Bioinformatics Study of Cancer-Related Mutations within p53 Phosphorylation Site Motifs



Abstract
:
1. Introduction
2. Results
2.1. Sequence Features of Phosphorylation Sites in p53
2.1.1. Consensus Motifs within the Phosphorylation Sites in p53

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| P−3 | P−2 | P−1 | pS/pT | P+1 | P+2 | P+3 | Kinases | Consensus Motif | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| E | P | Q | S6 | D | P | S | JNK2, CK1δ | P-X-S/T-P | [6,7] |
| S | D | P | S9 | V | E | P | CK1ε | pS-X-X-S/T | [6,7] |
| P | P | L | S15 | Q | E | T | CDK5, mTOR, ATM | P-L-S/T-P (CDK) D-S/T-Q-E (ATM)D | [14,15] |
| S | Q | E | T18 | F | S | D | Chk2, TTK, VRK1 | [14,15] | |
| E | T | F | S20 | D | L | W | Chk2, Plk3 | T-X-S/T-X-X-W (Chk2) | [14,15] |
| N | V | L | S33 | P | L | P | Cdk5/7/9, GSK3β, p38K | P-L-S/T-P (CDK) | [6,7] |
| P | L | P | S37 | Q | A | M | ATR, PRAK | L-P-S/T-Q-A (ATR) | [6,7] |
| L | M | L | S46 | P | D | D | Cdk5, p38K, PKC | P-L-S/T-P (CDK) | [20] |
| Q | W | F | T55 | E | D | P | ERK2, TAF1 | [21] | |
| A | A | P | T81 | P | A | A | JNK2 | P-X-S/T-P | [6,7] |
| S | V | P | S99 | Q | K | T | ATM, ATR | V-P-S/T-Q (ATR) | [6,7] |
| Y | Q | G | S106 | Y | G | F | Aurora A | [6,7] | |
| N | N | T | S313 | S | S | P | Chk1/2 | [6,7] | |
| N | T | S | S314 | S | P | Q | Chk1/2 | T-X-S/T (Chk2) | [6,7] |
| T | S | S | S315 | P | Q | P | STK15, Cdk9, CDK2 | P-L-S/T-P (CDK) | [24,25] |
| P | G | G | S362 | R | A | H | IKK2B | [6,7] | |
| R | A | H | S366 | S | H | L | IKK2, Chk2 | [6,7] | |
| K | G | Q | S376 | T | S | R | PKC, GSK3β | R-K/R-X-S/T-X-K-K/R (PKC) | [25,26] |
| G | Q | S | T377 | S | R | H | Chk1/2 | [6,7] | |
| Q | S | T | S378 | R | H | K | Chk1/2, PKC | R-K/R-X-S/T-X-K-K/R (PKC) | [6,7] |
| M | F | K | T387 | E | G | P | Chk1 | [6,7] | |
| G | P | D | S392 | D | Cdk9, PKR, FACT | [30] | |||
| W | V | D | S149 | T | P | P | CSN | [22,23] | |
| V | D | S | T150 | P | P | P | CSN | [22,23] | |
| P | P | G | T155 | R | V | R | CSN | [22,23] | |
| E | R | C | S183 | D | S | D | Aurora B | R/K-X-S/T (Aurara B) | [68] |
| D | R | N | T211 | F | R | H | Aurora B | R/K-X-S/T (Aurara B) | [68] |
| F | R | H | S215 | V | V | V | STK15, Aurora B | R/K-X-S/T (Aurara B) | [32] |
| G | R | N | S269 | F | E | V | Aurora B | R/K-X-S/T (Aurara B) | [68] |
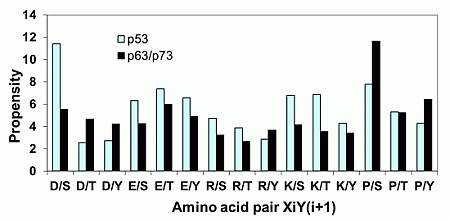
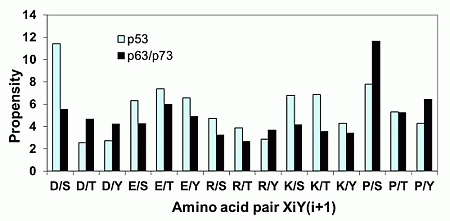
2.1.2. Acidic Residues Occur at Positions P−1 and P+1 Adjacent to Phosphorylation Sites in p53
| Amino Acid Pair | P−1-P(S/T)-P+1 | P−2-P(S/T)-P+2 | P−3-P(S/T)-P+3 |
|---|---|---|---|
| D/S | 6 | 2 | 2 |
| P/S | 6 | 6 | 8 |
| S/Q | 5 | 2 | 2 |
| S/T | 4 | 4 | 3 |
| L/S | 3 | 3 | 2 |
| E/T | 3 | 0 | 0 |
| P/T | 3 | 2 | 4 |
| S/S | 3 | 3 | 2 |
| F/T | 3 | 1 | 0 |
| V/S | 2 | 4 | 2 |
| F/S | 2 | 0 | 2 |
| R/S | 2 | 3 | 2 |
| S/H | 2 | 2 | 1 |
| S/G | 2 | 3 | 2 |
| K/T | 1 | 0 | 0 |
| T/G | 1 | 0 | 0 |
| R/T | 1 | 3 | 1 |
| S/N | 1 | 1 | 3 |
| N/T | 1 | 0 | 0 |
| S/Y | 1 | 0 | 1 |
| S/C | 1 | 0 | 0 |
| E/S | 0 | 3 | 3 |
| Q/T | 0 | 2 | 1 |
| A/S | 0 | 3 | 0 |
| M/S | 0 | 1 | 1 |
| W/T | 0 | 1 | 0 |
| D/T | 0 | 2 | 2 |
| A/T | 0 | 2 | 2 |
| T/G | 0 | 1 | 1 |
| V/T | 0 | 1 | 1 |
| W/S | 0 | 0 | 2 |
| K/S | 0 | 0 | 2 |
| T/H | 0 | 0 | 2 |
| M/T | 0 | 0 | 1 |
| S/K | 0 | 1 | 0 |
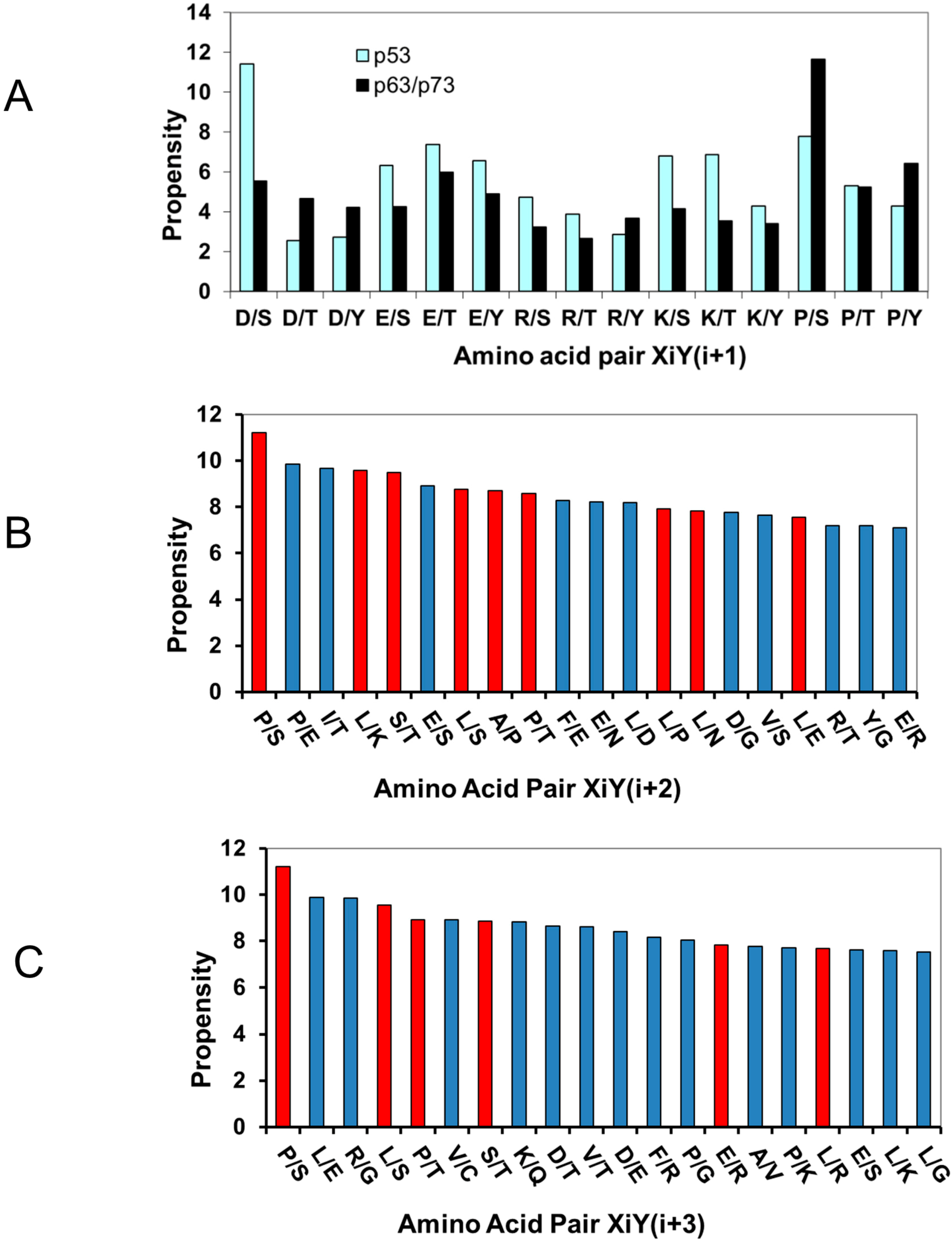

2.1.3. Proline Residue in P−2, P−3, P+2, and P+3 Positions in the Phosphorylation Motifs

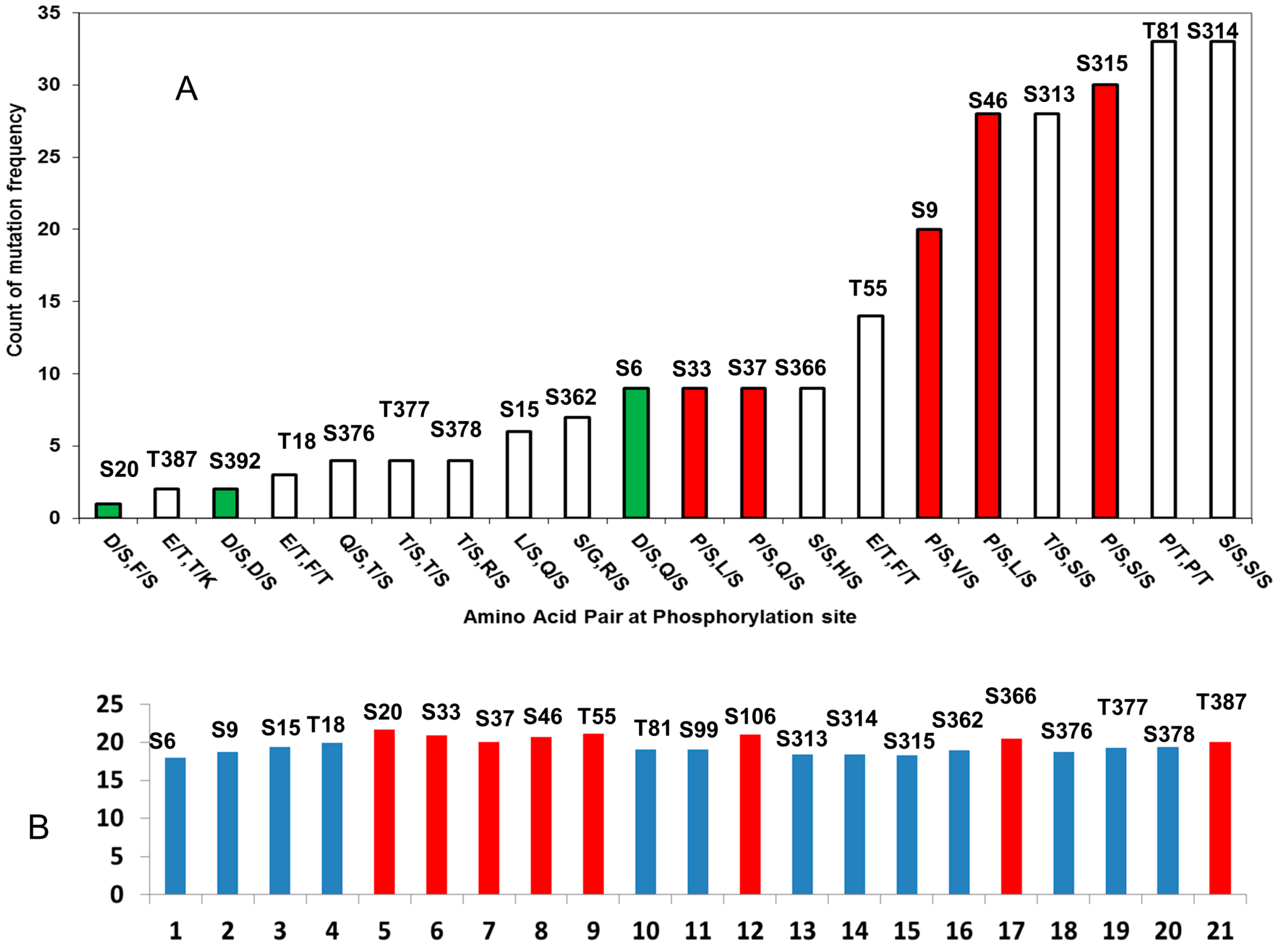
2.2. Cancer Mutations in p53 Phosphorylation Motif
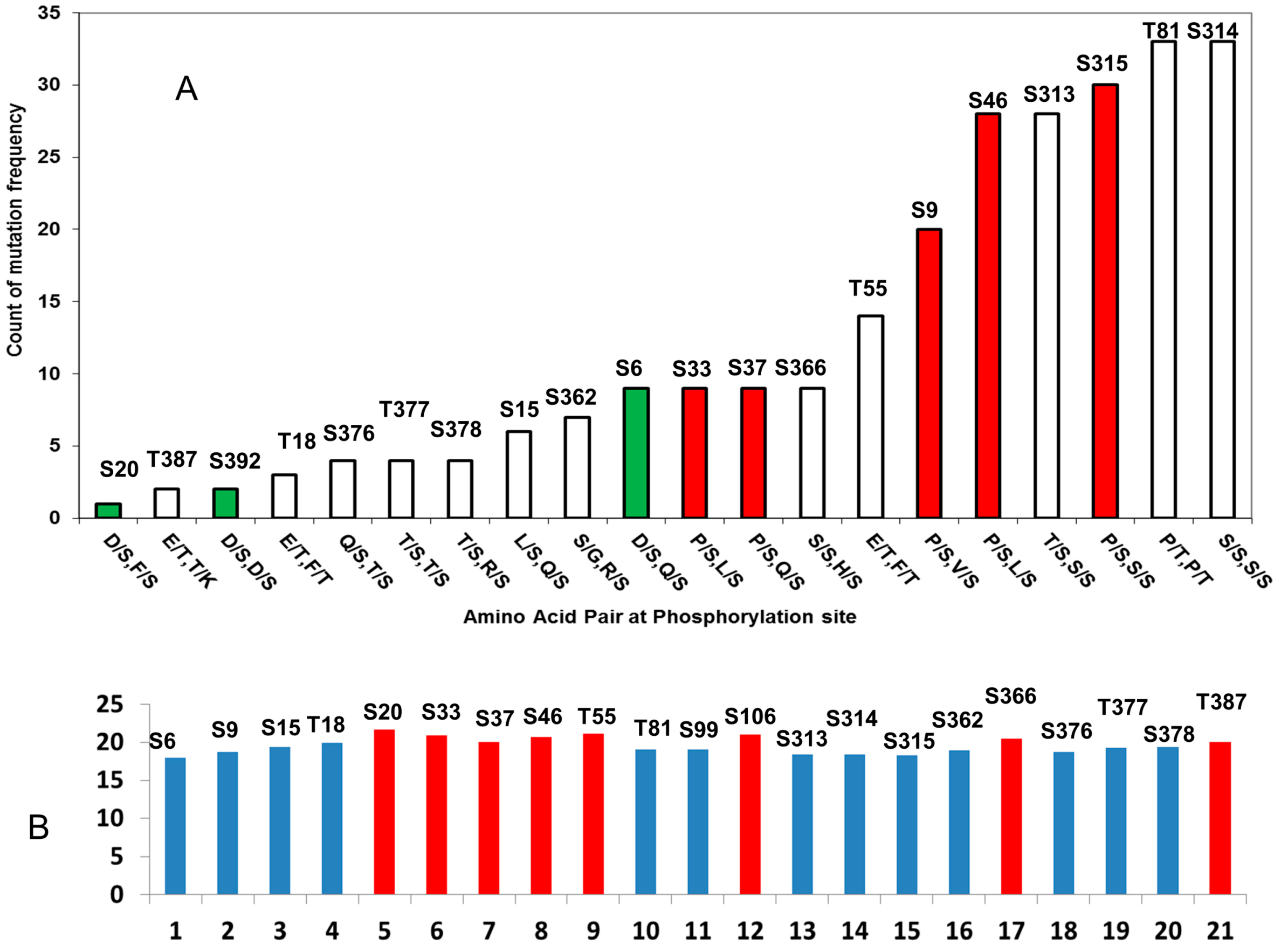
2.2.1. D/S (E/T) Pairs Decrease while P/S Pair Increase Mutation Counts in p53 Phosphorylation Motif
| pS/pT | P−3 | P−2 | P−1 | P0 | P +1 | P +2 | P +3 | Total | Amino Acid Pair |
|---|---|---|---|---|---|---|---|---|---|
| N- and C-Terminus Domains | |||||||||
| S6 | 0 | 3 | 1 | 2 | 1 | 2 | 0 | 9 | D/S, Q/S |
| S9 | 2 | 1 | 2 | 0 | 1 | 11 | 2 | 19 | P/S, V/S |
| S15 | 2 | 1 | 0 | 1 | 1 | 1 | 0 | 6 | L/S, Q/S |
| T18 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 3 | E/T, F/T |
| S20 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | D/S, F/S |
| S33 | 1 | 2 | 0 | 1 | 1 | 3 | 1 | 9 | P/S, L/S |
| S37 | 1 | 3 | 1 | 2 | 0 | 2 | 0 | 9 | P/S, Q/S |
| S46 | 1 | 4 | 1 | 5 | 8 | 1 | 8 | 28 | P/S, L/S |
| T55 | 1 | 4 | 2 | 0 | 5 | 0 | 2 | 14 | E/T, F/T |
| T81 | 3 | 4 | 2 | 2 | 9 | 3 | 10 | 33 | P/T, P/T |
| S99 | 6 | 3 | 6 | 4 | 1 | 4 | 7 | 31 | P/S, Q/S |
| S106 | 0 | 3 | 11 | 8 | 5 | 3 | 6 | 36 | S/G, S/Y |
| S313 | 4 | 6 | 8 | 4 | 1 | 3 | 2 | 28 | T/S, S/S |
| S314 | 6 | 8 | 4 | 1 | 3 | 2 | 9 | 33 | S/S, S/S |
| S315 | 8 | 4 | 1 | 3 | 2 | 9 | 3 | 30 | P/S, S/S |
| S362 | 0 | 1 | 0 | 0 | 1 | 3 | 2 | 7 | S/G, R/S |
| S366 | 2 | 3 | 2 | 2 | 0 | 0 | 0 | 9 | S/S, H/S |
| S376 | 0 | 0 | 0 | 2 | 0 | 0 | 2 | 4 | Q/S, T/S |
| T377 | 0 | 0 | 2 | 0 | 0 | 2 | 0 | 4 | S/S, S/S |
| S378 | 0 | 2 | 0 | 0 | 2 | 0 | 0 | 4 | T/S, R/S |
| T387 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 2 | E/T, T/K |
| S392 | 1 | 0 | 0 | 1 | 0 | 2 | D/S, D/S | ||
| Sum (N- and C-terminus) | 41 | 54 | 44 | 38 | 41 | 50 | 54 | ||
| Core Domain | |||||||||
| S149 | 14 | 32 | 16 | 16 | 14 | 190 | 141 | 423 | D/S, T/S |
| T150 | 32 | 16 | 16 | 14 | 190 | 141 | 36 | 445 | T/S, P/T |
| T155 | 141 | 36 | 85 | 79 | 85 | 228 | 247 | 901 | S/G, R/S |
| S183 | 18 | 88 | 13 | 6 | 39 | 16 | 15 | 195 | S/D, S/C |
| T211 | 39 | 15 | 14 | 30 | 16 | 81 | 87 | 282 | T/N, F/T |
| S215 | 16 | 81 | 87 | 86 | 94 | 16 | 45 | 425 | S/H, S/V |
| S269 | 187 | 57 | 17 | 20 | 102 | 54 | 169 | 606 | N/S, F/S |
| Sum (core domain) | 447 | 325 | 248 | 251 | 540 | 726 | 740 | ||
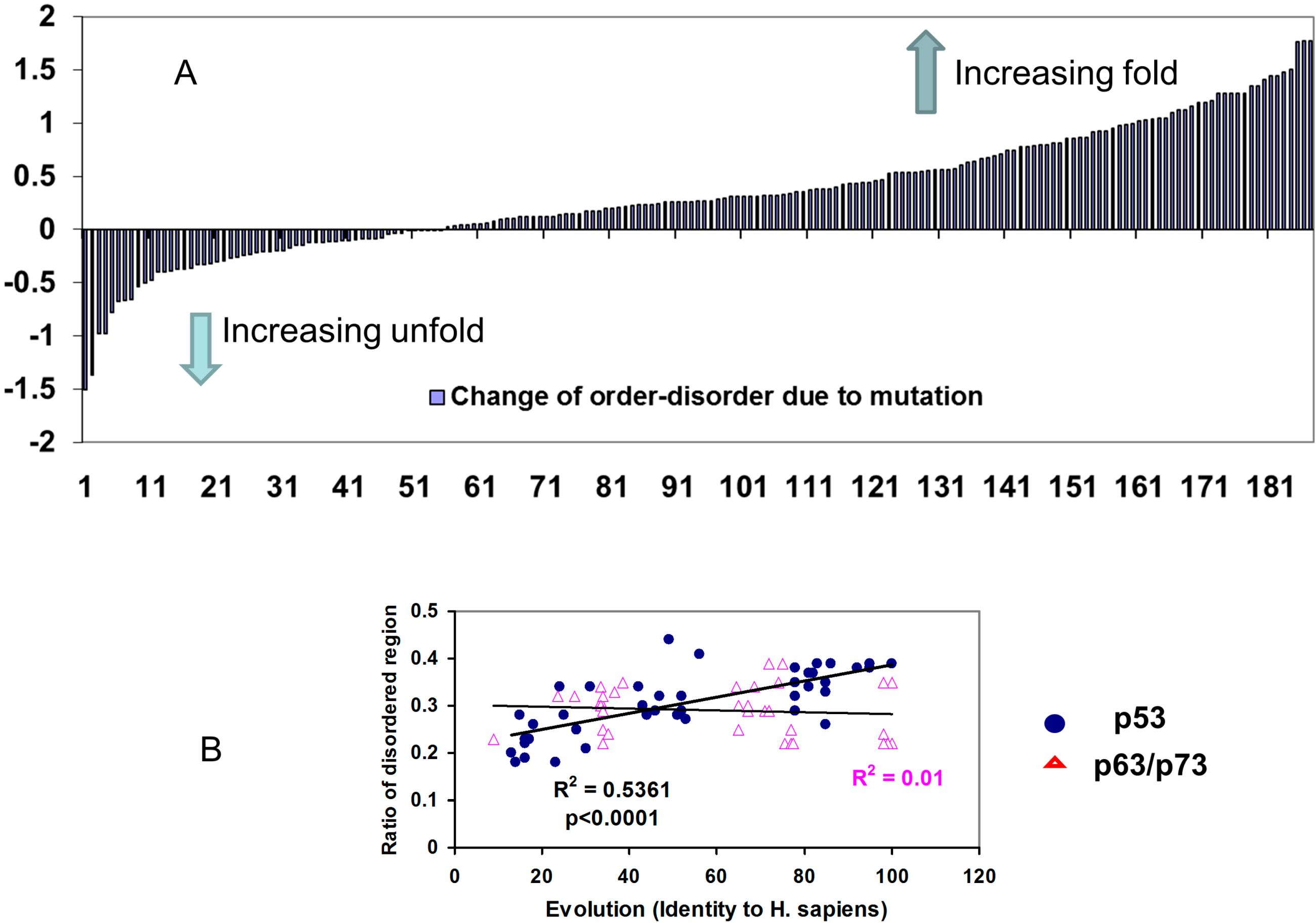
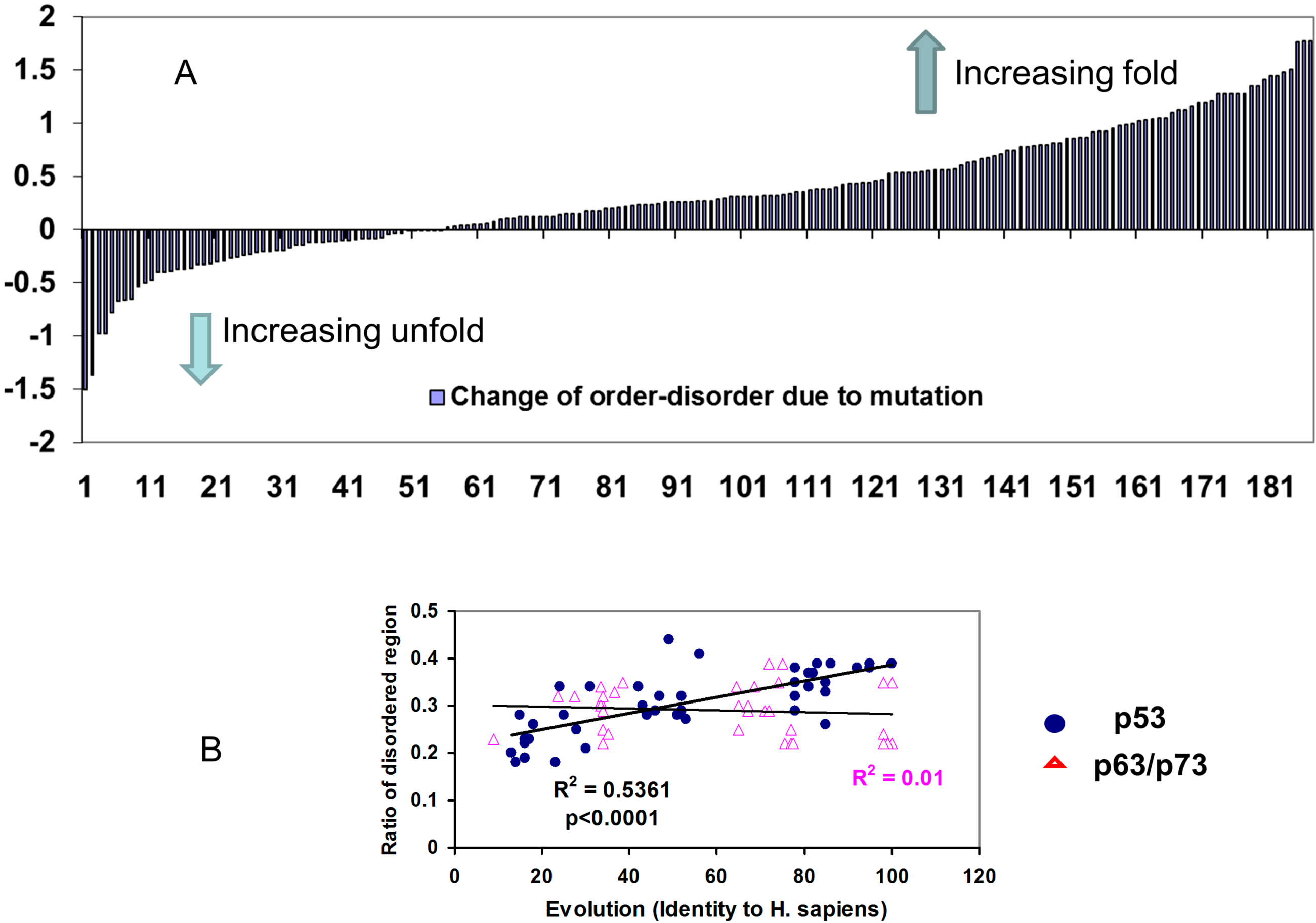
2.2.2. Mutations Decrease the Propensity of Disorder in p53 Phosphorylation Motifs
3. Discussion
3.1. Why Is P/S (or P/T) Association More Vulnerable to Cancer Mutation?
3.2. Acidic Amino Acids Adjacent to Phosphorylation Sites in p53 Protein Families Can Provide Phosphorylation Redundancy
| Peptides | Kd μm | Ref. | |||
|---|---|---|---|---|---|
| P300/TAZ1 | P300/TAZ2 | CBP/TAZ1 | CBP/TAZ2 | ||
| P73(10–40) wt | 39 | 4.5 | [51] | ||
| P73(10–40) pT14 | 4.6 | 0.47 | |||
| P53(1–57) wt | 0.77 | [95] | |||
| P53(1–57) pT18 | 0.11 | ||||
| P53(10–57) wt | 0.88 | ||||
| P53(10–57) pS15pS20 | 0.21 | ||||
| P53(13–61) wt | 0.9 | 0.026 | [96] | ||
| P53(13–57) pT18 | 0.5 | 0.05 | |||
| P53(13–57) pS15pT18pT20 | 0.07 | 0.08 | |||
| P53(1–39) wt | 0.43 (7.15) * | [97] * | |||
| P53(1–39) pS15 | 0.05 (1.83) * | ||||
| P53(1–39) pT18 | 0.05 (1.05) * | ||||
| P53(1–39) pS15pT18 | 0.05 (1.74) * | ||||
| Peptides | Kd μm | Ref. | |||
| TFB1 | P62 (wt) | P62 (K18E) | |||
| P53(25–65) wt | 0.39 | 3.18 | 3.63 | [98] | |
| P53(25–65) pS46 | 0.15 | 0.52 | |||
| P53(25–65) pT55 | 0.16 | 0.46 | |||
| P53(25–65) pS46pT55 | 0.07 | 0.10 | 4.44 | ||
3.3. The Natively Disordered Nature of p53 Phosphorylation Motifs: Vulnerable or Resistant to Mutations?
3.4. Implications of Targeting p53 Mutants in Cancer Therapy
4. Materials and Methods
4.1. Amino Acid Pair Correlation and Propensities

4.2. Disorder Propensities of Phosphorylation Motif and Proteins in the p53/p63/p73 Family
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef]
- Vousden, K.H.; Prives, C. Blinded by the light: The growing complexity of p53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef]
- Riley, T.; Sontag, E.; Chen, P.; Levine, A. Transcriptional control of human p53-regulated genes. Nat. Rev. Mol. Cell Biol. 2008, 9, 402–412. [Google Scholar] [CrossRef]
- Tozluoglu, M.; Karaca, E.; Haliloglu, T.; Nussinov, R. Cataloging and organizing p73 interactions in cell cycle arrest and apoptosis. Nucleic Acids Res. 2008, 36, 5033–5049. [Google Scholar] [CrossRef]
- Candi, E.; Agostini, M.; Melino, G.; Bernassola, F. How the TP53 family proteins TP63 and TP73 contribute to tumorigenesis: Regulators and effectors. Hum. Mutat. 2014, 35, 702–714. [Google Scholar] [CrossRef]
- Jenkins, L.M.; Durell, S.R.; Mazur, S.J.; Appella, E. p53 N-terminal phosphorylation: A defining layer of complex regulation. Carcinogenesis 2012, 33, 1441–1449. [Google Scholar] [CrossRef]
- MacLaine, N.J.; Hupp, T.R. How phosphorylation controls p53. Cell Cycle 2011, 10, 916–921. [Google Scholar] [CrossRef]
- Bode, A.M.; Dong, Z. Post-translational modification of p53 in tumorigenesis. Nat. Rev. Cancer 2004, 4, 793–805. [Google Scholar] [CrossRef]
- Kruse, J.P.; Gu, W. SnapShot: P53 posttranslational modifications. Cell 2008, 133, 930–30.e1. [Google Scholar] [CrossRef]
- Liang, S.H.; Clarke, M.F. Regulation of p53 localization. Eur. J. Biochem. 2001, 268, 2779–2783. [Google Scholar] [CrossRef]
- Flores-Lopez, L.A.; Diaz-Flores, M.; Garcia-Macedo, R.; Avalos-Rodriguez, A.; Vergara-Onofre, M.; Cruz, M.; Contreras-Ramos, A.; Konigsberg, M.; Ortega-Camarillo, C. High glucose induces mitochondrial p53 phosphorylation by p38 MAPK in pancreatic RINm5F cells. Mol. Biol. Rep. 2013, 40, 4947–4958. [Google Scholar] [CrossRef]
- Warnock, L.J.; Knox, A.; Mee, T.R.; Raines, S.A.; Milner, J. Influence of tetramerisation on site-specific post-translational modifications of p53: Comparison of human and murine p53 tumor suppressor protein. Cancer Biol. Ther. 2008, 7, 1481–1489. [Google Scholar] [CrossRef]
- Retzlaff, M.; Rohrberg, J.; Kupper, N.J.; Lagleder, S.; Bepperling, A.; Manzenrieder, F.; Peschek, J.; Kessler, H.; Buchner, J. The regulatory domain stabilizes the p53 tetramer by intersubunit contacts with the DNA binding domain. J. Mol. Biol. 2013, 425, 144–155. [Google Scholar] [CrossRef]
- Chehab, N.H.; Malikzay, A.; Stavridi, E.S.; Halazonetis, T.D. Phosphorylation of Ser-20 mediates stabilization of human p53 in response to DNA damage. Proc. Natl. Acad. Sci. USA 1999, 96, 13777–13782. [Google Scholar] [CrossRef]
- Sakaguchi, K.; Saito, S.; Higashimoto, Y.; Roy, S.; Anderson, C.W.; Appella, E. Damage-mediated phosphorylation of human p53 threonine 18 through a cascade mediated by a casein 1-like kinase. Effect on MDM2 binding. J. Biol. Chem. 2000, 275, 9278–9283. [Google Scholar]
- Al Rashid, S.T.; Dellaire, G.; Cuddihy, A.; Jalali, F.; Vaid, M.; Coackley, C.; Folkard, M.; Xu, Y.; Chen, B.P.; Chen, D.J.; et al. Evidence for the direct binding of phosphorylated p53 to sites of DNA breaks in vivo. Cancer Res. 2005, 65, 10810–10821. [Google Scholar] [CrossRef]
- Shouse, G.P.; Cai, X.; Liu, X. Serine 15 phosphorylation of p53 directs its interaction with B56γ and the tumor suppressor activity of B56γ-specific protein phosphatase 2A. Mol. Cell. Biol. 2008, 28, 448–456. [Google Scholar] [CrossRef]
- Taira, N.; Nihira, K.; Yamaguchi, T.; Miki, Y.; Yoshida, K. DYRK2 is targeted to the nucleus and controls p53 via Ser46 phosphorylation in the apoptotic response to DNA damage. Mol. Cell 2007, 25, 725–738. [Google Scholar]
- Feng, L.; Hollstein, M.; Xu, Y. Ser46 phosphorylation regulates p53-dependent apoptosis and replicative senescence. Cell Cycle 2006, 5, 2812–2819. [Google Scholar] [CrossRef]
- Rinaldo, C.; Prodosmo, A.; Mancini, F.; Iacovelli, S.; Sacchi, A.; Moretti, F.; Soddu, S. MDM2-regulated degradation of HIPK2 prevents p53Ser46 phosphorylation and DNA damage-induced apoptosis. Mol. Cell 2007, 25, 739–750. [Google Scholar] [CrossRef]
- Cai, X.; Liu, X. Inhibition of Thr-55 phosphorylation restores p53 nuclear localization and sensitizes cancer cells to DNA damage. Proc. Natl. Acad. Sci. USA 2008, 105, 16958–16963. [Google Scholar] [CrossRef]
- Bech-Otschir, D.; Kraft, R.; Huang, X.; Henklein, P.; Kapelari, B.; Pollmann, C.; Dubiel, W. COP9 signalosome-specific phosphorylation targets p53 to degradation by the ubiquitin system. EMBO J. 2001, 20, 1630–1639. [Google Scholar] [CrossRef]
- Lykke-Andersen, K.; Schaefer, L.; Menon, S.; Deng, X.W.; Miller, J.B.; Wei, N. Disruption of the COP9 signalosome CSN2 subunit in mice causes deficient cell proliferation, accumulation of p53 and cyclin E, and early embryonic death. Mol. Cell. Biol. 2003, 23, 6790–6797. [Google Scholar] [CrossRef]
- Fogal, V.; Hsieh, J.K.; Royer, C.; Zhong, S.; Lu, X. Cell cycle-dependent nuclear retention of p53 by E2F1 requires phosphorylation of p53 at Ser315. EMBO J. 2005, 24, 2768–27682. [Google Scholar] [CrossRef]
- Pluquet, O.; Qu, L.K.; Baltzis, D.; Koromilas, A.E. Endoplasmic reticulum stress accelerates p53 degradation by the cooperative actions of HDM2 and glycogen synthase kinase 3β. Mol. Cell. Biol. 2005, 25, 9392–9405. [Google Scholar]
- Katayama, H.; Sasai, K.; Kawai, H.; Yuan, Z.M.; Bondaruk, J.; Suzuki, F.; Fujii, S.; Arlinghaus, R.B.; Czerniak, B.A.; Sen, S. Phosphorylation by aurora kinase A induces MDM2-mediated destabilization and inhibition of p53. Nat. Genet. 2004, 36, 55–62. [Google Scholar] [CrossRef]
- Minamoto, T.; Buschmann, T.; Habelhah, H.; Matusevich, E.; Tahara, H.; Boerresen-Dale, A.L.; Harris, C.; Sidransky, D.; Ronai, Z. Distinct pattern of p53 phosphorylation in human tumors. Oncogene 2001, 20, 3341–3347. [Google Scholar] [CrossRef]
- Matsumoto, M.; Furihata, M.; Ohtsuki, Y. Posttranslational phosphorylation of mutant p53 protein in tumor development. Med. Mol. Morphol. 2006, 39, 79–87. [Google Scholar] [CrossRef]
- Reimand, J.; Bader, G.D. Systematic analysis of somatic mutations in phosphorylation signaling predicts novel cancer drivers. Mol. Syst. Biol. 2013, 9, 637. [Google Scholar] [CrossRef]
- Ullrich, S.J.; Sakaguchi, K.; Lees-Miller, S.P.; Fiscella, M.; Mercer, W.E.; Anderson, C.W.; Appella, E. Phosphorylation at Ser-15 and Ser-392 in mutant p53 molecules from human tumors is altered compared to wild-type p53. Proc. Natl. Acad. Sci. USA 1993, 90, 5954–5958. [Google Scholar] [CrossRef]
- Restle, A.; Farber, M.; Baumann, C.; Bohringer, M.; Scheidtmann, K.H.; Muller-Tidow, C.; Wiesmuller, L. Dissecting the role of p53 phosphorylation in homologous recombination provides new clues for gain-of-function mutants. Nucleic Acids Res. 2008, 36, 5362–5375. [Google Scholar] [CrossRef]
- Reimand, J.; Wagih, O.; Bader, G.D. The mutational landscape of phosphorylation signaling in cancer. Sci. Rep. 2013, 3, 2651. [Google Scholar]
- Okaichi, K.; Nose, K.; Kotake, T.; Izumi, N.; Kudo, T. Phosphorylation of p53 modifies sensitivity to ionizing radiation. Anticancer Res. 2011, 31, 2255–2258. [Google Scholar]
- Valenti, F.; Fausti, F.; Biagioni, F.; Shay, T.; Fontemaggi, G.; Domany, E.; Yaffe, M.B.; Strano, S.; Blandino, G.; di Agostino, S. Mutant p53 oncogenic functions are sustained by Plk2 kinase through an autoregulatory feedback loop. Cell Cycle 2011, 10, 4330–4340. [Google Scholar] [CrossRef]
- Zerbini, L.F.; Wang, Y.; Correa, R.G.; Cho, J.Y.; Libermann, T.A. Blockage of NF-κB induces serine 15 phosphorylation of mutant p53 by JNK kinase in prostate cancer cells. Cell Cycle 2005, 4, 1247–1253. [Google Scholar] [CrossRef]
- Gillotin, S.; Yap, D.; Lu, X. Mutation at Ser392 specifically sensitizes mutant p53H175 to MDM2-mediated degradation. Cell Cycle 2010, 9, 1390–1398. [Google Scholar]
- Lum, J.K.; Neuweiler, H.; Fersht, A.R. Long-range modulation of chain motions within the intrinsically disordered transactivation domain of tumor suppressor p53. J. Am. Chem. Soc. 2012, 134, 1617–1622. [Google Scholar]
- Craig, A.; Scott, M.; Burch, L.; Smith, G.; Ball, K.; Hupp, T. Allosteric effects mediate CHK2 phosphorylation of the p53 transactivation domain. EMBO Rep. 2003, 4, 787–792. [Google Scholar] [CrossRef]
- Kodama, M.; Otsubo, C.; Hirota, T.; Yokota, J.; Enari, M.; Taya, Y. Requirement of ATM for rapid p53 phosphorylation at Ser46 without Ser/Thr-Gln sequences. Mol. Cell. Biol. 2010, 30, 1620–1633. [Google Scholar] [CrossRef]
- Warnock, L.J.; Raines, S.A.; Milner, J. Aurora A mediates cross-talk between N- and C-terminal post-translational modifications of p53. Cancer Biol. Ther. 2011, 12, 1059–1068. [Google Scholar] [CrossRef]
- Moll, U.M.; Erster, S.; Zaika, A. p53, p63 and p73—Solos, alliances and feuds among family members. Biochim. Biophys. Acta 2001, 1552, 47–59. [Google Scholar]
- Levrero, M.; de Laurenzi, V.; Costanzo, A.; Gong, J.; Wang, J.Y.; Melino, G. The p53/p63/p73 family of transcription factors: Overlapping and distinct functions. J. Cell Sci 2000, 113, 1661–1670. [Google Scholar]
- Paliwal, P.; Radha, V.; Swarup, G. Regulation of p73 by Hck through kinase-dependent and independent mechanisms. BMC Mol. Biol. 2007, 8, 45. [Google Scholar] [CrossRef]
- Wang, X.; Zeng, L.; Wang, J.; Chau, J.F.; Lai, K.P.; Jia, D.; Poonepalli, A.; Hande, M.P.; Liu, H.; He, G.; et al. A positive role for c-Abl in ATM and ATR activation in DNA damage response. Cell Death Differ. 2011, 18, 5–15. [Google Scholar] [CrossRef]
- Tsai, K.K.; Yuan, Z.M. c-Abl stabilizes p73 by a phosphorylation-augmented interaction. Cancer Res. 2003, 63, 3418–3424. [Google Scholar]
- Oberst, A.; Rossi, M.; Salomoni, P.; Pandolfi, P.P.; Oren, M.; Melino, G.; Bernassola, F. Regulation of the p73 protein stability and degradation. Biochem. Biophys. Res. Commun. 2005, 331, 707–712. [Google Scholar] [CrossRef]
- Soond, S.M.; Barry, S.P.; Melino, G.; Knight, R.A.; Latchman, D.S.; Stephanou, A. p73-mediated transcriptional activity is negatively regulated by polo-like kinase 1. Cell Cycle 2008, 7, 1214–1223. [Google Scholar] [CrossRef]
- Koida, N.; Ozaki, T.; Yamamoto, H.; Ono, S.; Koda, T.; Ando, K.; Okoshi, R.; Kamijo, T.; Omura, K.; Nakagawara, A. Inhibitory role of Plk1 in the regulation of p73-dependent apoptosis through physical interaction and phosphorylation. J. Biol. Chem. 2008, 283, 8555–8563. [Google Scholar] [CrossRef]
- Jones, E.V.; Dickman, M.J.; Whitmarsh, A.J. Regulation of p73-mediated apoptosis by c-Jun N-terminal kinase. Biochem. J. 2007, 405, 617–623. [Google Scholar] [CrossRef]
- Vikhanskaya, F.; Toh, W.H.; Dulloo, I.; Wu, Q.; Boominathan, L.; Ng, H.H.; Vousden, K.H.; Sabapathy, K. p73 supports cellular growth through c-Jun-dependent AP-1 transactivation. Nat. Cell Biol. 2007, 9, 698–705. [Google Scholar] [CrossRef]
- Burge, S.; Teufel, D.P.; Townsley, F.M.; Freund, S.M.; Bycroft, M.; Fersht, A.R. Molecular basis of the interactions between the p73 N-terminus and p300: Effects on transactivation and modulation by phosphorylation. Proc. Natl. Acad. Sci. USA 2009, 106, 3142–3147. [Google Scholar] [CrossRef]
- Nyman, U.; Vlachos, P.; Cascante, A.; Hermanson, O.; Zhivotovsky, B.; Joseph, B. Protein kinase C-dependent phosphorylation regulates the cell cycle-inhibitory function of the p73 carboxy terminus transactivation domain. Mol. Cell. Biol. 2009, 29, 1814–1825. [Google Scholar] [CrossRef]
- Conforti, F.; Sayan, A.E.; Sreekumar, R.; Sayan, B.S. Regulation of p73 activity by post-translational modifications. Cell Death Dis. 2012, 3, e285. [Google Scholar] [CrossRef]
- Westfall, M.D.; Mays, D.J.; Sniezek, J.C.; Pietenpol, J.A. The δ Np63 α phosphoprotein binds the p21 and 14-3-3 σ promoters in vivo and has transcriptional repressor activity that is reduced by Hay–Wells syndrome-derived mutations. Mol. Cell. Biol. 2003, 23, 2264–2276. [Google Scholar] [CrossRef]
- Liao, J.M.; Zhang, Y.; Liao, W.; Zeng, S.X.; Su, X.; Flores, E.R.; Lu, H. IκB kinase β (IKKβ) inhibits p63 isoform γ (TAp63γ) transcriptional activity. J. Biol. Chem. 2013, 288, 18184–18193. [Google Scholar] [CrossRef]
- Westfall, M.D.; Joyner, A.S.; Barbieri, C.E.; Livingstone, M.; Pietenpol, J.A. Ultraviolet radiation induces phosphorylation and ubiquitin-mediated degradation of δNp63α. Cell Cycle 2005, 4, 710–716. [Google Scholar] [CrossRef]
- Livera, G.; Petre-Lazar, B.; Guerquin, M.J.; Trautmann, E.; Coffigny, H.; Habert, R. p63 null mutation protects mouse oocytes from radio-induced apoptosis. Reproduction 2008, 135, 3–12. [Google Scholar] [CrossRef]
- Suzuki, D.; Senoo, M. Expansion of epidermal progenitors with high p63 phosphorylation during wound healing of mouse epidermis. Exp. Dermatol. 2013, 22, 374–376. [Google Scholar] [CrossRef]
- Moretti, F.; Costanzo, A. A feedback regulatory loop between p63 and Dlx3: Implications for epidermal differentiation. Cell Cycle 2009, 8, 1113. [Google Scholar]
- Di Costanzo, A.; Festa, L.; Duverger, O.; Vivo, M.; Guerrini, L.; la Mantia, G.; Morasso, M.I.; Calabro, V. Homeodomain protein Dlx3 induces phosphorylation-dependent p63 degradation. Cell Cycle 2009, 8, 1185–1195. [Google Scholar] [CrossRef]
- Hong, B.; van den Heuvel, A.P.; Prabhu, V.V.; Zhang, S.; El-Deiry, W.S. Targeting tumor suppressor p53 for cancer therapy: Strategies, challenges and opportunities. Curr. Drug Targets 2014, 15, 80–89. [Google Scholar] [CrossRef]
- Wesierska-Gadek, J.; Gueorguieva, M.; Herbacek, I.; Ranftler, C. Effect of distinct anticancer drugs on the phosphorylation of p53 protein at serine 46 in human MCF-7 breast cancer cells. Ann. N. Y. Acad. Sci. 2007, 1095, 45–52. [Google Scholar] [CrossRef]
- Ma, T.; Yamada, S.; Ichwan, S.J.; Iseki, S.; Ohtani, K.; Otsu, M.; Ikeda, M.A. Inability of p53-reactivating compounds Nutlin-3 and RITA to overcome p53 resistance in tumor cells deficient in p53Ser46 phosphorylation. Biochem. Biophys. Res. Commun. 2012, 417, 931–937. [Google Scholar] [CrossRef]
- Clementi, M.; Sanchez, C.; Benitez, D.A.; Contreras, H.R.; Huidobro, C.; Cabezas, J.; Acevedo, C.; Castellon, E.A. Gonadotropin releasing hormone analogs induce apoptosis by extrinsic pathway involving p53 phosphorylation in primary cell cultures of human prostatic adenocarcinomas. Prostate 2009, 69, 1025–1033. [Google Scholar] [CrossRef]
- Fraser, M.; Bai, T.; Tsang, B.K. Akt promotes cisplatin resistance in human ovarian cancer cells through inhibition of p53 phosphorylation and nuclear function. Int. J. Cancer 2008, 122, 534–546. [Google Scholar] [CrossRef]
- Shi, R.; Huang, Q.; Zhu, X.; Ong, Y.B.; Zhao, B.; Lu, J.; Ong, C.N.; Shen, H.M. Luteolin sensitizes the anticancer effect of cisplatin via c-Jun NH2-terminal kinase-mediated p53 phosphorylation and stabilization. Mol. Cancer Ther. 2007, 6, 1338–1347. [Google Scholar] [CrossRef]
- Nguyen, T.A.; Menendez, D.; Resnick, M.A.; Anderson, C.W. Mutant TP53 posttranslational modifications: Challenges and opportunities. Hum. Mutat. 2014, 35, 738–755. [Google Scholar] [CrossRef]
- Gully, C.P.; Velazquez-Torres, G.; Shin, J.H.; Fuentes-Mattei, E.; Wang, E.; Carlock, C.; Chen, J.; Rothenberg, D.; Adams, H.P.; Choi, H.H.; et al. Aurora-B kinase phosphorylates and instigates degradation of p53. Proc. Natl. Acad. Sci. USA 2012, 109, E1513–E1522. [Google Scholar] [CrossRef]
- Xia, Y.; Padre, R.C.; de Mendoza, T.H.; Bottero, V.; Tergaonkar, V.B.; Verma, I.M. Phosphorylation of p53 by IκB kinase 2 promotes its degradation by β-TrCP. Proc. Natl. Acad. Sci. USA 2009, 106, 2629–2634. [Google Scholar] [CrossRef]
- Dehart, C.J.; Chahal, J.S.; Flint, S.J.; Perlman, D.H. Extensive post-translational modification of active and inactivated forms of endogenous p53. Mol. Cell. Proteomics 2014, 13, 1–17. [Google Scholar] [CrossRef]
- Ubersax, J.A.; Ferrell, J.E., Jr. Mechanisms of specificity in protein phosphorylation. Nat. Rev. Mol. Cell Biol. 2007, 8, 530–541. [Google Scholar] [CrossRef]
- Brown, N.R.; Lowe, E.D.; Petri, E.; Skamnaki, V.; Antrobus, R.; Johnson, L.N. Cyclin B and cyclin A confer different substrate recognition properties on CDK2. Cell Cycle 2007, 6, 1350–1359. [Google Scholar] [CrossRef]
- Sheridan, D.L.; Kong, Y.; Parker, S.A.; Dalby, K.N.; Turk, B.E. Substrate discrimination among mitogen-activated protein kinases through distinct docking sequence motifs. J. Biol. Chem. 2008, 283, 19511–19520. [Google Scholar]
- Seo, G.J.; Kim, S.E.; Lee, Y.M.; Lee, J.W.; Lee, J.R.; Hahn, M.J.; Kim, S.T. Determination of substrate specificity and putative substrates of Chk2 kinase. Biochem. Biophys. Res. Commun. 2003, 304, 339–343. [Google Scholar] [CrossRef]
- 75. Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R., 3rd; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef]
- Hsueh, K.W.; Fu, S.L.; Chang, C.B.; Chang, Y.L.; Lin, C.H. A novel Aurora-A-mediated phosphorylation of p53 inhibits its interaction with MDM2. Biochim. Biophys. Acta 2013, 1834, 508–515. [Google Scholar] [CrossRef]
- Huang, Q.; Yu, L.; Levine, A.J.; Nussinov, R.; Ma, B. Dipeptide analysis of p53 mutations and evolution of p53 family proteins. Biochim. Biophys. Acta 2014, 1844, 198–206. [Google Scholar] [CrossRef]
- Galzitskaya, O.V.; Garbuzynskiy, S.O.; Lobanov, M.Y. FoldUnfold: Web server for the prediction of disordered regions in protein chain. Bioinformatics 2006, 22, 2948–2949. [Google Scholar]
- Nishikawa, K.; Toker, A.; Johannes, F.J.; Songyang, Z.; Cantley, L.C. Determination of the specific substrate sequence motifs of protein kinase C isozymes. J. Biol. Chem. 1997, 272, 952–960. [Google Scholar]
- Meggio, F.; Pinna, L.A. One-thousand-and-one substrates of protein kinase CK2? FASEB J. 2003, 17, 349–368. [Google Scholar] [CrossRef]
- Agami, R.; Blandino, G.; Oren, M.; Shaul, Y. Interaction of c-Abl and p73α and their collaboration to induce apoptosis. Nature 1999, 399, 809–813. [Google Scholar] [CrossRef]
- Hamer, G.; Gademan, I.S.; Kal, H.B.; de Rooij, D.G. Role for c-Abl and p73 in the radiation response of male germ cells. Oncogene 2001, 20, 4298–4304. [Google Scholar] [CrossRef]
- Warnock, L.J.; Adamson, R.; Lynch, C.J.; Milner, J. Crosstalk between site-specific modifications on p53 and histone H3. Oncogene 2008, 27, 1639–1644. [Google Scholar] [CrossRef]
- Saito, S.; Yamaguchi, H.; Higashimoto, Y.; Chao, C.; Xu, Y.; Fornace, A.J., Jr.; Appella, E.; Anderson, C.W. Phosphorylation site interdependence of human p53 post-translational modifications in response to stress. J. Biol. Chem. 2003, 278, 37536–37544. [Google Scholar] [CrossRef]
- Olivier, M.; Eeles, R.; Hollstein, M.; Khan, M.A.; Harris, C.C.; Hainaut, P. The IARC TP53 database: New online mutation analysis and recommendations to users. Hum. Mutat. 2002, 19, 607–614. [Google Scholar] [CrossRef]
- Iakoucheva, L.M.; Radivojac, P.; Brown, C.J.; O’Connor, T.R.; Sikes, J.G.; Obradovic, Z.; Dunker, A.K. The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res. 2004, 32, 1037–1049. [Google Scholar] [CrossRef]
- Gsponer, J.; Futschik, M.E.; Teichmann, S.A.; Babu, M.M. Tight regulation of unstructured proteins: From transcript synthesis to protein degradation. Science 2008, 322, 1365–1368. [Google Scholar] [CrossRef]
- Pagano, B.; Jama, A.; Martinez, P.; Akanho, E.; Bui, T.T.; Drake, A.F.; Fraternali, F.; Nikolova, P.V. Structure and stability insights into tumour suppressor p53 evolutionary related proteins. PLoS One 2013, 8, e76014. [Google Scholar] [CrossRef]
- Canadillas, J.M.; Tidow, H.; Freund, S.M.; Rutherford, T.J.; Ang, H.C.; Fersht, A.R. Solution structure of p53 core domain: Structural basis for its instability. Proc. Natl. Acad. Sci. USA 2006, 103, 2109–2114. [Google Scholar]
- Huang, Y.; Sen, T.; Nagpal, J.; Upadhyay, S.; Trink, B.; Ratovitski, E.; Sidransky, D. ATM kinase is a master switch for the δ Np63 α phosphorylation/degradation in human head and neck squamous cell carcinoma cells upon DNA damage. Cell Cycle 2008, 7, 2846–2855. [Google Scholar] [CrossRef]
- Yang, A.; Kaghad, M.; Caput, D.; McKeon, F. On the shoulders of giants: p63, p73 and the rise of p53. Trends Genet. 2002, 18, 90–95. [Google Scholar] [CrossRef]
- Petitjean, A.; Mathe, E.; Kato, S.; Ishioka, C.; Tavtigian, S.V.; Hainaut, P.; Olivier, M. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: Lessons from recent developments in the IARC TP53 database. Hum. Mutat. 2007, 28, 622–629. [Google Scholar] [CrossRef]
- Kruse, J.P.; Gu, W. Modes of p53 regulation. Cell 2009, 137, 609–622. [Google Scholar] [CrossRef]
- Shaywitz, A.J.; Dove, S.L.; Kornhauser, J.M.; Hochschild, A.; Greenberg, M.E. Magnitude of the CREB-dependent transcriptional response is determined by the strength of the interaction between the kinase-inducible domain of CREB and the KIX domain of CREB-binding protein. Mol. Cell. Biol. 2000, 20, 9409–9422. [Google Scholar] [CrossRef]
- Teufel, D.P.; Bycroft, M.; Fersht, A.R. Regulation by phosphorylation of the relative affinities of the N-terminal transactivation domains of p53 for p300 domains and MDM2. Oncogene 2009, 28, 2112–2118. [Google Scholar] [CrossRef]
- Ferreon, J.C.; Lee, C.W.; Arai, M.; Martinez-Yamout, M.A.; Dyson, H.J.; Wright, P.E. Cooperative regulation of p53 by modulation of ternary complex formation with CBP/p300 and HDM2. Proc. Natl. Acad. Sci. USA 2009, 106, 6591–6596. [Google Scholar]
- Feng, H.; Jenkins, L.M.; Durell, S.R.; Hayashi, R.; Mazur, S.J.; Cherry, S.; Tropea, J.E.; Miller, M.; Wlodawer, A.; Appella, E.; et al. Structural basis for p300 TAZ2–p53 TAD1 binding and modulation by phosphorylation. Structure 2009, 17, 202–210. [Google Scholar] [CrossRef]
- Di Lello, P.; Jenkins, L.M.; Jones, T.N.; Nguyen, B.D.; Hara, T.; Yamaguchi, H.; Dikeakos, J.D.; Appella, E.; Legault, P.; Omichinski, J.G. Structure of the TFB1/p53 complex: Insights into the interaction between the p62/TFB1 subunit of TFIIH and the activation domain of p53. Mol. Cell 2006, 22, 731–740. [Google Scholar] [CrossRef]
- Wells, M.; Tidow, H.; Rutherford, T.J.; Markwick, P.; Jensen, M.R.; Mylonas, E.; Svergun, D.I.; Blackledge, M.; Fersht, A.R. Structure of tumor suppressor p53 and its intrinsically disordered N-terminal transactivation domain. Proc. Natl. Acad. Sci. USA 2008, 105, 5762–5767. [Google Scholar] [CrossRef]
- Dyson, H.J.; Wright, P.E. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 2005, 6, 197–208. [Google Scholar] [CrossRef]
- Iakoucheva, L.M.; Brown, C.J.; Lawson, J.D.; Obradovic, Z.; Dunker, A.K. Intrinsic disorder in cell-signaling and cancer-associated proteins. J. Mol. Biol. 2002, 323, 573–584. [Google Scholar] [CrossRef]
- Ma, B.; Nussinov, R. Regulating highly dynamic unstructured proteins and their coding mRNAs. Genome Biol. 2009, 10, 204. [Google Scholar] [CrossRef]
- Ma, B.; Nussinov, R. Amplification of signaling via cellular allosteric relay and protein disorder. Proc. Natl. Acad. Sci. USA 2009, 106, 6887–6888. [Google Scholar] [CrossRef]
- Vonderviszt, F.; Matrai, G.; Simon, I. Characteristic sequential residue environment of amino acids in proteins. Int. J. Pept. Protein Res. 1986, 27, 483–492. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ji, X.; Huang, Q.; Yu, L.; Nussinov, R.; Ma, B. Bioinformatics Study of Cancer-Related Mutations within p53 Phosphorylation Site Motifs. Int. J. Mol. Sci. 2014, 15, 13275-13298. https://doi.org/10.3390/ijms150813275
Ji X, Huang Q, Yu L, Nussinov R, Ma B. Bioinformatics Study of Cancer-Related Mutations within p53 Phosphorylation Site Motifs. International Journal of Molecular Sciences. 2014; 15(8):13275-13298. https://doi.org/10.3390/ijms150813275
Chicago/Turabian StyleJi, Xiaona, Qiang Huang, Long Yu, Ruth Nussinov, and Buyong Ma. 2014. "Bioinformatics Study of Cancer-Related Mutations within p53 Phosphorylation Site Motifs" International Journal of Molecular Sciences 15, no. 8: 13275-13298. https://doi.org/10.3390/ijms150813275
APA StyleJi, X., Huang, Q., Yu, L., Nussinov, R., & Ma, B. (2014). Bioinformatics Study of Cancer-Related Mutations within p53 Phosphorylation Site Motifs. International Journal of Molecular Sciences, 15(8), 13275-13298. https://doi.org/10.3390/ijms150813275