
Targeting Mitochondrial Function to Treat Quiescent Tumor Cells in Solid Tumors

Abstract
:
{kind=link}
{kind=link}
{kind=link}
1. Solid Tumors Contain Cell Populations with Limited Sensitivity to Treatment
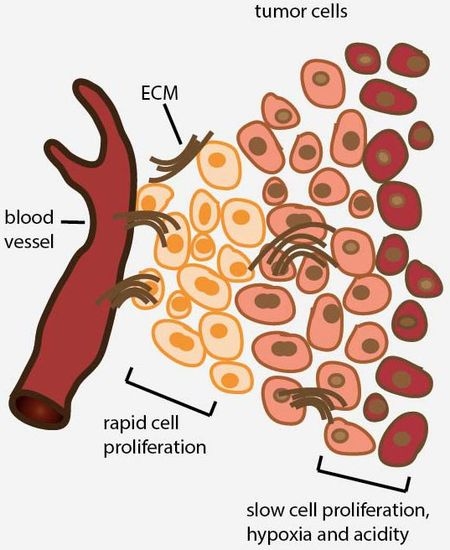
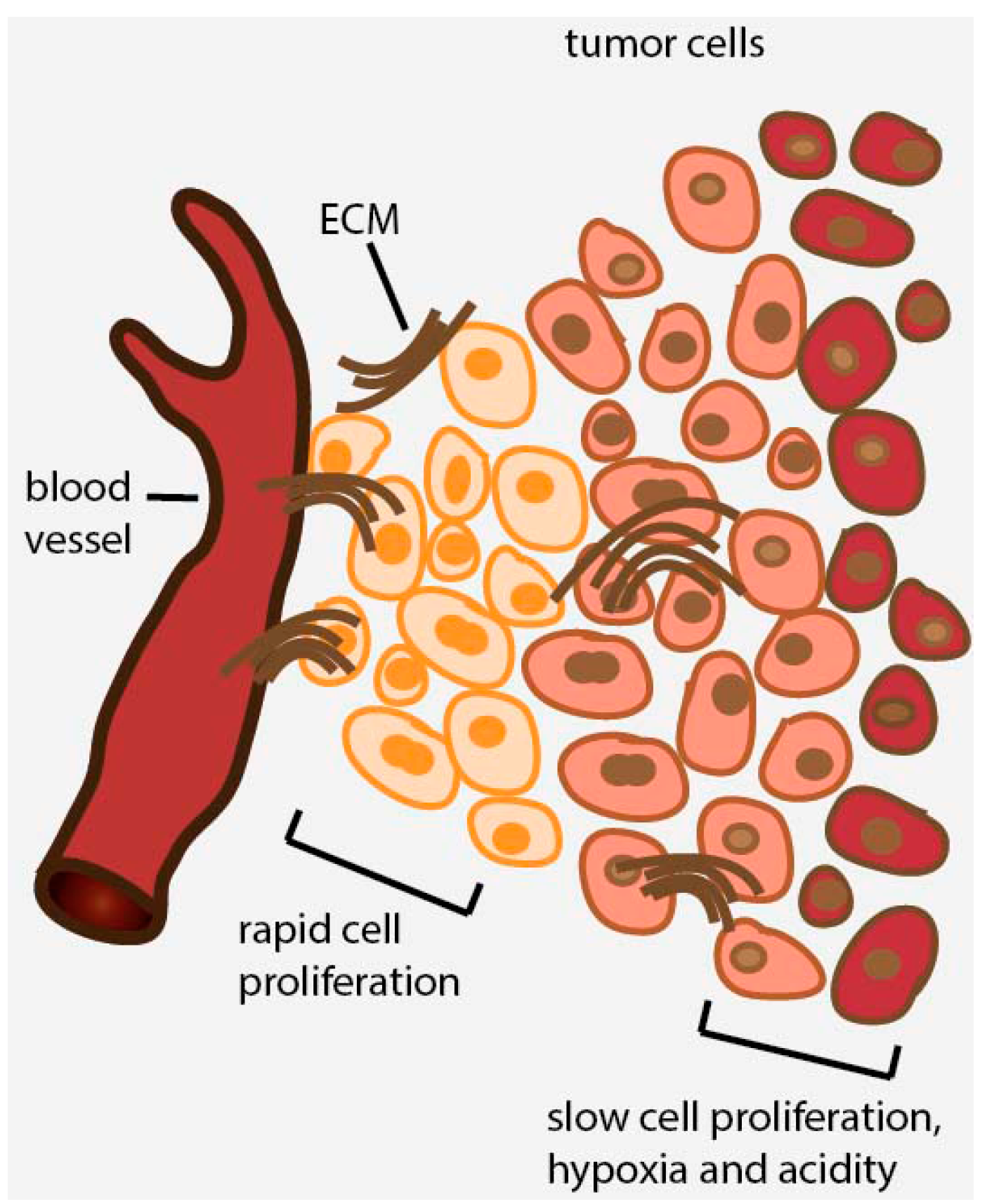
2. Avascular Areas of Solid Tumors Contain Quiescent Cell Populations
3. Conditional Drug Screening Aimed at Targeting Glucose-Starved Tumor Cells
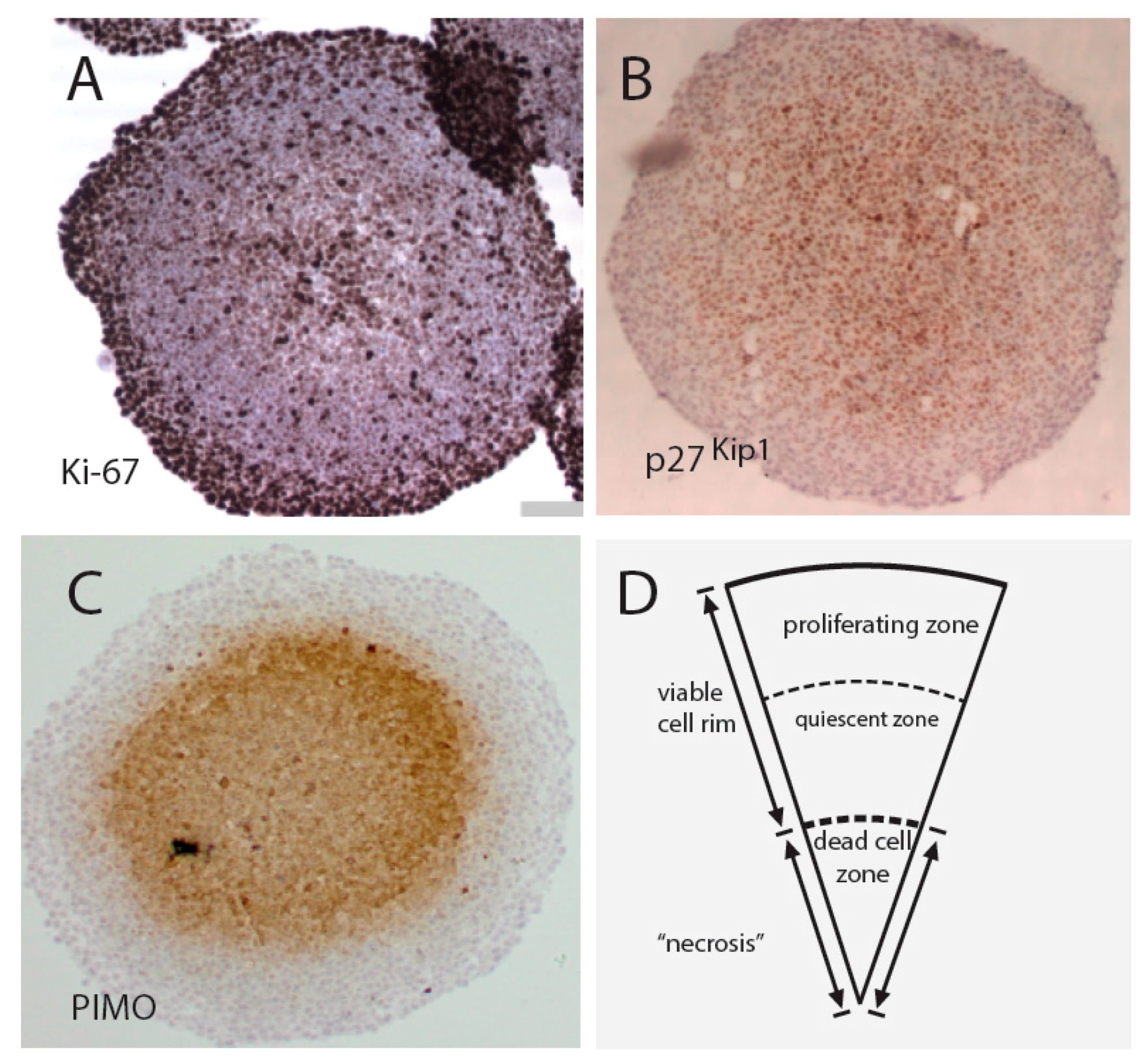
4. Drug Screening Efforts Using Spheroid Models Mimicking the Tumor Microenvironment and Heterogeneity

5. Drug Screening Using Cancer Stem Cells
6. Tumor Cell Metabolism Is Dependent on Oxidative Phosphorylation (OXPHOS)
7. Mitochondrial OXPHOS as a Therapeutic Opportunity to Target Non-Proliferating Tumor Cells
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Durand, R.E. Distribution and activity of antineoplastic drugs in a tumor model. J. Natl. Cancer Inst. 1989, 81, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.J.; Tannock, I.F. Repopulation of cancer cells during therapy: An important cause of treatment failure. Nat. Rev. Cancer 2005, 5, 516–525. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, S.; Sugiyama, Y.; Mitsuhashi, J.; Inaba, M. Kinetic analysis of cell killing effect induced by cytosine arabinoside and cisplatin in relation to cell cycle phase specificity in human colon cancer and Chinese hamster cells. Cancer Res. 1989, 49, 3823–3828. [Google Scholar] [PubMed]
- Malaise, E.; Tubiana, M. Growth of the cells of an experimental irradiated fibrosarcoma in the C3H mouse. C. R. Acad. Sci. Hebd. Seances Acad. Sci. D 1966, 263, 292–295. [Google Scholar] [PubMed]
- Saggar, J.K.; Tannock, I.F. Chemotherapy rescues hypoxic tumor cells and induces their reoxygenation and repopulation—An effect that is inhibited by the hypoxia-activated prodrug TH-302. Clin. Cancer Res. 2015, 21, 2107–2114. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.A.; Schwartz, G.K. The relevance of drug sequence in combination chemotherapy. Drug Resist. Update 2000, 3, 335–356. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Birle, D.C.; Tannock, I.F. Effects of the mammalian target of rapamycin inhibitor CCI-779 used alone or with chemotherapy on human prostate cancer cells and xenografts. Cancer Res. 2005, 65, 2825–2831. [Google Scholar] [CrossRef] [PubMed]
- Hernlund, E.; Olofsson, M.H.; Fayad, W.; Fryknas, M.; Lesiak-Mieczkowska, K.; Zhang, X.; Brnjic, S.; Schmidt, V.; D’Arcy, P.; Sjoblom, T.; et al. The phosphoinositide 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235 is effective in inhibiting regrowth of tumour cells after cytotoxic therapy. Eur. J. Cancer 2012, 48, 396–406. [Google Scholar] [CrossRef] [PubMed]
- Kurtova, A.V.; Xiao, J.; Mo, Q.; Pazhanisamy, S.; Krasnow, R.; Lerner, S.P.; Chen, F.; Roh, T.T.; Lay, E.; Ho, P.L.; et al. Blocking PGE2-induced tumour repopulation abrogates bladder cancer chemoresistance. Nature 2015, 517, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Thomlinson, R.H.; Gray, L.H. The histological structure of some human lung cancers and the possible implications for radiotherapy. Br. J. Cancer 1955, 9, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Less, J.R.; Skalak, T.C.; Sevick, E.M.; Jain, R.K. Microvascular architecture in a mammary carcinoma: Branching patterns and vessel dimensions. Cancer Res. 1991, 51, 265–273. [Google Scholar] [PubMed]
- Padera, T.P.; Stoll, B.R.; Tooredman, J.B.; Capen, D.; di Tomaso, E.; Jain, R.K. Pathology: Cancer cells compress intratumour vessels. Nature 2004, 427, 695. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.S.; Gillies, R.D.; Gatenby, R.A. Adaptation to hypoxia and acidosis in carcinogenesis and tumor progression. Semin. Cancer Biol. 2008, 18, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.M.; Wilson, W.R. Exploiting tumour hypoxia in cancer treatment. Nat. Rev. Cancer 2004, 4, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Man, S.; Graham, C.H.; Kapitain, S.J.; Teicher, B.A.; Kerbel, R.S. Acquired multicellular-mediated resistance to alkylating agents in cancer. Proc. Natl. Acad. Sci. USA 1993, 90, 3294–3298. [Google Scholar] [CrossRef] [PubMed]
- Tannock, I.F.; Lee, C.M.; Tunggal, J.K.; Cowan, D.S.; Egorin, M.J. Limited penetration of anticancer drugs through tumor tissue: A potential cause of resistance of solid tumors to chemotherapy. Clin. Cancer Res. 2002, 8, 878–884. [Google Scholar] [PubMed]
- Minchinton, A.I.; Tannock, I.F. Drug penetration in solid tumours. Nat. Rev. Cancer 2006, 6, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Tannock, I.F. Tumor physiology and drug resistance. Cancer Metastasis Rev. 2001, 20, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Yan, C.H.; Yuan, M.; Wei, Y.; Hu, G.; Kang, Y. In vivo dynamics and distinct functions of hypoxia in primary tumor growth and organotropic metastasis of breast cancer. Cancer Res. 2010, 70, 3905–3914. [Google Scholar] [CrossRef] [PubMed]
- Halle, C.; Andersen, E.; Lando, M.; Aarnes, E.K.; Hasvold, G.; Holden, M.; Syljuasen, R.G.; Sundfor, K.; Kristensen, G.B.; Holm, R.; et al. Hypoxia-induced gene expression in chemoradioresistant cervical cancer revealed by dynamic contrast-enhanced MRI. Cancer Res. 2012, 72, 5285–5295. [Google Scholar] [CrossRef] [PubMed]
- Kolosenko, I.; Fryknas, M.; Forsberg, S.; Johnsson, P.; Cheon, H.; Holvey-Bates, E.G.; Edsbacker, E.; Pellegrini, P.; Rassoolzadeh, H.; Brnjic, S.; et al. Cell crowding induces interferon regulatory factor 9, which confers resistance to chemotherapeutic drugs. Int. J. Cancer 2015, 136, E51–E61. [Google Scholar] [CrossRef] [PubMed]
- Shoemaker, R.H.; Scudiero, D.A.; Melillo, G.; Currens, M.J.; Monks, A.P.; Rabow, A.A.; Covell, D.G.; Sausville, E.A. Application of high-throughput, molecular-targeted screening to anticancer drug discovery. Curr. Top. Med. Chem. 2002, 2, 229–246. [Google Scholar] [CrossRef] [PubMed]
- Hirayama, A.; Kami, K.; Sugimoto, M.; Sugawara, M.; Toki, N.; Onozuka, H.; Kinoshita, T.; Saito, N.; Ochiai, A.; Tomita, M.; et al. Quantitative metabolome profiling of colon and stomach cancer microenvironment by capillary electrophoresis time-of-flight mass spectrometry. Cancer Res. 2009, 69, 4918–4925. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Kunimoto, S.; Yamazaki, Y.; Kaminishi, M.; Esumi, H. Kigamicin D, a novel anticancer agent based on a new anti-austerity strategy targeting cancer cells’ tolerance to nutrient starvation. Cancer Sci. 2004, 95, 547–552. [Google Scholar] [CrossRef] [PubMed]
- Awale, S.; Lu, J.; Kalauni, S.K.; Kurashima, Y.; Tezuka, Y.; Kadota, S.; Esumi, H. Identification of arctigenin as an antitumor agent having the ability to eliminate the tolerance of cancer cells to nutrient starvation. Cancer Res. 2006, 66, 1751–1757. [Google Scholar] [PubMed]
- Momose, I.; Ohba, S.; Tatsuda, D.; Kawada, M.; Masuda, T.; Tsujiuchi, G.; Yamori, T.; Esumi, H.; Ikeda, D. Mitochondrial inhibitors show preferential cytotoxicity to human pancreatic cancer PANC-1 cells under glucose-deprived conditions. Biochem. Biophys. Res. Commun. 2010, 392, 460–466. [Google Scholar] [CrossRef] [PubMed]
- Tomitsuka, E.; Kita, K.; Esumi, H. An anticancer agent, pyrvinium pamoate inhibits the NADH-fumarate reductase system—A unique mitochondrial energy metabolism in tumour microenvironments. J. Biochem. 2012, 152, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Qi, C.; Sun, X.; Ma, X.; Zhang, H.; Hu, L.; Yuan, J.; Yu, Q. Arctigenin preferentially induces tumor cell death under glucose deprivation by inhibiting cellular energy metabolism. Biochem. Pharmacol. 2012, 84, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Cross, R.L.; Kohlbrenner, W.E. The mode of inhibition of oxidative phosphorylation by efrapeptin (A23871). Evidence for an alternating site mechanism for ATP synthesis. J. Biol. Chem. 1978, 253, 4865–4873. [Google Scholar] [PubMed]
- Esumi, H.; Lu, J.; Kurashima, Y.; Hanaoka, T. Antitumor activity of pyrvinium pamoate, 6-(dimethylamino)-2-[2-(2,5-dimethyl-1-phenyl-1H-pyrrol-3-yl)ethenyl]-1-me thyl-quinolinium pamoate salt, showing preferential cytotoxicity during glucose starvation. Cancer Sci. 2004, 95, 685–690. [Google Scholar] [CrossRef] [PubMed]
- Tomitsuka, E.; Kita, K.; Esumi, H. The NADH-fumarate reductase system, a novel mitochondrial energy metabolism, is a new target for anticancer therapy in tumor microenvironments. Ann. N. Y. Acad. Sci. 2010, 1201, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Senkowski, W.; Zhang, X.; Olofsson, M.H.; Isacson, R.; Hoglund, U.; Gustafsson, M.; Nygren, P.; Linder, S.; Larsson, R.; Fryknas, M. Three-dimensional cell culture-based screening identifies the anthelmintic drug nitazoxanide as a candidate for treatment of colorectal cancer. Mol. Cancer Ther. 2015. [Google Scholar] [CrossRef] [PubMed]
- Izuishi, K.; Kato, K.; Ogura, T.; Kinoshita, T.; Esumi, H. Remarkable tolerance of tumor cells to nutrient deprivation: Possible new biochemical target for cancer therapy. Cancer Res. 2000, 60, 6201–6207. [Google Scholar] [PubMed]
- Kato, K.; Ogura, T.; Kishimoto, A.; Minegishi, Y.; Nakajima, N.; Miyazaki, M.; Esumi, H. Critical roles of AMP-activated protein kinase in constitutive tolerance of cancer cells to nutrient deprivation and tumor formation. Oncogene 2002, 21, 6082–6090. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, R.M. Cell and environment interactions in tumor microregions: The multicell spheroid model. Science 1988, 240, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Hirschhaeuser, F.; Menne, H.; Dittfeld, C.; West, J.; Mueller-Klieser, W.; Kunz-Schughart, L.A. Multicellular tumor spheroids: An underestimated tool is catching up again. J. Biotechnol. 2010, 148, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Motti, M.L.; De Marco, C.; Califano, D.; De Gisi, S.; Malanga, D.; Troncone, G.; Persico, A.; Losito, S.; Fabiani, F.; Santoro, M.; et al. Loss of p27 expression through RAS→BRAF→MAP kinase-dependent pathway in human thyroid carcinomas. Cell Cycle 2007, 6, 2817–2825. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mueller-Klieser, W. Multicellular spheroids. A review on cellular aggregates in cancer research. J. Cancer Res. Clin. Oncol. 1987, 113, 101–122. [Google Scholar] [CrossRef] [PubMed]
- Mueller-Klieser, W.; Freyer, J.P.; Sutherland, R.M. Influence of glucose and oxygen supply conditions on the oxygenation of multicellular spheroids. Br. J. Cancer 1986, 53, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Bredel-Geissler, A.; Karbach, U.; Walenta, S.; Vollrath, L.; Mueller-Klieser, W. Proliferation-associated oxygen consumption and morphology of tumor cells in monolayer and spheroid culture. J. Cell. Physiol. 1992, 153, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Kunz-Schughart, L.A.; Habbersett, R.C.; Freyer, J.P. Impact of proliferative activity and tumorigenic conversion on mitochondrial function of fibroblasts in 2D and 3D culture. Cell Biol. Int. 2001, 25, 919–930. [Google Scholar] [CrossRef] [PubMed]
- Casciari, J.J.; Sotirchos, S.V.; Sutherland, R.M. Glucose diffusivity in multicellular tumor spheroids. Cancer Res. 1988, 48, 3905–3909. [Google Scholar] [PubMed]
- Teutsch, H.F.; Goellner, A.; Mueller-Klieser, W. Glucose levels and succinate and lactate dehydrogenase activity in EMT6/Ro tumor spheroids. Eur. J. Cell Biol. 1995, 66, 302–307. [Google Scholar] [PubMed]
- Li, C.K. The role of glucose in the growth of 9L multicell tumor spheroids. Cancer 1982, 50, 2074–2078. [Google Scholar] [CrossRef]
- Li, C.K. The glucose distribution in 9L rat brain multicell tumor spheroids and its effect on cell necrosis. Cancer 1982, 50, 2066–2073. [Google Scholar] [CrossRef]
- Kunz-Schughart, L.A.; Doetsch, J.; Mueller-Klieser, W.; Groebe, K. Proliferative activity and tumorigenic conversion: Impact on cellular metabolism in 3-D culture. Am. J. Physiol. Cell Physiol. 2000, 278, C765–C780. [Google Scholar] [PubMed]
- Acker, H.; Carlsson, J.; Mueller-Klieser, W.; Sutherland, R.M. Comparative pO2 measurements in cell spheroids cultured with different techniques. Br. J. Cancer 1987, 56, 325–327. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, J.; Ebner, R.; Kunz-Schughart, L.A. Experimental anti-tumor therapy in 3-D: Spheroids—Old hat or new challenge? Int. J. Radiat. Biol. 2007, 83, 849–871. [Google Scholar] [CrossRef] [PubMed]
- Fayad, W.; Rickardson, L.; Haglund, C.; Olofsson, M.H.; D’Arcy, P.; Larsson, R.; Linder, S.; Fryknas, M. Identification of agents that induce apoptosis of multicellular tumour spheroids: Enrichment for mitotic inhibitors with hydrophobic properties. Chem. Biol. Drug Des. 2011, 78, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Kelm, J.M.; Timmins, N.E.; Brown, C.J.; Fussenegger, M.; Nielsen, L.K. Method for generation of homogeneous multicellular tumor spheroids applicable to a wide variety of cell types. Biotechnol. Bioeng. 2003, 83, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, R.; Fayad, W.; Schwarz, S.; Berndtsson, M.; Linder, S. Screening for compounds that induce apoptosis of cancer cells grown as multicellular spheroids. J. Biomol. Screen. 2008, 13, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, J.; Seidel, C.; Ebner, R.; Kunz-Schughart, L.A. Spheroid-based drug screen: Considerations and practical approach. Nat. Protoc. 2009, 4, 309–324. [Google Scholar] [CrossRef] [PubMed]
- Chan, H.F.; Zhang, Y.; Ho, Y.P.; Chiu, Y.L.; Jung, Y.; Leong, K.W. Rapid formation of multicellular spheroids in double-emulsion droplets with controllable microenvironment. Sci. Rep. 2013, 3, 3462. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, J.; Eder, W.; Castaneda, J.; Doss, M.; Huber, E.; Ebner, R.; Kunz-Schughart, L.A. A reliable tool to determine cell viability in complex 3-D culture: The acid phosphatase assay. J. Biomol. Screen. 2007, 12, 925–937. [Google Scholar] [CrossRef] [PubMed]
- Mellor, H.R.; Ferguson, D.J.; Callaghan, R. A model of quiescent tumour microregions for evaluating multicellular resistance to chemotherapeutic drugs. Br. J. Cancer 2005, 93, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, C.; Riefke, B.; Grundemann, S.; Krebs, A.; Christian, S.; Prinz, F.; Osterland, M.; Golfier, S.; Rase, S.; Ansari, N.; et al. 3D high-content screening for the identification of compounds that target cells in dormant tumor spheroid regions. Exp. Cell Res. 2014, 323, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Fryknas, M.; Hernlund, E.; Fayad, W.; De Milito, A.; Olofsson, M.H.; Gogvadze, V.; Dang, L.; Pahlman, S.; Schughart, L.A.; et al. Induction of mitochondrial dysfunction as a strategy for targeting tumour cells in metabolically compromised microenvironments. Nat. Commun. 2014, 5, 3295. [Google Scholar] [CrossRef] [PubMed]
- De Carvalho, L.P.; Darby, C.M.; Rhee, K.Y.; Nathan, C. Nitazoxanide disrupts membrane potential and intrabacterial pH homeostasis of Mycobacterium tuberculosis. ACS Med. Chem. Lett. 2011, 2, 849–854. [Google Scholar] [CrossRef] [PubMed]
- Jurgeit, A.; McDowell, R.; Moese, S.; Meldrum, E.; Schwendener, R.; Greber, U.F. Niclosamide is a proton carrier and targets acidic endosomes with broad antiviral effects. PLoS Pathog. 2012, 8, e1002976. [Google Scholar] [CrossRef] [PubMed]
- Skuce, P.J.; Fairweather, I. The effect of the hydrogen ionophore closantel upon the pharmacology and ultrastructure of the adult liver fluke Fasciola hepatica. Parasitol. Res. 1990, 76, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.B.; Onder, T.T.; Jiang, G.; Tao, K.; Kuperwasser, C.; Weinberg, R.A.; Lander, E.S. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell 2009, 138, 645–659. [Google Scholar] [CrossRef] [PubMed]
- Mitani, M.; Yamanishi, T.; Miyazaki, Y.; Otake, N. Salinomycin effects on mitochondrial ion translocation and respiration. Antimicrob. Agents Chemother. 1976, 9, 655–660. [Google Scholar] [CrossRef] [PubMed]
- Clarke, M.F.; Dick, J.E.; Dirks, P.B.; Eaves, C.J.; Jamieson, C.H.; Jones, D.L.; Visvader, J.; Weissman, I.L.; Wahl, G.M. Cancer stem cells—Perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006, 66, 9339–9344. [Google Scholar] [CrossRef] [PubMed]
- Costello, R.T.; Mallet, F.; Gaugler, B.; Sainty, D.; Arnoulet, C.; Gastaut, J.A.; Olive, D. Human acute myeloid leukemia CD34+/CD38− progenitor cells have decreased sensitivity to chemotherapy and Fas-induced apoptosis, reduced immunogenicity, and impaired dendritic cell transformation capacities. Cancer Res. 2000, 60, 4403–4411. [Google Scholar] [PubMed]
- Dean, M.; Fojo, T.; Bates, S. Tumour stem cells and drug resistance. Nat. Rev. Cancer 2005, 5, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Hegde, G.V.; de la Cruz, C.; Eastham-Anderson, J.; Zheng, Y.; Sweet-Cordero, E.A.; Jackson, E.L. Residual tumor cells that drive disease relapse after chemotherapy do not have enhanced tumor initiating capacity. PLoS ONE 2012, 7, e45647. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Suda, T. Metabolic requirements for the maintenance of self-renewing stem cells. Nat. Rev. Mol. Cell Biol. 2014, 15, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.; Shim, J.S. Existing drugs and their application in drug discovery targeting cancer stem cells. Arch. Pharm. Res. 2015, 38, 1617–1626. [Google Scholar] [CrossRef] [PubMed]
- Story, P.; Doube, A. A case of human poisoning by salinomycin, an agricultural antibiotic. N. Z. Med. J. 2004, 117, 1190. [Google Scholar]
- Yo, Y.T.; Lin, Y.W.; Wang, Y.C.; Balch, C.; Huang, R.L.; Chan, M.W.; Sytwu, H.K.; Chen, C.K.; Chang, C.C.; Nephew, K.P.; et al. Growth inhibition of ovarian tumor-initiating cells by niclosamide. Mol. Cancer Ther. 2012, 11, 1703–1712. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.C.; Chao, T.K.; Chang, C.C.; Yo, Y.T.; Yu, M.H.; Lai, H.C. Drug screening identifies niclosamide as an inhibitor of breast cancer stem-like cells. PLoS ONE 2013, 8, e74538. [Google Scholar] [CrossRef] [PubMed]
- Sztiller-Sikorska, M.; Koprowska, K.; Majchrzak, K.; Hartman, M.; Czyz, M. Natural compounds’ activity against cancer stem-like or fast-cycling melanoma cells. PLoS ONE 2014, 9, e90783. [Google Scholar] [CrossRef] [PubMed]
- Inouye, Y.; Okada, H.; Uno, J.; Arai, T.; Nakamura, S. Effects of streptonigrin derivatives and sakyomicin A on the respiration of isolated rat liver mitochondria. J. Antibiot. 1986, 39, 550–556. [Google Scholar] [CrossRef] [PubMed]
- Brunmair, B.; Staniek, K.; Gras, F.; Scharf, N.; Althaym, A.; Clara, R.; Roden, M.; Gnaiger, E.; Nohl, H.; Waldhausl, W.; et al. Thiazolidinediones, like metformin, inhibit respiratory complex I: A common mechanism contributing to their antidiabetic actions? Diabetes 2004, 53, 1052–1059. [Google Scholar] [CrossRef] [PubMed]
- Owen, M.R.; Doran, E.; Halestrap, A.P. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J. 2000, 348 Pt 3, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Andrzejewski, S.; Gravel, S.P.; Pollak, M.; St-Pierre, J. Metformin directly acts on mitochondria to alter cellular bioenergetics. Cancer Metab. 2014, 2, 12. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Wondisford, F.E. Metformin action: Concentrations matter. Cell Metab. 2015, 21, 159–162. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Madiraju, A.K.; Erion, D.M.; Rahimi, Y.; Zhang, X.M.; Braddock, D.T.; Albright, R.A.; Prigaro, B.J.; Wood, J.L.; Bhanot, S.; MacDonald, M.J.; et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature 2014, 510, 542–546. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, E.; Cioffi, M.; Sancho, P.; Sanchez-Ripoll, Y.; Trabulo, S.M.; Dorado, J.; Balic, A.; Hidalgo, M.; Heeschen, C. Metformin targets the metabolic achilles heel of human pancreatic cancer stem cells. PLoS ONE 2013, 8, e76518. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.M.; Donnelly, L.A.; Emslie-Smith, A.M.; Alessi, D.R.; Morris, A.D. Metformin and reduced risk of cancer in diabetic patients. BMJ 2005, 330, 1304–1305. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, H.A.; Iliopoulos, D.; Tsichlis, P.N.; Struhl, K. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res. 2009, 69, 7507–7511. [Google Scholar] [CrossRef] [PubMed]
- Rocha, G.Z.; Dias, M.M.; Ropelle, E.R.; Osorio-Costa, F.; Rossato, F.A.; Vercesi, A.E.; Saad, M.J.; Carvalheira, J.B. Metformin amplifies chemotherapy-induced AMPK activation and antitumoral growth. Clin. Cancer Res. 2011, 17, 3993–4005. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Qian, Y.; Wu, S. The Warburg effect: Evolving interpretations of an established concept. Free Radic. Biol. Med. 2015, 79, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Christofk, H.R.; Vander Heiden, M.G.; Harris, M.H.; Ramanathan, A.; Gerszten, R.E.; Wei, R.; Fleming, M.D.; Schreiber, S.L.; Cantley, L.C. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 2008, 452, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Ko, Y.H.; Pedersen, P.L.; Geschwind, J.F. Glucose catabolism in the rabbit VX2 tumor model for liver cancer: Characterization and targeting hexokinase. Cancer Lett. 2001, 173, 83–91. [Google Scholar] [CrossRef]
- Shoshan, M.C. 3-Bromopyruvate: Targets and outcomes. J. Bioenerg. Biomembr. 2012, 44, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, S.; Archer, S.L.; Allalunis-Turner, J.; Haromy, A.; Beaulieu, C.; Thompson, R.; Lee, C.T.; Lopaschuk, G.D.; Puttagunta, L.; Bonnet, S.; et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 2007, 11, 37–51. [Google Scholar] [CrossRef] [PubMed]
- Dunbar, E.M.; Coats, B.S.; Shroads, A.L.; Langaee, T.; Lew, A.; Forder, J.R.; Shuster, J.J.; Wagner, D.A.; Stacpoole, P.W. Phase 1 trial of dichloroacetate (DCA) in adults with recurrent malignant brain tumors. Investig. New Drugs 2014, 32, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.S.; Thompson, C.B. Metabolic reprogramming: A cancer hallmark even warburg did not anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Parks, S.K.; Chiche, J.; Pouyssegur, J. Disrupting proton dynamics and energy metabolism for cancer therapy. Nat. Rev. Cancer 2013, 13, 611–623. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Kepp, O.; Vander Heiden, M.G.; Kroemer, G. Metabolic targets for cancer therapy. Nat. Rev. Drug Discov. 2013, 12, 829–846. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar] [PubMed]
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, S.E.; Chandel, N.S. Targeting mitochondria metabolism for cancer therapy. Nat. Chem. Biol. 2015, 11, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Zu, X.L.; Guppy, M. Cancer metabolism: Facts, fantasy, and fiction. Biochem. Biophys. Res. Commun. 2004, 313, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Mandujano-Tinoco, E.A.; Gallardo-Pérez, J.C.; Marín-Hernández, A.; Moreno-Sánchez, R.; Rodríguez-Enríquez, S. Anti-mitochondrial therapy in human breast cancer multi-cellular spheroids. Biochim. Biophys. Acta 2013, 1833, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Mullen, A.R.; DeBerardinis, R.J. Genetically-defined metabolic reprogramming in cancer. Trends Endocrinol. Metab. 2012, 23, 552–559. [Google Scholar] [CrossRef] [PubMed]
- Zinkewich-Peotti, K.; Parent, M.; Morais, R. On the tumorigenicity of mitochondrial DNA-depleted avian cells. Cancer Lett. 1991, 59, 119–124. [Google Scholar] [CrossRef]
- Morais, R.; Zinkewich-Peotti, K.; Parent, M.; Wang, H.; Babai, F.; Zollinger, M. Tumor-forming ability in athymic nude mice of human cell lines devoid of mitochondrial DNA. Cancer Res. 1994, 54, 3889–3896. [Google Scholar] [PubMed]
- Hayashi, J.; Takemitsu, M.; Nonaka, I. Recovery of the missing tumorigenicity in mitochondrial DNA-less HeLa cells by introduction of mitochondrial DNA from normal human cells. Somat. Cell Mol. Genet. 1992, 18, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Han, B.; Izumi, H.; Yasuniwa, Y.; Akiyama, M.; Yamaguchi, T.; Fujimoto, N.; Matsumoto, T.; Wu, B.; Tanimoto, A.; Sasaguri, Y.; et al. Human mitochondrial transcription factor A functions in both nuclei and mitochondria and regulates cancer cell growth. Biochem. Biophys. Res. Commun. 2011, 408, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Bhalla, K.; Hwang, B.J.; Dewi, R.E.; Ou, L.; Twaddel, W.; Fang, H.B.; Vafai, S.B.; Vazquez, F.; Puigserver, P.; Boros, L.; et al. PGC1α promotes tumor growth by inducing gene expression programs supporting lipogenesis. Cancer Res. 2011, 71, 6888–6898. [Google Scholar] [CrossRef] [PubMed]
- Eskey, C.J.; Koretsky, A.P.; Domach, M.M.; Jain, R.K. Role of oxygen vs. glucose in energy metabolism in a mammary carcinoma perfused ex vivo: Direct measurement by 31P NMR. Proc. Natl. Acad. Sci. USA 1993, 90, 2646–2650. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V.; Semenza, G.L. Oncogenic alterations of metabolism. Trends Biochem. Sci. 1999, 24, 68–72. [Google Scholar] [CrossRef]
- Metallo, C.M.; Gameiro, P.A.; Bell, E.L.; Mattaini, K.R.; Yang, J.; Hiller, K.; Jewell, C.M.; Johnson, Z.R.; Irvine, D.J.; Guarente, L.; et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 2012, 481, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Corbet, C.; Draoui, N.; Polet, F.; Pinto, A.; Drozak, X.; Riant, O.; Feron, O. The SIRT1/HIF2α axis drives reductive glutamine metabolism under chronic acidosis and alters tumor response to therapy. Cancer Res. 2014, 74, 5507–5519. [Google Scholar] [CrossRef] [PubMed]
- Whitaker-Menezes, D.; Martinez-Outschoorn, U.E.; Flomenberg, N.; Birbe, R.C.; Witkiewicz, A.K.; Howell, A.; Pavlides, S.; Tsirigos, A.; Ertel, A.; Pestell, R.G.; et al. Hyperactivation of oxidative mitochondrial metabolism in epithelial cancer cells in situ: Visualizing the therapeutic effects of metformin in tumor tissue. Cell Cycle 2011, 10, 4047–4064. [Google Scholar] [CrossRef] [PubMed]
- Sotgia, F.; Whitaker-Menezes, D.; Martinez-Outschoorn, U.E.; Salem, A.F.; Tsirigos, A.; Lamb, R.; Sneddon, S.; Hulit, J.; Howell, A.; Lisanti, M.P. Mitochondria "fuel" breast cancer metabolism: Fifteen markers of mitochondrial biogenesis label epithelial cancer cells, but are excluded from adjacent stromal cells. Cell Cycle 2012, 11, 4390–4401. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Qiao, M.; Zhang, Y.; Jiang, Y.; Wei, P.; Yao, J.; Gu, B.; Wang, Y.; Lu, J.; Wang, Z.; et al. Quantitative proteomics study of breast cancer cell lines isolated from a single patient: Discovery of TIMM17A as a marker for breast cancer. Proteomics 2010, 10, 1374–1390. [Google Scholar] [CrossRef] [PubMed]
- Aleskandarany, M.A.; Negm, O.H.; Rakha, E.A.; Ahmed, M.A.; Nolan, C.C.; Ball, G.R.; Caldas, C.; Green, A.R.; Tighe, P.J.; Ellis, I.O. TOMM34 expression in early invasive breast cancer: A biomarker associated with poor outcome. Breast Cancer Res. Treat. 2012, 136, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Salhab, M.; Patani, N.; Jiang, W.; Mokbel, K. High TIMM17A expression is associated with adverse pathological and clinical outcomes in human breast cancer. Breast Cancer 2012, 19, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Bernal, S.D.; Lampidis, T.J.; McIsaac, R.M.; Chen, L.B. Anticarcinoma activity in vivo of rhodamine 123, a mitochondrial-specific dye. Science 1983, 222, 169–172. [Google Scholar] [CrossRef] [PubMed]
- Lampidis, T.J.; Bernal, S.D.; Summerhayes, I.C.; Chen, L.B. Selective toxicity of rhodamine 123 in carcinoma cells in vitro. Cancer Res. 1983, 43, 716–720. [Google Scholar] [PubMed]
- Abou-Khalil, S.; Abou-Khalil, W.H.; Planas, L.; Tapiero, H.; Lampidis, T.J. Interaction of rhodamine 123 with mitochondria isolated from drug-sensitive and -resistant Friend leukemia cells. Biochem. Biophys. Res. Commun. 1985, 127, 1039–1044. [Google Scholar] [CrossRef]
- Cheng, G.; Zielonka, J.; Dranka, B.P.; McAllister, D.; Mackinnon, A.C., Jr.; Joseph, J.; Kalyanaraman, B. Mitochondria-targeted drugs synergize with 2-deoxyglucose to trigger breast cancer cell death. Cancer Res. 2012. [Google Scholar] [CrossRef] [PubMed]
- Schimmer, A.D.; Skrtic, M. Therapeutic potential of mitochondrial translation inhibition for treatment of acute myeloid leukemia. Expert Rev. Hematol. 2012, 5, 117–119. [Google Scholar] [CrossRef] [PubMed]
- Skrtic, M.; Sriskanthadevan, S.; Jhas, B.; Gebbia, M.; Wang, X.; Wang, Z.; Hurren, R.; Jitkova, Y.; Gronda, M.; Maclean, N.; et al. Inhibition of mitochondrial translation as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell 2011, 20, 674–688. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; De Milito, A.; Olofsson, M.H.; Gullbo, J.; D’Arcy, P.; Linder, S. Targeting Mitochondrial Function to Treat Quiescent Tumor Cells in Solid Tumors. Int. J. Mol. Sci. 2015, 16, 27313-27326. https://doi.org/10.3390/ijms161126020
Zhang X, De Milito A, Olofsson MH, Gullbo J, D’Arcy P, Linder S. Targeting Mitochondrial Function to Treat Quiescent Tumor Cells in Solid Tumors. International Journal of Molecular Sciences. 2015; 16(11):27313-27326. https://doi.org/10.3390/ijms161126020
Chicago/Turabian StyleZhang, Xiaonan, Angelo De Milito, Maria Hägg Olofsson, Joachim Gullbo, Padraig D’Arcy, and Stig Linder. 2015. "Targeting Mitochondrial Function to Treat Quiescent Tumor Cells in Solid Tumors" International Journal of Molecular Sciences 16, no. 11: 27313-27326. https://doi.org/10.3390/ijms161126020
APA StyleZhang, X., De Milito, A., Olofsson, M. H., Gullbo, J., D’Arcy, P., & Linder, S. (2015). Targeting Mitochondrial Function to Treat Quiescent Tumor Cells in Solid Tumors. International Journal of Molecular Sciences, 16(11), 27313-27326. https://doi.org/10.3390/ijms161126020