
Homologous Recombination-Independent Large Gene Cassette Knock-in in CHO Cells Using TALEN and MMEJ-Directed Donor Plasmids



 and
and
Abstract
:
1. Introduction
2. Results and Discussion
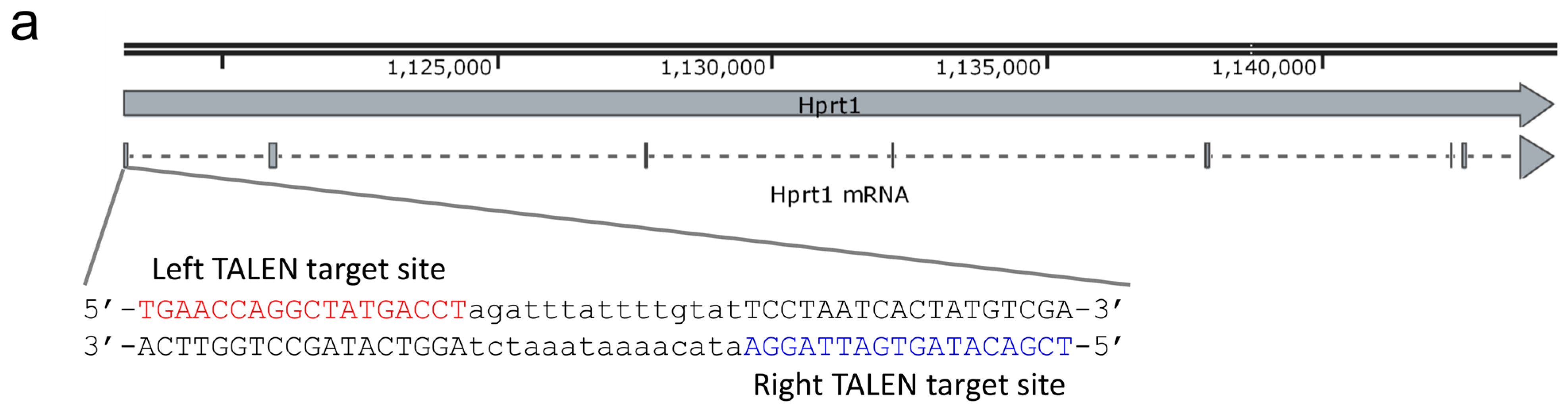
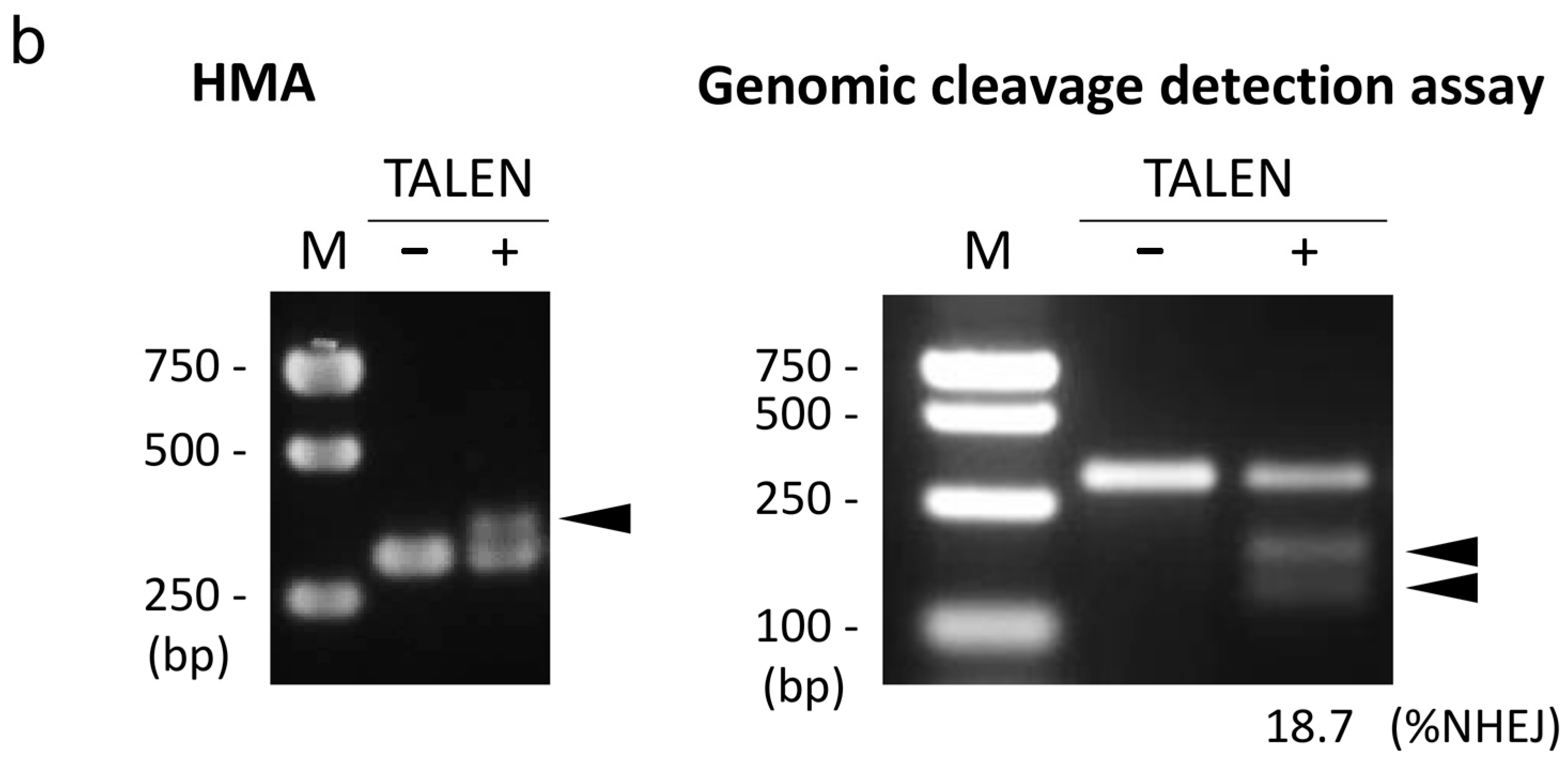
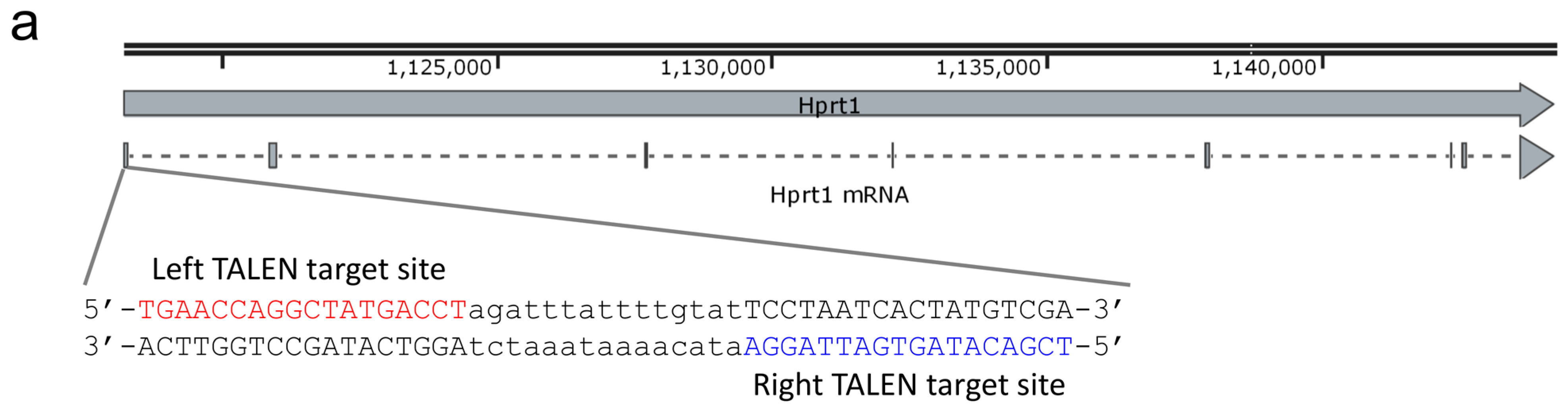
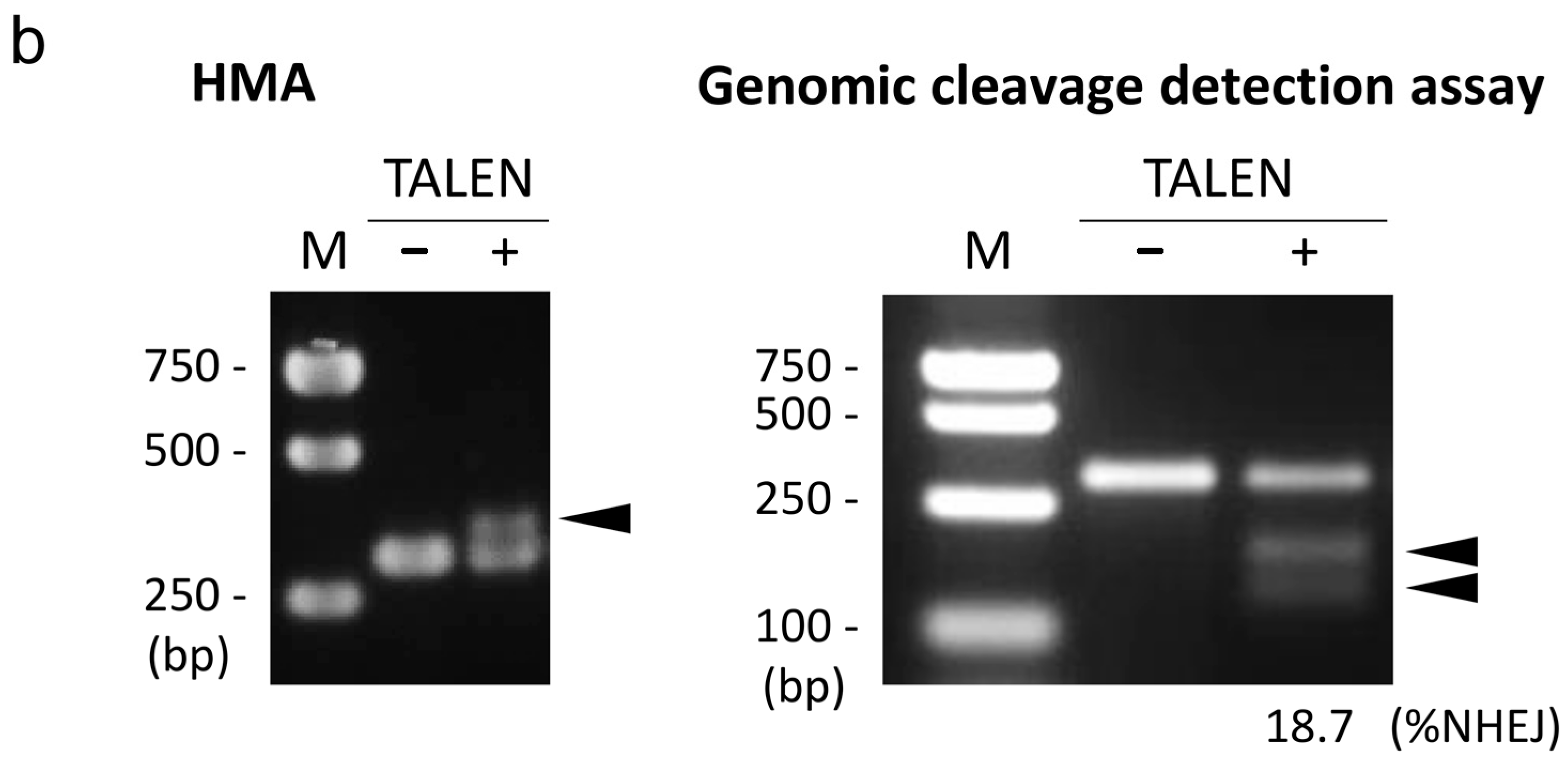
2.1. Design and Activity Validation of TALEN Targeting the HPRT1 Gene
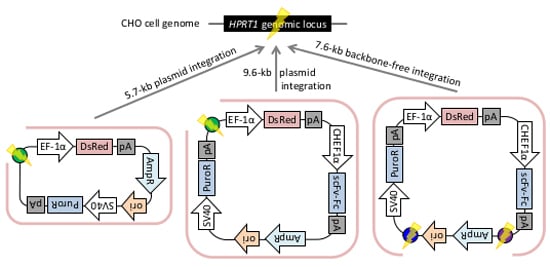
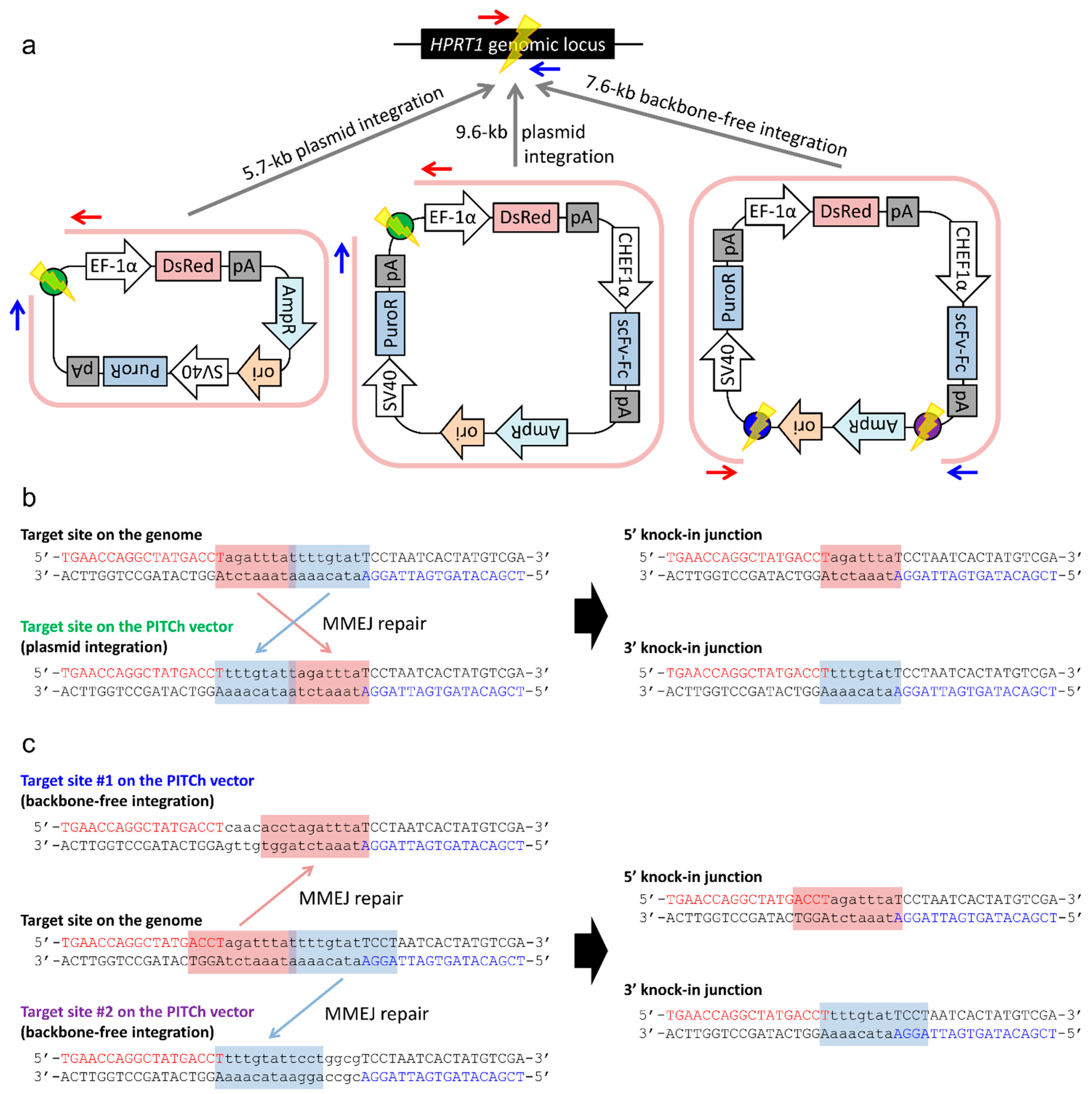
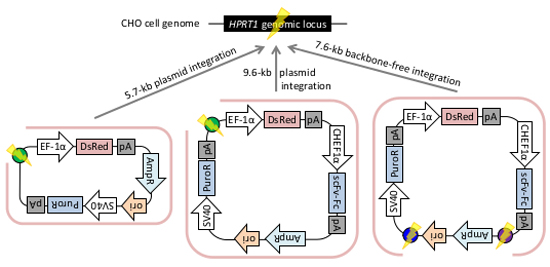
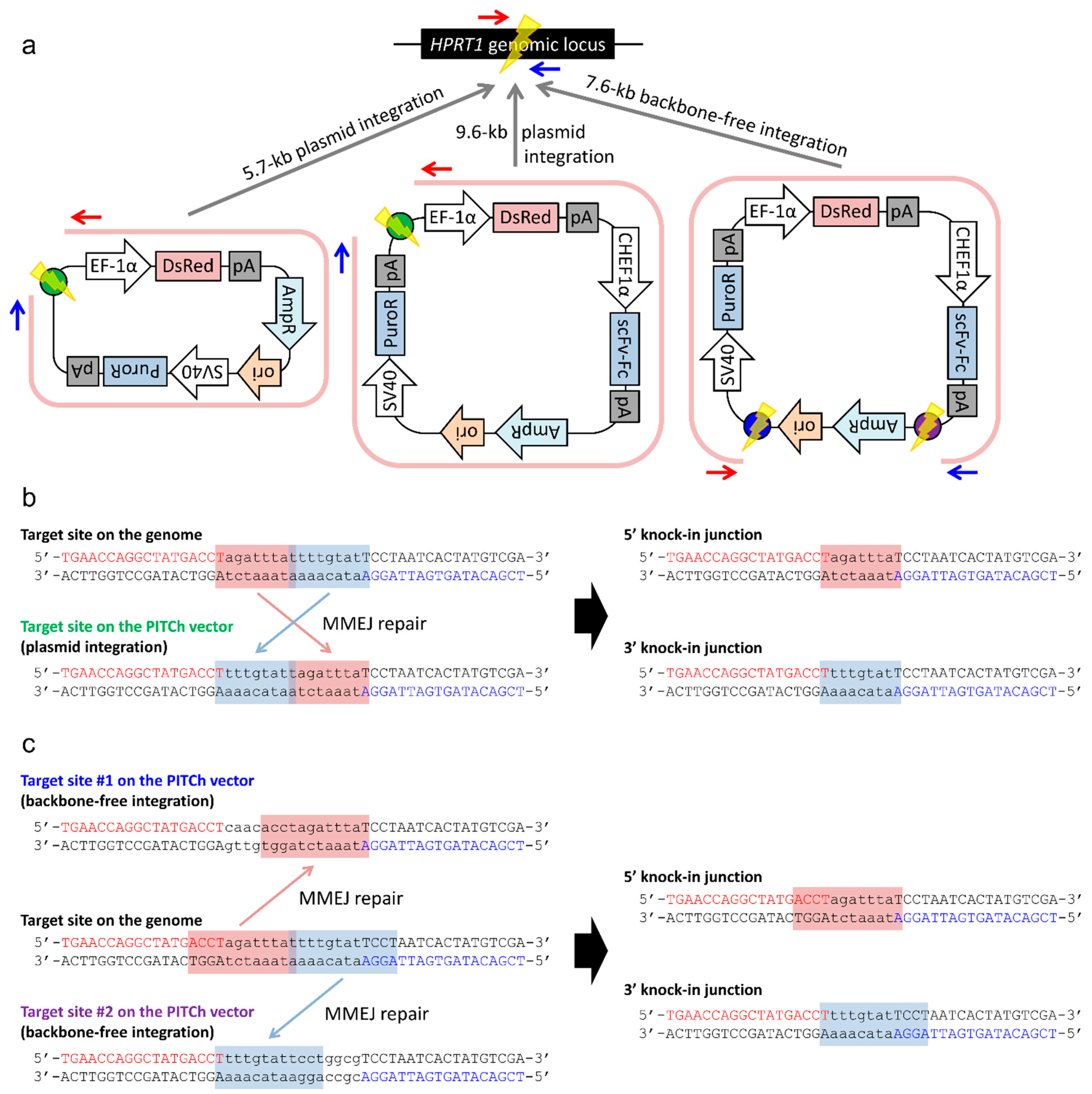
2.2. Gene Knock-in into the HPRT1 Locus Using the PITCh System
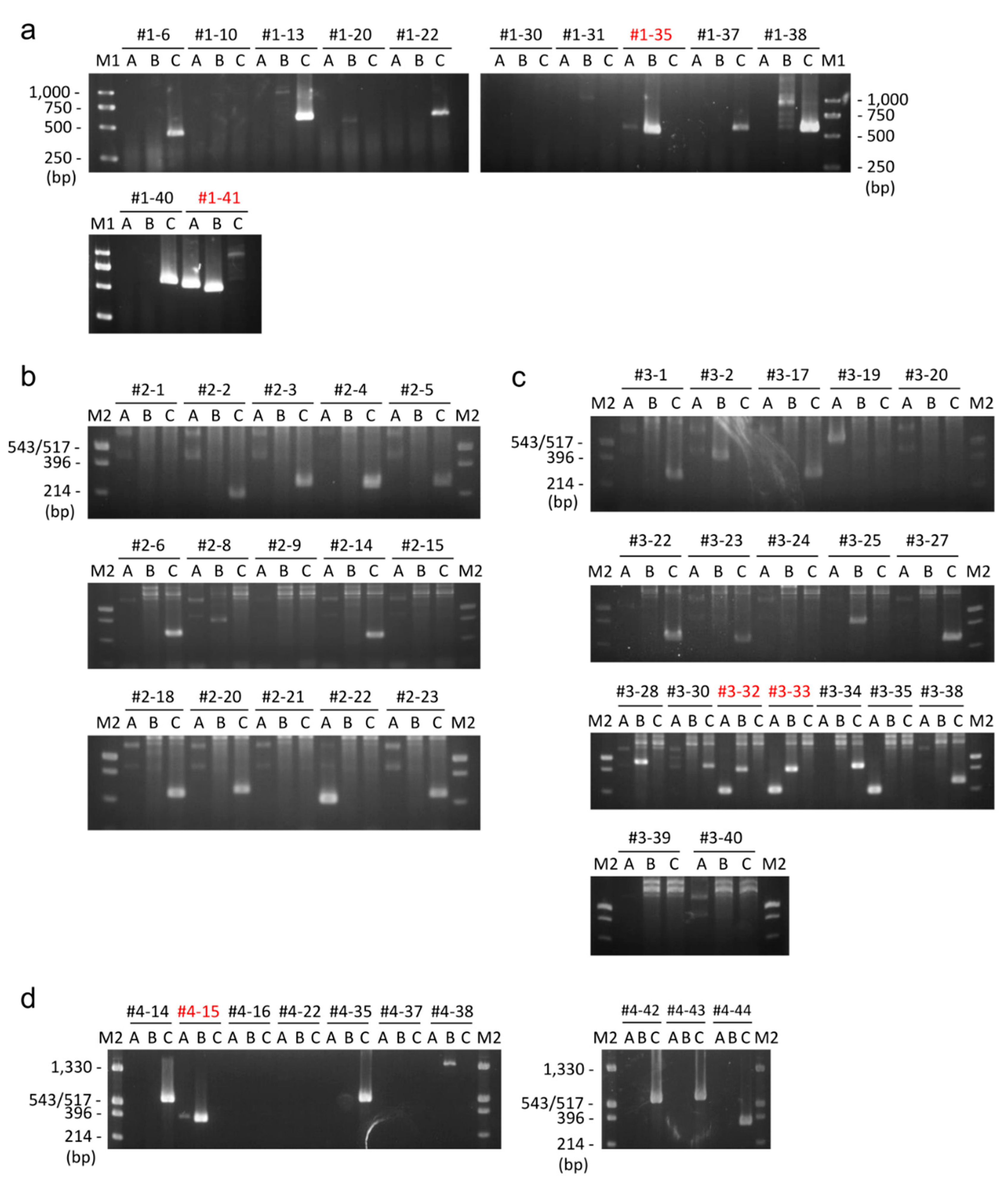
2.2.1. Whole Plasmid Integration Carrying DsRed and PuroR Gene Cassettes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Knock-in Type | No. of TALEN Sites on PITCh Vector | scFv-Fc Cassette | Knock-in Length | Puromycin Selection | No. of DsRed Expressed Clones (+; +/−; −) * | No. of Knock-in Clones/Analyzed Clones ** |
|---|---|---|---|---|---|---|
| Whole plasmid | 1 | − | 5.7 kb | From 72 h post-transfection | 17; 11; 5 | 2/12 (17%) |
| Whole plasmid | 1 | + | 9.6 kb | From 72 h post-transfection | 7; 8; 4 | 0/15 (0%) |
| Whole plasmid | 1 | + | 9.6 kb | From 24 h post-transfection | 6; 13; 4 | 2/19 (11%) |
| Cassette (backbone-free) | 2 | + | 7.6 kb | From 24 h post-transfection | 10; 4; 15 | 1/10 (10%) |
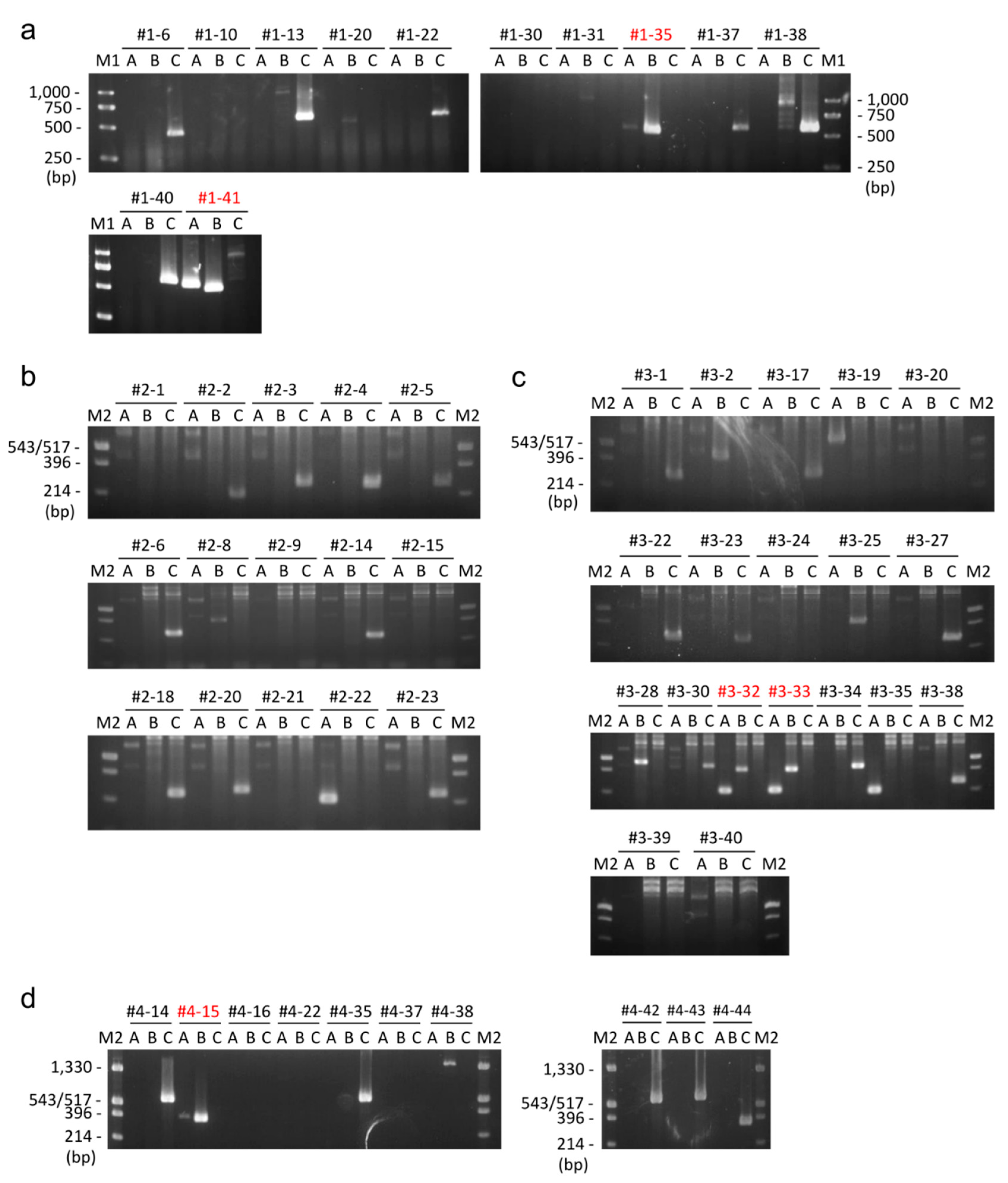
2.2.2. Whole Plasmid Integration Carrying an scFv-Fc Cassette along with DsRed and PuroR Cassettes
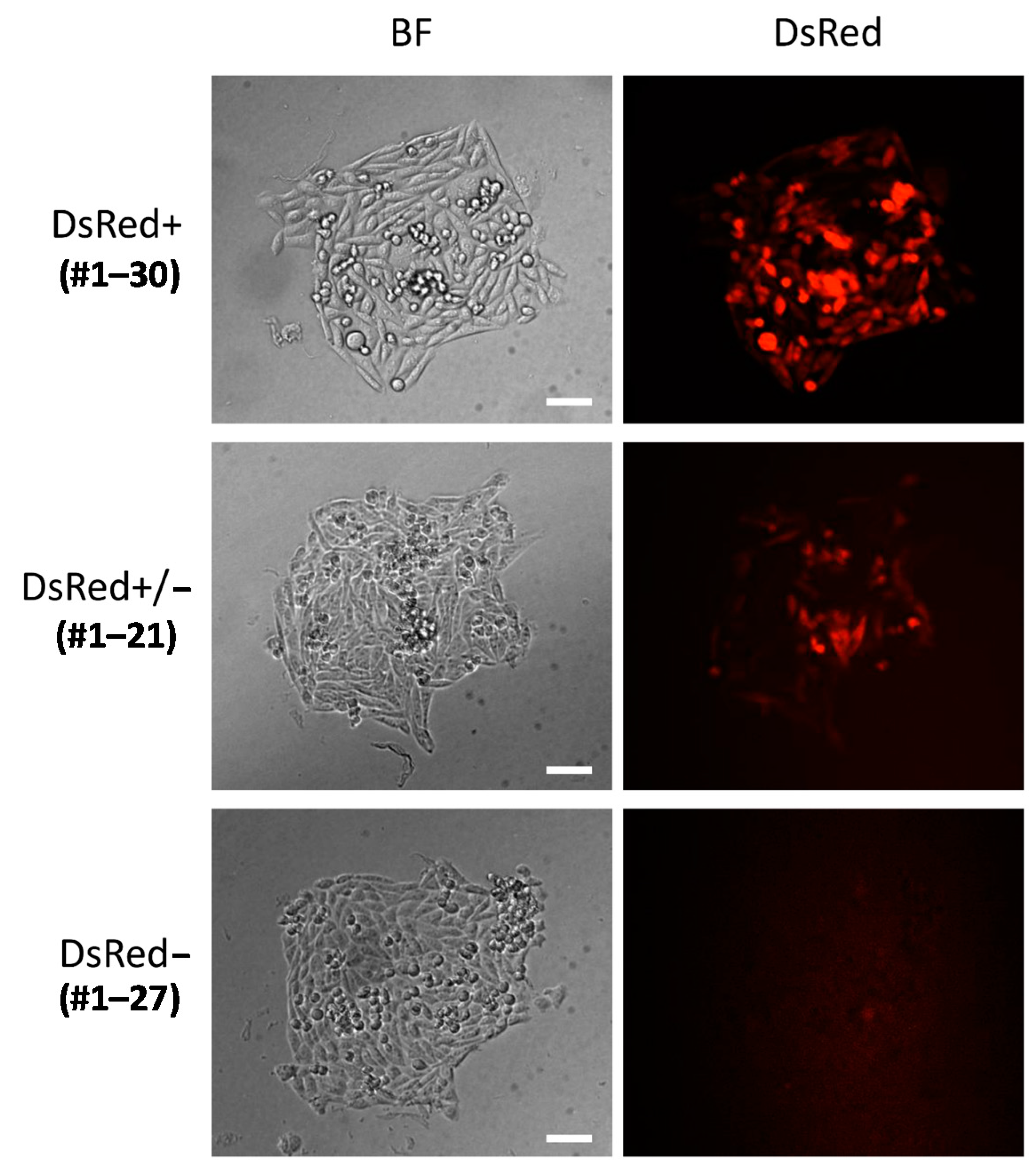
2.2.3. Backbone-Free Integration Carrying scFv-Fc, DsRed, and PuroR Cassettes

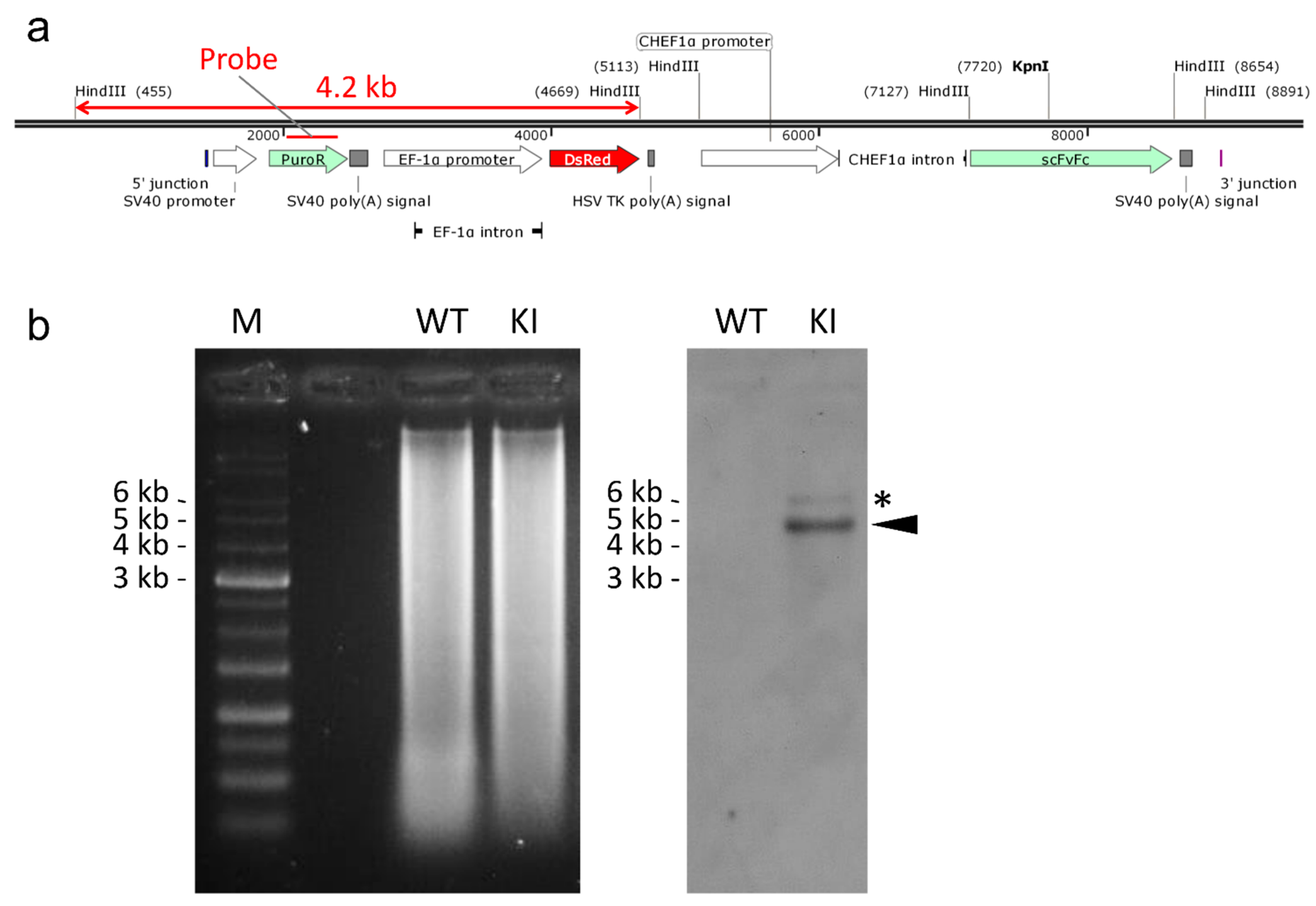
2.3. Confirmation of Genotype and Functionality of Knocked-in Cell Clone
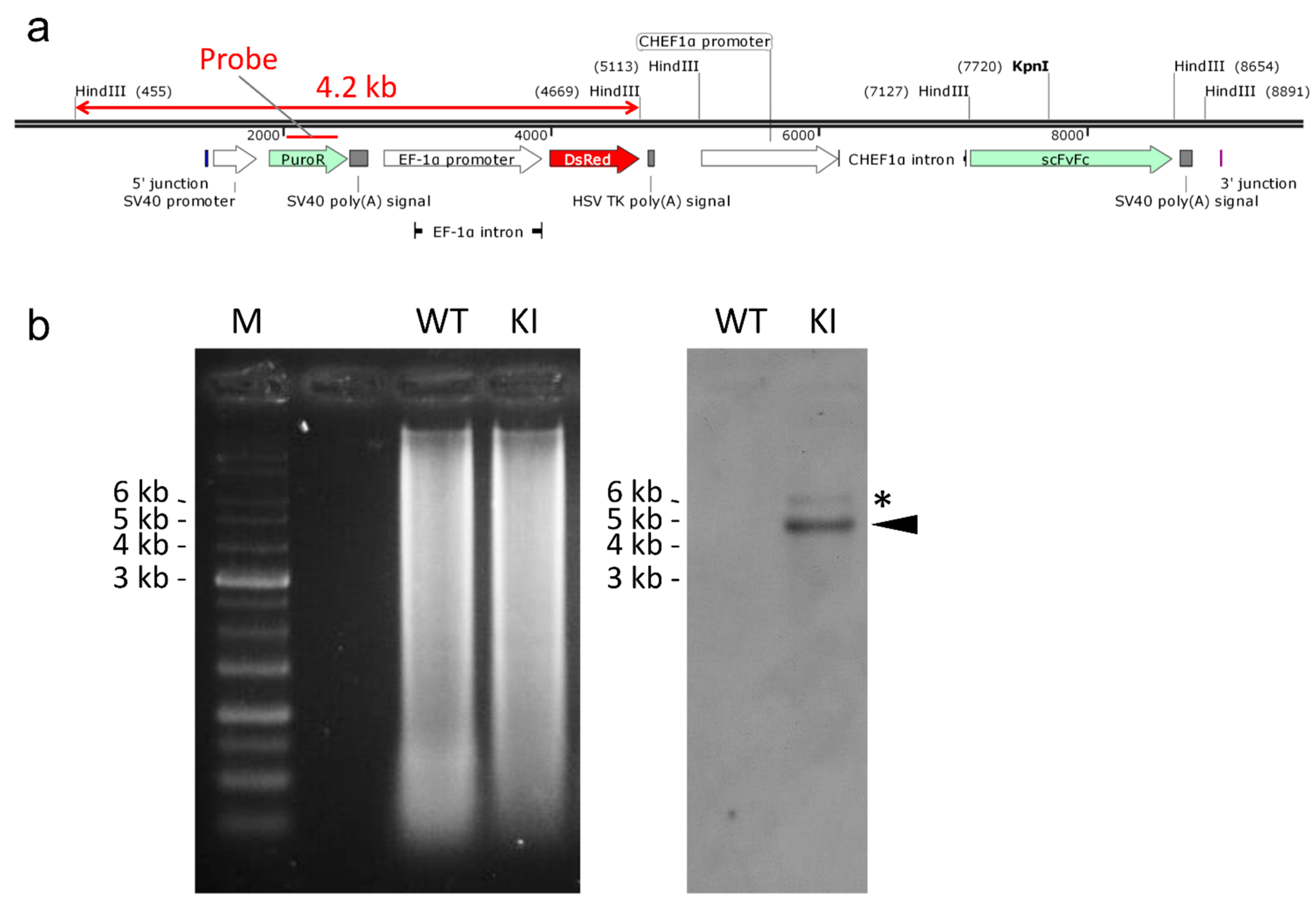
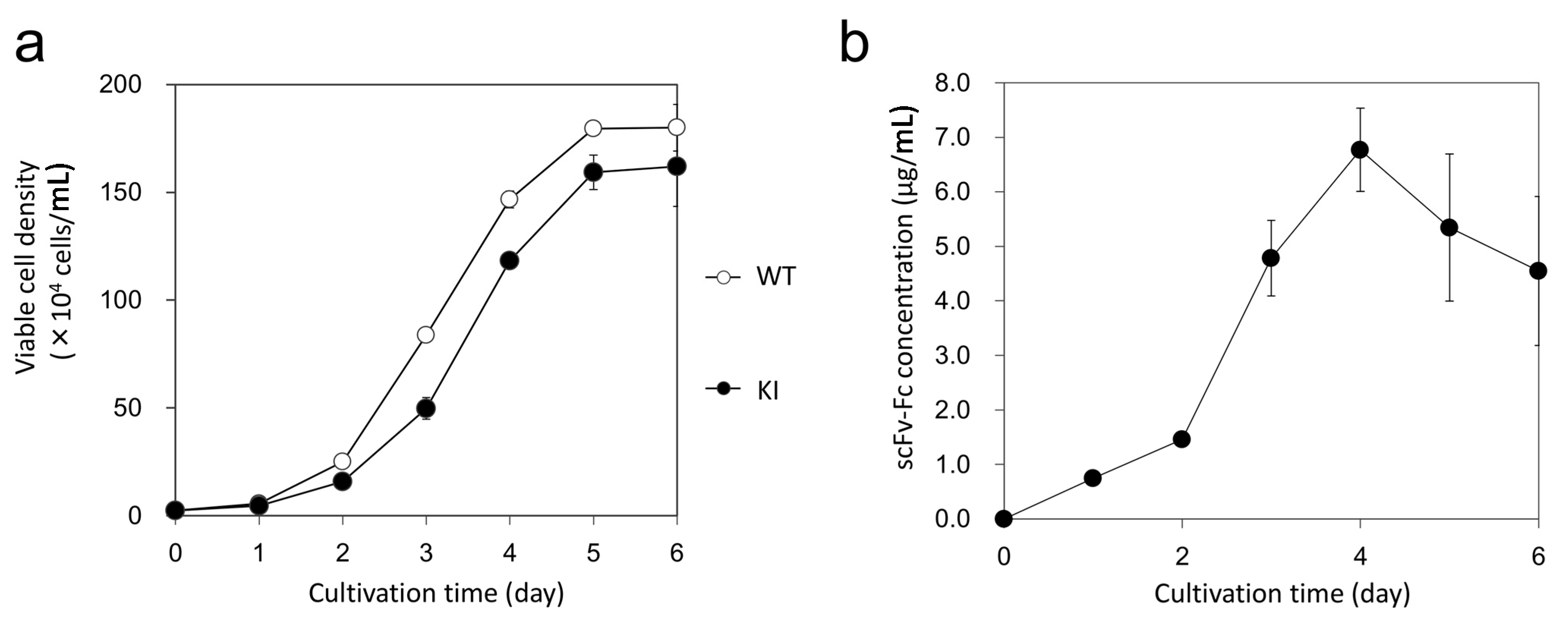
2.4. Off-Target Analysis of Knocked-in Cell Clone
3. Experimental Section
3.1. Construction of TALEN Plasmids and PITCh Vectors
| Name | Sequence * | Reference | TAL1 Score ** | TAL2 Score ** | Average Score | Mutation |
|---|---|---|---|---|---|---|
| On-target | 5ʹ-agatttattttgtatTCCTAATCACTATGTCGA-3ʹ 3ʹ-ACTTGGTCCGATACTGGAtctaaataaaacata-5ʹ | gi|351517617|ref|NW_003613932.1| | 5.79 | 6.61 | 6.2 | - |
| OT1 | 5ʹ-tttccaactggaattttctccaaccAGGTGAGTGTTTGATTTA-3ʹ 3ʹ-ATTTCGTCCGATACTGGAaaaggttgaccttaaaagaggttgg-5ʹ | gi|351517757|ref|NW_003613792.1| | 9.53 | 14.74 | 12.135 | Not detected |
| OT2 | 5ʹ-gcaacttaggagaaaAGGGTATATTTTGGTTTA-3ʹ 3ʹ-ATTTGTGTCACTGGTTTTcgttgaatcctcttt-5ʹ | gi|351517841|ref|NW_003613708.1| | 15.55 | 10.96 | 13.255 | Not detected |
| OT3 | 5ʹ-ccaacagtgagaatattaaaaTAAAAATTACTATGTTGA-3ʹ 3ʹ-ATTTGGTCCGATATTCTAggttgtcactcttataatttt-5ʹ | gi|351517438|ref|NW_003614111.1| | 12.05 | 14.61 | 13.33 | Not detected |
| OT4 | 5ʹ-cagattccttttgtTCCTGATGACTAGGTTGA-3ʹ 3ʹ-AGGTGGTTCGGTAATCCTgtctaaggaaaaca-5ʹ | gi|351516302|ref|NW_003615247.1| | 14.78 | 11.91 | 13.345 | Not detected |
| OT5 | 5ʹ-gttaattaattaattaaatcaatacaaatTTATATTCATTATGTAGA-3ʹ 3ʹ-AGTTTTTTTATTTATTTTcaattaattaattaatttagttatgttta-5ʹ | gi|351517764|ref|NW_003613785.1| | 13.95 | 13.1 | 13.525 | Not detected |
| OT6 | 5ʹ-aaatctttaaaaaaacaaAAGTTATAGTTTGGTTTA-3ʹ 3ʹ-ATATGTATTATTTATTTAtttagaaatttttttgtt-5ʹ | gi|351516670|ref|NW_003614879.1| | 17.3 | 9.84 | 13.57 | Not detected |
| OT7 | 5ʹ-aaataaataaatctttttaaaAAGTTATAGTTTGGTTTA-3ʹ 3ʹ-ATATGTATTATTTATTTAtttatttatttagaaaaattt-5ʹ | gi|351517952|ref|NW_003613597.1| | 17.3 | 9.84 | 13.57 | Not detected |
3.2. Cell Culture and Transfection
3.3. Genomic Cleavage Detection Assay
3.4. Puromycin Selection and Single Cell Cloning
3.5. Fluorescence Microscopy
3.6. Genotyping and Sequencing
3.7. Southern Blotting
| Genomic Cleavage Detection Assay | ||
|---|---|---|
| Locus | Forward primer (5′→3′) | Reverse primer (5′→3′) |
| HPRT1 | GCCCTTCATACCCTCTCATACCC | GCCATCTCTCCAGCCCCTTC |
| Genotyping of Knock-In Clones | ||
| Locus | Forward primer (5′→3′) | Reverse primer (5′→3′) |
| 5′ junction (in 2.2.1.) | GCATTTGCCCCCACAATGCTC | AGGCGGAGCCAGTACACGACATC |
| 3′ junction (in 2.2.1.) | GGTGCCTGAAGATCCAGACATGATAAG | TGTCCCTGCAGGCCAGAAGAG |
| Non-knock-in (in 2.2.1.) | GCATTTGCCCCCACAATGCTC | TGTCCCTGCAGGCCAGAAGAG |
| 5′ junction (in 2.2.2.) | TCATACCCAAATTCCTCTGGCGAAC | TGTGCGCTCTGCCCACTGAC |
| 3′ junction (in 2.2.2.) | GGTGCCTGAAGATCCAGACATGATAAG | AGCAGTCAGTGCTCTTAACCGCTGAG |
| Non-knock-in (in 2.2.2.) | TCATACCCAAATTCCTCTGGCGAAC | AGCAGTCAGTGCTCTTAACCGCTGAG |
| 5′ junction (in 2.2.3.) | GCATTTGCCCCCACAATGCTC | GGACTTTCCACACCTGGTTGCTGAC |
| 3′ junction (in 2.2.3.) | AAGCTTGTCGACATCGATGAATCAGG | ACTTCAATTAAGATCTGTTCTGTCTGCATGTGTC |
| Non-knock-in (in 2.2.3.) | GCATTTGCCCCCACAATGCTC | ACTTCAATTAAGATCTGTTCTGTCTGCATGTGTC |
| Southern Blot Analysis | ||
| Locus | Forward primer (5′→3′) | Reverse primer (5′→3′) |
| Probe | CCGAGCTGCAAGAACTCTTCCTCAC | GGTCCTTCGGGCACCTCGAC |
| Off-Target Analysis | ||
| Locus | Forward primer (5′→3′) | Reverse primer (5′→3′) |
| OT1 | TCACTAGCAGGTGTGACTCTCAGTACGC | GGAAACCTGAATAGACAGACACAAGCAAG |
| OT2 | CTAATTTGACCTTCCTGTTCACCAGTGCTAC | GATAGCATGAGAAAGCAAACTGAGCAAGC |
| OT3 | CAGTTCCCATGCTTACAGCATCAGTG | CTCATGCCTATGTGGCAAGTGCTTTAC |
| OT4 | GTCCCAAGCCTTTCTGAATTATTTCTACTTCC | AAAGCATCAAAATGTCCTCGCATTGAG |
| OT5 | CACACACCCTCTCACTTTCATTCTCTCTC | ATGAGGGAAGTTTGAAGAGAATAATATGGAAAGG |
| OT6 | GCAATACGGGTGATTGAAGAGCACTG | CTCCTCCAACTTCATTGTACTCTACGCATTC |
| OT7 | CTCTGTAGTTTGGTCTCTGATGGCAGTTTG | CCAAGGAAGCAGTCAGCTCTACCATAAAC |
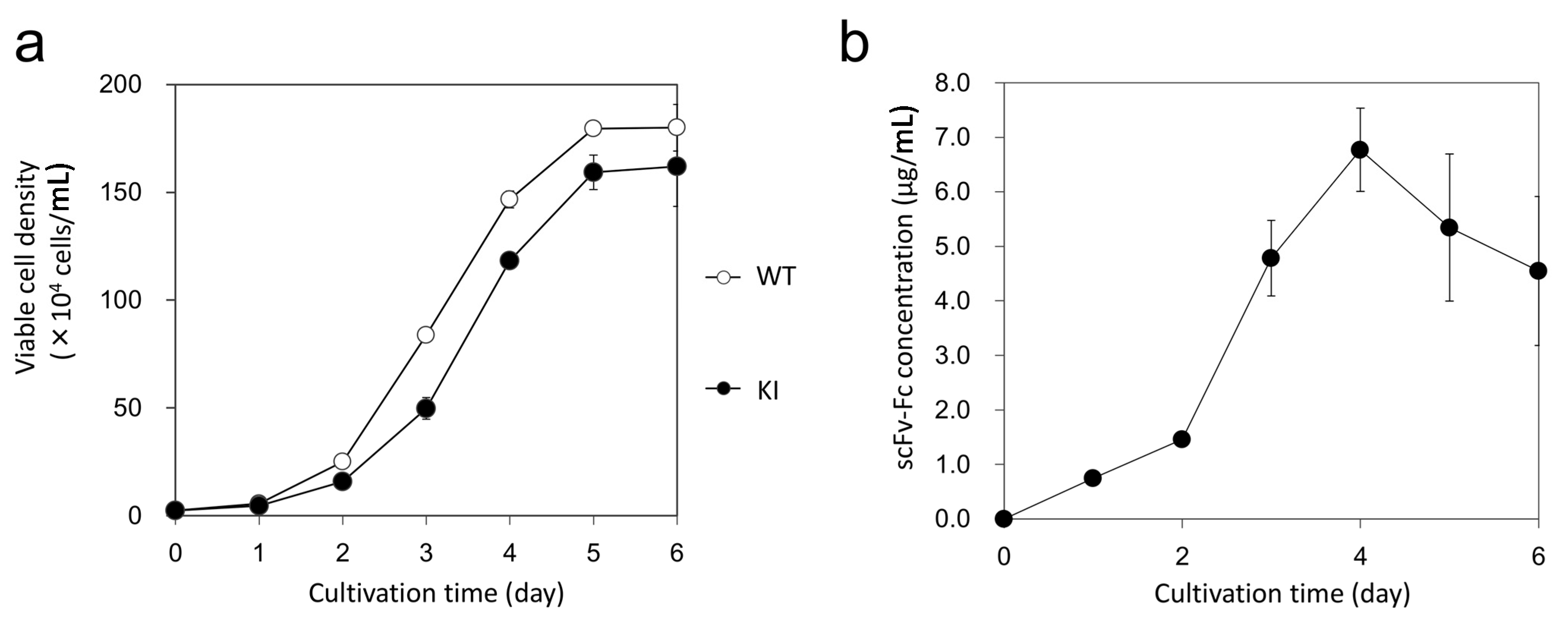
3.8. Cell Growth Analysis and Expression Analysis of scFv-Fc
3.9. Off-Target Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lee, J.S.; Grav, L.M.; Lewis, N.E.; Kildegaard, H.F. CRISPR/Cas9-mediated genome engineering of CHO cell factories: Application and perspectives. Biotechnol. J. 2015, 10, 979–994. [Google Scholar] [CrossRef] [PubMed]
- Sakuma, T.; Woltjen, K. Nuclease-mediated genome editing: At the front-line of functional genomics technology. Dev. Growth Differ. 2014, 56, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Orlando, S.J.; Santiago, Y.; DeKelver, R.C.; Freyvert, Y.; Boydston, E.A.; Moehle, E.A.; Choi, V.M.; Gopalan, S.M.; Lou, J.F.; Li, J.; et al. Zinc-finger nuclease-driven targeted integration into mammalian genomes using donors with limited chromosomal homology. Nucleic Acids Res. 2010, 38, e152. [Google Scholar] [CrossRef] [PubMed]
- Cristea, S.; Freyvert, Y.; Santiago, Y.; Holmes, M.C.; Urnov, F.D.; Gregory, P.D.; Cost, G.J. In vivo cleavage of transgene donors promotes nuclease-mediated targeted integration. Biotechnol. Bioeng. 2013, 110, 871–880. [Google Scholar] [CrossRef] [PubMed]
- Maresca, M.; Lin, V.G.; Guo, N.; Yang, Y. Obligate ligation-gated recombination (ObLiGaRe): custom-designed nuclease-mediated targeted integration through nonhomologous end joining. Genome Res. 2013, 23, 539–546. [Google Scholar] [CrossRef] [PubMed]
- Auer, T.O.; Duroure, K.; de Cian, A.; Concordet, J.P.; del Bene, F. Highly efficient CRISPR/Cas9-mediated knock-in in zebrafish by homology-independent DNA repair. Genome Res. 2014, 24, 142–153. [Google Scholar] [CrossRef] [PubMed]
- Bachu, R.; Bergareche, I.; Chasin, L.A. CRISPR-Cas targeted plasmid integration into mammalian cells via non-homologous end joining. Biotechnol. Bioeng. 2015, 112, 2154–2162. [Google Scholar] [CrossRef] [PubMed]
- Nakade, S.; Tsubota, T.; Sakane, Y.; Kume, S.; Sakamoto, N.; Obara, M.; Daimon, T.; Sezutsu, H.; Yamamoto, T.; Sakuma, T.; et al. Microhomology-mediated end-joining-dependent integration of donor DNA in cells and animals using TALENs and CRISPR/Cas9. Nat. Commun. 2014, 5, 5560. [Google Scholar] [CrossRef] [PubMed]
- Hisano, Y.; Sakuma, T.; Nakade, S.; Ohga, R.; Ota, S.; Okamoto, H.; Yamamoto, T.; Kawahara, A. Precise in-frame integration of exogenous DNA mediated by CRISPR/Cas9 system in zebrafish. Sci. Rep. 2015, 5, 8841. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Kallehauge, T.B.; Pedersen, L.E.; Kildegaard, H.F. Site-specific integration in CHO cells mediated by CRISPR/Cas9 and homology-directed DNA repair pathway. Sci. Rep. 2015, 5, 8572. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Foden, J.A.; Khayter, C.; Maeder, M.L.; Reyon, D.; Joung, J.K.; Sander, J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013, 31, 822–826. [Google Scholar] [CrossRef] [PubMed]
- Cradick, T.J.; Fine, E.J.; Antico, C.J.; Bao, G. CRISPR/Cas9 systems targeting β-globin and CCR5 genes have substantial off-target activity. Nucleic Acids Res. 2013, 41, 9584–9592. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Cradick, T.J.; Brown, M.T.; Deshmukh, H.; Ranjan, P.; Sarode, N.; Wile, B.M.; Vertino, P.M.; Stewart, F.J.; Bao, G. CRISPR/Cas9 systems have off-target activity with insertions or deletions between target DNA and guide RNA sequences. Nucleic Acids Res. 2014, 42, 7473–7485. [Google Scholar] [CrossRef] [PubMed]
- Koyama, Y.; Banzai, T.; Sonezaki, S.; Kusano, K. Stable expression of a heterogeneous gene introduced via gene targeting into the HPRT locus of human fibrosarcoma cells. Biotechnol. Bioeng. 2006, 95, 1052–1060. [Google Scholar] [CrossRef] [PubMed]
- Ota, S.; Hisano, Y.; Muraki, M.; Hoshijima, K.; Dahlem, T.J.; Grunwald, D.J.; Okada, Y.; Kawahara, A. Efficient identification of TALEN-mediated genome modifications using heteroduplex mobility assays. Genes Cells 2013, 18, 450–458. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, Y.; Yamamoto, T.; Suzuki, K.; Araki, K.; Takeda, N.; Ohmuraya, M.; Sakuma, T. Screening methods to identify TALEN-mediated knockout mice. Exp. Anim. 2014, 63, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Guschin, D.Y.; Waite, A.J.; Katibah, G.E.; Miller, J.C.; Holmes, M.C.; Rebar, E.J. A rapid and general assay for monitoring endogenous gene modification. Methods Mol. Biol. 2010, 649, 247–256. [Google Scholar] [PubMed]
- Hansen, K.; Coussens, M.J.; Sago, J.; Subramanian, S.; Gjoka, M.; Briner, D. Genome editing with CompoZr custom zinc finger nucleases (ZFNs). J. Vis. Exp. 2012, e3304. [Google Scholar] [CrossRef] [PubMed]
- Cricetulus griseus unplaced genomic scaffold, CriGri_1.0 scaffold1588, whole genome shotgun sequence. Available online: http://www.ncbi.nlm.nih.gov/nuccore/NW_003613932.1?from=1118242&to=1144234&report=genbank (accessed on 8 October 2015).
- Nehlsen, K.; Schucht, R.; da Gama-Norton, L.; Krömer, W.; Baer, A.; Cayli, A.; Hauser, H.; Wirth, D. Recombinant protein expression by targeting pre-selected chromosomal loci. BMC Biotechnol. 2009, 9, 100. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.; Kweon, J.; Kim, H.S.; Kim, J.S. Microhomology-based choice of Cas9 nuclease target sites. Nat. Methods 2014, 11, 705–706. [Google Scholar] [CrossRef] [PubMed]
- Li, H.L.; Fujimoto, N.; Sasakawa, N.; Shirai, S.; Ohkame, T.; Sakuma, T.; Tanaka, M.; Amano, N.; Watanabe, A.; Sakurai, H.; et al. Precise correction of the dystrophin gene in duchenne muscular dystrophy patient induced pluripotent stem cells by TALEN and CRISPR-Cas9. Stem Cell Rep. 2015, 4, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Yasue, A.; Mitsui, S.N.; Watanabe, T.; Sakuma, T.; Oyadomari, S.; Yamamoto, T.; Noji, S.; Mito, T.; Tanaka, E. Highly efficient targeted mutagenesis in one-cell mouse embryos mediated by the TALEN and CRISPR/Cas systems. Sci. Rep. 2014, 4, 5705. [Google Scholar] [CrossRef] [PubMed]
- Sakuma, T.; Ochiai, H.; Kaneko, T.; Mashimo, T.; Tokumasu, D.; Sakane, Y.; Suzuki, K.; Miyamoto, T.; Sakamoto, N.; Matsuura, S.; et al. Repeating pattern of non-RVD variations in DNA-binding modules enhances TALEN activity. Sci. Rep. 2013, 3, 3379. [Google Scholar] [CrossRef] [PubMed]
- Sakuma, T.; Yamamoto, T. Engineering customized TALENs using the Platinum Gate TALEN Kit. Methods Mol. Biol. 2016, 1338, 61–70. [Google Scholar] [PubMed]
- Kawabe, Y.; Makitsubo, H.; Kameyama, Y.; Huang, S.; Ito, A.; Kamihira, M. Repeated integration of antibody genes into a pre-selected chromosomal locus of CHO cells using an accumulative site-specific gene integration system. Cytotechnology 2012, 64, 267–279. [Google Scholar] [CrossRef] [PubMed]
- Doyle, E.L.; Booher, N.J.; Standage, D.S.; Voytas, D.F.; Brendel, V.P.; Vandyk, J.K.; Bogdanove, A.J. TAL Effector-Nucleotide Targeter (TALE-NT) 2.0: Tools for TAL effector design and target prediction. Nucleic Acids Res. 2012, 40, W117–W122. [Google Scholar] [CrossRef] [PubMed]
- Sakuma, T.; Nishikawa, A.; Kume, S.; Chayama, K.; Yamamoto, T. Multiplex genome engineering in human cells using all-in-one CRISPR/Cas9 vector system. Sci. Rep. 2014, 4, 5400. [Google Scholar] [CrossRef] [PubMed]
- Kamihira, M.; Ono, K.; Esaka, K.; Nishijima, K.; Kigaku, R.; Komatsu, H.; Yamashita, T.; Kyogoku, K.; Iijima, S. High-level expression of single-chain Fv-Fc fusion protein in serum and egg white of genetically manipulated chickens by using a retroviral vector. J. Virol. 2005, 79, 10864–10874. [Google Scholar] [CrossRef] [PubMed]
- Kamihira, M.; Kawabe, Y.; Shindo, T.; Ono, K.; Esaka, K.; Yamashita, T.; Nishijima, K.; Iijima, S. Production of chimeric monoclonal antibodies by genetically manipulated chickens. J. Biotechnol. 2009, 141, 18–25. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sakuma, T.; Takenaga, M.; Kawabe, Y.; Nakamura, T.; Kamihira, M.; Yamamoto, T. Homologous Recombination-Independent Large Gene Cassette Knock-in in CHO Cells Using TALEN and MMEJ-Directed Donor Plasmids. Int. J. Mol. Sci. 2015, 16, 23849-23866. https://doi.org/10.3390/ijms161023849
Sakuma T, Takenaga M, Kawabe Y, Nakamura T, Kamihira M, Yamamoto T. Homologous Recombination-Independent Large Gene Cassette Knock-in in CHO Cells Using TALEN and MMEJ-Directed Donor Plasmids. International Journal of Molecular Sciences. 2015; 16(10):23849-23866. https://doi.org/10.3390/ijms161023849
Chicago/Turabian StyleSakuma, Tetsushi, Mitsumasa Takenaga, Yoshinori Kawabe, Takahiro Nakamura, Masamichi Kamihira, and Takashi Yamamoto. 2015. "Homologous Recombination-Independent Large Gene Cassette Knock-in in CHO Cells Using TALEN and MMEJ-Directed Donor Plasmids" International Journal of Molecular Sciences 16, no. 10: 23849-23866. https://doi.org/10.3390/ijms161023849
APA StyleSakuma, T., Takenaga, M., Kawabe, Y., Nakamura, T., Kamihira, M., & Yamamoto, T. (2015). Homologous Recombination-Independent Large Gene Cassette Knock-in in CHO Cells Using TALEN and MMEJ-Directed Donor Plasmids. International Journal of Molecular Sciences, 16(10), 23849-23866. https://doi.org/10.3390/ijms161023849