Small Engine, Big Power: MicroRNAs as Regulators of Cardiac Diseases and Regeneration


Abstract
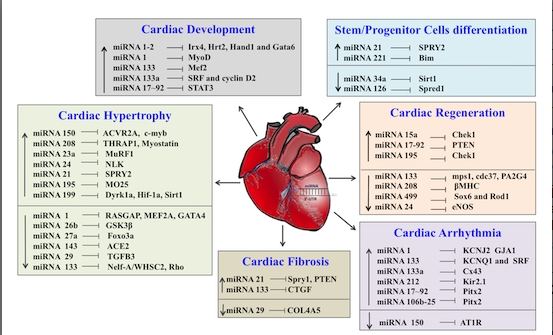
:
1. Introduction
2. Biogenesis of miRNA and Its Target Sites
3. Role of miRNAs in Cardiac Development and Diseases
3.1. miRNAs in Cardiac Development
3.2. miRNAs in Cardiac Hypertrophy
3.3. miRNAs in Cardiac Fibrosis
3.4. miRNAs in Cardiac Arrhythmias
3.5. miRNAs in Endothelial Homeostasis and Angiogenesis
4. miRNAs in Cardiac Regeneration and Stem Cell-Mediated Repair
5. Challenges of MicroRNA-Based Therapies in Cardiovascular Diseases
6. Clinical Perspectives and Conclusions

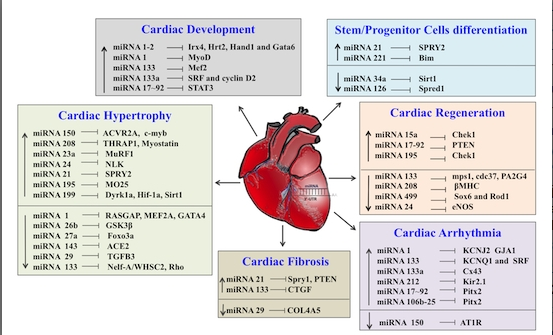{kind=link}
{kind=link}
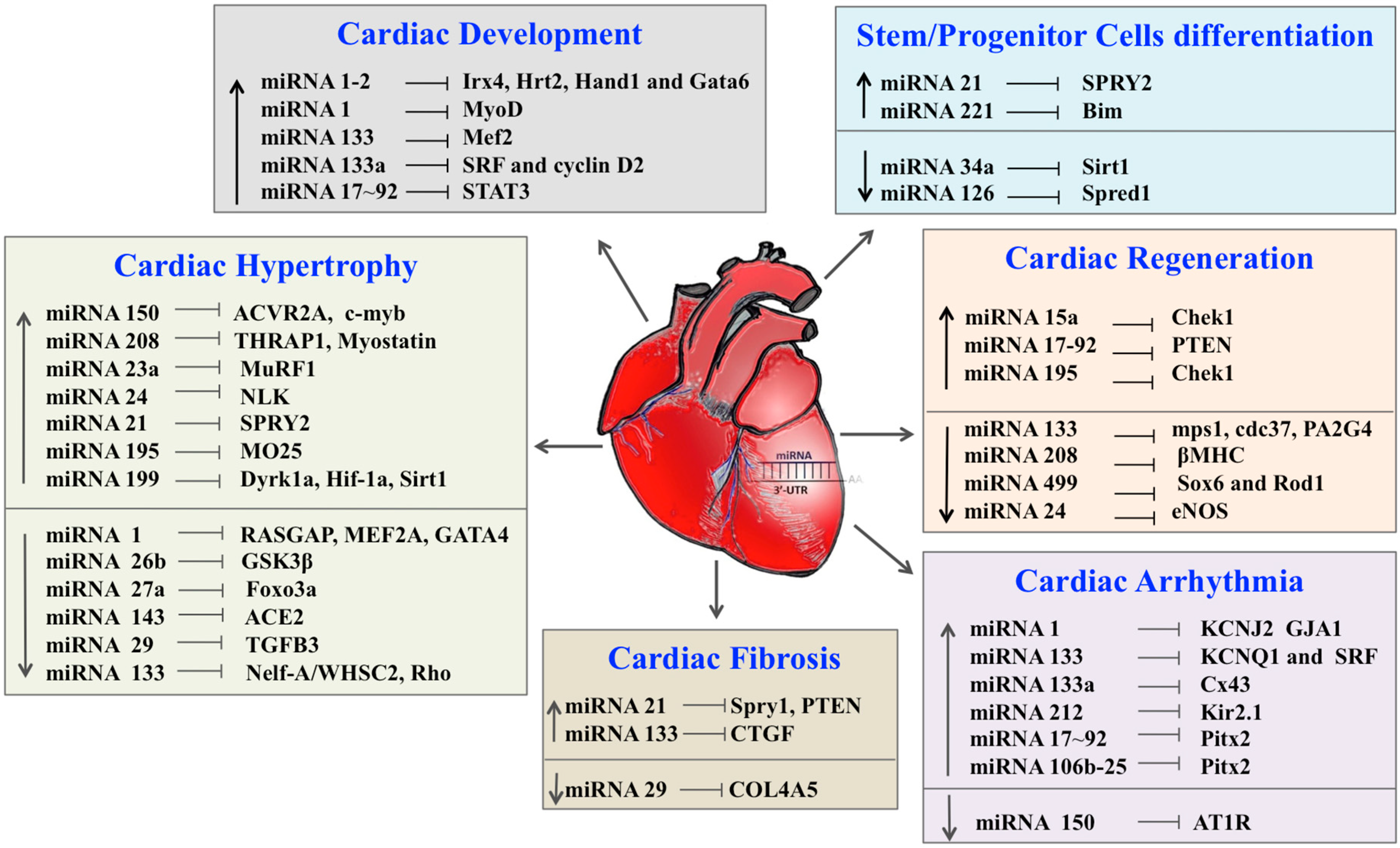
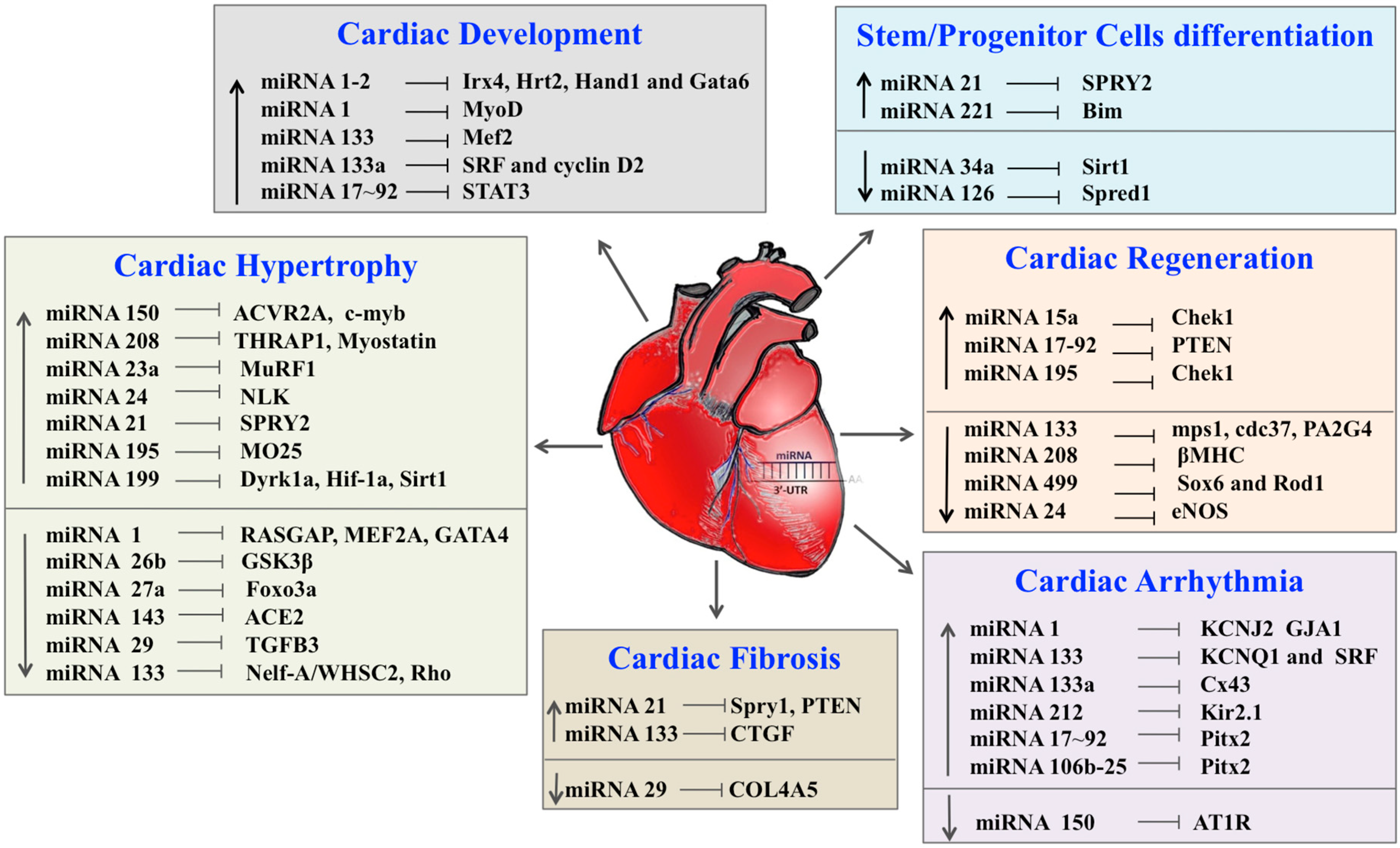
| miRNA | Targets | References |
|---|---|---|
| Cardiac Development | ||
| miRNA-1–2 | Irx4, Hrt2, Hand1 and Gata6 | [120] |
| miRNA-1 | MyoD, Hand2 | [41,42] |
| miRNA-133 | Mef2 | [41,42] |
| miRNA-133a | SRF and cyclin D2 | [43] |
| miRNA-17–92 | STAT3 | [44,121] |
| miRNA-20 | Egln3 | [122] |
| miRNA-23b | Rb phosphorylation | [123] |
| miRNA-24 | BIM and GATA | [94] |
| miRNA-30c | CTGF | [79] |
| miRNA-143 | Adducin3 | [124] |
| Cardiac Hypertrophy | ||
| miRNA-150 | ACVR2A, c-myb | [125] |
| miRNA-208 | THRAP1, Myostatin | [56] |
| miRNA-23a | MuRF1 | [126] |
| miRNA-24 | NLK | [127] |
| miRNA-21 | SPRY2 | [128] |
| miRNA-195 | MO25 | [63] |
| miRNA-199 | Dyrk1a, Hif-1a, Sirt1 | [129,130,131,132] |
| miRNA-1 | RASGAP, MEF2A, GATA4 | [60,133] |
| miRNA-26b | GSK3β | [134] |
| miRNA-27a | TGF-β1 | [135] |
| miRNA-143 | ACE2 | [136] |
| miRNA-29 | TGFB3 | [127] |
| miRNA-133 | Nelf-A/WHSC2, Rho | [137] |
| Cardiac Fibrosis | ||
| miRNA-21 | Spry1, PTEN | [77] |
| miRNA-133 | CTGF | [79] |
| miRNA-29 | COL4A5 | [16] |
| Cardiac Arrhythmia | ||
| miRNA-1 | KCNJ2 GJA1 | [75] |
| miRNA-133 | KCNQ1 and SRF | [138] |
| miRNA-133a | Cx43 | [82] |
| miRNA-212 | Kir2.1 | [83,84] |
| miRNA-17–92 | Pitx2 | [87] |
| miRNA-106b | Pitx2 | [87] |
| miRNA-150 | AT1R | [88] |
| Cardiac Regeneration | ||
| miRNA-15a | Chek1 | [102] |
| miRNA-17–92 | PTEN | [103] |
| miRNA-195 | Chek1 | [102] |
| miRNA-133 | mps1, cdc37, PA2G4, cx43, cldn5 | [139] |
| miRNA-208 | βMHC | [41] |
| miRNA-499 | Sox6 and Rod1 | [115] |
| miRNA-24 | eNOS | [94] |
| Stem/Progenitor Cells Differentiation | ||
| miRNA-21 | SPRY2 | [110] |
| miRNA-221 | Bim | [140] |
| miRNA-34a | Sirt1 | [112] |
| miRNA-126 | Spred1 | [109,141] |
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Go, A.S.; Mozaffarian, D.; Roger, V.L.; Benjamin, E.J.; Berry, J.D.; Blaha, M.J.; Dai, S.; Ford, E.S.; Fox, C.S.; Franco, S.; et al. Heart disease and stroke statistics—2014 update: A report from the American Heart Association. Circulation 2014, 129, e28–e292. [Google Scholar] [CrossRef]
- Mathers, C.D.; Loncar, D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med. 2006, 3, e442. [Google Scholar] [CrossRef]
- Lopez, A.D.; Mathers, C.D.; Ezzati, M.; Jamison, D.T.; Murray, C.J. Global and regional burden of disease and risk factors, 2001: Systematic analysis of population health data. Lancet 2006, 367, 1747–1757. [Google Scholar] [CrossRef]
- Chen, J.; Wang, D.Z. MicroRNAs in cardiovascular development. J. Mol. Cell. Cardiol. 2012, 52, 949–957. [Google Scholar] [CrossRef]
- Liu, N.; Olson, E.N. MicroRNA regulatory networks in cardiovascular development. Dev. Cell 2010, 18, 510–525. [Google Scholar] [CrossRef]
- Kishore, R.; Verma, S.K.; Mackie, A.R.; Vaughan, E.E.; Abramova, T.V.; Aiko, I.; Krishnamurthy, P. Bone marrow progenitor cell therapy-mediated paracrine regulation of cardiac miRNA-155 modulates fibrotic response in diabetic hearts. PLoS One 2013, 8, e60161. [Google Scholar]
- Liu, D.; Fan, J.; Zeng, W.; Zhou, Y.; Ingvarsson, S.; Chen, H. Quantitative analysis of miRNA expression in several developmental stages of human livers. Hepatol. Res. 2010, 40, 813–822. [Google Scholar] [CrossRef]
- Abdelmohsen, K.; Srikantan, S.; Kuwano, Y.; Gorospe, M. miR-519 reduces cell proliferation by lowering RNA-binding protein HuR levels. Proc. Natl. Acad. Sci. USA 2008, 105, 20297–20302. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Cascio, S.; D’Andrea, A.; Ferla, R.; Surmacz, E.; Gulotta, E.; Amodeo, V.; Bazan, V.; Gebbia, N.; Russo, A. miR-20b modulates VEGF expression by targeting HIF-1α and STAT3 in MCF-7 breast cancer cells. J. Cell. Physiol. 2010, 224, 242–249. [Google Scholar]
- El Ouaamari, A.; Baroukh, N.; Martens, G.A.; Lebrun, P.; Pipeleers, D.; van Obberghen, E. miR-375 targets 3'-phosphoinositide-dependent protein kinase-1 and regulates glucose-induced biological responses in pancreatic β-cells. Diabetes 2008, 57, 2708–2717. [Google Scholar] [CrossRef]
- Esau, C.; Davis, S.; Murray, S.F.; Yu, X.X.; Pandey, S.K.; Pear, M.; Watts, L.; Booten, S.L.; Graham, M.; McKay, R.; et al. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab. 2006, 3, 87–98. [Google Scholar] [CrossRef]
- Fish, J.E.; Srivastava, D. MicroRNAs: Opening a new vein in angiogenesis research. Sci. Signal. 2009, 2, pe1. [Google Scholar]
- Ivey, K.N.; Muth, A.; Arnold, J.; King, F.W.; Yeh, R.F.; Fish, J.E.; Hsiao, E.C.; Schwartz, R.J.; Conklin, B.R.; Bernstein, H.S.; et al. MicroRNA regulation of cell lineages in mouse and human embryonic stem cells. Cell Stem Cell 2008, 2, 219–229. [Google Scholar] [CrossRef]
- Urbich, C.; Kuehbacher, A.; Dimmeler, S. Role of microRNAs in vascular diseases, inflammation, and angiogenesis. Cardiovasc. Res. 2008, 79, 581–588. [Google Scholar] [CrossRef]
- Van Rooij, E.; Sutherland, L.B.; Thatcher, J.E.; DiMaio, J.M.; Naseem, R.H.; Marshall, W.S.; Hill, J.A.; Olson, E.N. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13027–13032. [Google Scholar] [CrossRef]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Wightman, B.; Ha, I.; Ruvkun, G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell 1993, 75, 855–862. [Google Scholar] [CrossRef]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef]
- Friedman, R.C.; Farh, K.K.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar]
- Lee, Y.; Kim, M.; Han, J.; Yeom, K.H.; Lee, S.; Baek, S.H.; Kim, V.N. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004, 23, 4051–4060. [Google Scholar] [CrossRef]
- Ha, M.; Kim, V.N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef]
- Kim, V.N.; Han, J.; Siomi, M.C. Biogenesis of small RNAs in animals. Nat. Rev. Mol. Cell Biol. 2009, 10, 126–139. [Google Scholar] [CrossRef]
- Guo, H.; Ingolia, N.T.; Weissman, J.S.; Bartel, D.P. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 2010, 466, 835–840. [Google Scholar] [CrossRef]
- Vasudevan, S.; Tong, Y.; Steitz, J.A. Switching from repression to activation: MicroRNAs can up-regulate translation. Science 2007, 318, 1931–1934. [Google Scholar] [CrossRef]
- Chi, S.W.; Zang, J.B.; Mele, A.; Darnell, R.B. Argonaute HITS-CLIP decodes microRNA–mRNA interaction maps. Nature 2009, 460, 479–486. [Google Scholar]
- Corrado, C.; Raimondo, S.; Chiesi, A.; Ciccia, F.; de Leo, G.; Alessandro, R. Exosomes as intercellular signaling organelles involved in health and disease: Basic science and clinical applications. Int. J. Mol. Sci. 2013, 14, 5338–5366. [Google Scholar] [CrossRef]
- Vo, N.K.; Dalton, R.P.; Liu, N.; Olson, E.N.; Goodman, R.H. Affinity purification of microRNA-133a with the cardiac transcription factor, Hand2. Proc. Natl. Acad. Sci. USA 2010, 107, 19231–19236. [Google Scholar] [CrossRef]
- Hendrickson, D.G.; Hogan, D.J.; McCullough, H.L.; Myers, J.W.; Herschlag, D.; Ferrell, J.E.; Brown, P.O. Concordant regulation of translation and mRNA abundance for hundreds of targets of a human microRNA. PLoS Biol. 2009, 7, e1000238. [Google Scholar] [CrossRef]
- Zhao, Y.; Ransom, J.F.; Li, A.; Vedantham, V.; von Drehle, M.; Muth, A.N.; Tsuchihashi, T.; McManus, M.T.; Schwartz, R.J.; Srivastava, D. Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking miRNA-1–2. Cell 2007, 129, 303–317. [Google Scholar] [CrossRef]
- Rao, P.K.; Toyama, Y.; Chiang, H.R.; Gupta, S.; Bauer, M.; Medvid, R.; Reinhardt, F.; Liao, R.; Krieger, M.; Jaenisch, R.; et al. Loss of cardiac microRNA-mediated regulation leads to dilated cardiomyopathy and heart failure. Circ. Res. 2009, 105, 585–594. [Google Scholar] [CrossRef]
- Olson, E.N.; Schneider, M.D. Sizing up the heart: Development redux in disease. Genes Dev. 2003, 17, 1937–1956. [Google Scholar] [CrossRef]
- Li, F.; Wang, X.; Capasso, J.M.; Gerdes, A.M. Rapid transition of cardiac myocytes from hyperplasia to hypertrophy during postnatal development. J. Mol. Cell. Cardiol. 1996, 28, 1737–1746. [Google Scholar] [CrossRef]
- Kajstura, J.; Zhang, X.; Reiss, K.; Szoke, E.; Li, P.; Lagrasta, C.; Cheng, W.; Darzynkiewicz, Z.; Olivetti, G.; Anversa, P. Myocyte cellular hyperplasia and myocyte cellular hypertrophy contribute to chronic ventricular remodeling in coronary artery narrowing-induced cardiomyopathy in rats. Circ. Res. 1994, 74, 383–400. [Google Scholar] [CrossRef]
- Paradis, A.; Xiao, D.; Zhou, J.; Zhang, L. Endothelin-1 promotes cardiomyocyte terminal differentiation in the developing heart via heightened DNA methylation. Int. J. Med. Sci. 2014, 11, 373–380. [Google Scholar] [CrossRef]
- Schupp, M.O.; Waas, M.; Chun, C.Z.; Ramchandran, R. Transcriptional inhibition of etv2 expression is essential for embryonic cardiac development. Dev. Biol. 2014, 393, 71–83. [Google Scholar] [CrossRef]
- Deb, A.; Ubil, E. Cardiac fibroblast in development and wound healing. J. Mol. Cell. Cardiol. 2014, 70, 47–55. [Google Scholar] [CrossRef]
- Ieda, M. Heart development and regeneration via cellular interaction and reprogramming. Keio J. Med. 2013, 62, 99–106. [Google Scholar] [CrossRef]
- Peralta, M.; Gonzalez-Rosa, J.M.; Marques, I.J.; Mercader, N. The epicardium in the embryonic and adult zebrafish. J. Dev. Biol. 2014, 2, 101–116. [Google Scholar] [CrossRef]
- Liu, N.; Williams, A.H.; Kim, Y.; McAnally, J.; Bezprozvannaya, S.; Sutherland, L.B.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. An intragenic MEF2-dependent enhancer directs muscle-specific expression of microRNAs 1 and 133. Proc. Natl. Acad. Sci. USA 2007, 104, 20844–20849. [Google Scholar] [CrossRef]
- Zhao, Y.; Samal, E.; Srivastava, D. Serum response factor regulates a muscle-specific microRNA that targets Hand2 during cardiogenesis. Nature 2005, 436, 214–220. [Google Scholar] [CrossRef]
- Liu, N.; Bezprozvannaya, S.; Williams, A.H.; Qi, X.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. microRNA-133a regulates cardiomyocyte proliferation and suppresses smooth muscle gene expression in the heart. Genes Dev. 2008, 22, 3242–3254. [Google Scholar] [CrossRef]
- Ventura, A.; Young, A.G.; Winslow, M.M.; Lintault, L.; Meissner, A.; Erkeland, S.J.; Newman, J.; Bronson, R.T.; Crowley, D.; Stone, J.R.; et al. Targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of miRNA clusters. Cell 2008, 132, 875–886. [Google Scholar] [CrossRef]
- Thum, T.; Galuppo, P.; Wolf, C.; Fiedler, J.; Kneitz, S.; van Laake, L.W.; Doevendans, P.A.; Mummery, C.L.; Borlak, J.; Haverich, A.; et al. MicroRNAs in the human heart: A clue to fetal gene reprogramming in heart failure. Circulation 2007, 116, 258–267. [Google Scholar] [CrossRef]
- Watanabe, K.; Thandavarayan, R.A.; Harima, M.; Sari, F.R.; Gurusamy, N.; Veeraveedu, P.T.; Mito, S.; Arozal, W.; Sukumaran, V.; Laksmanan, A.P.; et al. Role of differential signaling pathways and oxidative stress in diabetic cardiomyopathy. Curr. Cardiol. Rev. 2010, 6, 280–290. [Google Scholar] [CrossRef]
- Watanabe, K.; Thandavarayan, R.A.; Gurusamy, N.; Zhang, S.; Muslin, A.J.; Suzuki, K.; Tachikawa, H.; Kodama, M.; Aizawa, Y. Role of 14–3-3 protein and oxidative stress in diabetic cardiomyopathy. Acta Physiol. Hung. 2009, 96, 277–287. [Google Scholar] [CrossRef]
- Thandavarayan, R.A.; Watanabe, K.; Ma, M.; Gurusamy, N.; Veeraveedu, P.T.; Konishi, T.; Zhang, S.; Muslin, A.J.; Kodama, M.; Aizawa, Y. Dominant-negative p38α mitogen-activated protein kinase prevents cardiac apoptosis and remodeling after streptozotocin-induced diabetes mellitus. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H911–H919. [Google Scholar] [CrossRef]
- Thandavarayan, R.A.; Giridharan, V.V.; Sari, F.R.; Arumugam, S.; Veeraveedu, P.T.; Pandian, G.N.; Palaniyandi, S.S.; Ma, M.; Suzuki, K.; Gurusamy, N.; et al. Depletion of 14–3-3 protein exacerbates cardiac oxidative stress, inflammation and remodeling process via modulation of MAPK/NF-κB signaling pathways after streptozotocin-induced diabetes mellitus. Cell. Physiol. Biochem. 2011, 28, 911–922. [Google Scholar] [CrossRef]
- McCarthy, J.J.; Esser, K.A. MicroRNA-1 and microRNA-133a expression are decreased during skeletal muscle hypertrophy. J. Appl. Physiol. 2007, 102, 306–313. [Google Scholar] [CrossRef]
- Krishnamurthy, P.; Subramanian, V.; Singh, M.; Singh, K. β1 integrins modulate β-adrenergic receptor-stimulated cardiac myocyte apoptosis and myocardial remodeling. Hypertension 2007, 49, 865–872. [Google Scholar] [CrossRef]
- Verma, S.K.; Krishnamurthy, P.; Barefield, D.; Singh, N.; Gupta, R.; Lambers, E.; Thal, M.; Mackie, A.; Hoxha, E.; Ramirez, V.; et al. Interleukin-10 treatment attenuates pressure overload-induced hypertrophic remodeling and improves heart function via signal transducers and activators of transcription 3-dependent inhibition of nuclear factor-κB. Circulation 2012, 126, 418–429. [Google Scholar] [CrossRef]
- Rajabi, M.; Kassiotis, C.; Razeghi, P.; Taegtmeyer, H. Return to the fetal gene program protects the stressed heart: A strong hypothesis. Heart Fail. Rev. 2007, 12, 331–343. [Google Scholar] [CrossRef]
- Mooren, F.C.; Viereck, J.; Kruger, K.; Thum, T. Circulating microRNAs as potential biomarkers of aerobic exercise capacity. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H557–H563. [Google Scholar] [CrossRef]
- Martinelli, N.C.; Cohen, C.R.; Santos, K.G.; Castro, M.A.; Biolo, A.; Frick, L.; Silvello, D.; Lopes, A.; Schneider, S.; Andrades, M.E.; et al. An analysis of the global expression of microRNAs in an experimental model of physiological left ventricular hypertrophy. PLoS One 2014, 9, e93271. [Google Scholar] [CrossRef]
- Callis, T.E.; Pandya, K.; Seok, H.Y.; Tang, R.H.; Tatsuguchi, M.; Huang, Z.P.; Chen, J.F.; Deng, Z.; Gunn, B.; Shumate, J.; et al. MicroRNA-208a is a regulator of cardiac hypertrophy and conduction in mice. J. Clin. Investig. 2009, 119, 2772–2786. [Google Scholar] [CrossRef]
- Van Rooij, E.; Sutherland, L.B.; Liu, N.; Williams, A.H.; McAnally, J.; Gerard, R.D.; Richardson, J.A.; Olson, E.N. A signature pattern of stress-responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proc. Natl. Acad. Sci. USA 2006, 103, 18255–18260. [Google Scholar] [CrossRef]
- Chen, J.F.; Murchison, E.P.; Tang, R.; Callis, T.E.; Tatsuguchi, M.; Deng, Z.; Rojas, M.; Hammond, S.M.; Schneider, M.D.; Selzman, C.H.; et al. Targeted deletion of Dicer in the heart leads to dilated cardiomyopathy and heart failure. Proc. Natl. Acad. Sci. USA 2008, 105, 2111–2116. [Google Scholar] [CrossRef]
- Cheng, Y.; Ji, R.; Yue, J.; Yang, J.; Liu, X.; Chen, H.; Dean, D.B.; Zhang, C. MicroRNAs are aberrantly expressed in hypertrophic heart: Do they play a role in cardiac hypertrophy? Am. J. Pathol. 2007, 170, 1831–1840. [Google Scholar] [CrossRef]
- Sayed, D.; Hong, C.; Chen, I.Y.; Lypowy, J.; Abdellatif, M. MicroRNAs play an essential role in the development of cardiac hypertrophy. Circ. Res. 2007, 100, 416–424. [Google Scholar] [CrossRef]
- Ikeda, S.; Kong, S.W.; Lu, J.; Bisping, E.; Zhang, H.; Allen, P.D.; Golub, T.R.; Pieske, B.; Pu, W.T. Altered microRNA expression in human heart disease. Physiol. Genomics 2007, 31, 367–373. [Google Scholar] [CrossRef]
- Tatsuguchi, M.; Seok, H.Y.; Callis, T.E.; Thomson, J.M.; Chen, J.F.; Newman, M.; Rojas, M.; Hammond, S.M.; Wang, D.Z. Expression of microRNAs is dynamically regulated during cardiomyocyte hypertrophy. J. Mol. Cell. Cardiol. 2007, 42, 1137–1141. [Google Scholar] [CrossRef]
- Chen, H.; Untiveros, G.M.; McKee, L.A.; Perez, J.; Li, J.; Antin, P.B.; Konhilas, J.P. Micro-RNA-195 and -451 regulate the LKB1/AMPK signaling axis by targeting MO25. PLoS One 2012, 7, e41574. [Google Scholar]
- Montgomery, R.L.; Hullinger, T.G.; Semus, H.M.; Dickinson, B.A.; Seto, A.G.; Lynch, J.M.; Stack, C.; Latimer, P.A.; Olson, E.N.; van Rooij, E. Therapeutic inhibition of miR-208a improves cardiac function and survival during heart failure. Circulation 2011, 124, 1537–1547. [Google Scholar] [CrossRef]
- Bang, C.; Batkai, S.; Dangwal, S.; Gupta, S.K.; Foinquinos, A.; Holzmann, A.; Just, A.; Remke, J.; Zimmer, K.; Zeug, A.; et al. Cardiac fibroblast-derived microRNA passenger strand-enriched exosomes mediate cardiomyocyte hypertrophy. J. Clin. Investig. 2014, 124, 2136–2146. [Google Scholar] [CrossRef]
- Rossi, M.A. Pathologic fibrosis and connective tissue matrix in left ventricular hypertrophy due to chronic arterial hypertension in humans. J. Hypertens. 1998, 16, 1031–1041. [Google Scholar] [CrossRef]
- Swynghedauw, B. Molecular mechanisms of myocardial remodeling. Physiol. Rev. 1999, 79, 215–262. [Google Scholar]
- Manabe, I.; Shindo, T.; Nagai, R. Gene expression in fibroblasts and fibrosis: Involvement in cardiac hypertrophy. Circ. Res. 2002, 91, 1103–1113. [Google Scholar] [CrossRef]
- Brown, R.D.; Ambler, S.K.; Mitchell, M.D.; Long, C.S. The cardiac fibroblast: Therapeutic target in myocardial remodeling and failure. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 657–687. [Google Scholar] [CrossRef]
- Khan, R.; Sheppard, R. Fibrosis in heart disease: Understanding the role of transforming growth factor-β in cardiomyopathy, valvular disease and arrhythmia. Immunology 2006, 118, 10–24. [Google Scholar] [CrossRef]
- Martos, R.; Baugh, J.; Ledwidge, M.; O’Loughlin, C.; Conlon, C.; Patle, A.; Donnelly, S.C.; McDonald, K. Diastolic heart failure: Evidence of increased myocardial collagen turnover linked to diastolic dysfunction. Circulation 2007, 115, 888–895. [Google Scholar] [CrossRef]
- Benjamin, I.J.; Jalil, J.E.; Tan, L.B.; Cho, K.; Weber, K.T.; Clark, W.A. Isoproterenol-induced myocardial fibrosis in relation to myocyte necrosis. Circ. Res. 1989, 65, 657–670. [Google Scholar] [CrossRef]
- Buja, L.M.; Willerson, J.T. The role of coronary artery lesions in ischemic heart disease: Insights from recent clinicopathologic, coronary arteriographic, and experimental studies. Hum. Pathol. 1987, 18, 451–461. [Google Scholar] [CrossRef]
- Krishnamurthy, P.; Peterson, J.T.; Subramanian, V.; Singh, M.; Singh, K. Inhibition of matrix metalloproteinases improves left ventricular function in mice lacking osteopontin after myocardial infarction. Mol. Cell. Biochem. 2009, 322, 53–62. [Google Scholar] [CrossRef]
- Yang, B.; Lin, H.; Xiao, J.; Lu, Y.; Luo, X.; Li, B.; Zhang, Y.; Xu, C.; Bai, Y.; Wang, H.; et al. The muscle-specific microRNA miR-1 regulates cardiac arrhythmogenic potential by targeting GJA1 and KCNJ2. Nat. Med. 2007, 13, 486–491. [Google Scholar] [CrossRef]
- Ichimura, A.; Ruike, Y.; Terasawa, K.; Tsujimoto, G. miRNAs and regulation of cell signaling. FEBS J. 2011, 278, 1610–1618. [Google Scholar] [CrossRef]
- Thum, T.; Gross, C.; Fiedler, J.; Fischer, T.; Kissler, S.; Bussen, M.; Galuppo, P.; Just, S.; Rottbauer, W.; Frantz, S.; et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature 2008, 456, 980–984. [Google Scholar] [CrossRef]
- Roy, S.; Khanna, S.; Hussain, S.R.; Biswas, S.; Azad, A.; Rink, C.; Gnyawali, S.; Shilo, S.; Nuovo, G.J.; Sen, C.K. MicroRNA expression in response to murine myocardial infarction: miR-21 regulates fibroblast metalloprotease-2 via phosphatase and tensin homologue. Cardiovasc. Res. 2009, 82, 21–29. [Google Scholar] [CrossRef]
- Duisters, R.F.; Tijsen, A.J.; Schroen, B.; Leenders, J.J.; Lentink, V.; van der Made, I.; Herias, V.; van Leeuwen, R.E.; Schellings, M.W.; Barenbrug, P.; et al. miR-133 and miR-30 regulate connective tissue growth factor: Implications for a role of microRNAs in myocardial matrix remodeling. Circ. Res. 2009, 104, 170–178. [Google Scholar] [CrossRef]
- He, B.; Xiao, J.; Ren, A.J.; Zhang, Y.F.; Zhang, H.; Chen, M.; Xie, B.; Gao, X.G.; Wang, Y.W. Role of miR-1 and miR-133a in myocardial ischemic postconditioning. J. Biomed. Sci. 2011, 18, 22. [Google Scholar] [CrossRef]
- Danowski, N.; Manthey, I.; Jakob, H.G.; Siffert, W.; Peters, J.; Frey, U.H. Decreased expression of miR-133a but not of miR-1 is associated with signs of heart failure in patients undergoing coronary bypass surgery. Cardiology 2013, 125, 125–130. [Google Scholar] [CrossRef]
- Osbourne, A.; Calway, T.; Broman, M.; McSharry, S.; Earley, J.; Kim, G.H. Downregulation of connexin43 by microRNA-130a in cardiomyocytes results in cardiac arrhythmias. J. Mol. Cell. Cardiol. 2014, 74, 53–63. [Google Scholar] [CrossRef]
- Ucar, A.; Gupta, S.K.; Fiedler, J.; Erikci, E.; Kardasinski, M.; Batkai, S.; Dangwal, S.; Kumarswamy, R.; Bang, C.; Holzmann, A.; et al. The miRNA-212/132 family regulates both cardiac hypertrophy and cardiomyocyte autophagy. Nat. Commun. 2012, 3, 1078. [Google Scholar] [CrossRef]
- Xiao, J.; Liang, D.; Zhang, Y.; Liu, Y.; Zhang, H.; Liu, Y.; Li, L.; Liang, X.; Sun, Y.; Chen, Y.H. MicroRNA expression signature in atrial fibrillation with mitral stenosis. Physiol. Genomics 2011, 43, 655–664. [Google Scholar] [CrossRef]
- Chinchilla, A.; Daimi, H.; Lozano-Velasco, E.; Dominguez, J.N.; Caballero, R.; Delpon, E.; Tamargo, J.; Cinca, J.; Hove-Madsen, L.; Aranega, A.E.; et al. PITX2 insufficiency leads to atrial electrical and structural remodeling linked to arrhythmogenesis. Circ. Cardiovasc. Genet. 2011, 4, 269–279. [Google Scholar] [CrossRef]
- Wang, J.; Klysik, E.; Sood, S.; Johnson, R.L.; Wehrens, X.H.; Martin, J.F. Pitx2 prevents susceptibility to atrial arrhythmias by inhibiting left-sided pacemaker specification. Proc. Natl. Acad. Sci. USA 2010, 107, 9753–9758. [Google Scholar] [CrossRef]
- Wang, J.; Bai, Y.; Li, N.; Ye, W.; Zhang, M.; Greene, S.B.; Tao, Y.; Chen, Y.; Wehrens, X.H.; Martin, J.F. Pitx2-microRNA pathway that delimits sinoatrial node development and inhibits predisposition to atrial fibrillation. Proc. Natl. Acad. Sci. USA 2014, 111, 9181–9186. [Google Scholar] [CrossRef]
- Goren, Y.; Meiri, E.; Hogan, C.; Mitchell, H.; Lebanony, D.; Salman, N.; Schliamser, J.E.; Amir, O. Relation of reduced expression of miR-150 in platelets to atrial fibrillation in patients with chronic systolic heart failure. Am. J. Cardiol. 2014, 113, 976–981. [Google Scholar] [CrossRef]
- Xiao, J.; Luo, X.; Lin, H.; Zhang, Y.; Lu, Y.; Wang, N.; Zhang, Y.; Yang, B.; Wang, Z. MicroRNA miR-133 represses HERG K+ channel expression contributing to QT prolongation in diabetic hearts. J. Biol. Chem. 2007, 282, 12363–12367. [Google Scholar] [CrossRef]
- Bauters, C.; Lamblin, N.; Mc Fadden, E.P.; van Belle, E.; Millaire, A.; de Groote, P. Influence of diabetes mellitus on heart failure risk and outcome. Cardiovasc. Diabetol. 2003, 2, 1. [Google Scholar] [CrossRef]
- Li, Y.; Yang, C.M.; Xi, Y.; Wu, G.; Shelat, H.; Gao, S.; Cheng, J.; Geng, Y.J. MicroRNA-1/133 targeted dysfunction of potassium channels KCNE1 and KCNQ1 in human cardiac progenitor cells with simulated hyperglycemia. Int. J. Cardiol. 2013, 167, 1076–1078. [Google Scholar] [CrossRef]
- Fasanaro, P.; D’Alessandra, Y.; di Stefano, V.; Melchionna, R.; Romani, S.; Pompilio, G.; Capogrossi, M.C.; Martelli, F. MicroRNA-210 modulates endothelial cell response to hypoxia and inhibits the receptor tyrosine kinase ligand Ephrin-A3. J. Biol. Chem. 2008, 283, 15878–15883. [Google Scholar] [CrossRef]
- Icli, B.; Wara, A.K.; Moslehi, J.; Sun, X.; Plovie, E.; Cahill, M.; Marchini, J.F.; Schissler, A.; Padera, R.F.; Shi, J.; et al. MicroRNA-26a regulates pathological and physiological angiogenesis by targeting BMP/SMAD1 signaling. Circ. Res. 2013, 113, 1231–1241. [Google Scholar] [CrossRef]
- Meloni, M.; Marchetti, M.; Garner, K.; Littlejohns, B.; Sala-Newby, G.; Xenophontos, N.; Floris, I.; Suleiman, M.S.; Madeddu, P.; Caporali, A.; et al. Local inhibition of microRNA-24 improves reparative angiogenesis and left ventricle remodeling and function in mice with myocardial infarction. Mol. Ther. 2013, 21, 1390–1402. [Google Scholar] [CrossRef]
- Dentelli, P.; Rosso, A.; Orso, F.; Olgasi, C.; Taverna, D.; Brizzi, M.F. microRNA-222 controls neovascularization by regulating signal transducer and activator of transcription 5A expression. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1562–1568. [Google Scholar] [CrossRef]
- Liu, X.; Cheng, Y.; Zhang, S.; Lin, Y.; Yang, J.; Zhang, C. A necessary role of miR-221 and miR-222 in vascular smooth muscle cell proliferation and neointimal hyperplasia. Circ. Res. 2009, 104, 476–487. [Google Scholar] [CrossRef]
- Chen, Y.; Gorski, D.H. Regulation of angiogenesis through a microRNA (miR-130a) that down-regulates antiangiogenic homeobox genes GAX and HOXA5. Blood 2008, 111, 1217–1226. [Google Scholar] [CrossRef]
- Sessa, R.; Seano, G.; di Blasio, L.; Gagliardi, P.A.; Isella, C.; Medico, E.; Cotelli, F.; Bussolino, F.; Primo, L. The miR-126 regulates angiopoietin-1 signaling and vessel maturation by targeting p85β. Biochim. Biophys. Acta 2012, 1823, 1925–1935. [Google Scholar] [CrossRef]
- Porrello, E.R.; Mahmoud, A.I.; Simpson, E.; Hill, J.A.; Richardson, J.A.; Olson, E.N.; Sadek, H.A. Transient regenerative potential of the neonatal mouse heart. Science 2011, 331, 1078–1080. [Google Scholar] [CrossRef]
- Bergmann, O.; Bhardwaj, R.D.; Bernard, S.; Zdunek, S.; Barnabe-Heider, F.; Walsh, S.; Zupicich, J.; Alkass, K.; Buchholz, B.A.; Druid, H.; et al. Evidence for cardiomyocyte renewal in humans. Science 2009, 324, 98–102. [Google Scholar] [CrossRef]
- Mollova, M.; Bersell, K.; Walsh, S.; Savla, J.; Das, L.T.; Park, S.Y.; Silberstein, L.E.; Dos Remedios, C.G.; Graham, D.; Colan, S.; et al. Cardiomyocyte proliferation contributes to heart growth in young humans. Proc. Natl. Acad. Sci. USA 2013, 110, 1446–1451. [Google Scholar] [CrossRef]
- Porrello, E.R.; Johnson, B.A.; Aurora, A.B.; Simpson, E.; Nam, Y.J.; Matkovich, S.J.; Dorn, G.W., II; van Rooij, E.; Olson, E.N. MiR-15 family regulates postnatal mitotic arrest of cardiomyocytes. Circ. Res. 2011, 109, 670–679. [Google Scholar] [CrossRef]
- Chen, J.; Huang, Z.P.; Seok, H.Y.; Ding, J.; Kataoka, M.; Zhang, Z.; Hu, X.; Wang, G.; Lin, Z.; Wang, S.; et al. miR-17–92 cluster is required for and sufficient to induce cardiomyocyte proliferation in postnatal and adult hearts. Circ. Res. 2013, 112, 1557–1566. [Google Scholar] [CrossRef]
- Ieda, M.; Fu, J.D.; Delgado-Olguin, P.; Vedantham, V.; Hayashi, Y.; Bruneau, B.G.; Srivastava, D. Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell 2010, 142, 375–386. [Google Scholar] [CrossRef]
- Qian, L.; Huang, Y.; Spencer, C.I.; Foley, A.; Vedantham, V.; Liu, L.; Conway, S.J.; Fu, J.D.; Srivastava, D. In vivo reprogramming of murine cardiac fibroblasts into induced cardiomyocytes. Nature 2012, 485, 593–598. [Google Scholar] [CrossRef]
- Song, K.; Nam, Y.J.; Luo, X.; Qi, X.; Tan, W.; Huang, G.N.; Acharya, A.; Smith, C.L.; Tallquist, M.D.; Neilson, E.G.; et al. Heart repair by reprogramming non-myocytes with cardiac transcription factors. Nature 2012, 485, 599–604. [Google Scholar] [CrossRef]
- Jayawardena, T.M.; Egemnazarov, B.; Finch, E.A.; Zhang, L.; Payne, J.A.; Pandya, K.; Zhang, Z.; Rosenberg, P.; Mirotsou, M.; Dzau, V.J. MicroRNA-mediated in vitro and in vivo direct reprogramming of cardiac fibroblasts to cardiomyocytes. Circ. Res. 2012, 110, 1465–1473. [Google Scholar] [CrossRef]
- Van Solingen, C.; Seghers, L.; Bijkerk, R.; Duijs, J.M.; Roeten, M.K.; van Oeveren-Rietdijk, A.M.; Baelde, H.J.; Monge, M.; Vos, J.B.; de Boer, H.C.; et al. Antagomir-mediated silencing of endothelial cell specific microRNA-126 impairs ischemia-induced angiogenesis. J. Cell. Mol. Med. 2009, 13, 1577–1585. [Google Scholar] [CrossRef]
- Fish, J.E.; Santoro, M.M.; Morton, S.U.; Yu, S.; Yeh, R.F.; Wythe, J.D.; Ivey, K.N.; Bruneau, B.G.; Stainier, D.Y.; Srivastava, D. miR-126 regulates angiogenic signaling and vascular integrity. Dev. Cell 2008, 15, 272–284. [Google Scholar] [CrossRef]
- Fleissner, F.; Jazbutyte, V.; Fiedler, J.; Gupta, S.K.; Yin, X.; Xu, Q.; Galuppo, P.; Kneitz, S.; Mayr, M.; Ertl, G.; et al. Short communication: Asymmetric dimethylarginine impairs angiogenic progenitor cell function in patients with coronary artery disease through a microRNA-21-dependent mechanism. Circ. Res. 2010, 107, 138–143. [Google Scholar] [CrossRef]
- Minami, Y.; Satoh, M.; Maesawa, C.; Takahashi, Y.; Tabuchi, T.; Itoh, T.; Nakamura, M. Effect of atorvastatin on microRNA 221/222 expression in endothelial progenitor cells obtained from patients with coronary artery disease. Eur. J. Clin. Investig. 2009, 39, 359–367. [Google Scholar] [CrossRef]
- Zhao, T.; Li, J.; Chen, A.F. MicroRNA-34a induces endothelial progenitor cell senescence and impedes its angiogenesis via suppressing silent information regulator 1. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E110–E116. [Google Scholar] [CrossRef]
- Miranda, K.C.; Huynh, T.; Tay, Y.; Ang, Y.S.; Tam, W.L.; Thomson, A.M.; Lim, B.; Rigoutsos, I. A pattern-based method for the identification of microRNA binding sites and their corresponding heteroduplexes. Cell 2006, 126, 1203–1217. [Google Scholar] [CrossRef]
- Van Rooij, E.; Marshall, W.S.; Olson, E.N. Toward microRNA-based therapeutics for heart disease: The sense in antisense. Circ. Res. 2008, 103, 919–928. [Google Scholar] [CrossRef]
- Corsten, M.F.; Dennert, R.; Jochems, S.; Kuznetsova, T.; Devaux, Y.; Hofstra, L.; Wagner, D.R.; Staessen, J.A.; Heymans, S.; Schroen, B. Circulating microRNA-208b and microRNA-499 reflect myocardial damage in cardiovascular disease. Circ. Cardiovasc. Genet. 2010, 3, 499–506. [Google Scholar] [CrossRef]
- D’Alessandra, Y.; Devanna, P.; Limana, F.; Straino, S.; di Carlo, A.; Brambilla, P.G.; Rubino, M.; Carena, M.C.; Spazzafumo, L.; de Simone, M.; et al. Circulating microRNAs are new and sensitive biomarkers of myocardial infarction. Eur. Heart J. 2010, 31, 2765–2773. [Google Scholar] [CrossRef]
- Fukushima, Y.; Nakanishi, M.; Nonogi, H.; Goto, Y.; Iwai, N. Assessment of plasma miRNAs in congestive heart failure. Circ. J. 2011, 75, 336–340. [Google Scholar] [CrossRef]
- Van Empel, V.P.; de Windt, L.J.; da Costa Martins, P.A. Circulating miRNAs: Reflecting or affecting cardiovascular disease? Curr. Hypertens. Rep. 2012, 14, 498–509. [Google Scholar] [CrossRef]
- Qiang, L.; Hong, L.; Ningfu, W.; Huaihong, C.; Jing, W. Expression of miR-126 and miR-508–5p in endothelial progenitor cells is associated with the prognosis of chronic heart failure patients. Int. J. Cardiol. 2013, 168, 2082–2088. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, W.; Han, L.; Rayburn, E.R.; Hill, D.L.; Wang, H.; Zhang, R. Isolation, structural determination, and evaluation of the biological activity of 20(S)-25-methoxyl-dammarane-3β, 12β, 20-triol [20(S)-25-OCH3-PPD], a novel natural product from Panax notoginseng. Med. Chem. 2007, 3, 51–60. [Google Scholar] [CrossRef]
- Foshay, K.M.; Gallicano, G.I. miR-17 family miRNAs are expressed during early mammalian development and regulate stem cell differentiation. Dev. Biol. 2009, 326, 431–443. [Google Scholar] [CrossRef]
- Frank, D.; Gantenberg, J.; Boomgaarden, I.; Kuhn, C.; Will, R.; Jarr, K.U.; Eden, M.; Kramer, K.; Luedde, M.; Mairbaurl, H.; et al. MicroRNA-20a inhibits stress-induced cardiomyocyte apoptosis involving its novel target Egln3/PHD3. J. Mol. Cell. Cardiol. 2012, 52, 711–717. [Google Scholar] [CrossRef]
- Wang, K.C.; Garmire, L.X.; Young, A.; Nguyen, P.; Trinh, A.; Subramaniam, S.; Wang, N.; Shyy, J.Y.; Li, Y.S.; Chien, S. Role of microRNA-23b in flow-regulation of Rb phosphorylation and endothelial cell growth. Proc. Natl. Acad. Sci. USA 2010, 107, 3234–3239. [Google Scholar] [CrossRef]
- Deacon, D.C.; Nevis, K.R.; Cashman, T.J.; Zhou, Y.; Zhao, L.; Washko, D.; Guner-Ataman, B.; Burns, C.G.; Burns, C.E. The miR-143-adducin3 pathway is essential for cardiac chamber morphogenesis. Development 2010, 137, 1887–1896. [Google Scholar] [CrossRef]
- Li, X.; Kong, M.; Jiang, D.; Qian, J.; Duan, Q.; Dong, A. MicroRNA-150 aggravates H2O2-induced cardiac myocyte injury by down-regulating c-myb gene. Acta Biochim. Biophys. Sin. 2013, 45, 734–741. [Google Scholar] [CrossRef]
- Lin, Z.; Murtaza, I.; Wang, K.; Jiao, J.; Gao, J.; Li, P.F. miR-23a functions downstream of NFATc3 to regulate cardiac hypertrophy. Proc. Natl. Acad. Sci. USA 2009, 106, 12103–12108. [Google Scholar] [CrossRef]
- Diao, X.; Shen, E.; Wang, X.; Hu, B. Differentially expressed microRNAs and their target genes in the hearts of streptozotocin-induced diabetic mice. Mol. Med. Rep. 2011, 4, 633–640. [Google Scholar]
- Sayed, D.; Rane, S.; Lypowy, J.; He, M.; Chen, I.Y.; Vashistha, H.; Yan, L.; Malhotra, A.; Vatner, D.; Abdellatif, M. MicroRNA-21 targets Sprouty2 and promotes cellular outgrowths. Mol. Biol. Cell 2008, 19, 3272–3282. [Google Scholar] [CrossRef]
- Van Rooij, E.; Doevendans, P.A.; de Theije, C.C.; Babiker, F.A.; Molkentin, J.D.; de Windt, L.J. Requirement of nuclear factor of activated T-cells in calcineurin-mediated cardiomyocyte hypertrophy. J. Biol. Chem. 2002, 277, 48617–48626. [Google Scholar]
- Arron, J.R.; Winslow, M.M.; Polleri, A.; Chang, C.P.; Wu, H.; Gao, X.; Neilson, J.R.; Chen, L.; Heit, J.J.; Kim, S.K.; et al. NFAT dysregulation by increased dosage of DSCR1 and DYRK1A on chromosome 21. Nature 2006, 441, 595–600. [Google Scholar] [CrossRef]
- Song, X.W.; Li, Q.; Lin, L.; Wang, X.C.; Li, D.F.; Wang, G.K.; Ren, A.J.; Wang, Y.R.; Qin, Y.W.; Yuan, W.J.; et al. MicroRNAs are dynamically regulated in hypertrophic hearts, and miR-199a is essential for the maintenance of cell size in cardiomyocytes. J. Cell. Physiol. 2010, 225, 437–443. [Google Scholar] [CrossRef]
- Rane, S.; He, M.; Sayed, D.; Vashistha, H.; Malhotra, A.; Sadoshima, J.; Vatner, D.E.; Vatner, S.F.; Abdellatif, M. Downregulation of miR-199a derepresses hypoxia-inducible factor-1α and Sirtuin 1 and recapitulates hypoxia preconditioning in cardiac myocytes. Circ. Res. 2009, 104, 879–886. [Google Scholar] [CrossRef]
- Ikeda, S.; He, A.; Kong, S.W.; Lu, J.; Bejar, R.; Bodyak, N.; Lee, K.H.; Ma, Q.; Kang, P.M.; Golub, T.R.; et al. MicroRNA-1 negatively regulates expression of the hypertrophy-associated calmodulin and Mef2a genes. Mol. Cell Biol. 2009, 29, 2193–2204. [Google Scholar] [CrossRef]
- Zhang, Z.H.; Li, J.; Liu, B.R.; Luo, C.F.; Dong, Q.; Zhao, L.N.; Zhong, Y.; Chen, W.Y.; Chen, M.S.; Liu, S.M. MicroRNA-26 was decreased in rat cardiac hypertrophy model and may be a promising therapeutic target. J. Cardiovasc. Pharmacol. 2013, 62, 312–319. [Google Scholar] [CrossRef]
- Wang, J.; Song, Y.; Zhang, Y.; Xiao, H.; Sun, Q.; Hou, N.; Guo, S.; Wang, Y.; Fan, K.; Zhan, D.; et al. Cardiomyocyte overexpression of miR-27b induces cardiac hypertrophy and dysfunction in mice. Cell Res. 2012, 22, 516–527. [Google Scholar] [CrossRef]
- Boettger, T.; Beetz, N.; Kostin, S.; Schneider, J.; Kruger, M.; Hein, L.; Braun, T. Acquisition of the contractile phenotype by murine arterial smooth muscle cells depends on the Mir143/145 gene cluster. J. Clin. Investig. 2009, 119, 2634–2647. [Google Scholar] [CrossRef]
- Care, A.; Catalucci, D.; Felicetti, F.; Bonci, D.; Addario, A.; Gallo, P.; Bang, M.L.; Segnalini, P.; Gu, Y.; Dalton, N.D.; et al. MicroRNA-133 controls cardiac hypertrophy. Nat. Med. 2007, 13, 613–618. [Google Scholar] [CrossRef]
- Luo, X.; Xiao, J.; Lin, H.; Li, B.; Lu, Y.; Yang, B.; Wang, Z. Transcriptional activation by stimulating protein 1 and post-transcriptional repression by muscle-specific microRNAs of IKs-encoding genes and potential implications in regional heterogeneity of their expressions. J. Cell. Physiol. 2007, 212, 358–367. [Google Scholar] [CrossRef]
- Yin, V.P.; Lepilina, A.; Smith, A.; Poss, K.D. Regulation of zebrafish heart regeneration by miR-133. Dev. Biol. 2012, 365, 319–327. [Google Scholar] [CrossRef]
- Hu, S.; Huang, M.; Nguyen, P.K.; Gong, Y.; Li, Z.; Jia, F.; Lan, F.; Liu, J.; Nag, D.; Robbins, R.C.; et al. Novel microRNA prosurvival cocktail for improving engraftment and function of cardiac progenitor cell transplantation. Circulation 2011, 124, S27–S34. [Google Scholar] [CrossRef]
- Jakob, P.; Landmesser, U. Role of microRNAs in stem/progenitor cells and cardiovascular repair. Cardiovasc. Res. 2012, 93, 614–622. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Joladarashi, D.; Thandavarayan, R.A.; Babu, S.S.; Krishnamurthy, P. Small Engine, Big Power: MicroRNAs as Regulators of Cardiac Diseases and Regeneration. Int. J. Mol. Sci. 2014, 15, 15891-15911. https://doi.org/10.3390/ijms150915891
Joladarashi D, Thandavarayan RA, Babu SS, Krishnamurthy P. Small Engine, Big Power: MicroRNAs as Regulators of Cardiac Diseases and Regeneration. International Journal of Molecular Sciences. 2014; 15(9):15891-15911. https://doi.org/10.3390/ijms150915891
Chicago/Turabian StyleJoladarashi, Darukeshwara, Rajarajan A. Thandavarayan, Sahana Suresh Babu, and Prasanna Krishnamurthy. 2014. "Small Engine, Big Power: MicroRNAs as Regulators of Cardiac Diseases and Regeneration" International Journal of Molecular Sciences 15, no. 9: 15891-15911. https://doi.org/10.3390/ijms150915891
APA StyleJoladarashi, D., Thandavarayan, R. A., Babu, S. S., & Krishnamurthy, P. (2014). Small Engine, Big Power: MicroRNAs as Regulators of Cardiac Diseases and Regeneration. International Journal of Molecular Sciences, 15(9), 15891-15911. https://doi.org/10.3390/ijms150915891