
Minimizing off-Target Mutagenesis Risks Caused by Programmable Nucleases

Abstract
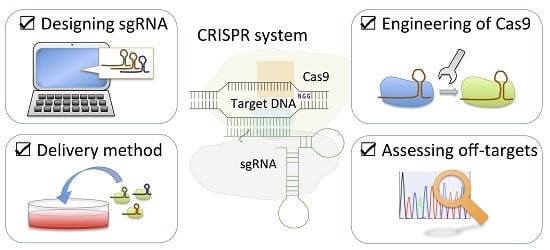
:
1. Introduction
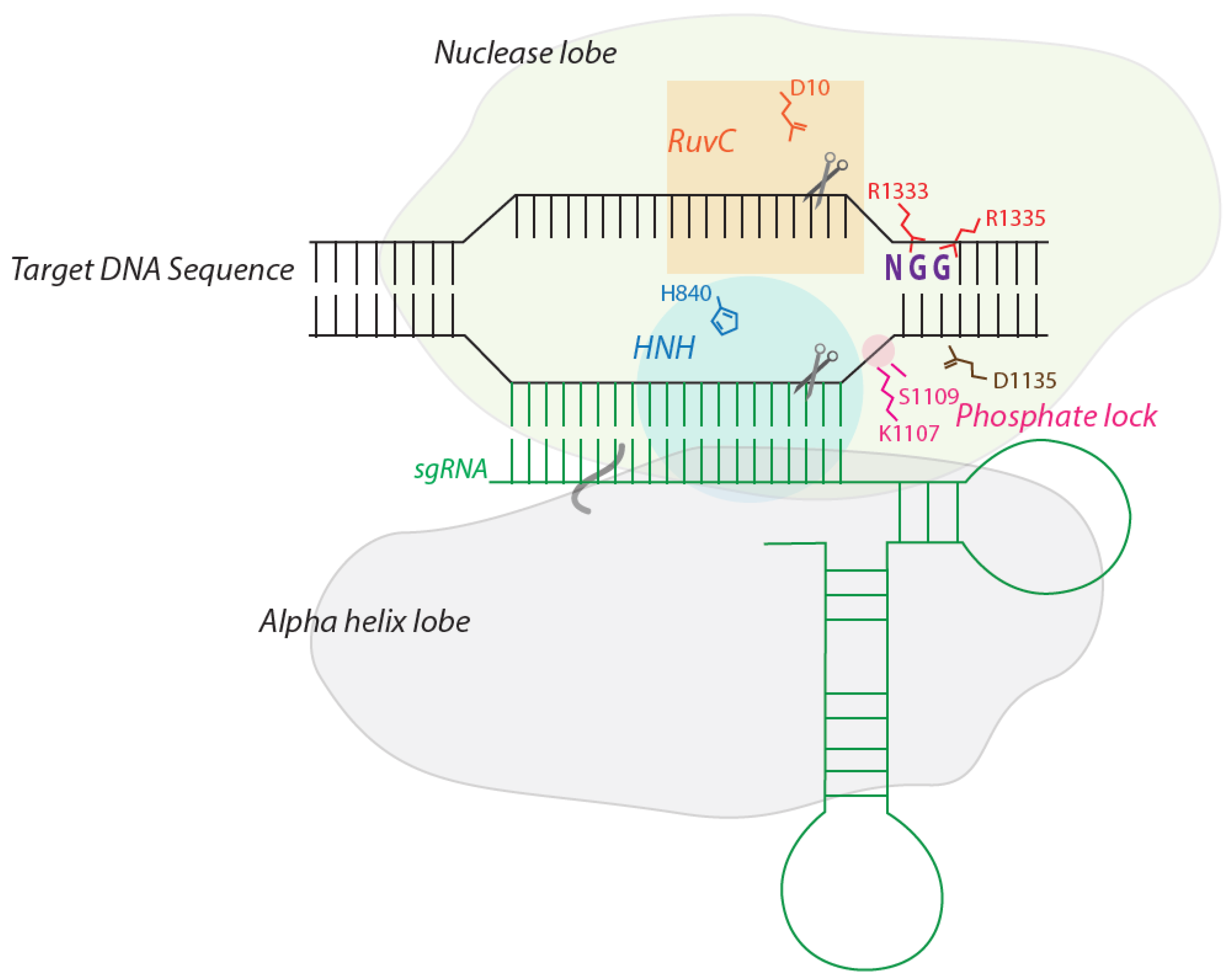
2. The Structure of Cas9: How it Recognizes and Cleaves DNA
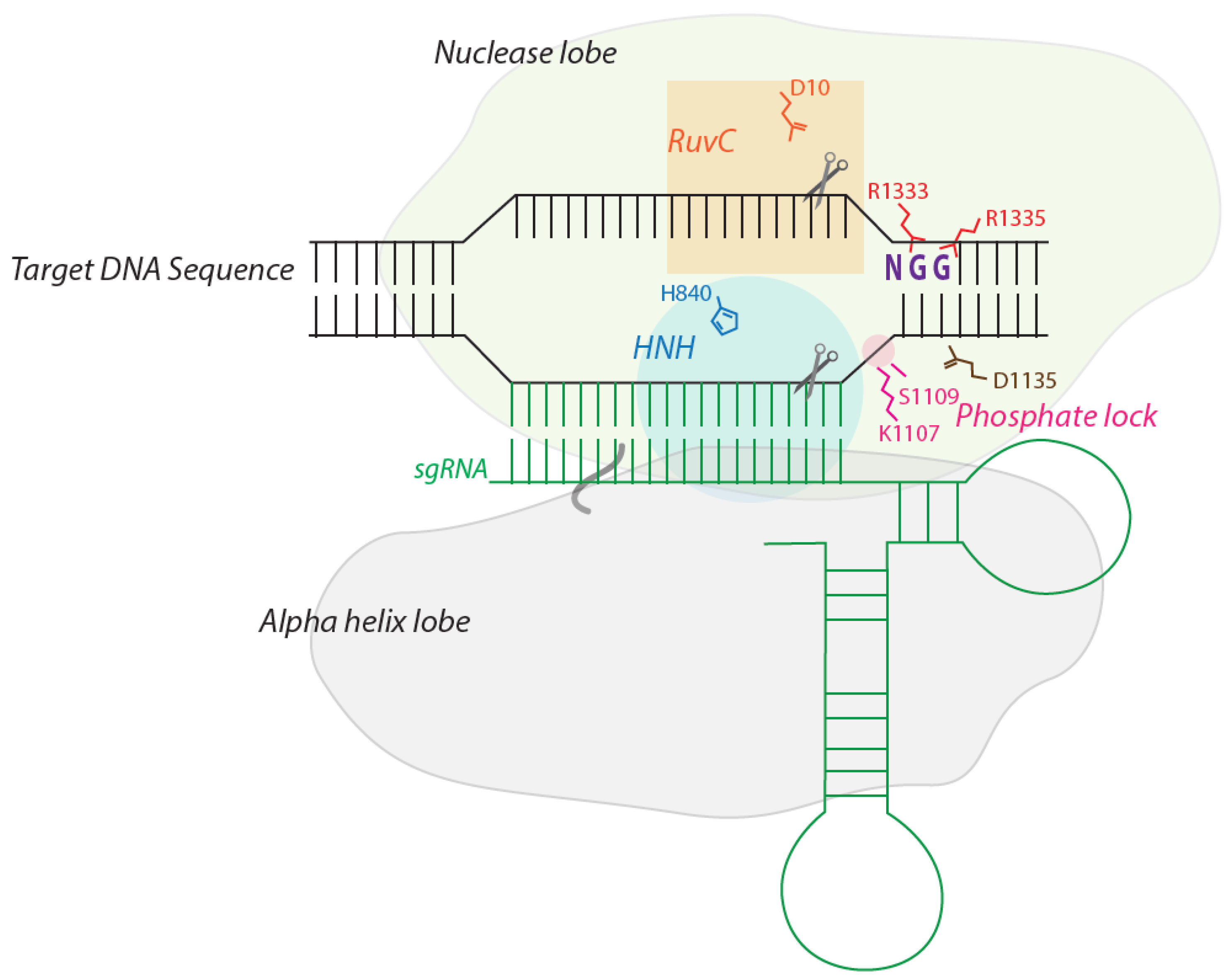
3. Designing sgRNA to Target a Genomic Target Site
4. Tools to Design Highly Specific sgRNA

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tool | Organism | Input (Length) | off-Target Sites | Reference |
|---|---|---|---|---|
| ZiFiT | Hs, Rn, Mm, Dr, Dm, Ce, Aa, Ec | Target sequence (<1000 bp) | Mismatches | [48] |
| CRISPR Design | >15 species | Target sequence (<250 bp) | Mismatches | [6] |
| Cas9 design | >10 species | Target sequence (>10 kbp) | Mismatches | [50] |
| E-CRISP | >15 species | Target sequence (>10 kbp), gene name | Mismatches | [52] |
| CasOT | Any species | Target sequence (>10 kbp) | Mismatches | [55] |
| Cas-OFFinder | >10 species | Designed sgRNA (10–25 nt) | Mismatches, insertion and deletions | [56] |
| CHOPCHOP | >20 species | Target sequence (>10 kbp) | Mismatches | [54] |
| GT-Scan | >20 species | Target sequence (<4000 bp) | Mismatches | [57] |
| sgRNAcas9 | Any species | Target sequence (>10 kbp) | Mismatches | [58] |
| CRISPR-P | >20 plants | Target sequence (<5 kbp) | Mismatches | [59] |
| COSMID | Hs, Mm, Rn, Ce, Mam, Dr | Designed sgRNA (10–55 nt) | Mismatches, insertions and deletions | [60] |
| sgRNA Designer | Hs, Mm | Target sequence (<10 kbp), gene ID | N.A. | [61] |
| iGEATs | Hs, Mm | Chromosomal locus, target sequence (<25 kb), gene name, gRNA | N.A. | [17] |
| CRISPRdirect | >15 species | Target sequence (<10 kbp) | Mismatches, insertions and deletions | [46] |
| CRISPR-ERA | Hs, Mm, Rn, Dr, Dm, Ce, Sc, Ec, Bs | Target sequence (<5 kbp), gene name | Mismatches | [62] |
| Protospacer Workbench | Any species | Target sequence (>10 kbp), gene name | Mismatches | [63] |

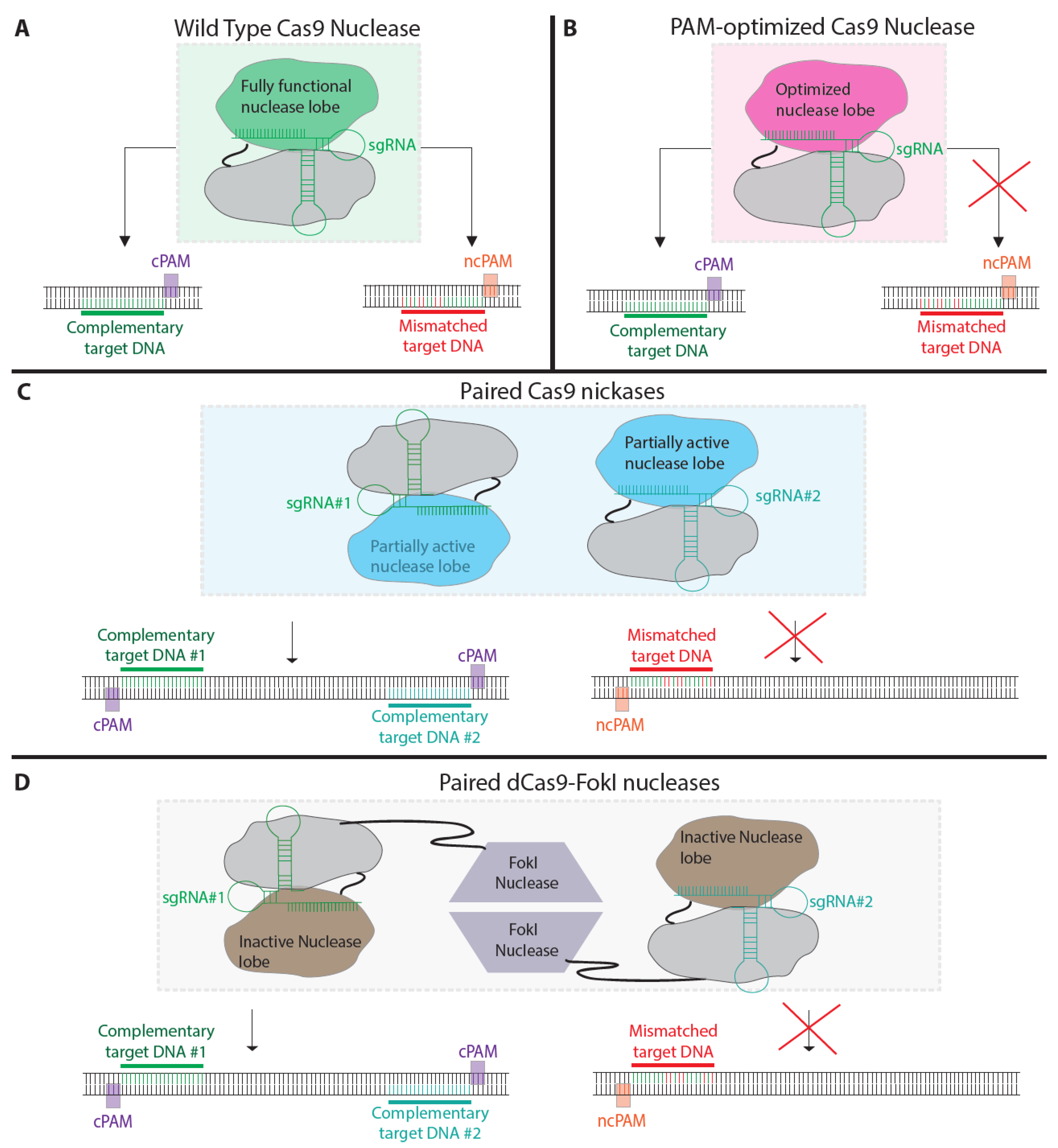
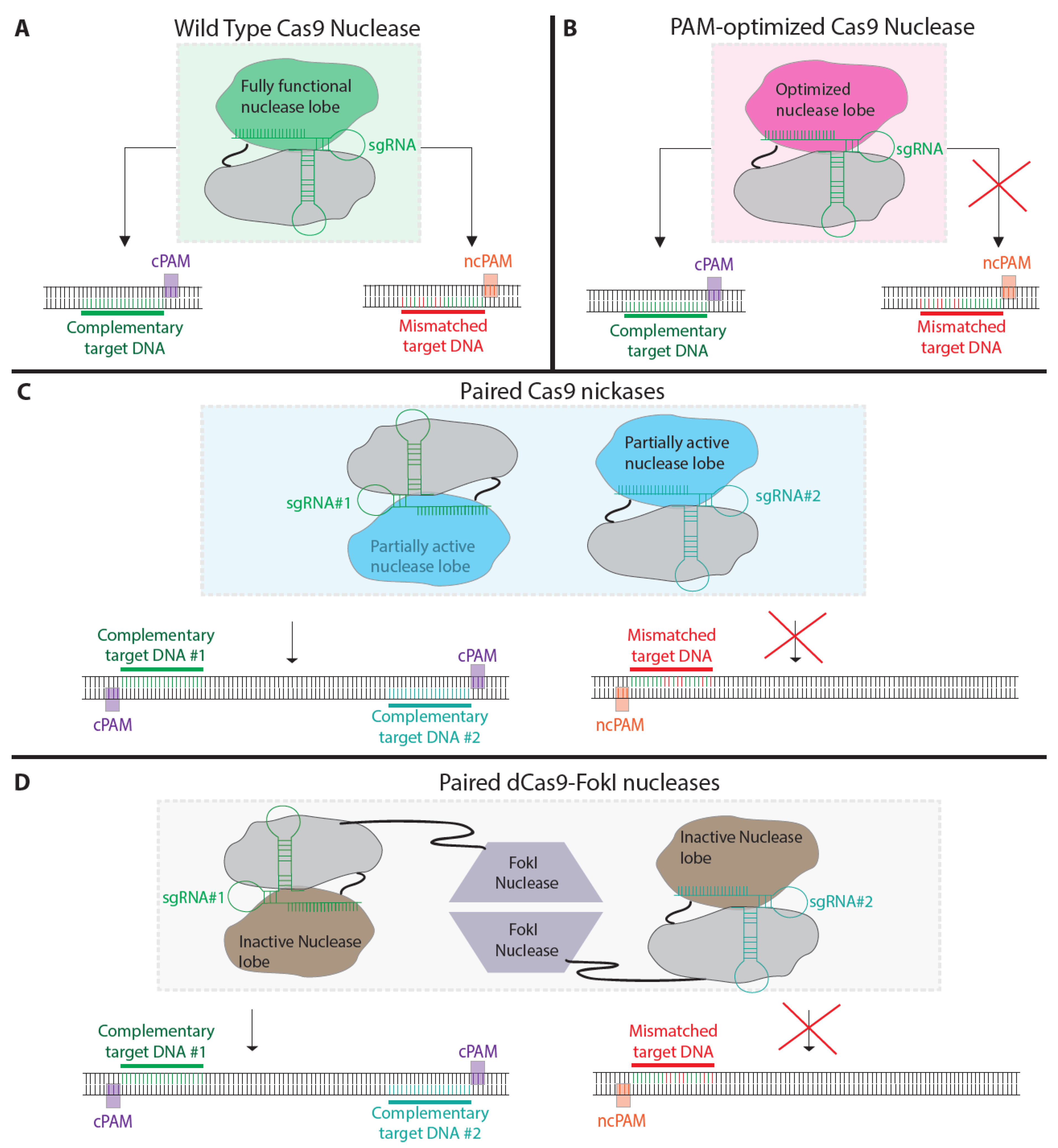
5. Modified Nucleases to Reduce off-Target Mutagenesis
6. Cas9 Delivery and Expression
7. Assessing off-Target Mutagenesis
8. Off-Target Analysis Based on Genome Editing Objectives
9. Target Cells Matter
| Cell Types | Target Gene | Programmable Nuclease Used | Detection Assay of off-Target Mutagenesis | off-Target Mutagenesis Detected | Ref. |
|---|---|---|---|---|---|
| U2OS, HEK293 and K562 cells | VEGFA, EMX1, RNF2, FANCF | CRISPR | EGFP reporter, T7EI | Yes | [42] |
| 293T | EMX1 | TALENs, CRISPR | amplicon seq, Surveyor assay | Yes | [6] |
| 293T | HBB, CCR5 | CRISPR | T7EI, Sanger sequencing | Yes | [105] |
| 293T | HBB, CCR5 | CRISPR | T7EI, Sanger sequencing | Yes | [69] |
| 293 and U2OS cells | VEFGA, EMX1, FANCF, RNF2 | CRISPR | GUIDE-seq, amplicon seq (AMP-based seq) | Yes | [97] |
| HAP1 cells, K562 cells, | HBB, VEFGA | CRISPR | Digenome-seq | Yes | [98] |
| 293T cells, A549 cells | RAG1, C-MYC, ATM | TALENs, CRISPR | HTGTS method | Yes | [94] |
| iPS cells | A1AT | ZFNs | CGH, SNP array, exome seq | No | [106] |
| myoblasts | DMD | TALENs | exome seq | No | [107] |
| iPS cells | PPP1R12C | TALENs, CRISPR | whole genome seq | No | [100] |
| ES cells, iPS cells | SORT1, LINC00116 | TALENs, CRISPR | whole genome seq | No | [102] |
| iPS cells | HBB | TALENs, HDAdV mediated HR | whole genome seq | No | [101] |
| 293FT cells, iPS cells | PPP1R12C, AKT2, CDK19, ATP6AP2, SLC35A2 | CRISPR | amplicon seq | No | [108] |
| iPS cells | DMD | TALENs and CRISPR | T7EI, amplicon seq, Karyotyping, CNV analysis, exome seq | No | [17] |
| iPS cells | PLN | TALEN | exome seq | No | [109] |
| iPS cells | F8 | CRISPR | amplicon seq | No | [21] |
10. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Bhaya, D.; Davison, M.; Barrangou, R. CRISPR-Cas systems in bacteria and archaea: versatile small RNAs for adaptive defense and regulation. Ann. Rev. Genet. 2011, 45, 273–297. [Google Scholar] [CrossRef] [PubMed]
- Wiedenheft, B.; Sternberg, S.H.; Doudna, J.A. RNA-guided genetic silencing systems in bacteria and archaea. Nature 2012, 482, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.W.; Kim, S.; Kim, J.M.; Kim, J.-S. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat. Biotechnol. 2013, 31, 230–232. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.D.; Scott, D.A.; Weinstein, J.A.; Ran, F.A.; Konermann, S.; Agarwala, V.; Li, Y.; Fine, E.J.; Wu, X.; Shalem, O.; et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013, 31, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Hotta, A.; Yamanaka, S. From Genomics to Gene Therapy: iPS Cells Meet Genome Editing. Annu. Rev. Genet. 2015, 49. [Google Scholar] [CrossRef] [PubMed]
- Tzur, Y.B.; Friedland, A.E.; Nadarajan, S.; Church, G.M.; Calarco, J.A.; Colaiácovo, M.P. Heritable custom genomic modifications in Caenorhabditis elegans via a CRISPR-Cas9 system. Genetics 2013, 195, 1181–1185. [Google Scholar] [CrossRef] [PubMed]
- Friedland, A.E.; Tzur, Y.B.; Esvelt, K.M.; Colaiácovo, M.P.; Church, G.M.; Calarco, J.A. Heritable genome editing in C. elegans via a CRISPR-Cas9 system. Nat. Methods 2013, 10, 741–743. [Google Scholar] [CrossRef] [PubMed]
- Chang, N.; Sun, C.; Gao, L.; Zhu, D.; Xu, X.; Zhu, X.; Xiong, J.-W.; Xi, J.J. Genome editing with RNA-guided Cas9 nuclease in zebrafish embryos. Cell Res. 2013, 23, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Platt, R.J.; Chen, S.; Zhou, Y.; Yim, M.J.; Swiech, L.; Kempton, H.R.; Dahlman, J.E.; Parnas, O.; Eisenhaure, T.M.; Jovanovic, M.; et al. CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell 2014, 159, 440–455. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yang, H.; Shivalila, C.S.; Dawlaty, M.M.; Cheng, A.W.; Zhang, F.; Jaenisch, R. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 2013, 153, 910–918. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Qiu, Z.; Shao, Y.; Chen, Y.; Guan, Y.; Liu, M.; Li, Y.; Gao, N.; Wang, L.; Lu, X.; et al. Heritable gene targeting in the mouse and rat using a CRISPR-Cas system. Nat. Biotechnol. 2013, 31, 681–683. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Teng, F.; Li, T.; Zhou, Q. Simultaneous generation and germline transmission of multiple gene mutations in rat using CRISPR-Cas systems. Nat. Biotechnol. 2013, 31, 684–686. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Xu, J.; Zhu, T.; Fan, J.; Lai, L.; Zhang, J.; Chen, Y.E. Effective gene targeting in rabbits using RNA-guided Cas9 nucleases. J. Mol. Cell Biol. 2014, 6, 97–99. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Shen, B.; Cui, Y.; Chen, Y.; Wang, J.; Wang, L.; Kang, Y.; Zhao, X.; Si, W.; Li, W.; et al. Generation of gene-modified cynomolgus monkey via Cas9/RNA-mediated gene targeting in one-cell embryos. Cell 2014, 156, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Li, H.L.; Fujimoto, N.; Sasakawa, N.; Shirai, S.; Ohkame, T.; Sakuma, T.; Tanaka, M.; Amano, N.; Watanabe, A.; Sakurai, H.; et al. Precise correction of the dystrophin gene in duchenne muscular dystrophy patient induced pluripotent stem cells by TALEN and CRISPR-Cas9. Stem Cell Rep. 2015, 4, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Li, H.L.; Nakano, T.; Hotta, A. Genetic correction using engineered nucleases for gene therapy applications. Dev. Growth Differ. 2014, 56, 63–77. [Google Scholar] [PubMed]
- Xie, F.; Ye, L.; Chang, J.C.; Beyer, A.I.; Wang, J.; Muench, M.O.; Kan, Y.W. Seamless gene correction of β-thalassemia mutations in patient-specific iPSCs using CRISPR/Cas9 and piggyBac. Genome Res. 2014, 24, 1526–1533. [Google Scholar] [CrossRef] [PubMed]
- Schwank, G.; Koo, B.-K.; Sasselli, V.; Dekkers, J.F.; Heo, I.; Demircan, T.; Sasaki, N.; Boymans, S.; Cuppen, E.; van der Ent, C.K.; et al. Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell 2013, 13, 653–658. [Google Scholar] [CrossRef] [PubMed]
- Park, C.-Y.; Kim, D.H.; Son, J.S.; Sung, J.J.; Lee, J.; Bae, S.; Kim, J.-H.; Kim, D.-W.; Kim, J.-S. Functional correction of large factor VIII gene chromosomal inversions in hemophilia A patient-derived iPSCs using CRISPR-Cas9. Cell Stem Cell 2015, 17, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Liang, D.; Wang, Y.; Bai, M.; Tang, W.; Bao, S.; Yan, Z.; Li, D.; Li, J. Correction of a genetic disease in mouse via use of CRISPR-Cas9. Cell Stem Cell 2013, 13, 659–662. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Xue, W.; Chen, S.; Bogorad, R.L.; Benedetti, E.; Grompe, M.; Koteliansky, V.; Sharp, P.A.; Jacks, T.; Anderson, D.G. Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nat. Biotechnol. 2014, 32, 551–553. [Google Scholar] [CrossRef] [PubMed]
- Long, C.; McAnally, J.R.; Shelton, J.M.; Mireault, A.A.; Bassel-Duby, R.; Olson, E.N. Prevention of muscular dystrophy in mice by CRISPR/Cas9-mediated editing of germline DNA. Science 2014, 345, 1184–1188. [Google Scholar] [CrossRef] [PubMed]
- Ramanan, V.; Shlomai, A.; Cox, D.B.T.; Schwartz, R.E.; Michailidis, E.; Bhatta, A.; Scott, D.A.; Zhang, F.; Rice, C.M.; Bhatia, S.N. CRISPR/Cas9 cleavage of viral DNA efficiently suppresses hepatitis B virus. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Seeger, C.; Sohn, J.A. Targeting hepatitis B virus with CRISPR/Cas9. Mol. Ther. Nucleic Acids 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Ebina, H.; Misawa, N.; Kanemura, Y.; Koyanagi, Y. Harnessing the CRISPR/Cas9 system to disrupt latent HIV-1 provirus. Sci. Rep. 2013, 3. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Wang, J.; Beyer, A.I.; Teque, F.; Cradick, T.J.; Qi, Z.; Chang, J.C.; Bao, G.; Muench, M.O.; Yu, J.; et al. Seamless modification of wild-type induced pluripotent stem cells to the natural CCR5Δ32 mutation confers resistance to HIV infection. Proc. Natl. Acad. Sci. USA 2014, 111, 9591–9596. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.-K.; Gu, Y.; Diaz, A.; Marlett, J.; Takahashi, Y.; Li, M.; Suzuki, K.; Xu, R.; Hishida, T.; Chang, C.-J.; et al. Use of the CRISPR/Cas9 system as an intracellular defense against HIV-1 infection in human cells. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Cornu, T.I.; Thibodeau-Beganny, S.; Guhl, E.; Alwin, S.; Eichtinger, M.; Joung, J.K.; Joung, J.K.; Cathomen, T. DNA-binding specificity is a major determinant of the activity and toxicity of zinc-finger nucleases. Mol. Ther. 2008, 16, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Porteus, M.H.; Baltimore, D. Chimeric nucleases stimulate gene targeting in human cells. Science 2003, 300, 763–763. [Google Scholar] [CrossRef] [PubMed]
- Guilinger, J.P.; Pattanayak, V.; Reyon, D.; Tsai, S.Q.; Sander, J.D.; Joung, J.K.; Liu, D.R. Broad specificity profiling of TALENs results in engineered nucleases with improved DNA-cleavage specificity. Nat. Methods 2014, 11, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Huang, S.; Zhao, X.; Wright, D.A.; Carpenter, S.; Spalding, M.H.; Weeks, D.P.; Yang, B. Modularly assembled designer TAL effector nucleases for targeted gene knockout and gene replacement in eukaryotes. Nucleic Acids Res. 2011, 39, 6315–6325. [Google Scholar] [CrossRef] [PubMed]
- Mussolino, C.; Morbitzer, R.; Lütge, F.; Dannemann, N.; Lahaye, T.; Cathomen, T. A novel TALE nuclease scaffold enables high genome editing activity in combination with low toxicity. Nucleic Acids Res. 2011, 39, 9283–9293. [Google Scholar] [CrossRef] [PubMed]
- Pattanayak, V.; Guilinger, J.P.; Liu, D.R. Determining the specificities of TALENs, Cas9, and other genome-editing enzymes. Methods Enzymol. 2014, 546, 47–78. [Google Scholar] [PubMed]
- Jinek, M.; Jiang, F.; Taylor, D.W.; Sternberg, S.H.; Kaya, E.; Ma, E.; Anders, C.; Hauer, M.; Zhou, K.; Lin, S.; et al. Structures of Cas9 Endonucleases Reveal RNA-Mediated Conformational Activation. Science 2014, 343. [Google Scholar] [CrossRef] [PubMed]
- Nishimasu, H.; Ran, F.A.; Hsu, P.D.; Konermann, S.; Shehata, S.I.; Dohmae, N.; Ishitani, R.; Zhang, F.; Nureki, O. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell 2014, 156, 935–949. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Zhou, K.; Ma, L.; Gressel, S.; Doudna, J.A. STRUCTURAL BIOLOGY. A Cas9-guide RNA complex preorganized for target DNA recognition. Science 2015, 348, 1477–1481. [Google Scholar] [CrossRef] [PubMed]
- Anders, C.; Niewoehner, O.; Duerst, A.; Jinek, M. Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature 2014, 513, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ge, X.; Yang, F.; Zhang, L.; Zheng, J.; Tan, X.; Jin, Z.-B.; Qu, J.; Gu, F. Comparison of non-canonical PAMs for CRISPR/Cas9-mediated DNA cleavage in human cells. Sci. Rep. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Kuscu, C.; Arslan, S.; Singh, R.; Thorpe, J.; Adli, M. Genome-wide analysis reveals characteristics of off-target sites bound by the Cas9 endonuclease. Nat. Biotechnol. 2014, 32, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Foden, J.A.; Khayter, C.; Maeder, M.L.; Reyon, D.; Joung, J.K.; Sander, J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013, 31, 822–826. [Google Scholar] [CrossRef] [PubMed]
- Sternberg, S.H.; Redding, S.; Jinek, M.; Greene, E.C.; Doudna, J.A. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 2014, 507, 62–67. [Google Scholar] [CrossRef] [PubMed]
- CRISPR Design. Available online: http://crispr.mit.edu (accessed on 14 October 2015).
- CRISPRdirect. Available online: http://crispr.dbcls.jp/ (accessed on 14 October 2015).
- Naito, Y.; Hino, K.; Bono, H.; Ui-Tei, K. CRISPRdirect: Software for designing CRISPR/Cas guide RNA with reduced off-target sites. Bioinformatics 2015, 31, 1120–1123. [Google Scholar] [CrossRef] [PubMed]
- ZiFit. Available online: http://zifit.partners.org/ZiFiT/ (accessed on 14 October 2015).
- Sander, J.D.; Maeder, M.L.; Reyon, D.; Voytas, D.F.; Joung, J.K.; Dobbs, D. ZiFiT (Zinc Finger Targeter): An updated zinc finger engineering tool. Nucleic Acids Res. 2010, 38, W462–W468. [Google Scholar] [CrossRef] [PubMed]
- Cas9 Design. Available online: http://cas9.cbi.pku.edu.cn (accessed on 14 October 2015).
- Ma, M.; Ye, A.Y.; Zheng, W.; Kong, L. A guide RNA sequence design platform for the CRISPR/Cas9 system for model organism genomes. BioMed. Res. Int. 2013, 2013, 1–4. [Google Scholar] [CrossRef] [PubMed]
- E-CRISP. Available online: http://www.e-crisp.org/E-CRISP/ (accessed on 14 October 2015).
- Heigwer, F.; Kerr, G.; Boutros, M. E-CRISP: Fast CRISPR target site identification. Nat. Methods 2014, 11, 122–123. [Google Scholar] [CrossRef] [PubMed]
- CHOPCHOP. Available online: https://chopchop.rc.fas.harvard.edu (accessed on 14 October 2015).
- Montague, T.G.; Cruz, J.M.; Gagnon, J.A.; Church, G.M.; Valen, E. CHOPCHOP: A CRISPR/Cas9 and TALEN web tool for genome editing. Nucleic Acids Res. 2014, 42, W401–W407. [Google Scholar] [CrossRef] [PubMed]
- Xiao, A.; Cheng, Z.; Kong, L.; Zhu, Z.; Lin, S.; Gao, G.; Zhang, B. CasOT: A genome-wide Cas9/gRNA off-target searching tool. Bioinformatics 2014, 30, 1180–1182. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.; Park, J.; Kim, J.-S. Cas-OFFinder: A fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 2014, 30, 1473–1475. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, A.; Bailey, T.L. GT-Scan: Identifying unique genomic targets. Bioinformatics 2014, 30, 2673–2675. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.; Shen, B.; Zhang, C.; Huang, X.; Zhang, Y. sgRNAcas9: A software package for designing CRISPR sgRNA and evaluating potential off-target cleavage sites. PLoS ONE 2014, 9, e100448–e100449. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Lu, L.; Liu, H.-Y.; Li, Sen; Xing, F.; Chen, L.-L. CRISPR-P: A web tool for synthetic single-guide RNA design of CRISPR-system in plants. Mol. Plant 2014, 7, 1494–1496. [Google Scholar] [CrossRef] [PubMed]
- Cradick, T.J.; Qiu, P.; Lee, C.M.; Fine, E.J.; Bao, G. COSMID: A web-based tool for identifying and validating CRISPR/Cas off-target sites. Mol. Ther. Nucleic Acids 2014, 3, e214–10. [Google Scholar] [CrossRef] [PubMed]
- Doench, J.G.; Hartenian, E.; Graham, D.B.; Tothova, Z.; Hegde, M.; Smith, I.; Sullender, M.; Ebert, B.L.; Xavier, R.J.; Root, D.E. Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nat. Biotechnol. 2014, 32, 1262–1267. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wei, Z.; Dominguez, A.; Li, Y.; Wang, X.; Qi, L.S. CRISPR-ERA: A comprehensive design tool for CRISPR-mediated gene editing, repression and activation. Bioinformatics 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- MacPherson, C.R.; Scherf, A. Flexible guide-RNA design for CRISPR applications using Protospacer Workbench. Nat. Biotechnol. 2015, 33, 805–806. [Google Scholar] [CrossRef] [PubMed]
- CasOT. Available online: http://eendb.zfgenetics.org/casot/ (accessed on 14 October 2015).
- sgRNAcas9. Available online: http://www.biootools.com (accessed on 14 October 2015).
- Wang, T.; Wei, J.J.; Sabatini, D.M.; Lander, E.S. Genetic screens in human cells using the CRISPR-Cas9 system. Science 2014, 343, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Cas-OFFinder. Available online: http://www.rgenome.net/cas-offinder/ (accessed on 14 October 2015).
- COSMID. Available online: https://crispr.bme.gatech.edu (accessed on 14 October 2015).
- Lin, Y.; Cradick, T.J.; Brown, M.T.; Deshmukh, H.; Ranjan, P.; Sarode, N.; Wile, B.M.; Vertino, P.M.; Stewart, F.J.; Bao, G. CRISPR/Cas9 systems have off-target activity with insertions or deletions between target DNA and guide RNA sequences. Nucleic Acids Res. 2014, 42, 7473–7485. [Google Scholar] [CrossRef] [PubMed]
- Gagnon, J.A.; Valen, E.; Thyme, S.B.; Huang, P.; Akhmetova, L.; Ahkmetova, L.; Pauli, A.; Montague, T.G.; Zimmerman, S.; Richter, C.; et al. Efficient mutagenesis by Cas9 protein-mediated oligonucleotide insertion and large-scale assessment of single-guide RNAs. PLoS ONE 2014, 9, e98186. [Google Scholar] [CrossRef] [PubMed]
- Chari, R.; Mali, P.; Moosburner, M.; Church, G.M. Unraveling CRISPR-Cas9 genome engineering parameters via a library-on-library approach. Nat. Methods 2015, 12, 823–826. [Google Scholar] [CrossRef] [PubMed]
- iGEATS. Available online: https://apps.cira.kyoto-u.ac.jp/igeats/index.html (accessed on 14 October 2015).
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10. [Google Scholar] [CrossRef] [PubMed]
- Kleinstiver, B.P.; Prew, M.S.; Tsai, S.Q.; Topkar, V.V.; Nguyen, N.T.; Zheng, Z.; Gonzales, A.P.W.; Li, Z.; Peterson, R.T.; Yeh, J.-R.J.; et al. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature 2015, 523, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Davis, K.M.; Pattanayak, V.; Thompson, D.B.; Zuris, J.A.; Liu, D.R. Small molecule-triggered Cas9 protein with improved genome-editing specificity. Nat. Chem. Biol. 2015, 11, 316–318. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Lin, C.-Y.; Gootenberg, J.S.; Konermann, S.; Trevino, A.E.; Scott, D.A.; Inoue, A.; Matoba, S.; Zhang, Y.; et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 2013, 154, 1380–1389. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Aach, J.; Stranges, P.B.; Esvelt, K.M.; Moosburner, M.; Kosuri, S.; Yang, L.; Church, G.M. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat. Biotechnol. 2013, 31, 833–838. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Zhang, W.; Zhang, J.; Zhou, J.; Wang, J.; Chen, L.; Wang, L.; Hodgkins, A.; Iyer, V.; Huang, X.; et al. Efficient genome modification by CRISPR-Cas9 nickase with minimal off-target effects. Nat. Methods 2014, 11, 399–402. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.Q.; Wyvekens, N.; Khayter, C.; Foden, J.A.; Thapar, V.; Reyon, D.; Goodwin, M.J.; Aryee, M.J.; Joung, J.K. Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nat. Biotechnol. 2014, 32, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Friedman, G.; Doyon, Y.; Wang, N.S.; Li, C.J.; Miller, J.C.; Hua, K.L.; Yan, J.J.; Babiarz, J.E.; Gregory, P.D.; et al. Targeted gene addition to a predetermined site in the human genome using a ZFN-based nicking enzyme. Genome Res. 2012, 22, 1316–1326. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Kim, S.; Kim, D.H.; Choi, B.-S.; Choi, I.-Y.; Kim, J.-S. Precision genome engineering with programmable DNA-nicking enzymes. Genome Res. 2012, 22, 1327–1333. [Google Scholar] [CrossRef] [PubMed]
- Guilinger, J.P.; Thompson, D.B.; Liu, D.R. Fusion of catalytically inactive Cas9 to FokI nuclease improves the specificity of genome modification. Nat. Biotechnol. 2014, 32, 577–582. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Cong, L.; Yan, W.X.; Scott, D.A.; Gootenberg, J.S.; Kriz, A.J.; Zetsche, B.; Shalem, O.; Wu, X.; Makarova, K.S.; et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature 2015, 520, 186–191. [Google Scholar] [CrossRef] [PubMed]
- González, F.; Zhu, Z.; Shi, Z.-D.; Lelli, K.; Verma, N.; Li, Q.V.; Huangfu, D. An iCRISPR platform for rapid, multiplexable, and inducible genome editing in human pluripotent stem cells. Stem Cell 2014, 15, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Moore, R.; Spinhirne, A.; Lai, M.J.; Preisser, S.; Li, Y.; Kang, T.; Bleris, L. CRISPR-based self-cleaving mechanism for controllable gene delivery in human cells. Nucleic Acids Res. 2014, 43, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kim, D.; Cho, S.W.; Kim, J.; Kim, J.-S. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 2014, 24, 1012–1019. [Google Scholar] [CrossRef] [PubMed]
- Hendel, A.; Bak, R.O.; Clark, J.T.; Kennedy, A.B.; Ryan, D.E.; Roy, S.; Steinfeld, I.; Lunstad, B.D.; Kaiser, R.J.; Wilkens, A.B.; et al. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nat. Biotechnol. 2015, 33, 985–989. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Sander, J.D.; Reyon, D.; Cascio, V.M.; Joung, J.K. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat. Biotechnol. 2014, 32, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.W.; Kim, S.; Kim, Y.; Kweon, J.; Kim, H.S.; Bae, S.; Kim, J.S. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 2014, 24, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Koo, T.; Lee, J.; Kim, J.-S. Measuring and reducing off-target activities of programmable nucleases including CRISPR-Cas9. Mol. Cells 2015, 38, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, R.; Lombardo, A.; Arens, A.; Miller, J.C.; Genovese, P.; Kaeppel, C.; Nowrouzi, A.; Bartholomae, C.C.; Wang, J.; Friedman, G.; et al. An unbiased genome-wide analysis of zinc-finger nuclease specificity. Nat. Biotechnol. 2011, 29, 816–823. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Scott, D.A.; Kriz, A.J.; Chiu, A.C.; Hsu, P.D.; Dadon, D.B.; Cheng, A.W.; Trevino, A.E.; Konermann, S.; Chen, S.; et al. Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nat. Biotechnol. 2014, 32, 670–676. [Google Scholar] [CrossRef] [PubMed]
- Frock, R.L.; Hu, J.; Meyers, R.M.; Ho, Y.-J.; Kii, E.; Alt, F.W. Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nat. Biotechnol. 2014, 33, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, Y.; Wu, X.; Wang, J.; Wang, Y.; Qiu, Z.; Chang, T.; Huang, H.; Lin, R.-J.; Yee, J.K. Unbiased detection of off-target cleavage by CRISPR-Cas9 and TALENs using integrase-defective lentiviral vectors. Nat. Biotechnol. 2015, 33, 175–178. [Google Scholar] [CrossRef] [PubMed]
- Crosetto, N.; Mitra, A.; Silva, M.J.; Bienko, M.; Dojer, N.; Wang, Q.; Karaca, E.; Chiarle, R.; Skrzypczak, M.; Ginalski, K.; et al. Nucleotide-resolution DNA double-strand break mapping by next-generation sequencing. Nat. Methods 2013, 10, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.Q.; Zheng, Z.; Nguyen, N.T.; Liebers, M.; Topkar, V.V.; Thapar, V.; Wyvekens, N.; Khayter, C.; Iafrate, A.J.; Le, L.P.; et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 2014, 33, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Bae, S.; Park, J.; Kim, E.; Kim, S.; Yu, H.R.; Hwang, J.; Kim, J.-I.; Kim, J.-S. Digenome-seq: Genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nat. Methods 2015, 12, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Canver, M.C.; Bauer, D.E.; Dass, A.; Yien, Y.Y.; Chung, J.; Masuda, T.; Maeda, T.; Paw, B.H.; Orkin, S.H. Characterization of genomic deletion efficiency mediated by clustered regularly interspaced palindromic repeats (CRISPR)/Cas9 nuclease system in mammalian cells. J. Biol. Chem. 2014, 289, 21312–21324. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.; Gore, A.; Yan, W.; Abalde-Atristain, L.; Li, Z.; He, C.; Wang, Y.; Brodsky, R.A.; Zhang, K.; Cheng, L.; et al. Whole-Genome Sequencing Analysis Reveals High Specificity of CRISPR/Cas9 and TALEN-Based Genome Editing in Human iPSCs. Stem Cell 2014, 15, 12–13. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Yu, C.; Qu, J.; Li, M.; Yao, X.; Yuan, T.; Goebl, A.; Tang, S.; Ren, R.; Aizawa, E.; et al. Targeted gene correction minimally impacts whole-genome mutational load in human-disease-specific induced pluripotent stem cell clones. Cell Stem Cell 2014, 15, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Veres, A.; Gosis, B.S.; Ding, Q.; Collins, R.; Ragavendran, A.; Brand, H.; Erdin, S.; Cowan, C.A.; Talkowski, M.E.; Musunuru, K. Low incidence of off-target mutations in individual CRISPR-Cas9 and TALEN targeted human stem cell clones detected by whole-genome sequencing. Cell Stem Cell 2014, 15, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Iyer, V.; Shen, B.; Zhang, W.; Hodgkins, A.; Keane, T.; Huang, X.; Skarnes, W.C. Off-target mutations are rare in Cas9-modified mice. Nat. Methods 2015, 12. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Lv, W.; Ye, X.; Wang, L.; Zhang, M.; Yang, H.; Okuka, M.; Zhou, C.; Zhang, X.; Liu, L.; et al. Zscan4 promotes genomic stability during reprogramming and dramatically improves the quality of iPS cells as demonstrated by tetraploid complementation. Cell Res. 2013, 23, 92–106. [Google Scholar] [CrossRef] [PubMed]
- Cradick, T.J.; Fine, E.J.; Antico, C.J.; Bao, G. CRISPR/Cas9 systems targeting-globin and CCR5 genes have substantial off-target activity. Nucleic Acids Res. 2013, 41, 9584–9592. [Google Scholar] [CrossRef] [PubMed]
- Yusa, K.; Rashid, S.T.; Strick-Marchand, H.; Varela, I.; Liu, P.-Q.; Paschon, D.E.; Miranda, E.; Ordóñez, A.; Hannan, N.R.F.; Rouhani, F.J.; et al. Targeted gene correction of α1-antitrypsin deficiency in induced pluripotent stem cells. Nature 2011, 478, 391–394. [Google Scholar] [CrossRef] [PubMed]
- Ousterout, D.G.; Perez-Pinera, P.; Thakore, P.I.; Kabadi, A.M.; Brown, M.T.; Qin, X.; Fedrigo, O.; Mouly, V.; Tremblay, J.P.; Gersbach, C.A. Reading frame correction by targeted genome editing restores dystrophin expression in cells from Duchenne muscular dystrophy patients. Mol. Ther. 2013, 21, 1718–1726. [Google Scholar] [CrossRef] [PubMed]
- Tan, E.-P.; Li, Y.; Velasco-Herrera, M.D.C.; Yusa, K.; Bradley, A. Off-target assessment of CRISPR-Cas9 guiding RNAs in human iPS and mouse ES cells. Genesis 2015, 53, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Karakikes, I.; Stillitano, F.; Nonnenmacher, M.; Tzimas, C.; Sanoudou, D.; Termglinchan, V.; Kong, C.-W.; Rushing, S.; Hansen, J.; Ceholski, D.; et al. Correction of human phospholamban R14del mutation associated with cardiomyopathy using targeted nucleases and combination therapy. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ishida, K.; Gee, P.; Hotta, A. Minimizing off-Target Mutagenesis Risks Caused by Programmable Nucleases. Int. J. Mol. Sci. 2015, 16, 24751-24771. https://doi.org/10.3390/ijms161024751
Ishida K, Gee P, Hotta A. Minimizing off-Target Mutagenesis Risks Caused by Programmable Nucleases. International Journal of Molecular Sciences. 2015; 16(10):24751-24771. https://doi.org/10.3390/ijms161024751
Chicago/Turabian StyleIshida, Kentaro, Peter Gee, and Akitsu Hotta. 2015. "Minimizing off-Target Mutagenesis Risks Caused by Programmable Nucleases" International Journal of Molecular Sciences 16, no. 10: 24751-24771. https://doi.org/10.3390/ijms161024751
APA StyleIshida, K., Gee, P., & Hotta, A. (2015). Minimizing off-Target Mutagenesis Risks Caused by Programmable Nucleases. International Journal of Molecular Sciences, 16(10), 24751-24771. https://doi.org/10.3390/ijms161024751