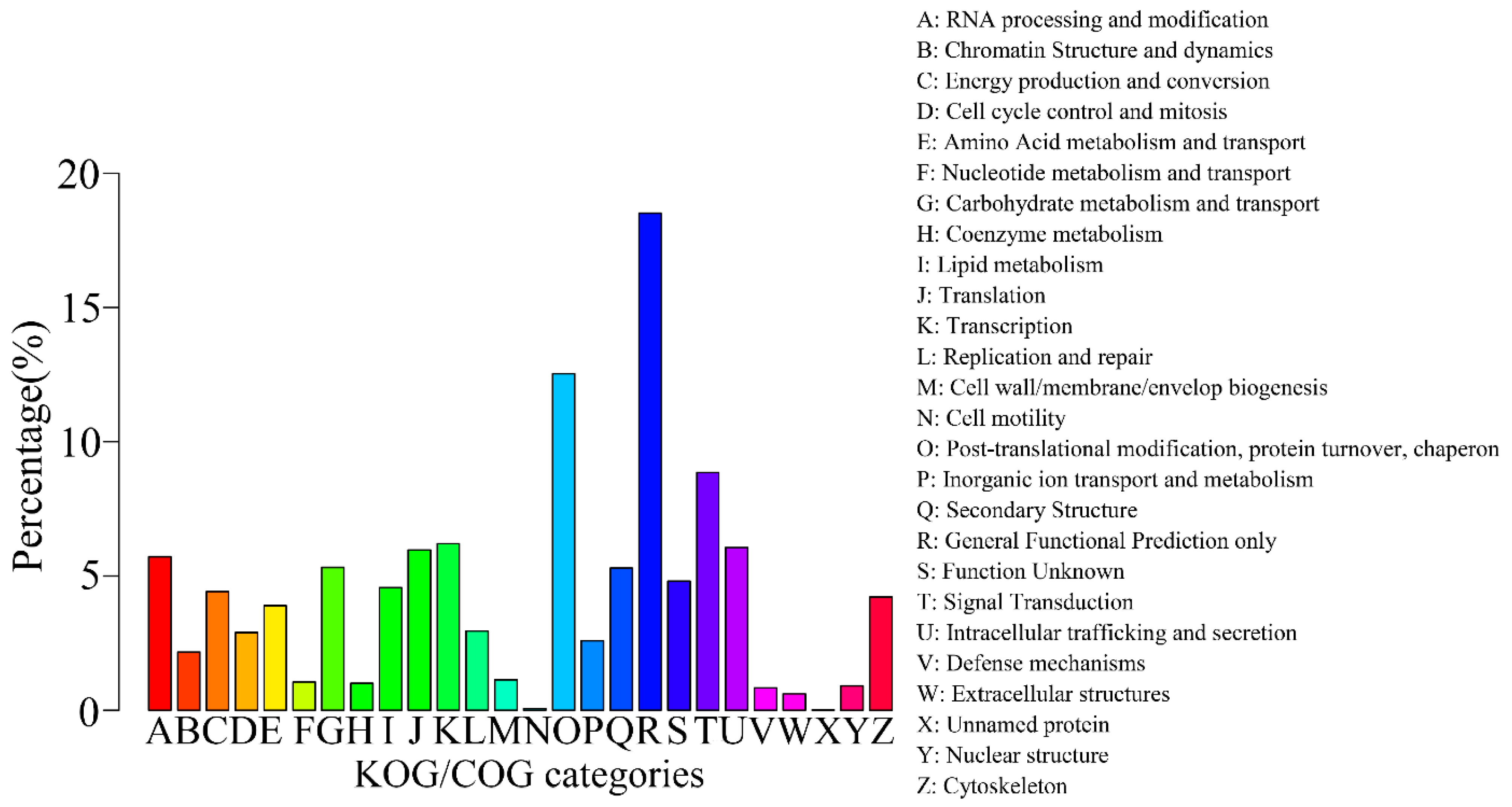
2.3. Gene Function Annotation and Classification
| Item | Number of Unigenes (n) | Percentage (%) |
|---|---|---|
| Annotated in NR | 26,686 | 34.78 |
| Annotated in NT | 8158 | 10.64 |
| Annotated in TAIR10 | 24,371 | 31.77 |
| Annotated in KEGG | 7998 | 10.43 |
| Annotated in SwissProt | 18,627 | 24.28 |
| Annotated in PFAM | 23,287 | 30.35 |
| Annotated in GO | 26,494 | 34.53 |
| Annotated in KOG/COG | 10,524 | 13.72 |
| Annotated in all Databases | 3019 | 3.94 |
| Annotated in at least one Database | 33,855 | 44.13 |
| Total queries/unigenes | 76,717 | 100 |
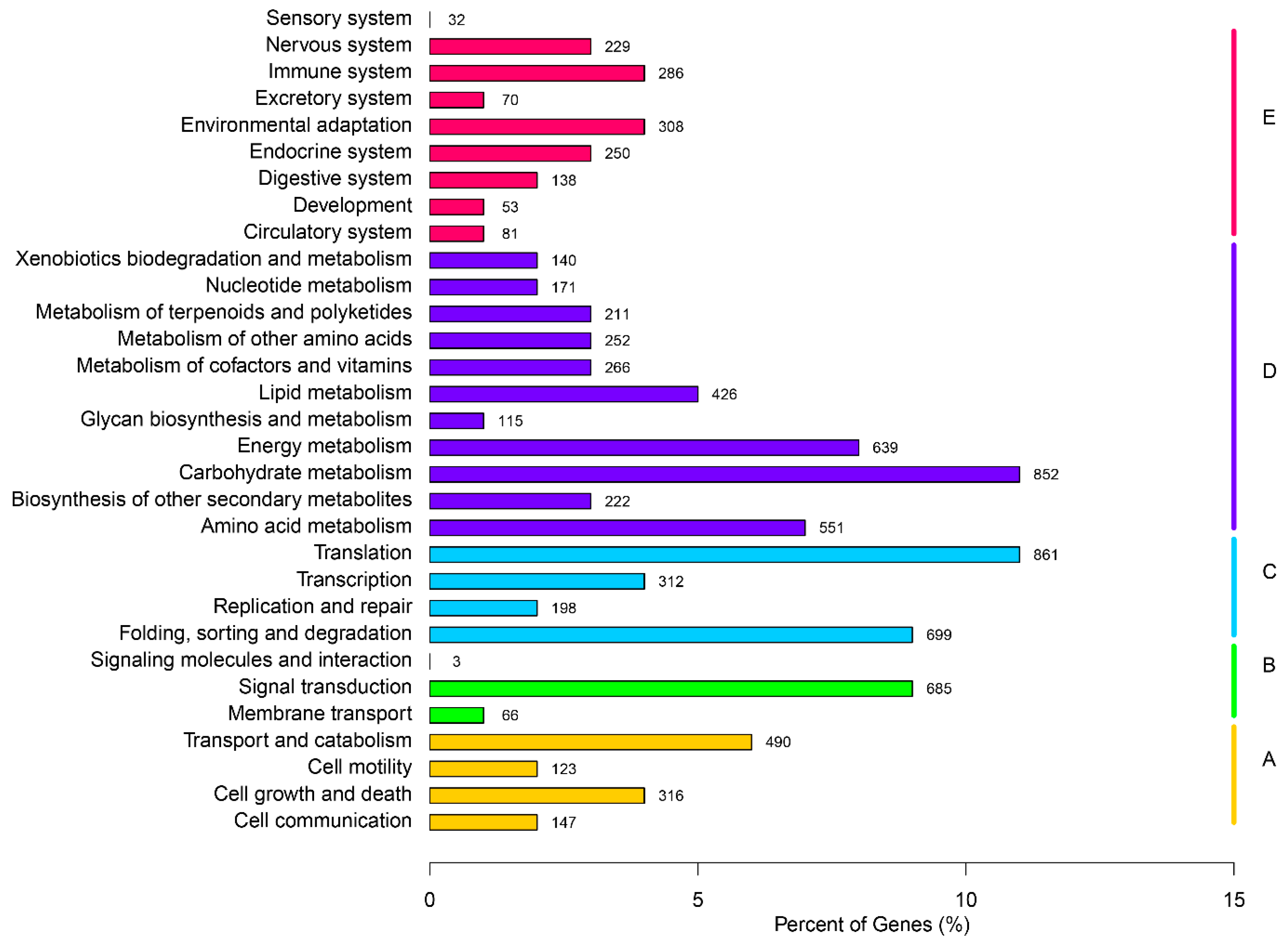
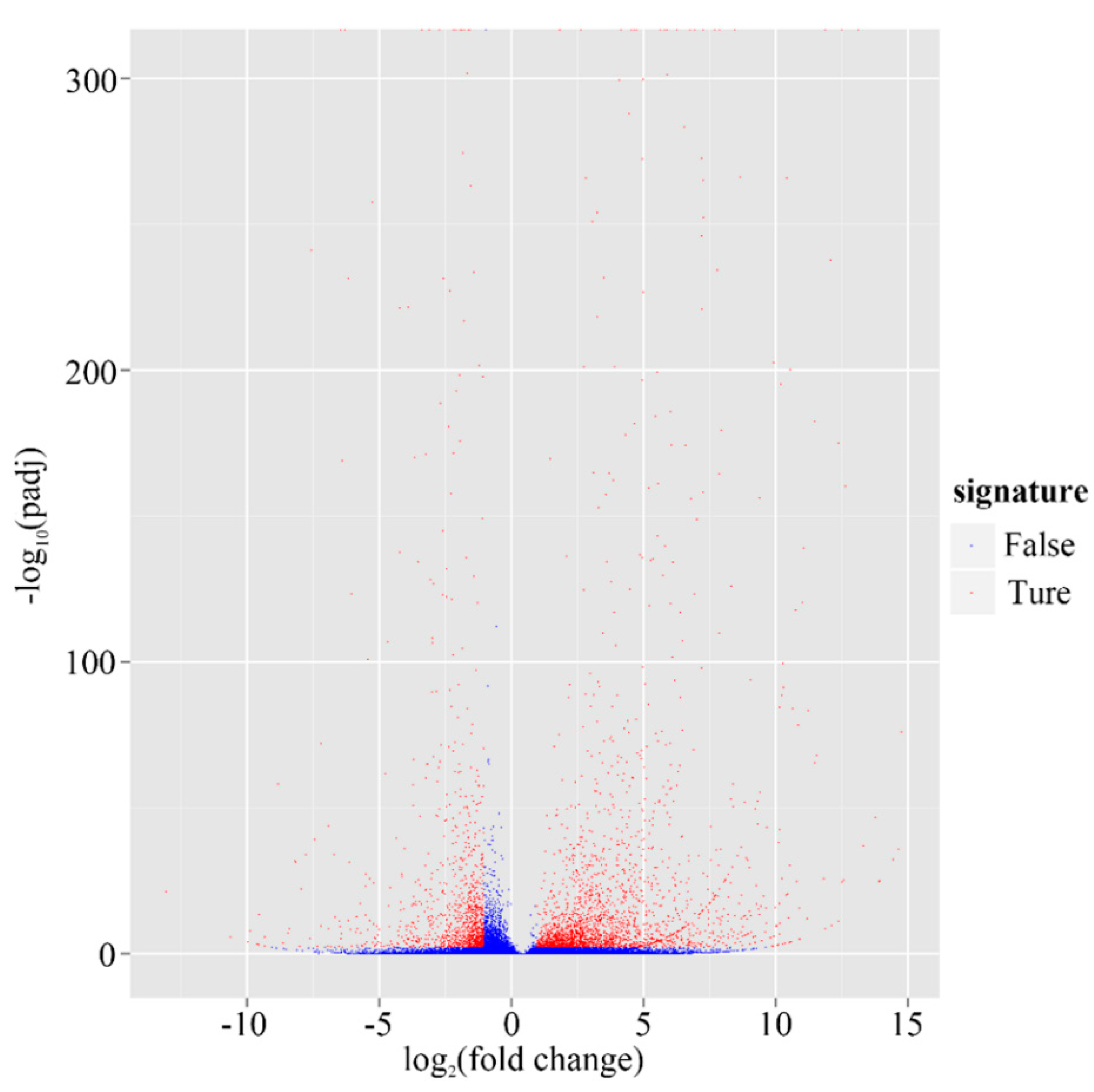
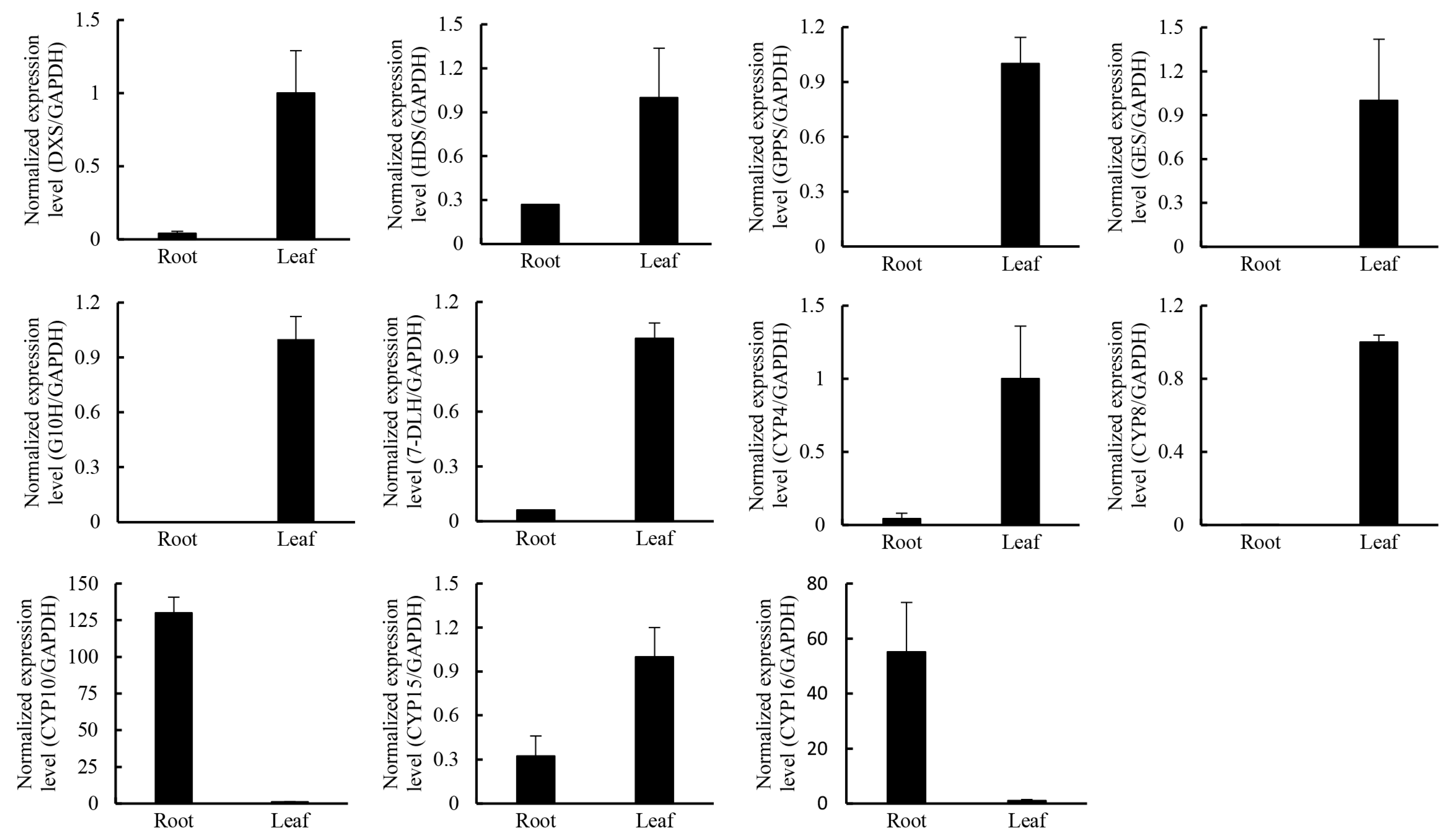
2.4. Putative Genes Involved in the Terpenoid Backbone Biosynthesis and Gentiopicroside Biosynthetic Pathways
| Pathway | Gene Name | Unigene | RPKM in Leaf | RPKM in Root |
|---|---|---|---|---|
| MVA | AACT1 | comp81670_c0 | 50.65 | 52.76 |
| AACT2 | comp86403_c0 | 13.16 | 38.43 | |
| HMGS | comp1196622_c0 | 0.00 | 0.49 | |
| HMGR1 | comp87249_c0 | 27.14 | 3.53 | |
| HMGR2 | comp92954_c0 | 35.56 | 17.08 | |
| HMGR3 | comp4296_c0 | 0.25 | 0.57 | |
| HMGR4 | comp25979_c0 | 0.57 | 0.04 | |
| HMGR5 | comp114241_c0 | 5.72 | 0.00 | |
| MK | comp83300_c0 | 20.74 | 16.19 | |
| PMK1 | comp371052_c0 | 0.55 | 0.48 | |
| PMK2 | comp82309_c1 | 2.21 | 1.23 | |
| PMK3 | comp82309_c0 | 3.94 | 2.21 | |
| PMK4 | comp92698_c0 | 8.08 | 11.20 | |
| MVD1 | comp86107_c0 | 51.01 | 39.76 | |
| MVD2 | comp73189_c0 | 0.55 | 0.58 | |
| IDI1 | comp81822_c0 | 114.79 | 89.95 | |
| IDI2 | comp67360_c0 | 37.65 | 31.50 | |
| IDI3 | comp92050_c0 | 49.18 | 2.79 | |
| MEP | DXS1 * | comp87916_c0 | 45.34 | 1.45 |
| DXS2 | comp89290_c0 | 10.85 | 3.64 | |
| DXS3 | comp93517_c0 | 48.69 | 35.56 | |
| DXR | comp92087_c3 | 123.96 | 107.04 | |
| MCT | comp67067_c0 | 36.76 | 8.28 | |
| MCS | comp91375_c0 | 56.61 | 36.13 | |
| HDS * | comp94424_c0 | 151.28 | 77.99 | |
| HDR1 | comp87777_c0 | 114.07 | 97.97 | |
| HDR2 | comp509208_c0 | 0.00 | 0.91 | |
| HDR3 | comp1116482_c0 | 0.00 | 0.36 |
| Gene Name | Unigene | RPKMs in Leaf | RPKMs in Root |
|---|---|---|---|
| GPPS1 * | comp57663_c0 | 47.61 | 0.04 |
| GPPS2 | comp79818_c0 | 53.25 | 8.76 |
| GES * | comp45416_c0 | 66.71 | 0.06 |
| G10H | comp95013_c1 | 304.45 | 1075.00 |
| G10H * | comp59018_c0 | 128.37 | 0.15 |
| G10H | comp67411_c0 | 6.01 | 10.68 |
| G10H | comp84881_c0 | 41.33 | 89.38 |
| G10H | comp74631_c0 | 31.33 | 70.85 |
| G10H | comp64598_c0 | 15.04 | 42.75 |
| G10H | comp42518_c0 | 3.07 | 13.76 |
| G10H | comp89824_c0 | 14.26 | 28.73 |
| G10H | comp67522_c0 | 11.48 | 15.46 |
| G10H | comp92644_c0 | 164.26 | 389.80 |
| G10H | comp67745_c0 | 13.73 | 25.95 |
| G10H | comp77398_c0 | 4.41 | 7.13 |
| G10H | comp51247_c0 | 0.00 | 2.77 |
| G10H | comp67165_c0 | 16.78 | 27.36 |
| G10H | comp67799_c0 | 0.00 | 0.90 |
| G10H | comp63189_c0 | 0.10 | 1.55 |
| G10H | comp76700_c0 | 1.03 | 2.28 |
| G10H | comp42518_c0 | 3.07 | 13.76 |
| 8HGO | comp93669_c0 | 161.97 | 2.80 |
| 8HGO | comp53753_c1 | 81.29 | 1.37 |
| 8HGO | comp53753_c2 | 121.86 | 0.92 |
| 8HGO | comp76718_c0 | 65.89 | 77.05 |
| 8HGO | comp90961_c0 | 5.52 | 2.34 |
| 8HGO | comp92998_c0 | 219.14 | 245.73 |
| SLS | comp94595_c0 | 504.62 | 20.96 |
| SLS | comp94064_c5 | 368.11 | 50.11 |
| SLS | comp84511_c0 | 27.90 | 35.43 |
| SLS | comp81016_c0 | 0.59 | 0.46 |
| SLS | comp85876_c0 | 318.25 | 345.13 |
| SLS | comp67629_c0 | 2.53 | 3.00 |
| SLS | comp54852_c0 | 0.08 | 1.74 |
| SLS | comp55055_c0 | 0.56 | 2.47 |
| SLS | comp61732_c0 | 0.75 | 2.06 |
| SLS | comp93282_c0 | 143.24 | 185.59 |
| SLS | comp281520_c0 | 0.49 | 0.75 |
| SLS | comp87446_c0 | 22.83 | 39.78 |
| SLS | comp41718_c0 | 0.09 | 4.01 |
| SLS | comp167742_c0 | 0.43 | 1.74 |
| SLS | comp94107_c0 | 121.99 | 222.06 |
| SLS | comp49781_c0 | 0.00 | 0.70 |
| SLS | comp90874_c0 | 14.95 | 24.36 |
| SLS | comp212851_c0 | 1.23 | 0.83 |
| SLS | comp73409_c0 | 0.00 | 0.72 |
| SLS | comp87446_c0 | 22.83 | 39.78 |
| SLS | comp73685_c0 | 0.95 | 2.11 |
| SLS | comp76988_c0 | 4.19 | 4.57 |
| SLS | comp103080_c0 | 4.73 | 6.98 |
| SLS | comp81659_c0 | 3.97 | 6.59 |
| IS | comp85292_c0 | 64.52 | 0.00 |
| IO | comp84741_c0 | 361.42 | 0.00 |
| 7-DLGT | comp82018_c0 | 65.59 | 0.00 |
| 7-DLH * | comp94064_c5 | 368.11 | 50.11 |
| CYP1 | comp84741_c0 | 361.42 | 0.00 |
| CYP2 | comp89478_c0 | 33.35 | 2.23 |
| CYP3 | comp108293_c0 | 10.24 | 0.09 |
| CYP4 * | comp92783_c1 | 27.39 | 8.86 |
| CYP5 | comp80146_c0 | 17.02 | 3.45 |
| CYP6 | comp83496_c0 | 46.67 | 160.59 |
| CYP7 | comp94595_c0 | 504.62 | 20.96 |
| CYP8 * | comp97650_c0 | 29.44 | 2.77 |
| CYP9 | comp90225_c0 | 16.82 | 1.16 |
| CYP10 * | comp80525_c0 | 0.06 | 15.35 |
| CYP11 | comp92026_c0 | 60.41 | 4.19 |
| CYP12 | comp68870_c0 | 3.04 | 31.70 |
| CYP13 | comp85931_c0 | 59.63 | 6.58 |
| CYP14 | comp79921_c0 | 75.72 | 33.66 |
| CYP15 * | comp80492_c0 | 19.18 | 6.02 |
| CYP16 * | comp95479_c0 | 99.29 | 14.68 |
| CYP17 | comp90874_c0 | 14.95 | 24.36 |
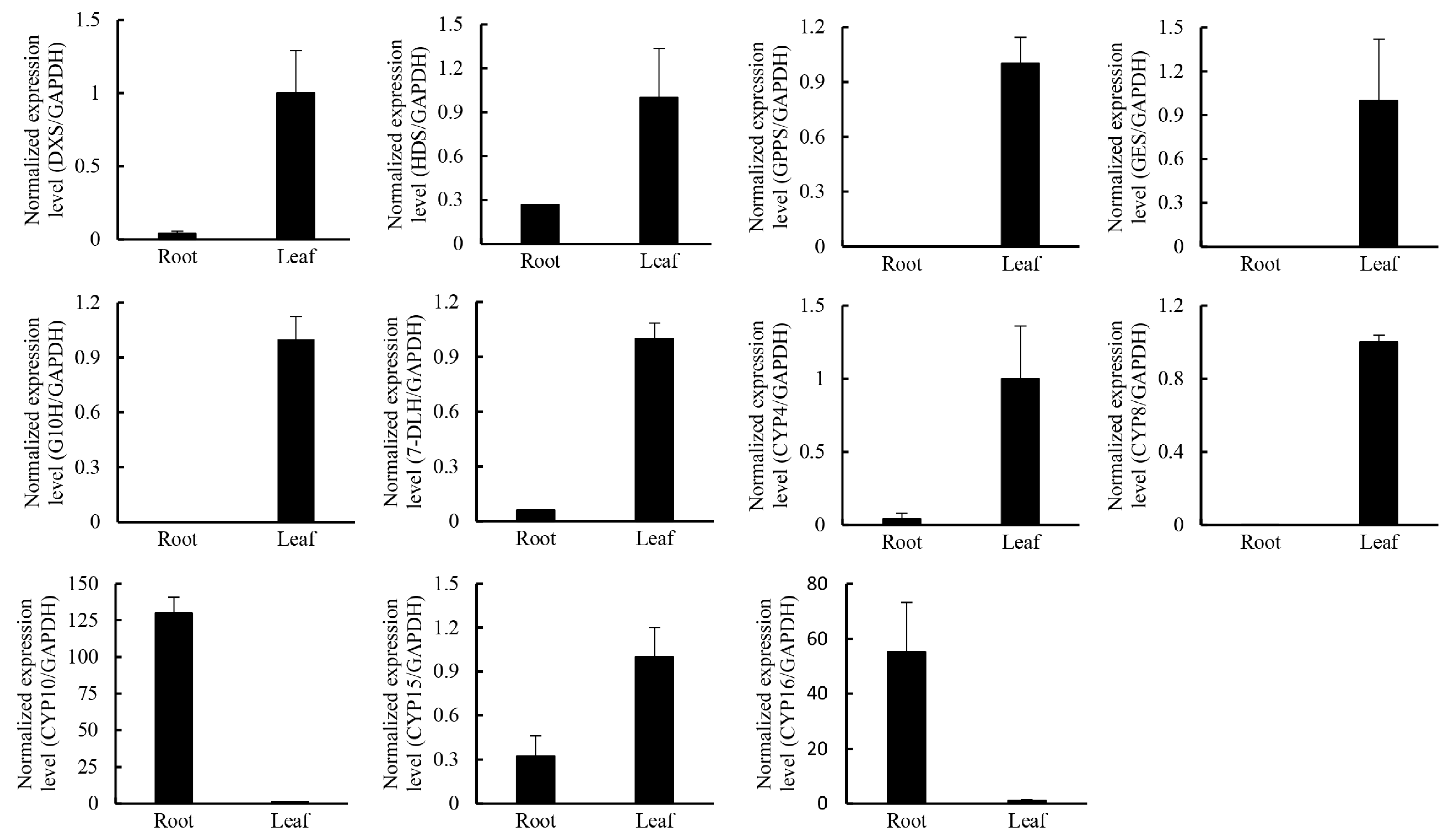

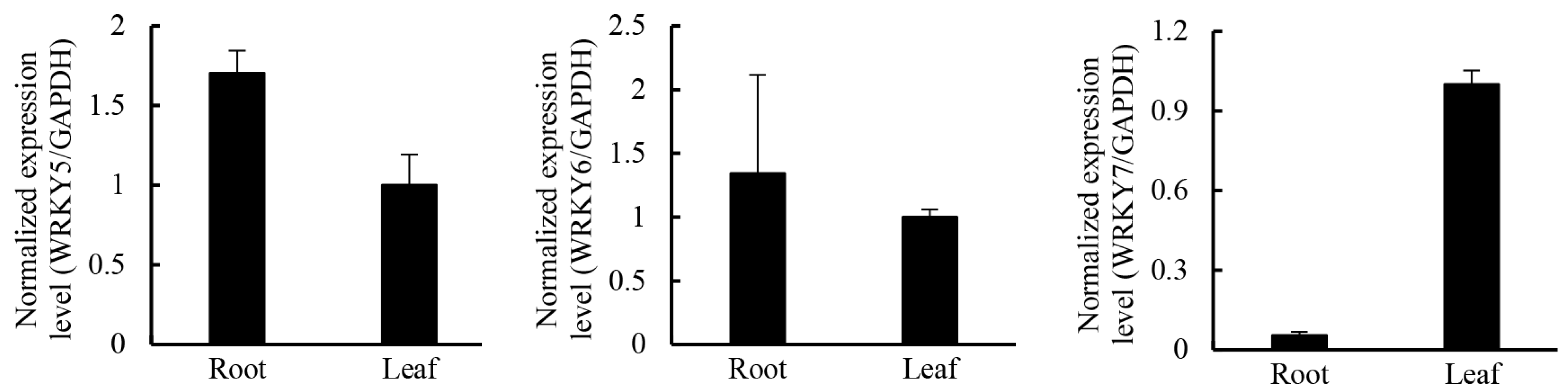
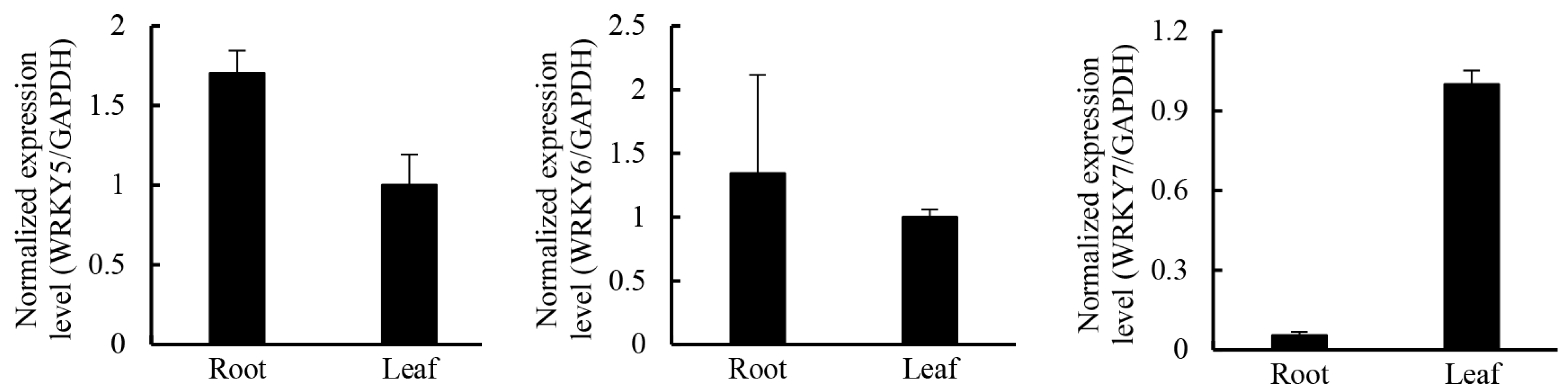
2.5. Candidate Transcription Factors Involved in Regulating the Terpenoid Biosynthetic Pathway
| TF Family | Number of Genes Detected | Up-Regulated in Leaves (log2(Fold_Change) > 2) | Up-Regulated in Roots (log2(Fold_Change) > 2) |
|---|---|---|---|
| HLH | 349 | 26 | 5 |
| AP2-EREBP | 172 | 20 | 4 |
| WRKY | 141 | 17 | 1 |
| MYB | 129 | 7 | 2 |
| bZIP | 115 | 3 | 4 |
| GRAS | 94 | 7 | 1 |
| Total | 1000 | 80 | 17 |
3. Experimental Section
3.1. Plant Materials and RNA Isolation
3.2. Transcriptome Sample Preparation for Sequencingc

3.3. Data Filtering
3.4. Transcriptome Assembly and Contamination Sequences Filtering
3.5. Gene Functional Annotation
3.6. Differential Expression Analysis
3.7. KEGG Enrichment Analysis
3.8. Real-Time PCR Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Yang, Y.; Shao, A.; Jin, H.; Ou, X.; Chen, M.; Liu, D.; Huang, L. Variation of botanical morphologic characteristics between wild and cultivated populations of Gentiana rigescens in Yunnan-Guizhou Plateau. Chin. Tradit. Herb. Drugs 2012, 43, 1604–1610. [Google Scholar]
- Chinanews. Available online: http://www.chinanews.com/gn/2013/07-12/5037538.shtml (accessed on 13 July 2013).
- Zheng, P.; Zhang, K.; Wang, Z. Genetic diversity and gentiopicroside content of four Gentiana species in China revealed by ISSR and HPLC methods. Biochem. Syst. Ecol. 2011, 39, 704–710. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, Y.; Yang, T.; Jin, H.; Zhang, J. Use of gibberellic acid to overcome the allelopathic effect of a range of species on the germination of seeds of Gentiana rigescens, a medicinal herb. Seed Sci. Technol. 2012, 40, 443–447. [Google Scholar] [CrossRef]
- Keller, F. Gentiopicroside is located in the vacuoles of root protoplasts of Gentiana lutea. J. Plant Physiol. 1986, 122, 473–476. [Google Scholar] [CrossRef]
- Zhu, H.; Zheng, C.; Zhao, P.; Li, Y.; Yang, C.; Zhang, Y. Contents analysis of gentiopicroside in wild and tissue culture seedlings of Gentiana rigescens. Nat. Prod. Res. Dev. 2011, 23, 482–485. [Google Scholar]
- Shen, T. Studies on Breeding Traits and Accumulation of Effective Components in Cultivation Conditions in Gentiana rigescens. Master Thesis, Yunnan University, Kunming, China, 2011. [Google Scholar]
- Yuan, T.; Wang, Y.; Zhao, Y.; Zhang, J.; Jin, H.; Zhang, J. The common and variation peak ratio dual index sequence analysis in UV fingerprint spectra of Gentiana Rigescens. Spectrosc. Spectr. Anal. 2011, 31, 2161–2165. [Google Scholar]
- Tasheva, K.; Kosturkova, G. Role of biotechnology for protection of endangered medicinal plants. In Environmental Biotechnology—New Approaches and Prospective Applications; Petre, M., Ed.; InTech: Rijeka, Croatia, 2013; pp. 235–238. [Google Scholar]
- Nakatsuka, T.; Yamada, E.; Saito, M.; Hikage, T.; Ushiku, Y.; Nishihara, M. Construction of the first genetic linkage map of Japanese gentian (Gentianaceae). BMC Genomics 2012, 13. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Li, S.; Cong, W. Karyotype analysis of chromosomes in Gentiana manshurica. J. Qiqihar Norm. Univ.: Nat. Sci. Ed. 1996, 16, 61–62. [Google Scholar]
- Nakatsuka, T.; Saito, M.; Yamada, E.; Nishihara, M. Production of picotee-type flowers in Japanese gentian by CRES-T. Plant Biotechnol. Nar. 2011, 28, 173–180. [Google Scholar] [CrossRef]
- Nishihara, M.; Hikage, T.; Yamada, E.; Nakatsuka, T. A single-base substitution suppresses flower color mutation caused by a novel miniature inverted-repeat transposable element in gentian. Mol. Genet. Genomics 2011, 286, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Nakatsuka, T.; Mishiba, K.; Kubota, A.; Abe, Y.; Yamamura, S.; Nakamura, N.; Tanaka, Y.; Nishihara, M. Genetic engineering of novel flower colour by suppression of anthocyanin modification genes in gentian. J. Plant Physiol. 2010, 167, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, J.; Wang, Y.; Yang, S.; Yang, M.; Jin, H. Effects of tree species on seed germination and seedlings growth of Chinese medicinal herb Gentiana rigescens. Allelopath. J. 2012, 29, 325–332. [Google Scholar]
- Zhang, J.; Yuan, T.; Wang, Y.; Zhao, Y.; Zhang, J.; Jin, H. Determination of mineral elements in Gentiana rigescens from different zones of Yunnan, China. Biol. Trace Elem. Res. 2012, 147, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Shen, T.; Yang, M.Q.; Zhao, Z.L.; Zhang, Z.H.; Wang, Y.Z.; Jin, H.; Zhang, J.Y.; Wang, Y.H. Dynamic changes in terpenoid contents in Gentiana rigescens. Bull. Bot. 2011, 46, 652–657. [Google Scholar] [CrossRef]
- Suyama, Y.; Kurimoto, S.; Kawazoe, K.; Murakami, K.; Sun, H.D.; Li, S.L.; Takaishi, Y.; Kashiwada, Y. Rigenolide A, a new secoiridoid glucoside with a cyclobutane skeleton, and three new acylated secoiridoid glucosides from Gentiana rigescens Franch. Fitoterapia 2013, 91, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Sun, C.; Chen, S.; H., L.; Li, Y.; Sun, Y.; Niu, Y. Application of transcriptomics in the studies of medicinal plants. World Sci. Technol. Mod. Tradit. Chin. Med. 2010, 3, 457–462. [Google Scholar]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Abdullayev, I.; Kirkham, M.; Björklund, Å.K.; Simon, A.; Sandberg, R. A reference transcriptome and inferred proteome for the salamander Notophthalmus viridescens. Exp. Cell Res. 2013, 319, 1187–1197. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Xu, M.; Luo, Q.; Wang, J.; Li, H. De novo transcriptome analysis of Liriodendron chinense petals and leaves by Illumina sequencing. Gene 2014, 534, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Tourova, T.P.; Kovaleva, O.L.; Sorokin, D.Y.; Muyzer, G. Ribulose-1,5-bisphosphate carboxylase/oxygenase genes as a functional marker for chemolithoautotrophic halophilic sulfur-oxidizing bacteria in hypersaline habitats. Microbiology 2010, 156, 2016–2025. [Google Scholar] [CrossRef] [PubMed]
- Amara, I.; Zaidi, I.; Masmoudi, K.; Ludevid, M.D.; Pagès, M.; Goday, A.; Brini, F. Insights into Late Embryogenesis Abundant (LEA) Proteins in Plants: From Structure to the Functions. Am. J. Plant. Sci. 2014, 5, 3440–3455. [Google Scholar] [CrossRef]
- Zhao, P.; Liu, F.; Zheng, G.; Liu, H. Group 3 late embryogenesis abundant protein in Arabidopsis: Structure, regulation, and function. Acta Physiol. Plant 2011, 33, 1063–1073. [Google Scholar] [CrossRef]
- Jia, F.; Qi, S.; Li, H.; Liu, P.; Li, P.; Wu, C.; Zheng, C.; Huang, J. Overexpression of Late Embryogenesis Abundant 14 enhances Arabidopsis salt stress tolerance. Biochem. Biophys. Res. Commun. 2014, 454, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, L.; Jiang, S.; Pan, J.; Cai, G.; Li, D. Group 5 LEA protein, ZmLEA5C, enhance tolerance to osmotic and low temperature stresses in transgenic tobacco and yeast. Plant Physiol. Biochem. 2014, 84, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Kiselev, K.V.; Turlenko, A.V.; Zhuravlev, Y.N. Structure and expression profiling of a novel calcium-dependent protein kinase gene PgCDPK1a in roots, leaves, and cell cultures of Panax ginseng. Plant Cell Tissue Organ 2010, 103, 197–204. [Google Scholar] [CrossRef]
- Kang, H.; Zhu, H.; Chu, X.; Yang, Z.; Yuan, S.; Yu, D.; Wang, C.; Hong, Z.; Zhang, Z. A novel interaction between CCaMK and a protein containing the Scythe_N ubiquitin-like domain in Lotus japonicus. Plant Physiol. 2011, 155, 1312–1324. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Banba, M.; Shimoda, Y.; Kouchi, H.; Hayashi, M.; Imaizumi-Anraku, H. A dominant function of CCaMK in intracellular accommodation of bacterial and fungal endosymbionts. Plant J. 2010, 63, 141–154. [Google Scholar] [PubMed]
- Takeda, N.; Maekawa, T.; Hayashi, M. Nuclear-localized and deregulated calcium- and calmodulin-dependent protein kinase activates rhizobial and mycorrhizal responses in Lotus japonicus. Plant Cell 2012, 24, 810–822. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.B.; Pratap, A.; Miyahara, A.; Zhou, L.; Bornemann, S.; Morris, R.J.; Oldroyd, G.E. Calcium/Calmodulin-dependent protein kinase is negatively and positively regulated by calcium, providing a mechanism for decoding calcium responses during symbiosis signaling. Plant Cell 2013, 25, 5053–5066. [Google Scholar] [CrossRef] [PubMed]
- Shimoda, Y.; Han, L.; Yamazaki, T.; Suzuki, R.; Hayashi, M.; Imaizumi-Anraku, H. Rhizobial and fungal symbioses show different requirements for calmodulin binding to calcium calmodulin-dependent protein kinase in Lotus japonicus. Plant Cell 2012, 24, 304–321. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Parniske, M. Activation of calcium- and calmodulin-dependent protein kinase (CCaMK), the central regulator of plant root endosymbiosis. Curr. Opin. Plant Biol. 2012, 15, 444–453. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Lu, R.; Liu, H.; Shi, B.; Zhang, J.; Tan, M.; Zhang, A.; Jiang, M. Nitric oxide-activated calcium/calmodulin-dependent protein kinase regulates the abscisic acid-induced antioxidant defence in maize. J. Exp. Bot. 2012, 63, 4835–4847. [Google Scholar] [CrossRef] [PubMed]
- Kiba, T.; Feria-Bourrellier, A.B.; Lafouge, F.; Lezhneva, L.; Boutet-Mercey, S.; Orsel, M.; Brehaut, V.; Miller, A.; Daniel-Vedele, F.; Sakakibara, H.; et al. The Arabidopsis nitrate transporter NRT2.4 plays a double role in roots and shoots of nitrogen-starved plants. Plant Cell 2012, 24, 245–258. [Google Scholar] [CrossRef] [PubMed]
- Bagchi, R.; Salehin, M.; Adeyemo, O.S.; Salazar, C.; Shulaev, V.; Sherrier, D.J.; Dickstein, R. Functional assessment of the Medicago truncatula NIP/LATD protein demonstrates that it is a high-affinity nitrate transporter. Plant Physiol. 2012, 160, 906–916. [Google Scholar] [CrossRef] [PubMed]
- Kotur, Z.; Glass, A.D. A 150 kDa plasma membrane complex of AtNRT2.5 and AtNAR2.1 is the major contributor to constitutive high-affinity nitrate influx in Arabidopsis thaliana. Plant Cell Environ. 2014. [Google Scholar] [CrossRef]
- Sun, P.; Song, S.; Zhou, L.; Zhang, B.; Qi, J.; Li, X. Transcriptome analysis reveals putative genes involved in iridoid biosynthesis in Rehmannia glutinosa. Int. J. Mol. Sci. 2012, 13, 13748–13763. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Xu, H.; Ma, X.; Zhan, R.; Chen, W. Triterpenoid saponin biosynthetic pathway profiling and candidate gene mining of the Ilex asprella root using RNA-Seq. Int. J. Mol. Sci. 2014, 15, 5970–5987. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Ding, G.; Lin, H.; Cheng, H.; Kong, Y.; Wei, Y.; Fang, X.; Liu, R.; Wang, L.; Chen, X.; et al. Transcriptome analysis of medicinal plant Salvia miltiorrhiza and identification of genes related to tanshinone biosynthesis. PLoS ONE 2013, 8, e80464. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Li, Y.; Li, C.; Luo, H.; Wang, L.; Qian, J.; Luo, X.; Xiang, L.; Song, J.; Sun, C.; et al. Analysis of the Dendrobium officinale transcriptome reveals putative alkaloid biosynthetic genes and genetic markers. Gene 2013, 527, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Weitzel, C.; Simonsen, H.T. Cytochrome P450-enzymes involved in the biosynthesis of mono-and sesquiterpenes. Phytochem. Rev. 2015, 14, 7–24. [Google Scholar] [CrossRef]
- Hamberger, B.; Bak, S. Plant P450s as versatile drivers for evolution of species-specific chemical diversity. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2013, 368, 1–16. [Google Scholar] [CrossRef]
- Salim, V.; Wiens, B.; Masada-Atsumi, S.; Yu, F.; de Luca, V. 7-deoxyloganetic acid synthase catalyzes a key 3 step oxidation to form 7-deoxyloganetic acid in Catharanthus roseus iridoid biosynthesis. Phytochemistry 2014, 101, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, K.; Dong, L.; Navrot, N.; Schneider, T.; Burlat, V.; Pollier, J.; Woittiez, L.; van der Krol, S.; Lugan, R.; Ilc, T.; et al. The seco-iridoid pathway from Catharanthus roseus. Nat. Commun. 2014, 5, 3606–3616. [Google Scholar] [PubMed]
- Asada, K.; Salim, V.; Masada-Atsumi, S.; Edmunds, E.; Nagatoshi, M.; Terasaka, K.; Mizukami, H.; de Luca, V. A 7-deoxyloganetic acid glucosyltransferase contributes a key step in secologanin biosynthesis in Madagascar periwinkle. Plant Cell 2013, 25, 4123–4134. [Google Scholar] [CrossRef] [PubMed]
- Irmler, S.; Schröder, G.; St-Pierre, B.; Crouch, N.P.; Hotze, M.; Schmidt, J.; Strack, D.; Matern, U.; Schröder, J. Indole alkaloid biosynthesis in Catharanthus roseus: New enzyme activities and identification of cytochrome P450 CYP72A1 as secologanin synthase. Plant J. 2000, 24, 797–804. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.T.; Liu, H.; Gao, X.S.; Zhang, H.X. Overexpression of G10H and ORCA3 in the hairy roots of Catharanthus roseus improves catharanthine production. Plant Cell Rep. 2010, 29, 887–894. [Google Scholar] [CrossRef] [PubMed]
- Salim, V.; Yu, F.; Altarejos, J.; Luca, V. Virus-induced gene silencing identifies Catharanthus roseus 7-deoxyloganic acid-7-hydroxylase, a step in iridoid and monoterpene indole alkaloid biosynthesis. Plant J. 2013, 76, 754–765. [Google Scholar] [CrossRef] [PubMed]
- Guo, A.Y.; Chen, X.; Gao, G.; Zhang, H.; Zhu, Q.H.; Liu, X.C.; Zhong, Y.F.; Gu, X.; He, K.; Luo, J. PlantTFDB: A comprehensive plant transcription factor database. Nucleic Acids Res. 2008, 36, 966–969. [Google Scholar] [CrossRef]
- Vom Endt, D.; Kijne, J.W.; Memelink, J. Transcription factors controlling plant secondary metabolism: What regulates the regulators? Phytochemistry 2002, 61, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Crocoll, C. Biosynthesis of the Phenolic Monoterpenes, Thymol and Carvacrol, by Terpene Synthases and Cytochrome P450s in Oregano and Thyme. Ph.D. Thesis, Friedrich-Schiller-Universität, Jena, Germany, 2011. [Google Scholar]
- Yang, C.; Fang, X.; Wu, X.; Mao, Y.; Wang, L.; Chen, X. Transcriptional regulation of plant secondary metabolism. J. Integr. Plant Biol. 2012, 54, 703–712. [Google Scholar] [CrossRef] [PubMed]
- Van Schie, C.C.; Haring, M.A.; Schuurink, R.C. Regulation of terpenoid and benzenoid production in flowers. Curr. Opin. Plant Biol. 2006, 9, 203–208. [Google Scholar] [CrossRef]
- Li, C.; Li, D.; Shao, F.; Lu, S. Molecular cloning and expression analysis of WRKY transcription factor genes in Salvia miltiorrhiza. BMC Genomics 2015, 16. [Google Scholar] [CrossRef]
- Xu, Y.; Wang, J.; Wang, S.; Wang, J.; Chen, X. Characterization of GaWRKY1, a cotton transcription factor that regulates the sesquiterpene synthase gene (+)-δ-cadinene synthase-A. Plant Physiol. 2004, 135, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Kato, N.; Dubouzet, E.; Kokabu, Y.; Yoshida, S.; Taniguchi, Y.; Dubouzet, J.G.; Yazaki, K.; Sato, F. Identification of a WRKY protein as a transcriptional regulator of benzylisoquinoline alkaloid biosynthesis in Coptis japonica. Plant Cell Physiol. 2007, 48, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Spyropoulou, E.A.; Haring, M.A.; Schuurink, R.C. RNA sequencing on Solanum lycopersicum trichomes identifies transcription factors that activate terpene synthase promoters. BMC Genomics 2014, 15. [Google Scholar] [CrossRef] [PubMed]
- Suttipanta, N.; Pattanaik, S.; Kulshrestha, M.; Patra, B.; Singh, S.K.; Yuan, L. The transcription factor CrWRKY1 positively regulates the terpenoid indole alkaloid biosynthesis in Catharanthus roseus. Plant Physiol. 2011, 157, 2081–2093. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Patra, B.; Li, R.; Pattanaik, S.; Yuan, L. Promoter analysis reveals cis-regulatory motifs associated with the expression of the WRKY transcription factor CrWRKY1 in Catharanthus roseus. Planta 2013, 238, 1039–1049. [Google Scholar] [CrossRef]
- Skibbe, M.; Qu, N.; Galis, I.; Baldwin, I.T. Induced plant defenses in the natural environment: Nicotiana attenuata WRKY3 and WRKY6 coordinate responses to herbivory. Plant Cell 2008, 20, 1984–2000. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Pu, G.; Lei, C.; Ma, L.; Wang, H.; Guo, Y.; Chen, J.; Du, Z.; Wang, H.; Li, G. Isolation and characterization of AaWRKY1, an Artemisia annua transcription factor that regulates the amorpha-4,11-diene synthase gene, a key gene of artemisinin biosynthesis. Plant Cell Physiol. 2009, 50, 2146–2161. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Li, B.; Mu, S.; Han, B.; Cui, R.; Xu, M.; You, Z.; Dong, H. TTG2-regulated development is related to expression of putative AUXIN RESPONSE FACTOR genes in tobacco. BMC Genomics 2013, 14. [Google Scholar] [CrossRef] [PubMed]
- Shu, S.; Chen, B.; Zhou, M.; Zhao, X.; Xia, H.; Wang, M. De novo sequencing and transcriptome analysis of Wolfiporia cocos to reveal genes related to biosynthesis of triterpenoids. PLoS ONE 2013, 8, e71350. [Google Scholar] [CrossRef] [PubMed]
- Krasileva, K.V.; Buffalo, V.; Bailey, P.; Pearce, S.; Ayling, S.; Tabbita, F.; Soria, M.; Wang, S.; Akhunov, E.; Uauy, C. Separating homeologs by phasing in the tetraploid wheat transcriptome. Genome Biol. 2013, 14, R66. [Google Scholar] [CrossRef] [PubMed]
- Schmieder, R.; Edwards, R. Fast identification and removal of sequence contamination from genomic and metagenomic datasets. PLoS ONE 2011, 6, e17288. [Google Scholar] [CrossRef] [PubMed]
- Korf, I.; Yandell, M.; Bedell, J. Blast; OʼReilly Media, Inc.: Sebastopol, CA, USA, 2003. [Google Scholar]
- Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talon, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Lv, J.; Liu, P.; Wang, Y.; Gao, B.; Chen, P.; Li, J. Transcriptome Analysis of Portunus trituberculatus in response to salinity stress Provides Insights into the Molecular Basis of Osmoregulation. PLoS ONE 2013, 8, e82155. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhao, L.; Larson-Rabin, Z.; Li, D.; Guo, Z. De novo sequencing and characterization of the floral transcriptome of Dendrocalamus latiflorus (Poaceae: Bambusoideae). PLoS ONE 2012, 7, e42082. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef] [PubMed]
- McGettigan, P.A. Transcriptomics in the RNA-seq era. Curr. Opin. Chem. Biol. 2013, 17, 4–11. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Allan, A.C.; Li, C.; Wang, Y.; Yao, Q. De Novo Assembly and Characterization of the Transcriptome of the Chinese Medicinal Herb, Gentiana rigescens. Int. J. Mol. Sci. 2015, 16, 11550-11573. https://doi.org/10.3390/ijms160511550
Zhang X, Allan AC, Li C, Wang Y, Yao Q. De Novo Assembly and Characterization of the Transcriptome of the Chinese Medicinal Herb, Gentiana rigescens. International Journal of Molecular Sciences. 2015; 16(5):11550-11573. https://doi.org/10.3390/ijms160511550
Chicago/Turabian StyleZhang, Xiaodong, Andrew C. Allan, Caixia Li, Yuanzhong Wang, and Qiuyang Yao. 2015. "De Novo Assembly and Characterization of the Transcriptome of the Chinese Medicinal Herb, Gentiana rigescens" International Journal of Molecular Sciences 16, no. 5: 11550-11573. https://doi.org/10.3390/ijms160511550
APA StyleZhang, X., Allan, A. C., Li, C., Wang, Y., & Yao, Q. (2015). De Novo Assembly and Characterization of the Transcriptome of the Chinese Medicinal Herb, Gentiana rigescens. International Journal of Molecular Sciences, 16(5), 11550-11573. https://doi.org/10.3390/ijms160511550