
The Role of Sulforaphane in Epigenetic Mechanisms, Including Interdependence between Histone Modification and DNA Methylation



 and
and
Abstract
:1. Introduction

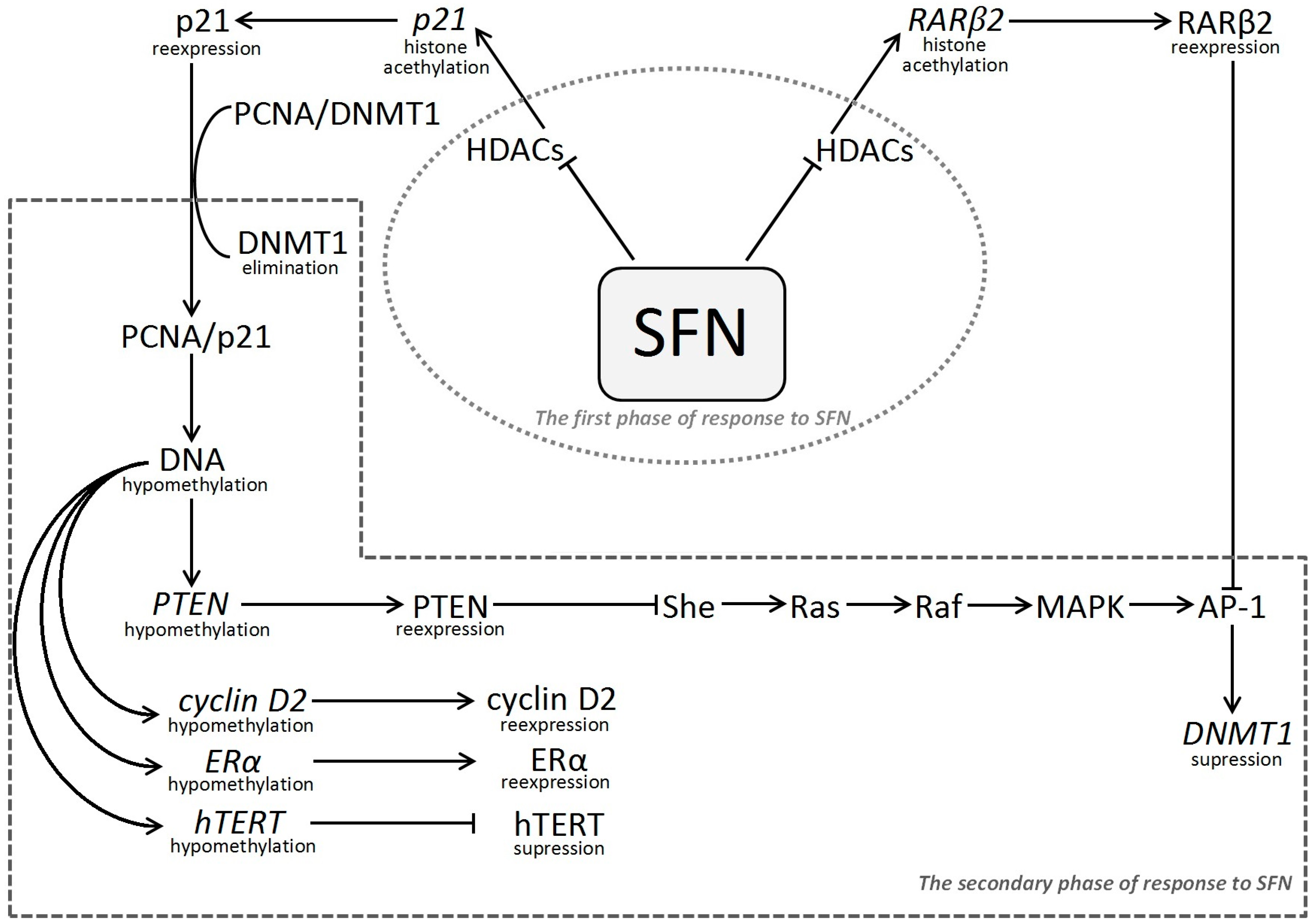{kind=link}
| Isothiocyanate | Chemical Structure | Glucosinolate—Isothiocyanate Precursor | Food Sources | Total Concentration (mg/100 g) |
|---|---|---|---|---|
| Sulforaphane | 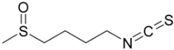 | Glucoraphanin | Broccoli | 61 |
| Brussels sprouts | 236 | |||
| Cabbage | 78 | |||
| Allyl isothiocyanate (AITC) |  | Sinigrin | Broccoli | 61 |
| Brussels sprouts | 236 | |||
| Cabbage | 78 | |||
| Mustard greens | 282 | |||
| Benzyl isothiocyanate (BITC) | 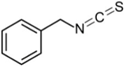 | Glucotropaeolin | Cabbage | 78 |
| Garden cress | 392 | |||
| Phenethyl isothiocyanate (PEITC) | 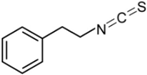 | Gluconasturtiin | Watercress | 94 |
2. Epigenetic Modifications Regulating Transcriptional Activity of Gene Promoters
2.1. Histone Deacetylation and the Role of Histone Deacetylase Inhibitors (HDIs)
2.2. Sulforaphane as HDI
2.3. DNA Methylation
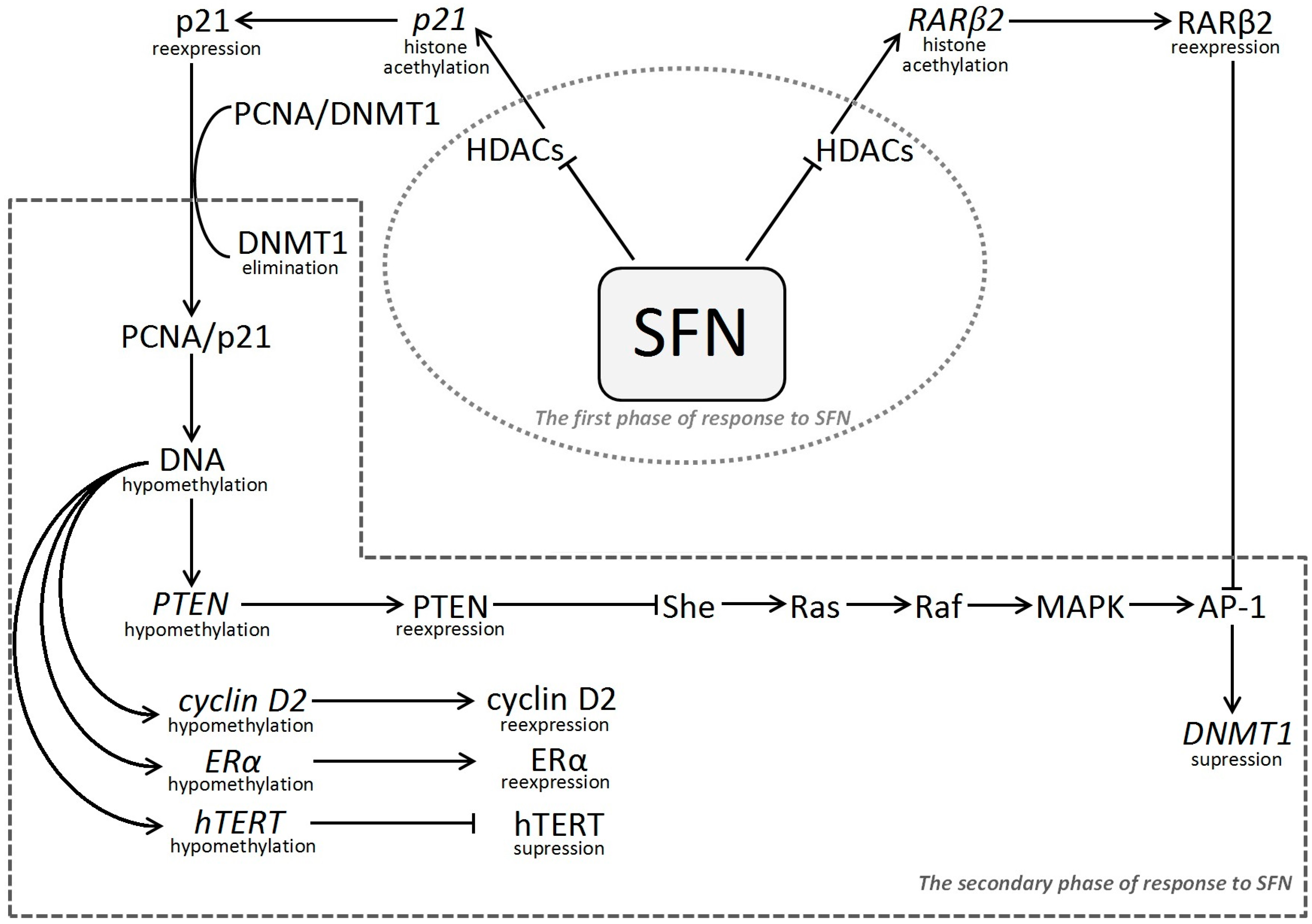
2.4. Sulforaphane as Indirect Regulator of Promoter Methylation
3. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Meeran, S.M.; Ahmed, A.; Tollefsbol, T.O. Epigenetic targets of bioactive dietary components for cancer prevention and therapy. Clin. Epigenet. 2010, 1, 101–116. [Google Scholar] [CrossRef] [PubMed]
- Higdon, J.V.; Delage, B.; Williams, D.E.; Dashwood, R.H. Cruciferous vegetables and human cancer risk: Epidemiologic evidence and mechanistic basis. Pharmacol. Res. 2007, 55, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Hussain, A.; Mohsin, J.; Prabhu, S.A.; Begum, S.; Nusri, Q.A.; Harish, G.; Javed, E.; Khan, M.A.; Sharma, C. Sulforaphane inhibits growth of human breast cancer cells and augments the therapeutic index of the chemotherapeutic drug, gemcitabine. Asian Pac. J. Cancer Prev. 2013, 14, 5855–5860. [Google Scholar] [CrossRef] [PubMed]
- Rudolf, K.; Cervinka, M.; Rudolf, E. Sulforaphane-induced apoptosis involves p53 and p38 in melanoma cells. Apoptosis 2014, 19, 734–747. [Google Scholar] [CrossRef] [PubMed]
- Ferreira de Oliveira, J.M.; Remédios, C.; Oliveira, H.; Pinto, P.; Pinho, F.; Pinho, S.; Costa, M.; Santos, C. Sulforaphane induces DNA damage and mitotic abnormalities in human osteosarcoma MG-63 cells: Correlation with cell cycle arrest and apoptosis. Nutr. Cancer 2014, 66, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Pledgie-Tracy, A.; Sobolewski, M.D.; Davidson, N.E. Sulforaphane induces cell type-specific apoptosis in human breast cancer cell lines. Mol. Cancer Ther. 2007, 6, 1013–1021. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhou, Y.; Peng, X.; Du, L.; Tian, H.; Yang, G.; Niu, J.; Wu, W. Sulforaphane inhibits invasion via activating ERK1/2 signaling in human glioblastoma U87MG and U373MG cells. PLoS ONE 2014, 9, e90520. [Google Scholar] [CrossRef] [PubMed]
- Suppipat, K.; Park, C.S.; Shen, Y.; Zhu, X.; Lacorazza, H.D. Sulforaphane induces cell cycle arrest and apoptosis in acute lymphoblastic leukemia cells. PLoS ONE 2012, 7, e51251. [Google Scholar] [CrossRef] [PubMed]
- Fimognari, C.; Turrini, E.; Sestili, P.; Caleabrini, C.; Carulli, G.; Fontanelli, G.; Hrelia, P. Antileukemic activity of sulforaphane in primary blasts from patients affected by myelo- and lympho-proliferative disorders and in hypoxic conditions. PLoS ONE 2014, 9, e101991. [Google Scholar] [CrossRef] [PubMed]
- Clarke, J.D.; Dashwood, R.H.; Ho, E. Multi-targeted prevention of cancer by sulforaphane. Cancer Lett. 2008, 269, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Feng, D.; Wu, J.; Tian, Y.; Zhou, H.; Hu, W.; Zhao, W.; Wei, H.; Ling, B.; Ma, C. Targeting of histone deacetylases to reactivate tumour suppressor genes and its therapeutic potential in a human cervical cancer xenograft model. PLoS ONE 2013, 8, e80657. [Google Scholar] [CrossRef] [PubMed]
- Seligson, D.B.; Horvath, S.; Shi, T.; Yu, H.; Grunstein, M.; Kurdistani, S.K. Global histone modification patterns predict risk of prostate cancer recurrence. Nature 2005, 435, 1262–1266. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.S.; Parmigiani, R.B.; Marks, P.A. Histone deacetylase inhibitors: Molecular mechanism of action. Oncogene 2007, 26, 5541–5552. [Google Scholar] [CrossRef] [PubMed]
- Frønsdal, K.; Saateioglu, F. Histone deacetylase inhibitors differentially mediate apoptosis in prostate cancer cells. Prostate 2005, 62, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Sirchia, S.M.; Ferguson, A.T.; Sironi, E.; Subramanyon, S.; Orlandi, R.; Sukumar, S.; Sacchi, N. Evidence of epigenetic changes affecting the chromatin state of the retinoic acid receptor β2 promoter in breast cancer cells. Oncogene 2000, 19, 1556–1563. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.F.; Qian, D.Z.; Ren, M.; Kato, Y.; Wei, Y.; Zhang, L.; Fransler, Z.; Clark, D.; Nakanishi, O.; Pili, R. Epigenetic modulation of retinoic acid receptor β2 by the histone deacetylase inhibitor MS-275 in human renal cell carcinoma. Clin. Cancer Res. 2005, 11, 3535–3542. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.F.; Shen, X.Q.; Shemshedini, L. Ligand-activated retinoic acid receptor inhibits AP-1 transactivation by disrupting c-Jun/c-Fos dimerization. Mol. Endocrinol. 1999, 13, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.Y.; Lu, Y.Y. Effects of histone acetylation and DNA methylation on p21WAF1 regulation. World J. Gastroenterol. 2002, 8, 400–405. [Google Scholar] [PubMed]
- Jung, Y.-S.; Qian, Y.; Chen, X. Examination of the expanding pathways for the regulation of p21 expression and activity. Cell Signal 2010, 22, 1003–1012. [Google Scholar]
- Pagliuca, A.; Gallo, P.; Lania, L. Differential role for Sp1/Sp3 transcription factor in the regulation of the promoter activity of multiple cyclin dependent kinase inhibitor genes. J Cell Biochem. 2000, 76, 360–367. [Google Scholar] [CrossRef]
- Ocker, M.; Schneider-Stock, R. Histone deacetylase inhibitors: Signalling towards p21cip1/waf1. Int. J. Biochem. Cell Biol. 2007, 39, 1367–1374. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Hasegawa, T.; Isobe, K. p300 collaborates with Sp1 and Sp3 in p21 promoter activation by histone deacetylase inhibitor. J. Biol. Chem. 2000, 275, 1371–1376. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.; Liu, B.; Gu, Q.; Su, L.; Yu, Y.; Zhu, A. STAT6 cooperates with Sp1 in controlling breast cancer cell proliferation by modulating the expression of p21Cip1/WAF1 and p27Kip1. Cell Oncol. 2013, 36, 79–93. [Google Scholar] [CrossRef] [PubMed]
- Piccolo, M.T.; Crispi, S. The dual role played by p21 may influence the apoptotic or anti-apoptotic fate in cancer. J. Cancer Res. Updates 2012, 1, 189–202. [Google Scholar]
- Shin, J.Y.; Kim, H.S.; Park, J.; Park, J.B.; Lee, J.Y. Mechanism for inactivation of the KIP family cyclin-dependent kinase inhibitor genes in gastric cancer cells. Cancer Res. 2000, 60, 262–265. [Google Scholar] [PubMed]
- Arzenani, M.K.; Zade, A.E.; Ming, Y.; Vijverberg, S.J.H.; Zhang, Z.; Khan, Z.; Sadique, S.; Kallenbach, L.; Hu, L.F.; Vukojević, V.; et al. Genomic DNA hypomethylation by histone deacetylase inhibition implicates DNMT1 nuclear dynamics. Mol. Cell. Biol. 2011, 31, 4119–4128. [Google Scholar] [CrossRef] [PubMed]
- Petrella, A.; Fontanella, B.; Carratu, A.; Bizzaro, V.; Rodriquez, M.; Parente, L. Histone deacetylase inhibitors in the treatment of hematological malignancies. Min. Rev. Med. Chem. 2011, 11, 519–527. [Google Scholar] [CrossRef]
- Ho, E.; Dashwood, R.H. Dietary manipulation of histone structure and function. J. Nutrigenet. Nutrigenomics 2010, 3, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Nian, H.; Delage, B.; Ho, E.; Dashwood, R.H. Modulation of histone deacetylase activity by dietary isothiocyanates and allyl sulfides: Studies with sulforaphane and garlic organosulfur compounds. Environ. Mol. Mutagen. 2009, 50, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Myzak, M.C.; Karplus, P.A.; Chung, F.L.; Dashwood, R.H. A novel mechanism of chemoprotection by sulforaphane: Inhibition of histone deacetylase. Cancer Res. 2004, 64, 5767–5774. [Google Scholar] [CrossRef] [PubMed]
- Ho, E.; Clarke, J.D.; Dashwood, R.H. Dietary sulforaphane a histone deacetylase inhibitor for cancer prevention. J. Nutr. 2009, 139, 2393–2396. [Google Scholar] [CrossRef] [PubMed]
- Myzak, M.C.; Hardin, K.; Wang, R.; Dashwood, R.H.; Ho, E. Sulforaphane inhibits histone deacetylase activity in BPH-1, LnCaP and PC-3 prostate epithelial cells. Carcinogenesis 2006, 27, 811–819. [Google Scholar] [CrossRef] [PubMed]
- Myzak, M.C.; Tong, P.; Dashwood, W.M.; Dashwood, R.H.; Ho, E. Sulforaphane retards the growth of human PC-3 xenografts and inhibits HDAC activity in human subjects. Exp. Biol. Med. 2007, 232, 227–234. [Google Scholar]
- Saw, C.L.; Huang, M.T.; Liu, Y.; Khor, T.O.; Conney, A.H.; Kong, A.N. Impact of Nrf2 on UVB-induced skin inflammation/photoprotection and photoprotective effect of sulforaphane. Mol. Carcinog. 2011, 50, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Motted, D.; Pirotte, S.; Lamour, V.; Hagedorn, M.; Javerzat, S.; Bikfalvi, A.; Bellahċene, A.; Verdin, E.; Castronovo, V. HDAC4 represses p21WAF1/Cip1 expression in human cancer cells though a Sp1-dependent, p53-independent mechanism. Oncogene 2009, 28, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Lubecka-Pietruszewska, K.; Kaufman-Szymczyk, A.; Stefańska, B.; Cebula-Obrzut, B.; Smolewski, P.; Fabianowska-Majewska, K. Sulforaphane alone and in combination with clofarabine epigenetically regulates the expression of DNA methylation-silenced tumour suppressor genes in human breast cancer cells. J. Nutrigenet. Nutrigenomics 2015, 8, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Ho, E.; Beaver, L.M.; Williams, D.E.; Dashwood, R.H. Dietary factors and epigenetic regulation for prostate cancer prevention. Adv. Nutr. 2011, 2, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Meeran, S.M.; Patel, S.N.; Tollefsbol, T.O. Sulforaphane causes epigenetic repression of hTERT expression in human breast cancer cell lines. PLoS ONE 2010, 5, e11457. [Google Scholar] [CrossRef] [PubMed]
- Feng, D.; Cao, Z.; Zhang, L.; Zhou, Y.; Ma, J.; Liu, R.; Zhou, H.; Zhao, W.; Wei, H.; Ling, B. Combination of valproic acid and ATRA restores RARβ2 expression and induces differentiation in cervical cancer through the PI3K/Akt pathway. Curr. Mol. Med. 2012, 12, 342–354. [Google Scholar] [CrossRef] [PubMed]
- D’Alessio, A.C.; Szyf, M. Epigenetic tête-à-tête: The bilateral relationship between chromatin modification and DNA methylation. Biochem. Cell Biol. 2006, 84, 463–476. [Google Scholar] [CrossRef] [PubMed]
- Daniel, F.I.; Cherubini, K.; Yurgel, L.S.; de Figueiredo, M.A.; Salum, F.G. The role of epigenetic transcription repression and DNA methyltransferases in cancer. Cancer 2011, 117, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Lubecka-Pietruszewska, K.; Kaufman-Szymczyk, A.; Stefanska, B.; Cebula-Obrzut, B.; Smolewski, P.; Fabianowska-Majewska, K. Clofarabine, a novel adenosine analogue, reactivates DNA methylation-silenced tumour suppressor genes and inhibits cell growth in breast cancer cells. Eur. J. Pharmacol. 2014, 723, 276–287. [Google Scholar] [CrossRef] [PubMed]
- Das, P.M.; Singal, R. DNA methylation and cancer. J. Clin. Oncol. 2004, 22, 4632–4642. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.H.; Porter, A.G. p21WAF1 Negatively regulates DNMT1 expression in mammalian cells. Biochem. Biophys. Res. Commun. 2009, 382, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Milutinovic, S.; Knox, J.D.; Szyf, M. DNA methyltransferase inhibition induces the transcription of the tumor suppressor p21WAF1/CIP1/sdi1. J. Biol. Chem. 2000, 275, 6353–6359. [Google Scholar] [CrossRef] [PubMed]
- Chuang, L.S.; Ian, H.I.; Koh, T.W.; Xu, G.; Li, B.F. Human DNA-(cytosine-5) methyltransferase-PCNA complex as a target for p21WAF1. Science 1997, 277, 1996–2000. [Google Scholar] [CrossRef] [PubMed]
- Ou, J.N.; Torrisani, J.; Unterberger, A.; Provençal, N.; Shikimi, K.; Karimi, M.; Ekström, T.J.; Szyf, M. Histone deacetylase inhibitor trichostatin A induces global and gene-specific DNA demethylation in human cancer cell lines. Biochem. Pharmacol. 2007, 73, 1297–1307. [Google Scholar] [CrossRef] [PubMed]
- Maass, N.; Biallek, M.; Rösel, F.; Schem, C.; Ohike, N.; Zhang, M.; Jonat, W.; Nagasaki, K. Hypermethylation and histone deacetylation lead to silencing of the maspin gene in human breast cancer. Biochem. Biophys. Res. Commun. 2002, 297, 125–128. [Google Scholar] [CrossRef]
- Januchowski, R.; Dabrowski, M.; Ofori, H.; Jagodzinski, P.P. Trichostatin A down-regulate DNA methyltransferase 1 in Jurkat T cells. Cancer Lett. 2007, 246, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Traka, M.; Gasper, A.V.; Smith, J.A.; Hawkey, C.J.; Bao, Y.; Mithen, R.F. Transcriptome analysis of human colon Caco-2 cells expose to sulforaphane. J. Nutr. 2005, 135, 1865–1872. [Google Scholar] [PubMed]
- Hsu, A.; Wong, C.P.; Yu, Z.; Williams, D.E.; Dashwood, R.H.; Ho, E. Promoter de-methylation of cyclin D2 by sulforaphane in prostate cancer cells. Clin. Epigenet. 2011, 3, 3. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.P.; Hsu, A.; Buchanan, A.; Palomera-Sanchez, Z.; Beaver, L.M.; Houseman, E.A.; Williams, D.E.; Daswood, R.H.; Ho, E. Effects of sulforaphane and 3,3′-diindolylmethane on genome-wide promoter methylation in normal prostate epithelial cells and prostate cancer cells. PLoS ONE 2014, 9, e86787. [Google Scholar] [CrossRef] [PubMed]
- Meeran, S.M.; Patel, S.N.; Li, Y.; Shukla, S.; Tollefsbol, T.O. Bioactive dietary supplements reactive ER expression in ER-negative breast cancer cells by active chromatin modifications. PLoS ONE 2012, 7, e37748. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Su, Z.Y.; Khor, T.O.; Shu, L.; Kong, A.N. Sulforaphane enhances Nrf2 expression in prostate cancer TRAMP C1 cells through epigenetic regulation. Biochem. Pharmacol. 2013, 85, 1398–1404. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.Y.; Zhang, C.; Lee, J.H.; Shu, L.; Wu, T.Y.; Khor, T.O.; Conney, A.H.; Lu, Y.P.; Kong, A.N. Requirement and epigenetics reprogramming of Nrf2 in suppression of tumor promoter TPA-induced mouse skin cell transformation by sulforaphane. Cancer Prev. Res. 2014, 7, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Tamura, M.; Yamada, K.M. Tumor suppressor PTEN inhibits integrin- and growth factor-mediated mitogen-activated protein (MAP) kinase signaling pathways. J. Cell Biol. 1998, 143, 1375–1383. [Google Scholar] [CrossRef] [PubMed]
- Espada, J.; Ballestar, E.; Fraga, M.F.; Villar-Garea, A.; Juarranz, A.; Stockert, J.C.; Robertson, K.D.; Fuks, F.; Esteller, M. Human DNA methyltransferase 1 is required for maintenance of the histone H3 modification pattern. J. Biol. Chem. 2004, 279, 37175–37184. [Google Scholar] [CrossRef] [PubMed]
- Atwell, L.L.; Hsu, A.; Wong, C.P.; Stevens, J.F.; Bella, D.; Yu, T.W.; Pereira, C.B.; Lohr, C.V.; Christensen, J.M.; Dashwood, R.H.; et al. Absorption and chemopreventive targets of sulforaphane in humans following consumption of broccoli sprouts or a myrosinase-treated broccoli sprout extract. Mol. Nutr. Food Res. 2015, 59, 424–433. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaufman-Szymczyk, A.; Majewski, G.; Lubecka-Pietruszewska, K.; Fabianowska-Majewska, K. The Role of Sulforaphane in Epigenetic Mechanisms, Including Interdependence between Histone Modification and DNA Methylation. Int. J. Mol. Sci. 2015, 16, 29732-29743. https://doi.org/10.3390/ijms161226195
Kaufman-Szymczyk A, Majewski G, Lubecka-Pietruszewska K, Fabianowska-Majewska K. The Role of Sulforaphane in Epigenetic Mechanisms, Including Interdependence between Histone Modification and DNA Methylation. International Journal of Molecular Sciences. 2015; 16(12):29732-29743. https://doi.org/10.3390/ijms161226195
Chicago/Turabian StyleKaufman-Szymczyk, Agnieszka, Grzegorz Majewski, Katarzyna Lubecka-Pietruszewska, and Krystyna Fabianowska-Majewska. 2015. "The Role of Sulforaphane in Epigenetic Mechanisms, Including Interdependence between Histone Modification and DNA Methylation" International Journal of Molecular Sciences 16, no. 12: 29732-29743. https://doi.org/10.3390/ijms161226195
APA StyleKaufman-Szymczyk, A., Majewski, G., Lubecka-Pietruszewska, K., & Fabianowska-Majewska, K. (2015). The Role of Sulforaphane in Epigenetic Mechanisms, Including Interdependence between Histone Modification and DNA Methylation. International Journal of Molecular Sciences, 16(12), 29732-29743. https://doi.org/10.3390/ijms161226195